
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDeactivation and regeneration of carbon nanotubes and nitrogen-doped carbon nanotubes in catalytic peroxymonosulfate activation for phenol degradation: variation of surface functionalities†
Jifei Hou,
Lixia Xu,
Yuxiang Han,
Yuqiong Tang,
Haiqin Wan,
Zhaoyi Xu and
Shourong Zheng*
and
Shourong Zheng*
State Key Laboratory of Pollution Control and Resource Reuse, Jiangsu Key Laboratory of Vehicle Emissions Control, School of the Environment, Nanjing University, Nanjing 210093, P. R. China. E-mail: srzheng@nju.edu.cn; Fax: +86-25-89680596; Tel: +86-25-89680373
First published on 9th January 2019
Abstract
The reuse, deactivation and regeneration of carbon nanotubes (CNT) and N-doped carbon nanotubes (NCNT) were studied in catalytic peroxymonosulfate (PMS) activation for phenol degradation. The results showed that for catalytic PMS activation, marked deactivation was observed on both CNT and NCNT, resulting in marked variation of the surface functionalities of the catalysts. Catalytic PMS activation led to markedly increased oxygen-containing functionalities and decreased points of zero charge (PZCs) of CNT and NCNT. The catalytic activity of CNT was strongly dependent on the initial PMS concentration but was independent of the initial phenol concentration. Furthermore, the dependency of the CNT activity on the initial PMS concentration closely followed the Langmuir–Hinshelwood model, indicating that the catalytic activation of adsorbed PMS was the rate controlling step. For the used CNT and NCNT, chemical reduction by NaBH4 or thermal treatment regeneration under inert atmosphere could effectively remove surface O-containing functionalities and enhance PZCs, restoring their catalytic activities; meanwhile, the N-containing functionalities of NCNT decreased with regeneration treatment, resulting in a negative impact on catalyst regeneration. The present findings indicate that surface functionalities are closely correlated with catalyst deactivation and regeneration, playing crucial roles in the catalytic activation of PMS.
1. Introduction
Radical-based advanced oxidation processes (AOPs) are considered to be effective approaches for the abatement of recalcitrant organic pollutants in water. Among AOPs, the Fenton reaction has been widely used in the removal of organic pollutants via an oxidation pathway involving highly reactive hydroxyl radicals (·OH). Notably, hydroxyl radicals are generated only under acidic pH conditions (3 < pH < 4) in the Fenton reaction system,1 imposing strong restrictions for its wide application in wastewater treatment. In contrast, sulfate radical (SO4˙−)-based AOPs (SR-AOPs), which are capable of degrading organic contaminants by highly reactive sulfate radicals in a very wide pH range of 3 to 10, are gaining increasing attention.2,3Different methods have been applied to activate persulfates (peroxymonosulfate/PMS or peroxodisulfate/PDS) to produce SO4˙−, such as heat,4 ultraviolet light,5,6 ultrasound,7 and catalysis. Among these methods, catalysis is extensively used because of its high efficiency and feasibility. In general, metal oxides and carbonaceous materials have been usually used as effective catalysts for persulfate activation. For persulfate activation over metal oxides, Co3O4, Fe2O3 and MnO2 were reported to be active,8–10 whereas metal ion leaching concurrently happened during degradation processes, leading to serious secondary pollution. In contrast, carbonaceous materials displayed much stronger advantages over metal oxides due to their high stability toward acid and base in water. Accordingly, different carbonaceous catalysts have been explored for the activation of persulfate. For example, Wang's group systematically explored the catalytic activation on different carbonaceous materials, including carbon nanotubes, hexagonal ordered mesoporous carbon (CMK-3), graphene, nanodiamonds, S- and N-doped graphenes and N-doped nanocarbon materials.11–16 A variety of active sites of carbon nanomaterials were believed to be capable of activating PMS, such as graphitic carbon structure defect sites, ketonic (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) groups and N-doped functional groups.17–22 In heterogeneous catalysis, catalyst reuse is essential for practical application. Despite their high catalytic activities, carbonaceous materials usually undergo deactivation during SR-AOPs processes. For example, Duan et al.17 observed remarkable deactivation of reduced graphene oxide for phenol degradation; they ascribed the catalyst deactivation to changes in the pore structure and surface chemistry as well as coverage of the catalyst surface by organic intermediates. In parallel, catalyst deactivation was also observed on carbon nanotubes, mesoporous carbon, and N-doped carbon nanotubes.12,16,17 However, few studies have systematically discussed the mechanism of catalyst deactivation and regeneration of carbon-based materials in persulfate activation. Accordingly, the variation of the surface functionalities and their roles during catalyst deactivation and regeneration remain unclear.
O) groups and N-doped functional groups.17–22 In heterogeneous catalysis, catalyst reuse is essential for practical application. Despite their high catalytic activities, carbonaceous materials usually undergo deactivation during SR-AOPs processes. For example, Duan et al.17 observed remarkable deactivation of reduced graphene oxide for phenol degradation; they ascribed the catalyst deactivation to changes in the pore structure and surface chemistry as well as coverage of the catalyst surface by organic intermediates. In parallel, catalyst deactivation was also observed on carbon nanotubes, mesoporous carbon, and N-doped carbon nanotubes.12,16,17 However, few studies have systematically discussed the mechanism of catalyst deactivation and regeneration of carbon-based materials in persulfate activation. Accordingly, the variation of the surface functionalities and their roles during catalyst deactivation and regeneration remain unclear.
In the present study, we used carbon nanotubes (CNTs) and N-doped carbon nanotubes (NCNTs) with stable structures as model catalysts to systematically study the roles of surface functionalities in the deactivation and regeneration of carbon-based catalysts during catalytic PMS activation. The fresh, used and regenerated catalysts were characterized using N2 adsorption, Raman spectroscopy, X-ray photoelectron spectroscopy (XPS), Boehm titration and point of zero charge (PZC) measurements. The catalytic performance of the fresh, used and regenerated catalysts was evaluated via activating PMS for phenol degradation.
2. Experimental
2.1. Catalyst preparation
To prepare N-doped CNT, 2.0 g of pretreated CNT was suspended in 300 mL of mixed acid consisting of concentrated H2SO4 and HNO3 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v); the CNT was refluxed at 80 °C in a water bath for 5 h. After cooling to room temperature, the sample was collected by filtration, washed with distilled water until neutral, and dried at 80 °C for 12 h in vacuum. Then, 2.0 g of acid-treated sample was suspended in 20 mL of distilled water containing 20 g of urea; the suspension was subjected to ultrasonic treatment for 20 min. After water evaporation at 60 °C, the resulting composite material was heat-treated under the same conditions as CNT-X. After cooling to room temperature, NCNT-X was washed using distilled water to remove unreacted impurities and dried at 80 °C for 12 h in vacuum.
