
Electrochemical Minisci-type trifluoromethylation of electron-deficient heterocycles mediated by bromide ions†
Gui-Yuan
Dou
,
Yang-Ye
Jiang
,
Kun
Xu
and
Cheng-Chu
Zeng
 *
*
Beijing Key Laboratory of Environmental and Viral Oncology, College of Life Science & Bioengineering, Beijing University of Technology, Beijing 100124, China. E-mail: zengcc@bjut.edu.cn
First published on 15th May 2019
Abstract
An electrochemical methodology for the Minisci-type trifluoromethylation of electron-deficient heterocycles mediated by cheap and easily available bromide ions has been developed. By virtue of the in situ generated sulfonyl hypobromite intermediate, the CF3 radical can be regulated and controlled at a low concentration, thereby improving the reaction efficiency over direct electrolysis. Also, this indirect electrochemical process is performed in a beaker-type undivided cell under galvanostatic conditions, without using external expensive supporting electrolytes. This protocol provides an alternative electrochemical trifluoromethylation methodology for the late-stage functionalization of biologically important molecules.
Introduction
Trifluoromethylation reactions have attracted increasing attention in pharmaceutic, agrochemical and materials sciences since the incorporation of a CF3 group into organic molecules can dramatically alter the physical, chemical and biological properties of the mother molecules in lipophilicity, acidity, dipole moment, metabolic stability or bioavailability.1 As a result, various trifluoromethylation strategies have been developed, among which the radical trifluoromethylation initiated by redox chemical reagents or photocatalysts has been proved to be the main and powerful means.2,3 Nevertheless, stoichiometric amounts of chemical redox oxidants or expensive Ir- or Ru-based photoredox catalysts are needed in most cases. Therefore, cheap and sustainable strategies for the trifluoromethylation of organic molecules are highly desirable.Electrochemical organic synthesis, making use of electrons instead of chemical redox reagents, thereby provides an atom economical, sustainable and versatile tool for organic chemists.4 To this end, an electrogenerated CF3 radical has been widely used for the trifluoromethylation of organic compounds, mainly in the difunctionalization of alkenes. For example, Lin reported electrocatalytic chlorotrifluoromethylation of alkenes using Mn(II) as a mediator.5 Later on, Lei6 and Xu7 independently reported the aminotrifluoromethylation and oxytrifluoromethylation of alkenes. Moreover, the electrochemical synthesis of functionalized oxindoles initiated from the anodically generated CF3 radical followed by intramolecular radical addition with arenes was achieved by Mo,8 Ackermann9 and our group.10 More recently, Wang et al. also described an electrochemical fluoroalkylation-migration reaction of unactivated olefins to afford fluorinated (hetero)aryl ketones.11 On the other hand, the CF3 radical generated from cathodic reduction of Togni's reagent could also undergo intramolecular addition with the isonitrile group to give phenanthridines.12 Mechanically, this chemistry starts from the addition of the electrochemically generated CF3 radical (from anodic oxidation or cathodic reduction of the CF3 radical precursor) to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds to give a new carbon-centered radical, which then undergoes three types of conversions, including radical coupling with other radicals, further oxidation followed by coupling with a nucleophile or radical addition to intramolecular arenes (Scheme 1a).
C bonds to give a new carbon-centered radical, which then undergoes three types of conversions, including radical coupling with other radicals, further oxidation followed by coupling with a nucleophile or radical addition to intramolecular arenes (Scheme 1a).
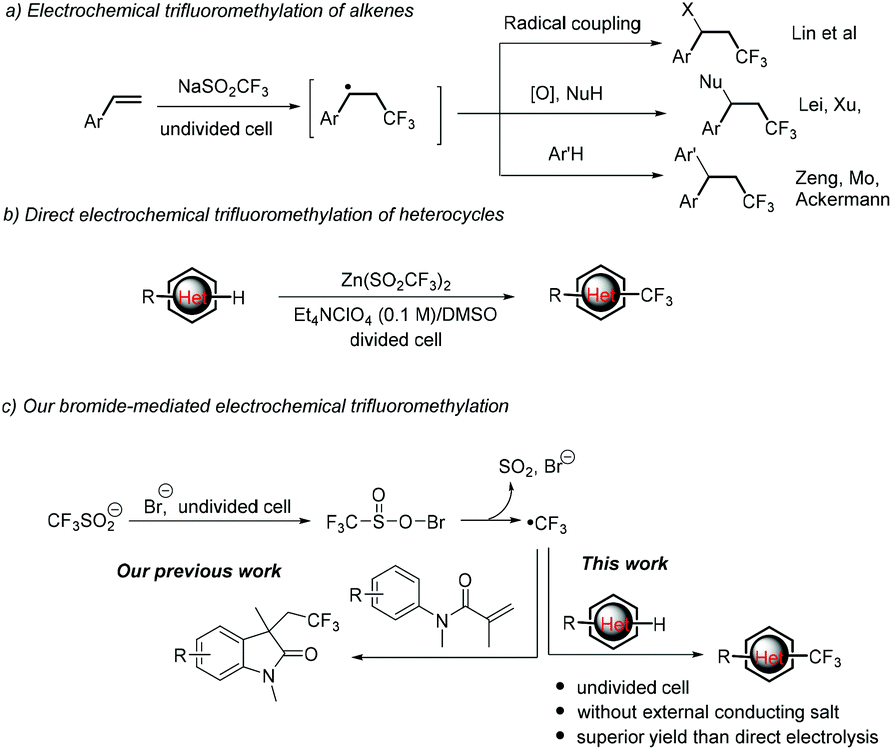 | ||
| Scheme 1 Electrochemical trifluoromethylation of organic molecules. | ||
Compared with trifluoromethylation of alkenes, the Minisci-type electrochemical trifluoromethylation of electron-deficient heterocycles is less studied.13 As far as we know, there is only one example on this case.14 In 2014, Baran, Blackmond, and co-workers reported an elegant synthesis of trifluoromethylated heterocyclic pharmacophores (Scheme 1b). The electrochemical variant improved the efficiency and enhanced the yields for 20 out of 24 examples over a conventional TBHP oxidant. The improvement is supposed to result from the controlling of the CF3 radical to a low concentration. Notably, complicated divided cells, along with an expensive Et4NClO4/DMSO supporting electrolyte, were required to give acceptable yields.