:1, v/v); the CNT was refluxed at 80 °C in a water bath for 5 h. After cooling to room temperature, the sample was collected by filtration, washed with distilled water until neutral, and dried at 80 °C for 12 h in vacuum. Then, 2.0 g of acid-treated sample was suspended in 20 mL of distilled water containing 20 g of urea; the suspension was subjected to ultrasonic treatment for 20 min. After water evaporation at 60 °C, the resulting composite material was heat-treated under the same conditions as CNT-X. After cooling to room temperature, NCNT-X was washed using distilled water to remove unreacted impurities and dried at 80 °C for 12 h in vacuum.
2.2. Catalyst characterization
The specific surface areas of the samples were measured using the N2 adsorption method on a Micromeritics ASAP 2020 instrument (Micromeritics Instrument Co., Norcross, GA), and calculated by the Brunauer–Emmett–Teller (BET) equation in the relative pressure (P/P0) range of 0.05 to 0.3. Raman spectra of the CNT and NCNT samples were recorded on a Jobin Yvon LabRam spectrometer (HR-800) with a laser excitation wavelength of 514 nm. The compositions and chemical states of the samples were determined by XPS performed on an ESCALAB 250 instrument (Thermo-VG Scientific, USA) equipped with a monochromatized Al Kα excitation source (hν = 1486.6 eV). The binding energy values were calibrated using the C1s peak (284.6 eV).The points of zero charge (PZCs) of the fresh, used and regenerated catalysts were measured using the potentiometric mass titration method.24,25 Briefly, 0.1 g of the sample was mixed with 20 mL of 0.01 M NaCl solution at 25 °C, to which 0.2 mL of 1.0 M NaOH was added. The mixture was equilibrated for 24 h and was then titrated with 0.5 M HNO3 solution under continuous bubbling with a N2 flow (100 mL min−1). The pH was recorded after each addition of the acidic solution. A blank solution was titrated using the above described procedure.
The concentrations of oxygen-containing groups of the samples were quantified using Boehm titration. Briefly, 0.5 g of sample was added to 25 mL of 0.025 M base or acid solution (e.g., NaHCO3, Na2CO3, NaOH, and HCl), and the mixture was stirred for 48 h. After filtration using a 0.45 µm filter, 10 mL of filtrate was placed in a beaker. The base solutions of NaHCO3 and NaOH were acidified with 20 mL of 0.025 M HCl solution, and Na2CO3 solution was acidified with 30 mL of 0.025 M HCl. The acidified solutions were back-titrated with 0.025 M NaOH solution using a Metrohm 877 Titrino Plus automated titrator (Metrohm Ltd., Switzerland). All solutions were titrated in duplicate and the averages were used. The standardization of the NaOH solution was conducted with potassium hydrogen phthalate as the primary standard and phenolphthalein as the indicator. The concentration of the HCl solution was determined using standardized NaOH solution. All titration processes were carried out under N2 gas protection to exclude the effects of CO2 from the air.
2.3. PMS activation for phenol degradation
Phenol oxidation by PMS activation on the catalysts was conducted in a 250 mL three-necked flask reactor under atmospheric pressure at 25 ± 0.5 °C with the reaction temperature stabilized by a water-bath (SDC-6, Scientz Co., China). Briefly, 20 mg of catalyst was suspended in 198 mL of 10 mM phosphate buffer solution (pH 7.0). Then, 2 mL of 10.6 mM phenol stock solution was added, and the mixture was vigorously stirred for 40 min in order to reach adsorption equilibrium. Preliminary experiments showed that phenol adsorption on the catalysts varied by 1% to 2% (data not presented). The catalytic reaction was started by adding 8 mM of PMS. Samples were taken at preset intervals, and the catalyst particles were rapidly separated by filtration using a 0.45 µm filter. The filtrates were added to vials containing 0.5 mL of methanol. The residual concentration of phenol in the filtrate was analyzed by high performance liquid chromatography (Agilent 1200 Series HPLC system). Phenol was separated on an SB-C18 column (150 × 4.6 mm, 5 µm) with 30% acetonitrile and 70% water (v/v) as the mobile phase at a flow rate of 0.8 mL min−1 and detected by a UV detector with a wavelength of 270 nm. Standard curves with six concentrations were used to determine the phenol concentrations. The catalyst activity was evaluated in terms of the initial activity, which was defined as the specific removal rate of phenol within the initial 5 min. Preliminary experiments indicated that mass transfer limitations were not observed under our experimental conditions, and the experiments had very high repeatability with experimental errors below 5%, as confirmed by two separate runs of phenol degradation on CNT-750 (the results are presented in Fig. S1 and S2, ESI†).3. Results and discussion
3.1. Catalyst characterization
The Raman spectra of the CNT and NCNT samples are compiled in Fig. 1. The samples exhibited very strong G and D bands at 1570 and 1331 cm−1, respectively. The G band is indicative of the stretching vibration from carbon atoms with sp2 bonds in a two-dimensional hexagonal lattice, whereas the D band is assigned to the vibration of carbon atoms with dangling bonds in plane terminations. The ratios of D band strength to G band strength (ID/IG) were calculated to be 1.06, 1.17 and 1.29 for CNT-750, U-CNT-1 and T-CNT-750, respectively, reflecting that the surface defect content of CNT increased after the PMS activation reaction and thermal regeneration. Similar trends were presented by the NCNT samples, with ID/IG ratios of 1.15, 1.21 and 1.23 for NCNT-750, U-NCNT-1 and T-NCNT-750, respectively.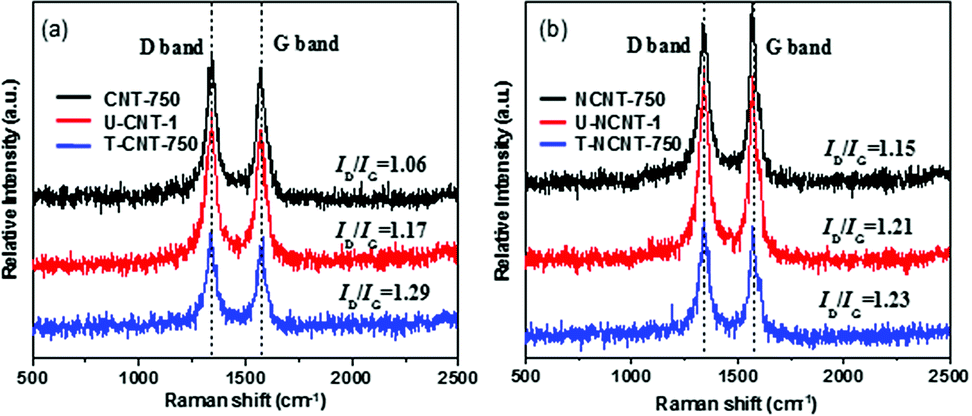 | ||
| Fig. 1 Raman spectra of fresh, used and regenerated samples: (a) CNT and (b) NCNT. | ||
The PZCs of the fresh, used and regenerated catalysts were determined using the potentiometric mass titration method, and the results are presented in Table 1. For the CNT series, the PZC values of CNT-750 and U-CNT-1 were determined to be 7.3 and 3.7, respectively. The markedly lower PZC of U-CNT-1 than of CNT-750 is likely due to the surface oxidation of CNT during the phenol degradation process. The PZCs of T-CNT-350, T-CNT-550 and T-CNT-750 were 5.9, 7.2 and 7.6, respectively, suggesting that thermal treatment at higher temperatures resulted in more markedly increased PZCs of the regenerated catalyst. It was previously reported that the O-containing groups of carbonaceous materials were susceptible to decomposition upon thermal treatment.26 Furthermore, Menendez et al.27 studied the influence of thermal treatment on the surface groups of activated carbon and concluded that base sites could be formed at the expense of acidic O-containing groups upon thermal treatment; increasing the thermal treatment temperature resulted in a high concentration of base sites. Hence, the gradually increased PZCs of the catalysts with increasing thermal treatment temperature can be ascribed to the removal of O-containing functionalities and the formation of surface base sites (see more results and discussion below). Notably, the catalyst regenerated at 750 °C had a slightly higher PZC than the fresh catalyst, despite their thermal treatment at identical temperatures. This can be attributed to the formation of new O-containing groups on the surface of the CNTs during PMS activation (see the XPS and Boehm titration results below), and these groups decomposed upon thermal regeneration, resulting in new Lewis base sites. In comparison with thermal treatment regeneration, reduction regeneration using NaBH4 led to a moderate increase of the catalyst PZC to 6.2.
| Sample | XPS | PZC | Sample | XPS | PZC | |||
|---|---|---|---|---|---|---|---|---|
| C (at%) | O (at%) | C (at%) | O (at%) | N (at%) | ||||
| a Below detection level.b Not determined. | ||||||||
| CNT-750 | 97.55 | 2.44 | 7.3 | NCNT-750 | 94.79 | 2.66 | 2.56 | 7.9 |
| U-CNT-1 | 96.50 | 3.50 | 3.7 | U-NCNT-1 | 93.08 | 4.54 | 2.38 | 5.4 |
| T-CNT-350 | 97.00 | 2.99 | 5.9 | T-NCNT-350 | 93.65 | 4.05 | 2.30 | 6.6 |
| T-CNT-550 | 97.26 | 2.73 | 7.2 | T-NCNT-550 | 94.80 | 3.30 | 1.90 | 7.4 |
| T-CNT-750 | 97.61 | 2.39 | 7.6 | T-NCNT-750 | 97.15 | 2.85 | BDLa | 8.4 |
| R-CNT | 97.09 | 2.90 | 6.2 | R-NCNT | 94.05 | 4.23 | 1.73 | 6.6 |
| NCNT-550 | 88.76 | 3.24 | 7.98 | NDb | ||||
| NCNT-950 | 95.86 | 2.08 | 2.05 | NDb | ||||
Similar results were observed on the NCNT samples. The PZC of fresh NCNT-750 was 7.9, higher than that of fresh CNT-750, likely due to the presence of N-containing base groups (e.g., pyridinic nitrogen). Application of NCNT-750 in PMS activation also led to a marked decrease of PZC from 7.9 to 5.4 after 1 catalyst reuse cycle. Increasing the thermal regeneration temperature from 350 °C to 750 °C led to an increase of the PZC of NCNT-750 from 6.6 to 8.4. In parallel, the PZC of the catalyst increased to 6.6 upon chemical reduction by NaBH4.
XPS analysis was conducted to elucidate the surface properties of the catalysts, and the contents of C, O and N are listed in Table 1. For the CNT samples, the surface O content in U-CNT-1 was determined to be 3.50 at%, higher than that of CNT-750 (2.44 at%); this is indicative of increased surface O-containing groups due to surface oxidation after the reaction. For the regenerated CNTs, the O contents of T-CNT-350, T-CNT-550 and T-CNT-750 were 2.99 at%, 2.73 at%, and 2.39 at%, respectively, indicating that thermal regeneration of the used catalysts led to marked decreases in their surface O contents. In comparison with NaBH4 reduction, a more marked decrease of O-content could be achieved upon thermal regeneration. Additionally, thermal regeneration at a higher temperature resulted in a lower O content. Notably, the C1s spectra of all the samples displayed very strong non-symmetry, implying that their carbon surfaces are composed of multiple species. Hence, the C1s spectra of the fresh, used, and regenerated CNTs were deconvoluted, and the results are compared in Fig. 2a and Table 2. For CNT-750, the C1s spectrum consisted of four peaks with binding energies of 284.3, 285.5, 287.3 and 288.7 eV, assigned to carbon atoms from aromatic rings and hydroxyl/ether, carbonyl and carboxyl groups, respectively. In comparison with CNT-750, increased contents of C atoms with O-functionalities were observed on U-CNT-1 except for the carbon atoms with carbonyl groups, which can be attributed to the conversion of carbonyl carbons to carboxyl carbons via surface oxidation during PMS activation.28 Thermal regeneration of U-CNT-1 led to a marked increase of the carbon atom content from aromatic rings and decreases of the carbon atom contents from hydroxyl/ether and carboxyl groups. Furthermore, the content of carbonyl carbon atoms in T-CNT-750 was slightly higher than that of CNT-750, which is indicative of the formation of additional carbonyl groups after the oxidation and regeneration cycle.