We are interested in the halide-mediated anodic oxidation for the C–H bond functionalization through ion or radical pathways.10 More recently, we reported the electrochemical trifluoromethylation/cyclization of N-arylacrylamides (Scheme 1c, left).10d In the reaction, the initially generated trifluoromethylsulfinyl hypohalite is proposed to undergo cathodic reduction to give the corresponding sulfonyl radical, followed by evolution of SO2 to give the CF3 radical. In this way, the formation and concentration of the CF3 radical can be well-manipulated and tuned, therefore providing an alternative means to address the challenge of controlling CF3 radical concentration. Inspired by the advantage described above, we envisioned that the Minisci-type electrochemical trifluoromethylation of electrondeficient heterocycles may also achieve using bromide as the redox mediator to initiate generating the CF3 radical (Scheme 1c, right). The realization of this hypothesis is the subject of the present communication. It is worth noting that this bromide-mediated electrochemical trifluoromethylation of heterocycles can be conducted in a simple beaker-type undivided cell employing cheap and easily available bromide ions as the redox mediator without utilization of an external large amount of supporting electrolytes. Moreover, randomly selected examples prove that this mediated electrolysis is superior to the direct electrolysis process.
Results and discussion
We commenced our studies by using quinoxaline-2(1H)-ones, 1a, and more readily available sodium trifluoromethylsulfite, 2a, as the model substrates to optimize the reaction conditions. As shown in Table 1, the desired product 3a was obtained in 12% yield when the reaction of 1a with 2a was carried out in an undivided cell equipped with a platinum net as the anode and a graphite plate as the cathode at a constant current of 5 mA cm−2 with NaBr (1.0 equiv.) as the mediator and CH3CN as the solvent at 50 °C. Consequent solvent screening proved that CH3CN was preferable for the electrochemical trifluoromethylation reaction of 1a (entries 2–5). When Baran's reagent, Zn(CF3SO2)2 (2b) was employed as the CF3 radical precursor, the yield of 3a increased to 24% (entry 6). The evaluation of the supporting electrolyte disclosed that Et4NBF4 was superior and the desired product 3a could be obtained in 33% yield (entries 7–11). In addition, Et4NBr was demonstrated to be the best redox catalyst among the redox mediators screened and 37% yield of 3a was afforded (entries 12–15). Notably, when the electrochemical trifluoromethylation of 1a with 2b was performed in the absence of a bromide redox mediator, only 21% yield of 3a was isolated, which indicates that bromide ions play an important role in the electrochemical trifluoromethylation reaction. To our delight, the desired product 3a could also be isolated in 40% yield in the absence of an external supporting electrolyte (entry 16); therefore the utilization of conducting salt, which generally is regarded as waste in the workup process, could be avertable. The yield of 3a decreased successively when the loading amount of Et4NBr was reduced (entries 17–19). Based on these results described above, we concluded that the electrochemical trifluoromethylation of quinoxaline-2(1H)-ones prefers the use of Et4NBr in CH3CN as the redox mediator and conducting salt, carried out in an undivided cell under constant current electrolysis.|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Electrolyte (0.1 M) | Additive (equiv.) | Yieldb (%) |
| a Reaction conditions: 1a (0.3 mmol) and 2b (0.6 mmol) in 3 mL of solvent, undivided cell, 50 °C, current density of 5 mA cm−2, graphite plate anode and Pt net cathode (working area: 1 cm2). b Isolated yields. c CF3SO2Na as the CF3 radical precursor. | ||||
| 1c | CH3CN | LiClO4 | NaBr (1.0) | 12 |
| 2c | DMSO | LiClO4 | NaBr (1.0) | Trace |
| 3c | DMF | LiClO4 | NaBr (1.0) | Trace |
| 4c | CH3OH | LiClO4 | NaBr (1.0) | n.r. |
| 5c | CH3CN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DCE (5:1) :DCE (5:1) |
LiClO4 | NaBr (1.0) | 11 |
| 6 | CH3CN | LiClO4 | NaBr (1.0) | 24 |
| 7 | CH3CN | Et4NClO4 | NaBr (1.0) | 19 |
| 8 | CH3CN | Et4NBF4 | NaBr (1.0) | 33 |
| 9 | CH3CN | Bu4NBF4 | NaBr (1.0) | 28 |
| 10 | CH3CN | Bu4NPF6 | NaBr (1.0) | 23 |
| 11 | CH3CN | Bu4NClO4 | NaBr (1.0) | 18 |
| 12 | CH3CN | Et4NBF4 | NH4Br (1.0) | 27 |
| 13 | CH3CN | Et4NBF4 | KBr (1.0) | 31 |
| 14 | CH3CN | Et4NBF4 | Et4NBr (1.0) | 37 |
| 15 | CH3CN | Et4NBF4 | Bu4NBr (1.0) | 34 |
| 16 | CH3CN | Et4NBF4 | — | 21 |
| 17 | CH3CN | — | Et4NBr (1.0) | 40 |
| 18 | CH3CN | — | Et4NBr (0.5) | 31 |
| 19 | CH3CN | — | Et4NBr (0.2) | 27 |
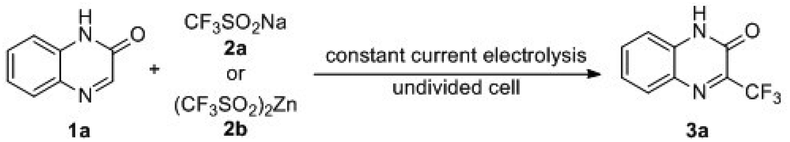
With the optimal reaction conditions in hand, we turned to examine the scope and generality of the protocol by examining the reactions of various heterocycles 1 with 2b. As shown in Table 2, the electrochemical trifluoromethylation of quinoxalinone derivatives proceeded smoothly under the standard conditions to give the corresponding 3-trifluoromethylated quinoxalinones 3b–3o in acceptable yields. For example, when N-methyl-(1b), N-benzyl-(1c–1j) and N-acetate-(1k) substituted quinoxalinones were subjected to the electrocatalytic trifluoromethylation reaction with 2b under the standard conditions, the corresponding products 3b–3k were afforded in 23–47% yields. In the cases of quinoxalinones 1l–1o, in which the substituents were on the aryl ring, the reactions also worked well to give corresponding 3l–3o.
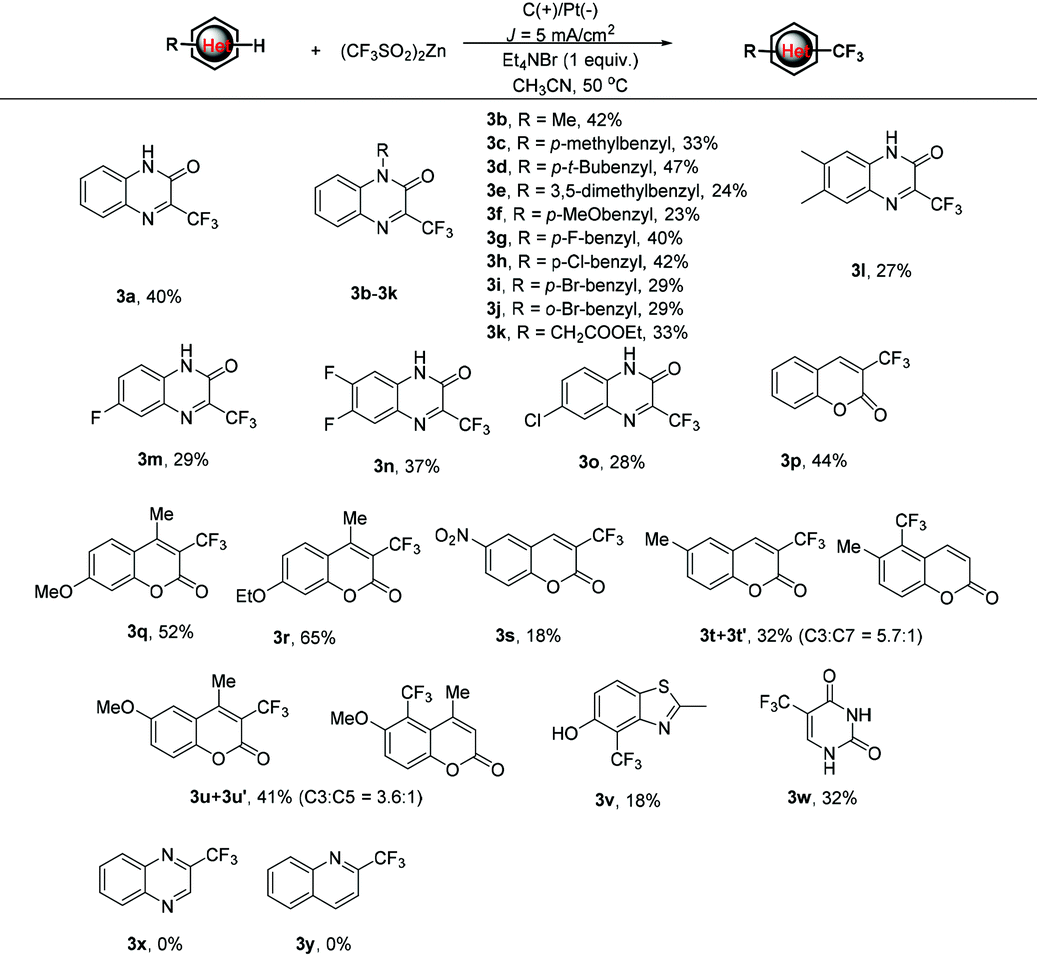
Other heterocyclic pharmacophores, such as coumarins, thiazole and pyrimidine, were also amenable to the protocol. For coumarin (1p), the corresponding 3p was isolated in 44% yield. It was observed that the electronic characters of the substituents on the coumarin skeleton dramatically influence the reaction efficiency. For example, when electron-donating methoxy or ethoxy substituted coumarins were subjected to the reaction, the corresponding 3q and 3r could be obtained in 52% and 65% yields. Conversely, the substitution of an electron-withdrawing nitro group, such as 