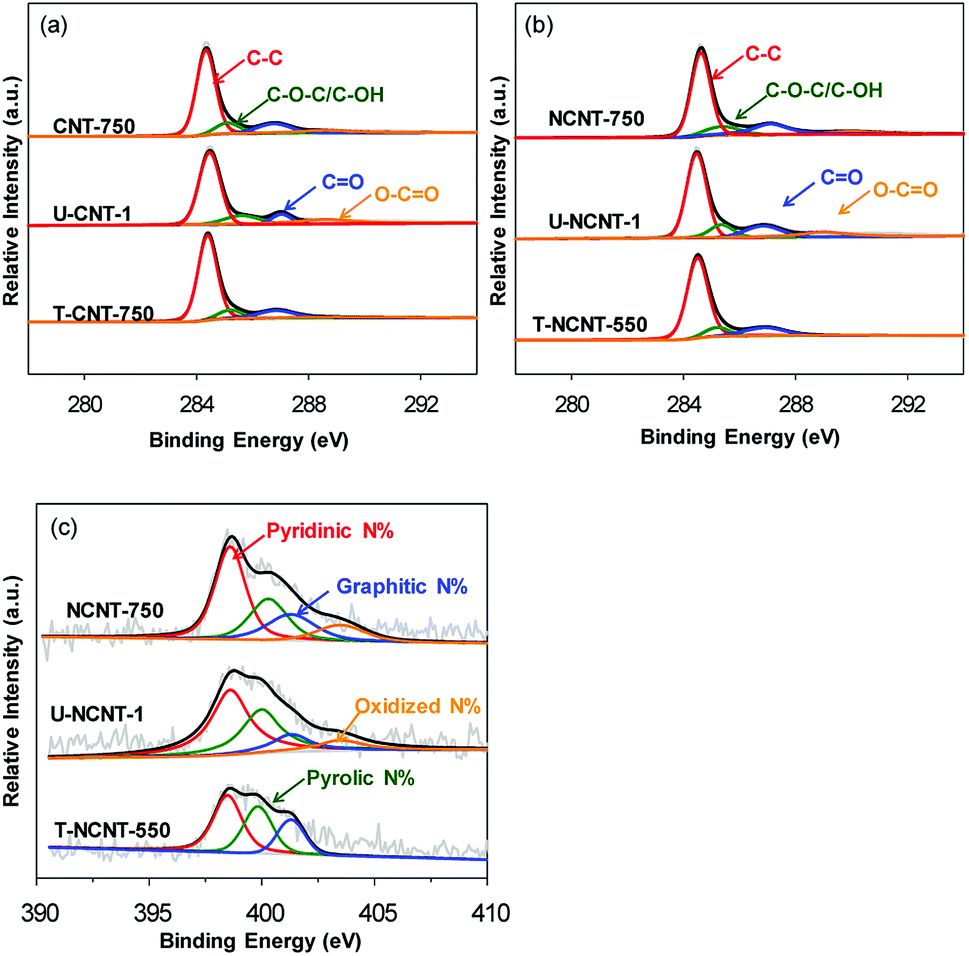 | ||
| Fig. 2 C1s spectra of (a) the CNT samples and (b) the NCNT samples; (c) N1s spectra of the NCNT samples. | ||
| Sample | C1s | N1s | ||||||
|---|---|---|---|---|---|---|---|---|
| C–C | C–O or C–N | CO or CN |
O–CO |
Pyridinic N% | Pyrrolic N% | Graphitic N% | Oxidized N% | |
| CNT-750 | 69.3 | 10.4 | 12.7 | 7.6 | — | — | — | — |
| U-CNT-1 | 63.2 | 12.7 | 7.9 | 16.2 | — | — | — | — |
| T-CNT-750 | 73.9 | 8.7 | 14.0 | 3.4 | — | — | — | — |
| NCNT-750 | 62.6 | 11.3 | 19.6 | 6.5 | 45.8 | 24.0 | 21.5 | 8.7 |
| U-NCNT-1 | 60.9 | 11.9 | 14.7 | 12.5 | 34.5 | 32.1 | 19.3 | 14.2 |
| T-NCNT-550 | 76.0 | 9.2 | 13.7 | 1.1 | 45.7 | 33.4 | 20.9 | — |
For NCNT-750, the surface N content was 2.56 at% (see Table 1), which is indicative of successful surface N-doping of CNT. Similar to CNT-750, catalytic PMS activation led to an increase of the surface O-content from 2.66 at% to 4.54 at% due to surface oxidation, whereas the surface N contents remained nearly constant, reflecting the relatively high stability of N-containing functionalities towards surface oxidation. Again, thermal regeneration resulted in decreased O-content, and the O-content decreased with increasing thermal treatment temperature. Notably, the N contents of T-NCNT-350 and T-NCNT-550 were 2.30 and 1.90 at%, respectively, much lower than those of NCNT-750. Additionally, the N content of T-NCNT-750 was even below the detection level, reflecting that surface N-containing functionalities were vulnerable to thermal treatment and that thermal treatment at 750 °C led to nearly complete loss of surface N. The C1s and N1s spectra of the NCNT samples were also deconvoluted, and the results are presented in Fig. 2b and c, and Table 2. Again, catalytic PMS activation led to increased contents of hydroxyl and carboxyl carbon atoms but also led to decreased carbonyl and aromatic carbon atoms. Thermal treatment regeneration at 750 °C resulted in markedly decreased surface functionalities. For fresh NCNT-750, pyridinic, pyrrolic, graphitic and oxidized N species were identified, and their contents were calculated to be 45.8%, 24.0%, 21.5% and 8.7% (see Table 2), respectively. Upon catalytic PMS activation, the contents of pyridinic and graphitic N decreased while the contents of pyrrolic and oxidized N increased. Thermal regeneration at 550 °C led to a marked decrease of oxidized N, and thermal treatment at 750 °C removed all N-containing functionalities.
These results were confirmed by Boehm titration (as listed in Table 3). For the CNT samples, CNT-750 had a very low concentration of O-containing groups (0.033 mmol g−1), while the concentration of O-containing groups of U-CNT-1 was 0.12 mmol g−1, much higher than that of CNT-750. Particularly, the concentration of carboxylic groups increased from 0.018 mmol g−1 to 0.051 mmol g−1 after one cycle of catalyst reuse, which is indicative of apparent surface oxidation of CNT. Concurrently, the content of base sites markedly decreased from 0.12 mmol g−1 to 0.054 mmol g−1. Upon thermal regeneration, substantial changes in the concentrations of O-containing groups were observed on used CNT-750. For U-CNT-1, the concentration of O-containing groups decreased from 0.12 mmol g−1 to 0.032 mmol g−1 and the concentration of base groups increased from 0.054 mmol g−1 to 0.078 mmol g−1 after thermal regeneration at 750 °C for 2 h. In comparison with U-CNT-1, markedly decreased carboxylic groups were observed on T-CNT-550 and T-CNT-750, whereas marked removal of lactone groups was only observed on T-CNT-750; this is indicative of the varied thermal stability of O-containing functionalities. Previous studies showed that after thermal treatment, carboxylic groups were vulnerable and decomposed below 250 °C, while hydroxyl and lactone groups were very stable and only decomposed above 600 °C.29–31 Additionally, thermal treatment led to the decomposition of O-containing functionalities by releasing CO or CO2, giving rise to surface defect sites and oxygen-free Lewis base sites.27 Compared with CNT-750, NCNT-750 had a much lower concentration of carboxylic groups but a much higher concentration of basic groups; this is likely due to the formation of pyridinic and pyrrolic N species at the expense of carboxylic groups during the N-doping process. Accordingly, Arrigo et al.32 studied the surface structure of N-doped CNT and concluded that N-doping occurred preferentially on carboxylic sites. Hence, the markedly higher PZC of NCNT-750 can be ascribed to its much lower content of carboxylic groups and higher content of basic groups compared to CNT-750. In parallel, regeneration of U-NCNT-1 led to markedly decreased acidic group content and to increased content of base groups.
| Sample | Carboxylic groups (mmol g−1) | Lactone groups (mmol g−1) | Hydroxy groups (mmol g−1) | Total acidic groups (mmol g−1) | Total basic groups/sites (mmol g−1) |
|---|---|---|---|---|---|
| CNT-750 | 0.018 | 0.013 | 0.002 | 0.033 | 0.120 |
| U-CNT-1 | 0.051 | 0.061 | 0.004 | 0.120 | 0.054 |
| T-CNT-350 | 0.020 | 0.053 | 0.005 | 0.078 | 0.059 |
| T-CNT-550 | 0.005 | 0.057 | 0.004 | 0.066 | 0.063 |
| T-CNT-750 | 0.006 | 0.026 | — | 0.032 | 0.078 |
| NCNT-750 | — | 0.025 | 0.031 | 0.056 | 0.890 |
| U-NCNT-1 | 0.073 | 0.150 | 0.009 | 0.230 | 0.093 |
| T-NCNT-350 | 0.028 | 0.130 | 0.003 | 0.160 | 0.100 |
| T-NCNT-550 | 0.013 | 0.120 | — | 0.130 | 0.120 |
| T-NCNT-750 | 0.009 | 0.054 | — | 0.063 | 0.130 |
Moreover, the specific surface areas (SBET) and pore volumes (Vt) of fresh, used and regenerated CNT and NCNT catalysts were detected by N2 adsorption curves and are shown in Table S1.† The SBET of the CNT series catalysts was 98 ± 4 m2 g−1, with a Vt of around 0.28 ± 0.05 cm3 g−1. The SBET of the NCNT series catalysts was 110 ± 5 m2 g−1 with Vt of around 0.28 ± 0.02 cm3 g−1. These results indicate that the reuse and regeneration processes play minor roles in the structures of CNT and NCNT.