1s, resulted in a very low yield (18%) of 3s. Notably, when 6-methyl coumarin was subjected to the trifluoromethylation reaction with 2b, a mixture of regioisomers 3t and 3t′ at a ratio of 5.7:1 was afforded in a 32% total yield. A similar case also occurred for 6-methoxy coumarin, 1u, wherein the mixture of regioisomers 3u/3u′ was isolated in 41% total yield and 3.6:1 ratio. The reaction also tolerated thiazole derivatives with a free hydroxyl group. For example, benzo[d]thiazol-5-ol, 1v, gave corresponding 3v, although in a bit lower yield. Finally, widely applied pharmaceutical and agrochemical, 5-(trifluoromethyl)pyrimidine-2,4(1H,3H)-dione 3w was also afforded in 32% yield under the standard conditions.15 However, when quinoxaline and quinoline were reacted under the standard conditions, the desired products 3x and 3y were not detected and the starting materials 1x and 1y were recovered completely.
As mentioned above, the bromide-mediated trifluoro-methylation reaction of 1a with 2b was found to be more efficient than that under direct electrolytic conditions (Scheme 2, 21% vs. 40%). To demonstrate the generality of this observation, we randomly selected several substrates and subjected them to electrolysis with 2b under the standard conditions and the results are listed in Scheme 2. In accordance with what we expected, the bromide-mediated trifluoromethylation of 1c, 1p and 1r gave slightly superior yields compared with that in the absence of Et4NBr. Considering that we use undivided cells without an external expensive supporting electrolyte, the bromide-mediated trifluoromethylation protocol should be more promising for potential industrial application.
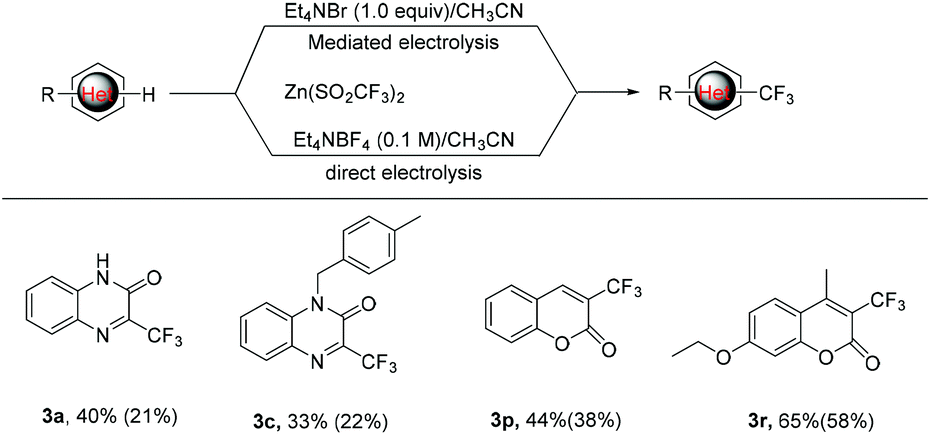 | ||
| Scheme 2 Comparison of electrochemical trifluoromethylation reactions under bromide-mediated indirect and direct electrolysis (the yield in parentheses). | ||
To demonstrate the practicability of the protocol, a preparative scale electrolysis was also conducted. As shown in Scheme 3, when 10 mmol of 1p was subjected to electrolysis with 2b under the standard conditions, the corresponding 3p was isolated in 31% yield (45% yield based on the recovered 1p), along with a mixture of regioisomers of 3p1 and 3p2 in 7% yield.16
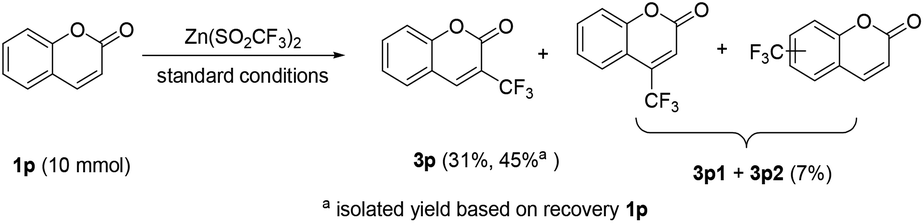 | ||
| Scheme 3 Scale up experiment. | ||
To understand the mechanism for the bromide-mediated trifluoromethylation reaction of heterocycles, a series of control experiments and cyclic voltammetric analysis were performed. As shown in Scheme 4, when the reaction of 1a with independently synthesized trifluoro-methanesulfonyl bromide, 4, was performed in the absence and presence of electricity, the corresponding 3a was afforded in 15% and 34% yields, respectively. These results indicate that compound 4 could be the key intermediate, and passing charge is very important. In addition, the reaction of 1-(1-cyclopropylvinyl)benzene, 5, with 2b gives compound 6 in 17% yield (see the ESI† for details); this radical clock experiment demonstrates that the CF3 radical might be involved.
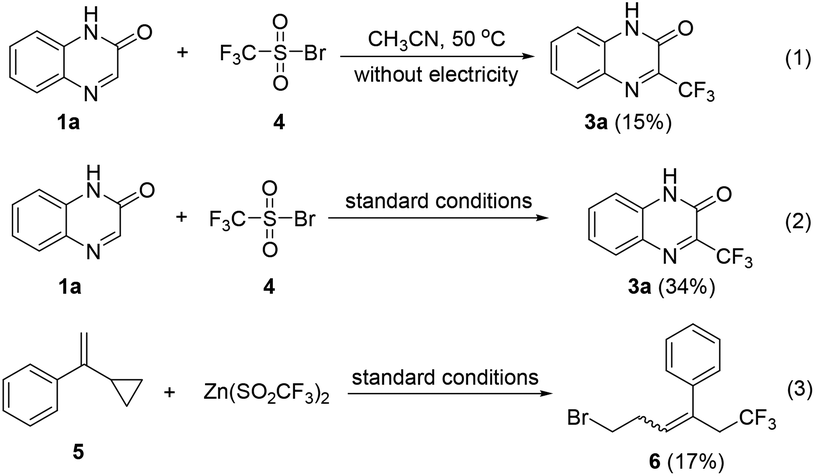 | ||
| Scheme 4 Control experiments. | ||
Cyclic voltammetric analysis was also performed to understand the possible mechanism. As shown in Fig. 1, quinoxalinone 1a is not oxidized up to 1.30 V (curve b), whereas Zn(CF3SO2)2 was found to be oxidized at 1.14 V (vs. Ag/AgNO3) (curve c). Et4NBr gave two obvious oxidation peaks at 0.49 V and 0.73 V (vs. Ag/AgNO3) and two reduction peaks at 0.3 V and 0.62 V vs. Ag/AgNO3 (curve d). The oxidation potential of Zn(CF3SO2)2 is higher than that of bromide ions (1.14 V vs. 0.49 V and 0.73 V); therefore, the bromide ion is easier to oxidize at the surface of the anode. The CV of Et4NBr exhibited an obvious catalytic current in the presence of 2b, along with a new reduction peak at about 0.14 V (being the reduction of CF3SO2Br10d) vs. Ag/AgNO3 (Fig. 1, curve e). This electrochemical behavior is quite different from that wherein the bromide ion mediates the C–H bond functionalization via an ionic pathway.10h–j This observation suggests that bromine-based active species, generated from the anodic oxidation of bromide ions, reacts with CF3SO2Na to afford intermediate CF3SO2Br, which could be reduced at the surface of the cathode.
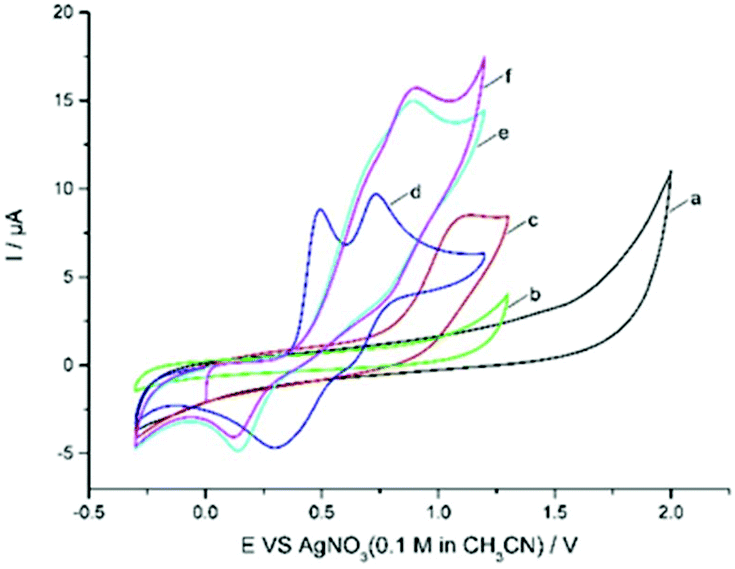 | ||
| Fig. 1 Cyclic voltammograms of Et4NBr and related compounds in 0.1 M LiClO4/CH3CN using a glass carbon working electrode, Pt wire, and Ag/AgNO3 (0.1 M in CH3CN) as counter and reference electrodes at 100 mV s−1 scan rate. (a) Background, (b) 1a (1.0 mmol L−1), (c) Zn(CF3SO2)2 (1.0 mmol L−1), (d) Et4NBr (1.0 mmol L−1), (e) Et4NBr (1.0 mmol L−1) and Zn(CF3SO2)2 (2.0 mmol L−1), (f) Et4NBr (1.0 mmol L−1), Zn(CF3SO2)2 (2.0 mmol L−1) and 1a (3.0 mmol L−1). | ||
Based on the above experiments and literature reports,10d,14 we proposed a mechanism for the electrochemical trifluoromethylation of heterocycles. As shown in Scheme 5, anodic oxidation of bromide gives molecular Br2, which then reacts with Zn(CF3SO2)2 to give sulfonyl hypobromite 4′ or sulfonyl bromide 4. Then, a cathodic reduction or a homolytic cleavage of the sulfonyl bromide 4, followed by rapid extrusion of a SO2 molecule, affords the key CF3 radical. Its Minisci-type radical addition to quinoxalinone 1a gives adduct 7, which undergoes further anodic oxidation to afford the desired product 3a after losing a proton.