3.2. Catalytic PMS activation for phenol degradation
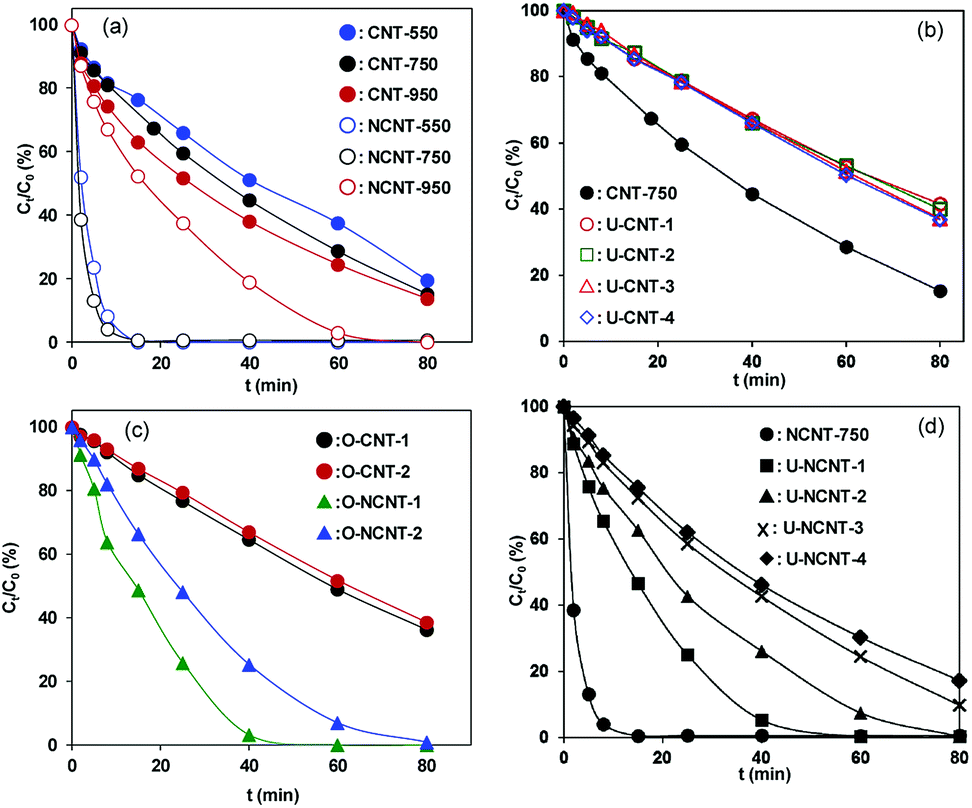
The catalytic PMS activation for phenol degradation on CNT and NCNT catalysts is shown in Fig. 3a. The catalytic activities of the fresh catalysts were found to be dependent on the activation temperature. For example, increasing the calcination temperature from 550 °C to 950 °C led to gradually increased initial activities from 0.028 to 0.041 mmol gCat−1 min−1 for the CNT catalysts. Previous studies showed that surface CO groups and sp2 hybrid and Lewis basic sites (defect sites) of carbonaceous catalysts are capable of acting as active sites for catalytic PMS activation.12,16 Increasing the thermal treatment temperature resulted in increased surface basicity and graphite content, which likely accounts for the enhanced catalytic activity of CNT for phenol degradation. For NCNT, increasing the calcination temperature to 950 °C resulted in sharply decreased catalytic activity due to the loss of nitrogen at this high temperature (see Table 1). Considering the balance between CNT and NCNT, all fresh catalysts were activated at 750 °C in our study.
 | ||
| Fig. 3 Phenol degradation on (a) CNT and NCNT catalysts with different calcination temperatures, (b) fresh and used CNT-750, (c) pre-oxidation CNT-750 and NCNT-750 catalysts prepared in the absence of phenol, and (d) fresh and reused NCNT-750. Reaction conditions: [PMS]0 = 8 mM; [phenol]0 = 0.106 mM; catalyst dosage = 0.1 g L−1. | ||
From the viewpoint of practical application, catalyst reuse is essential. Hence, the reuse of CNT-750 in catalytic PMS activation for phenol degradation was studied, and the results are presented in Fig. 3b. After reaction for 40 min, 55.3% phenol was removed on CNT-750 and 32.8% on U-CNT-1, which is indicative of higher catalytic activity of the fresh catalyst than of the reused catalyst. Accordingly, the initial activities were 0.031 mmol gCat−1 min−1 and 0.01 mmol gCat−1 min−1 for CNT-750 and U-CNT-1, respectively, reflecting marked catalyst deactivation after one cycle of catalyst reuse. For multiple-cycle reuses, however, the activities of the used catalysts remained almost constant and further deactivation was not observed. In parallel, catalyst deactivation was previously observed on other carbonaceous catalysts (e.g., activated carbon, mesoporous carbon and CNTs).16,17,19,33,34 The characterization results of XPS and Boehm titration revealed that the contents of O-containing groups substantially increased upon catalyst reuse. Accordingly, much lower PZCs were observed on the used catalysts than on CNT-750, confirming the surface oxidation of CNT-750 after phenol degradation. After one reuse cycle, the activity of U-CNT-1 was identical to those of U-CNT-2, U-CNT-3 and U-CNT-4. To exclude the role of residual organic species (i.e., intermediates of phenol degradation) on catalyst deactivation under our conditions, catalytic PMS activation on CNT-750 and NCNT-750 was conducted in the absence of phenol, and the results are presented in Fig. 3c. After PMS activation in the absence of phenol, the initial activity of O-CNT-1 was 0.009 mmol gCat−1 min−1, almost identical to that of U-CNT-1. Additionally, further deactivation of the catalyst within the consecutive catalytic cycles was not observed. This clearly suggests that the organic reactants and potential intermediate products had no influence on the used catalysts. Thus, excluding the influence of structure and residual organic species on catalyst deactivation, we studied the mechanism of surface functional groups in the process of deactivation and regeneration. Significant catalyst deactivation was also observed on NCNT-750, and the results are shown in Fig. 3d. NCNT-750 exhibited a much higher initial activity of 0.18 mmol gCat−1 min−1 than CNT-750, which can be attributed to the presence of additional N-containing active sites in NCNT.20,21,35 For catalyst reuse, the initial activity decreased from 0.18 mmol gCat−1 min−1 on NCNT-750 to 0.018 mmol gCat−1 min−1 on U-NCNT-4, which also displayed marked catalyst deactivation. Similarly, the initial activities of U-NCNT-1 and U-NCNT-2 were found to be nearly equal to those of O-NCNT-1 and O-NCNT-2 (results presented in Fig. 3c), confirming that the deactivation was predominantly due to surface oxidation of the catalyst by catalytic PMS activation. Characterization results revealed that catalytic PMS activation led to increased content of oxidized N species at the expense of pyridinic, graphitic and pyrrolic N species, reflecting decreased content of catalytic active sites. In contrast to CNT-750, however, NCNT-750 exhibited continuous deactivation within 4 test reuse cycles.
For catalytic PMS activation on carbonaceous catalysts, radical and non-radical mechanisms were previously reported. For example, Lee et al.2,36 observed a non-radical mechanism for the degradation of phenolic and pharmaceutical compounds on CNTs. In contrast, Duan et al.16 studied phenol degradation on N-doped single walled nanotubes and identified a combined radical and non-radical reaction mechanism. To gain more insight into the mechanisms of PMS activation and catalyst deactivation, 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) was used as a probe molecule, and the reaction was monitored using electron paramagnetic resonance (EPR). The EPR spectra under varied reaction conditions are compiled in Fig. 4a. In the absence of catalyst, characteristic peaks from active radicals (e.g., DMPO-OH adducts and DMPO-SO4 adducts) were not observed. In the presence of catalysts, characteristic peaks with an intensity ratio of 1:2:1:2:1:2:1 were clearly observed, assigned to the DMPO oxidation product, DMPO-X.2,37,38 In parallel, the signal of DMPO-X was also identified in PMS activation on the different catalytic reaction systems.2,38 Floyd et al.39 attributed the formation of DMPO-X to direct oxidation by single electron sources. Very recently, many studies have indicated that the appearance of DMPO-X can be ascribed to the proliferation of free radicals.40,41 For PMS activation on NCNTs, similar EPR signals were also observed, while the intensities of the EPR peaks were much stronger; this confirms that PMS activation was more effective on NCNT-750 than on CNT-750. To clarify the impact of catalyst surface oxidation on the activation efficiency, the EPR spectra of PMS activation on the reused catalysts (U-CNT-1 and U-NCNT-1) were also determined. In comparison with fresh catalysts, the intensities of DMPO-X were much weaker, clearly confirming the higher catalytic activities of the fresh catalysts compared to the used catalysts. Additionally, the absence of phenol led to the formation of similar but much stronger EPR peaks than those in the presence of phenol, which can be ascribed to the competitive reaction of phenol with the active species, consequently suppressing the formation of DMPO-X. The results were in good agreement with the data of catalytic PMS activation for phenol degradation. TEMP (2,2,6,6-tetramethyl-4-piperidinol) was used as a spin-trapping agent to identify other potential active radicals. As shown in Fig. 4b, a three-line spectrum with equal intensities was obtained, which is characteristic of TEMP-1O2 adducts (TEMPO);42,43 this is indicative of the presence of 1O2 in the PMS/CNT-750 system.
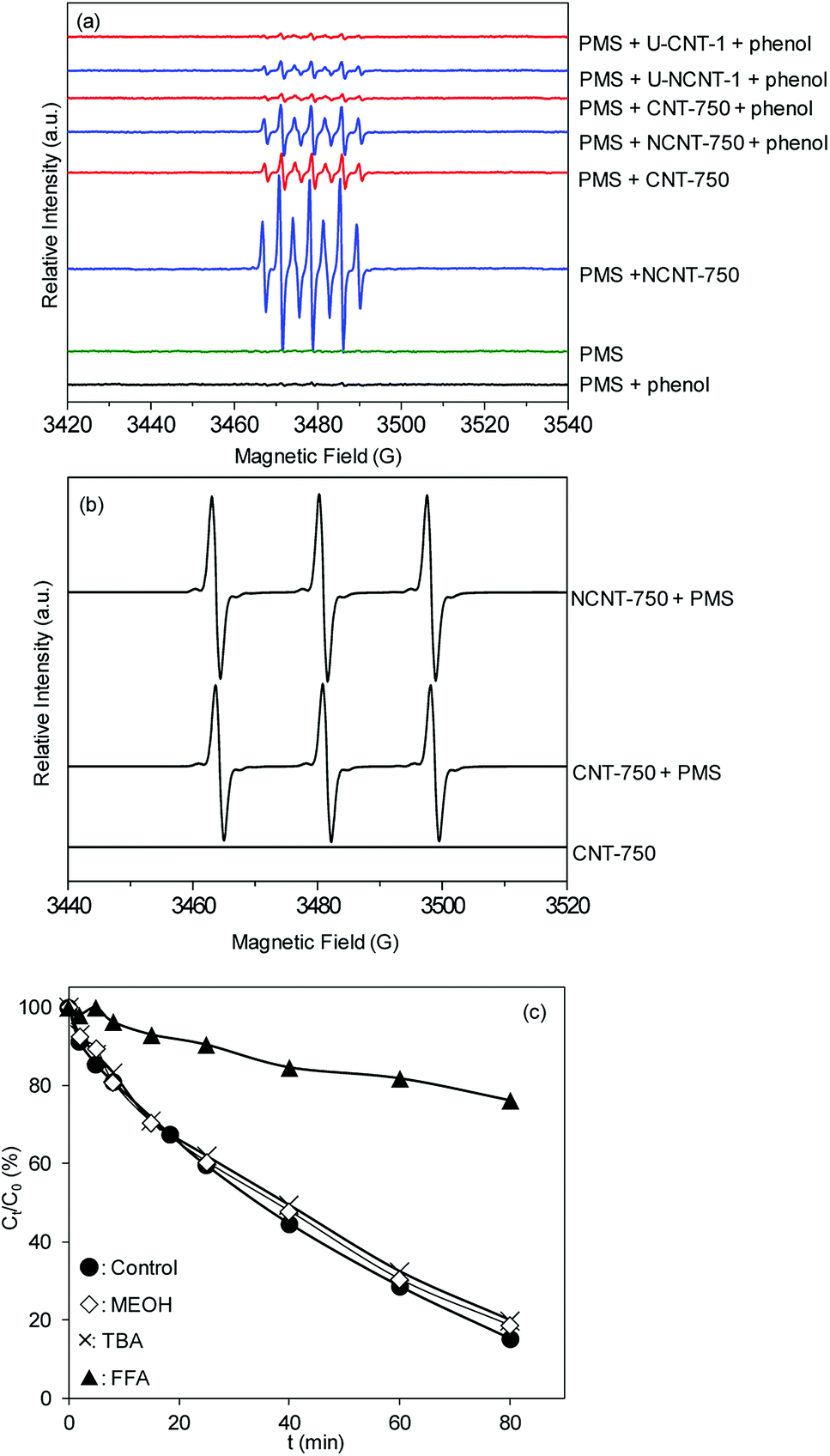 | ||
| Fig. 4 EPR spectra obtained by trapping with (a) DMPO and (b) TEMP under different conditions and (c) catalytic PMS activation for phenol degradation on CNT-750 in the presence of radical scavengers. EPR conditions: [DMPO]0 = [TEMP]0 = 100 mM; [catalyst]0 = 0.1 mg L−1; [PMS]0 = 8 mM; pH = 7.4 ± 0.2 (100 mM phosphate buffer); reaction time = 10 min; [phenol]0 = 0.106 mM; [MEOH]0 = [TBA]0 = [FFA]0 = 1.6 M. | ||
To further verify the activation mechanisms, methanol, t-butanol (TBA) and furfuralcohol (FFA) were used as radical scavengers to detect the effects on the degradation of phenol, and the results are presented in Fig. 4c. In parallel, the presence of neither MEOH nor TBA affected the phenol degradation. Furthermore, the addition of FFA to the PMS/CNT-750 reaction system resulted in decreased phenol removal from 84.7% to 23.8% within 80 min, confirming the importance of 1O2 in the reaction.