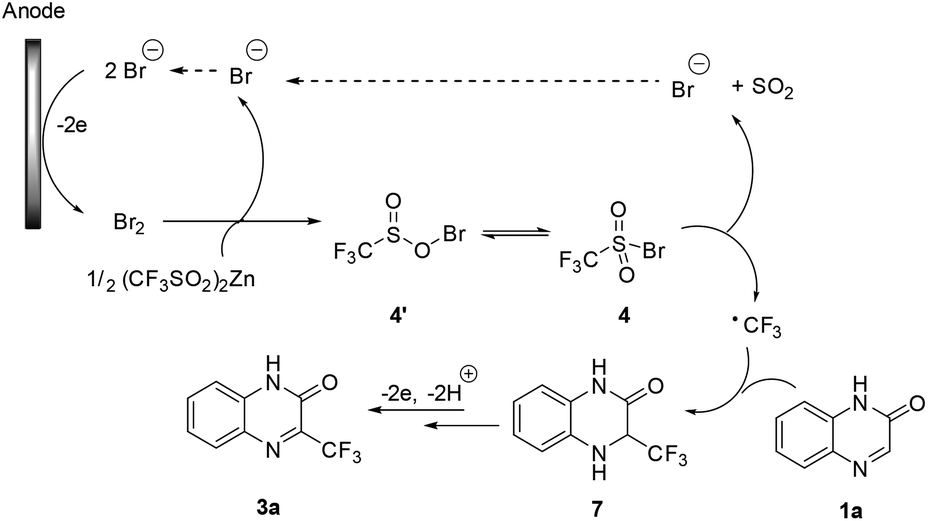 | ||
| Scheme 5 A proposed Mechanism for the trifluoromethylation of quinoxalinone. | ||
Conclusion
In summary, electrocatalyzed Minisci-type trifluoromethylation of electron-deficient heterocycles has been developed using cheap and easily available bromide ions as the redox mediator. The method efficiently controls the concentration of the CF3 radical by virtue of the in situ generated sulfonyl hypobromite as the potential CF3 precursor. The advantage of the protocol features performing it in the simple beaker-type undivided cell, without using a large amount of external supporting electrolyte, as well as achieving superior yields to that from direct electrolysis processes, therefore providing an alternative electrochemical trifluoromethylation methodology for the late-stage functionalization of biologically important molecules.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from the National Key Technology R&D Program (2017YFB0307502) and the National Natural Science Foundation of China (No. 21871019).Notes and references
- (a) W. Jud, C. O. Kappe and D. Cantillo, Org. Biomol. Chem., 2019, 17, 3529–3537 RSC; (b) H. I. Jung, Y. Kim and D. Y. Kim, Org. Biomol. Chem., 2019, 17, 3319–3323 RSC; (c) S. Kolanu, M. Atif, F. Natalia and G. Zeev, Dalton Trans., 2019, 48, 4798–4810 RSC; (d) J. C. Wang, K. Sun, X. L. Chen, T. Chen, Y. Liu, L. B. Qu, Y. F. Zhao and B. Yu, Org. Lett., 2019, 21, 1863–1867 CrossRef CAS PubMed; (e) S. L. Zhang, C. Xiao, H. X. Wan and X. M. Zhang, Chem. Commun., 2019, 55, 4099–4102 RSC; (f) X. H. He, Y. L. Ji, C. Peng and B. Han, Adv. Synth. Catal., 2019, 361, 1923–1957 CrossRef CAS; (g) C. Barrett and G. K. S. Prakash, Org. Lett., 2019, 21, 1526–1529 CrossRef PubMed.