It should be emphasized that in liquid phase catalytic reactions, the adsorption of reactants on the catalyst surface is a prerequisite step and thus plays a crucial role in the catalytic activity of the catalyst. For example, Chen et al.44 compared liquid phase catalytic hydrogenation of bromate on Pd catalysts supported on different supports (e.g., Al2O3, activated carbon (AC) and SiO2) and observed a much higher catalytic activity for Pd/Al2O3 than for the other catalysts due to strong electrostatic attractive interactions. Inspired by previous results, we assumed that the adsorption of PMS or/and phenol played important roles in phenol degradation. Hence, the influences of the initial concentrations of PMS and phenol on the initial reaction rate of phenol degradation were investigated. For the influence of PMS adsorption, phenol degradation was conducted by varying the initial PMS concentration from 2.5 to 12.0 mM while fixing the phenol concentration at 0.106 mM, and the results are compared in Fig. 5a. Increasing the PMS concentration led to an increased initial rate from 0.019 to 0.031 mmol gCat−1 min−1; meanwhile, at PMS concentrations above 8.0 mM, the phenol degradation rate remained nearly constant (Fig. 5b, inset). In the low PMS concentration range, the increased reaction rate can be ascribed to the increased concentration of adsorbed PMS on catalyst surface with increasing PMS concentration. At PMS concentrations above 8.0 mM, the adsorbed PMS concentration remained constant because saturated PMS adsorption was reached, which resulted in nearly identical reaction rates even with further increasing the initial PMS concentration. To confirm the reaction mechanism, the dependency of the initial reaction rate on the initial PMS concentration was further fitted to the Langmuir–Hinshelwood model,45
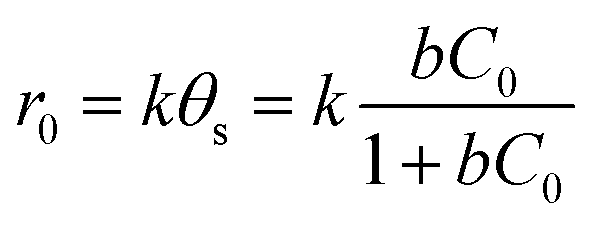 | (1) |
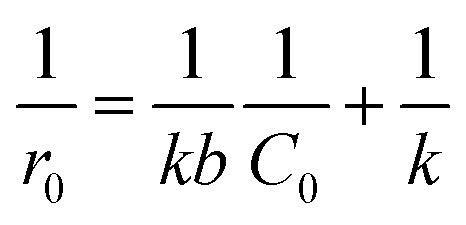 | (2) |
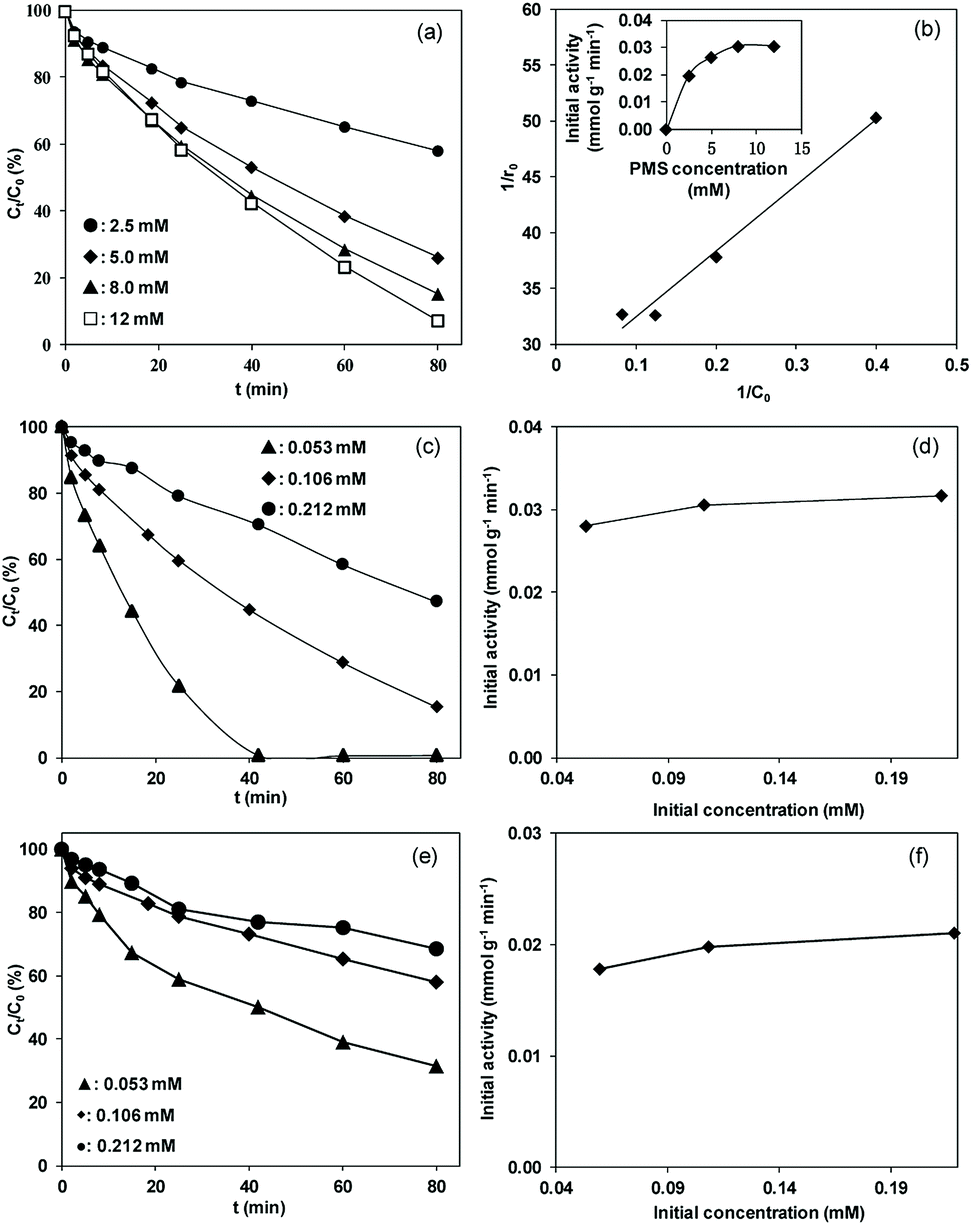 | ||
| Fig. 5 (a) Effects of initial PMS concentration on phenol degradation, (b) the plot of 1/r0 versus 1/C0 (inset: effects of initial PMS concentration on the initial activity of CNT); effects of initial phenol concentration on (c) phenol degradation and (d) the initial activity of CNT at a PMS concentration of 8 mM; effects of initial phenol concentration on (e) phenol degradation and (f) the initial activity of CNT at a low PMS concentration of 2.5 mM. Reaction conditions: catalyst dosage = 0.1 g L−1. | ||
Fig. 5b shows the plot of 1/r0 versus 1/C0. The linear plot of 1/r0 versus 1/C0 with R2 higher than 0.98 showed that phenol degradation on the catalyst obeyed the Langmuir-Hinshelwood model, reflecting that the reaction rate of phenol degradation is controlled by the catalytic activation of pre-adsorbed PMS on the catalyst surface.