- (a) P. Zhang, H. G. Shen, L. Zhu, W. G. Cao and C. Z. Li, Org. Lett., 2018, 20, 7062–7065 CrossRef CAS PubMed; (b) D. Q. Liang, Q. S. Dong, P. H. Xu, Y. Dong, W. L. Li and Y. H. Ma, J. Org. Chem., 2018, 83, 11978–11986 CrossRef CAS PubMed; (c) H. Yu, M. D. Jiao, X. W. Fang and P. F. Xuan, RSC Adv., 2018, 8, 23919–23923 RSC; (d) V. Krishnamurti, S. B. Munoz, X. I. Rodriguez, J. Vickerman, T. Mathew and G. K. S. Prakash, Chem. Commun., 2018, 54, 10574–20577 RSC; (e) S. Mandal, T. Bera, G. Dubey, J. Saha and J. K. Laha, ACS Catal., 2018, 8, 5085–5144 CrossRef CAS; (f) J. Li, X. F. Zhang, H. Y. Xiang, L. J. Tong, F. Feng, H. Xie, J. Ding and C. H. Yang, J. Org. Chem., 2017, 82, 6795–6800 CrossRef CAS PubMed; (g) A. Werf, M. Hribersek and N. Selander, Org. Lett., 2017, 19, 2374–2377 CrossRef PubMed; (h) W. X. Lv, Y. F. Zeng, Q. J. Li, Y. Y. Chen, D. H. Tan, L. Yang and H. G. Wang, Angew. Chem., Int. Ed., 2016, 55, 10069–10073 CrossRef CAS PubMed.
- (a) L. Zou, P. H. Li, B. Wang and L. Wang, Chem. Commun., 2019, 55, 3737–3740 RSC; (b) X. Yuan, M. W. Zheng, Z. C. Di, Y. S. Cui, K. Q. Zhuang, L. Z. Qin, Z. Fang, J. K. Qiu, G. G. Li and K. Guo, Adv. Synth. Catal., 2019, 361, 1835–1845 CrossRef CAS; (c) L. L. Zhao, P. H. Li, H. Zhang and L. Wang, Org. Chem. Front., 2019, 6, 87–93 RSC; (d) X. D. Zhang, Y. M. Li, X. Y. Hao, K. Jin, R. Zhang and C. Y. Duan, Tetrahedron, 2018, 74, 7358–7363 CrossRef CAS; (e) C. J. M. Frédéric, J. Cornil, M. Vandamme, L. Dumitrescu, A. Tikad, R. Robiette and S. P. Vincent, Org. Lett., 2018, 20, 6769–6773 CrossRef PubMed; (f) T. X. Zhang, X. Y. Guo, Y. S. Shi, C. He and C. Y. Duan, Nat. Commun., 2018, 9, 1–9 CrossRef PubMed; (g) Y. T. He, D. Kang, I. Kim and S. Hong, Green Chem., 2018, 20, 5209–5214 RSC; (h) L. H. Wu, J. K. Cheng, L. Shen, Z. L. She and T. Loh, Adv. Synth. Catal., 2018, 360, 3894–3899 CrossRef CAS; (i) C. Ghiazza, T. Billard and A. Tlili, Chem. – Eur. J., 2019, 25, 6482–6495 CrossRef CAS PubMed; (j) B. Yang, D. H. Yu, X. H. Xu and F. L. Qing, ACS Catal., 2018, 8, 2839–2843 CrossRef CAS; (k) D. A. Nagib, M. E. Scott and D. W. C. MacMillan, J. Am. Chem. Soc., 2009, 131, 10875–10877 CrossRef CAS PubMed; (l) D. A. Nagib and D. W. C. MacMillan, Nature, 2011, 480, 224–228 CrossRef CAS PubMed; (m) A. Studer, Angew. Chem., Int. Ed., 2012, 51, 8950–8958 CrossRef CAS PubMed; (n) Y. Ye and M. S. Sanford, J. Am. Chem. Soc., 2012, 134, 9034–9037 CrossRef CAS PubMed; (o) D. J. Wilger, N. J. Gesmundo and D. A. Nicewicz, Chem. Sci., 2013, 4, 3160–3165 RSC; (p) S. P. Pitre, C. D. McTiernan, H. Ismaili and J. C. Scaiano, ACS Catal., 2014, 4, 2530–2535 CrossRef CAS.
- (a) Y. Y. Jiang, K. Xu and C. C. Zeng, Chem. Rev., 2018, 118, 4485–4540 CrossRef CAS PubMed; (b) S. R. Waldvogel, S. Lips, M. Selt, B. Riehl and C. J. Kampf, Chem. Rev., 2018, 118, 6706–6765 CrossRef CAS PubMed; (c) M. L. Pegis, C. F. Wise, D. J. Martin and J. M. Mayer, Chem. Rev., 2018, 118, 2340–2391 CrossRef CAS PubMed; (d) S. Mçhle, M. Zirbes, E. Rodrigo, T. Gieshoff, A. Wiebe and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 6018–6041 CrossRef PubMed; (e) M. D. Kärkäs, Chem. Soc. Rev., 2018, 47, 5786–5865 RSC; (f) Y. Yu, Y. Yuan, H. L. Liu, M. He, M. Z. Yang, P. Liu, B. Y. Yu, X. C. Dong and A. W. Lei, Chem. Commun., 2019, 55, 1809–1812 RSC; (g) J. A. Marko, A. Durgham, S. L. Bretz and W. Liu, Chem. Commun., 2019, 55, 937–940 RSC; (h) J. Kim, K. Shin, S. Jin, D. Kim and S. Chang, J. Am. Chem. Soc., 2019, 141, 4137–4146 CrossRef CAS PubMed; (i) N. K. Fu, G. S. Sauer, A. Saha, A. Loo and S. Lin, Science, 2017, 357, 575–579 CrossRef CAS PubMed.
- K. Y. Ye, Z. D. Song, G. S. Sauer, J. H. Harenberg, N. K. Fu and S. Lin, Chem. – Eur. J., 2018, 24, 12274–12279 CrossRef CAS PubMed.
- L. L. Zhang, G. T. Zhang, P. Wang, Y. L. Li and A. W. Lei, Org. Lett., 2018, 20, 7396–7399 CrossRef CAS PubMed.
- S. Zhang, L. J. Li, J. J. Zhang, J. Q. Zhang, M. Y. Xue and K. Xu, Chem. Sci., 2019, 10, 3181–3185 RSC.
- Z. X. Zhang, L. Zhang, Y. Cao, F. Li, G. C. Bai, G. Q. Liu, Y. Yang and F. Y. Mo, Org. Lett., 2019, 21, 762–766 CrossRef CAS PubMed.
- Z. X. Ruan, Z. X. Huang, Z. N. Xu, G. Q. Mo, X. Tian, X. Y. Yu and L. Ackermann, Org. Lett., 2019, 21, 1237–1240 CrossRef CAS PubMed.
- (a) S. Liang, K. Xu, C. C. Zeng, H. Y. Tian and B. G. Sun, Adv. Synth. Catal., 2018, 360, 4266–4292 CrossRef CAS; (b) C. C. Sun, K. Xu and C. C. Zeng, ACS Sustainable Chem. Eng., 2019, 7, 2255–2261 CrossRef CAS; (c) M. Y. Lin, K. Xu, Y. Y. Jiang, Y. G. Liu, B. G. Sun and C. C. Zeng, Adv. Synth. Catal., 2018, 360, 1665–1672 CrossRef CAS; (d) Y. Y. Jiang, G. Y. Dou, K. Xu and C. C. Zeng, Org. Chem. Front., 2018, 5, 2573–2577 RSC; (e) S. Liang, C. C. Zeng, H. Y. Tian, B. G. Sun, X. G. Luo and F. Z. Ren, Adv. Synth. Catal., 2018, 360, 1444–1452 CrossRef CAS; (f) S. Zhang, L. J. Li, M. Y. Xue, R. K. Zhang, K. Xu and C. C. Zeng, Org. Lett., 2018, 20, 3443–3446 CrossRef CAS PubMed; (g) Q. Q. Wang, K. Xu, Y. Y. Jiang, Y. G. Liu, B. G. Sun and C. C. Zeng, Org. Lett., 2017, 73, 764–770 Search PubMed; (h) Y. Y. Jiang, Q. Q. Wang, S. Liang, L. M. Hu, R. D. Little and C. C. Zeng, J. Org. Chem., 2016, 81, 4713–4719 CrossRef CAS PubMed; (i) S. Liang, C. C. Zeng, H. Y. Tian, B. G. Sun, X. G. Luo and F. Z. Ren, J. Org. Chem., 2016, 81, 11565–11573 CrossRef CAS PubMed; (j) S. Liang, C. C. Zeng, X. G. Luo, F. Z. Ren, H. Y. Tian, B. G. Sun and R. D. Little, Green Chem., 2016, 18, 2222–2230 RSC; (k) Y. Y. Jiang, S. Liang, C. C. Zeng, L. M. Hu and B. G. Sun, Green Chem., 2016, 18, 6311–6319 RSC; (l) L. S. Kang, M. H. Luo, C. M. Lam, L. M. Hu, R. D. Little and C. C. Zeng, Green Chem., 2016, 18, 3767–3774 RSC; (m) J. Chen, W. Q. Yan, C. M. Lam, C. C. Zeng, L. M. Hu and R. D. Little, Org. Lett., 2015, 17, 986–989 CrossRef CAS PubMed.
- Z. L. Zou, W. G. Zhang, Y. Wang, L. Y. Kong, G. Karotsis, Y. Wang and Y. Pan, Org. Lett., 2019, 21, 1857–1862 CrossRef CAS PubMed.
- M. Lübbesmeyer, D. Leifert, H. Schäfer and A. Studer, Chem. Commun., 2018, 54, 2240–2243 RSC.
- R. S. J. Proctor and R. J. Phipps, Angew. Chem., Int. Ed. DOI:10.1002/anie.201900977.
- A. G. O'Brien, A. Maruyama, Y. Inokuma, M. Fujita, P. S. Baran and D. G. Blackmond, Angew. Chem., Int. Ed., 2014, 53, 11868–11871 CrossRef PubMed.
- Y. Huang, Y. Y. Lei, L. Zhao, J. W. Gu, Q. L. Yao, Z. Wang, X. F. Li, X. G. Zhang and C. Y. He, Chem. Commun., 2018, 54, 13662–13665 RSC.
- X. Yu, P. Dai, Y. C. Zhu, P. Teng, W. H. Zhan and C. Deng, ChemCatChem, 2018, 10, 5115–5118 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and characterization of all compounds. See DOI: 10.1039/c9qo00552h |
| This journal is © the Partner Organisations 2019 |