To determine the influence of initial phenol concentration, phenol degradation was performed by varying the phenol concentration from 0.053 to 0.212 mM at a fixed PMS concentration of 8.0 mM. The results are presented in Fig. 5c. The initial reaction rates of phenol degradation remained nearly constant with increasing phenol concentration from 0.053 to 0.212 mM, indicating that the phenol degradation was independent of the initial phenol concentration under our experimental conditions. In parallel, nearly identical reaction rates were observed for phenol degradation with varied initial concentrations at a lower PMS concentration of 2.5 mM (results presented in Fig. 5e and f), confirming the minor impact of the initial phenol concentration on phenol degradation.
In conclusion, the catalytic PMS activation for phenol degradation led to decreased contents of active sites on CNT-750 and NCNT-750 due to the increased acidic O-containing groups and decreased basic groups/sites resulting from surface oxidation. Additionally, the interaction between PMS and the catalyst played a crucial role in phenol degradation because reactive species were formed by the catalytic activation of adsorbed PMS. The PZCs of the fresh CNT-750 and NCNT-750 samples were 7.3 and 7.9, respectively. Considering that PMS is present as anions under the experimental conditions, electrostatic attractive interactions between PMS and the fresh catalysts can be expected, leading to strong PMS adsorption and, thus, high catalytic activity. In contrast, the PZCs of U-CNT-1 and U-NCNT-1 were 3.7 and 5.4, respectively. Hence, electrostatic repulsion interactions occurred between PMS and the used catalysts, which suppressed PMS adsorption and the access of PMS to the active sites; this also accounts for the markedly decreased catalytic activities of the used catalysts.
3.3. Catalyst regeneration
The catalytic PMS activation led to surface oxidation and formation of O-containing functionalities on both CNT-750 and NCNT-750, as reflected by XPS analysis, PZC determination and Boehm titration. As a result, decreased catalytic activities for phenol degradation were obtained on used CNT-750 and NCNT-750 due to their substantially decreased active sites and suppressed PMS adsorption. Accordingly, removal of the O-containing functionalities may restore the catalytic activity. Hence, the catalysts were regenerated by thermal treatment and chemical reduction, and the results are presented in Fig. 6a. For CNT, regeneration of U-CNT-1 by NaBH4 reduction led to an increase of the initial activity from 0.01 to 0.018 mmol gCat−1 min−1. More effective regeneration was achieved by thermal regeneration, and the catalytic activity of the regenerated CNT was found to be strongly dependent on the thermal regeneration temperature. For example, increasing the thermal treatment temperature from 350 °C to 750 °C caused an increase of the reaction rate from 0.021 to 0.059 mmol gCat−1 min−1.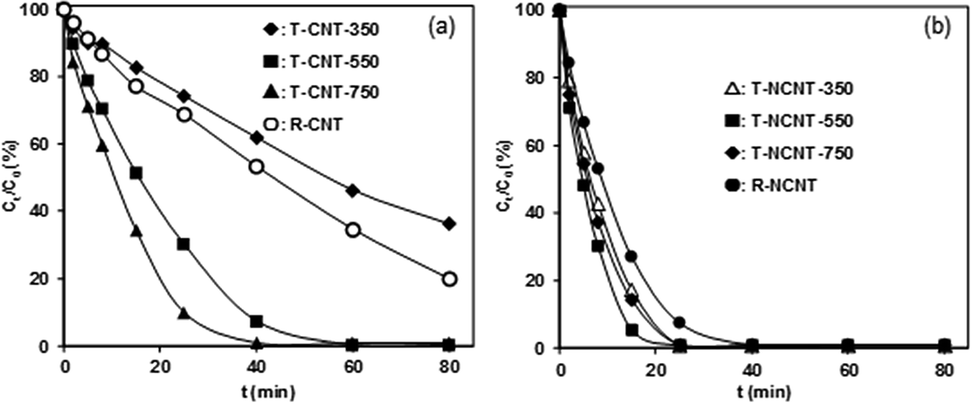 | ||
| Fig. 6 Phenol degradation on regenerated (a) CNT and (b) NCNT catalysts. Reaction conditions: [PMS]0 = 8 mM; [phenol]0 = 0.106 mM; catalyst dosage = 0.1 g L−1. | ||
The effective restoration of the catalytic activity can be readily explained in terms of the surface chemistry of the regenerated catalysts. After thermal regeneration, the PZCs were determined to be 5.9, 7.2, and 7.6 for T-CNT-350, T-CNT-550, and T-CNT-750, respectively, which are much higher than that of the used CNT. The increased PZC of CNT with increasing thermal treatment temperature clearly reveals the effective removal of negatively charged O-containing groups. Accordingly, XPS characterization showed that thermal treatment of used CNT at 750 °C led to a decrease of the O content from 3.50 to 2.39 at%, mainly contributed by the removal of surface O-containing groups. In parallel, Boehm titration results revealed that thermal treatment resulted in increased content of basic groups at the expense of acidic groups. The effective removal of O-containing groups and the formation of Lewis basic sites restored the active sites and improved PMS adsorption on the catalyst surface, giving rise to enhanced catalytic activity.
In comparison with CNT, different regeneration behavior of NCNT was observed (see Fig. 6b). In general, chemical reduction or thermal treatment of used NCNT-750 was capable of restoring its catalytic activity. For example, chemical reduction of used NCNT-750 by NaBH4 led to an increase of the initial activity from 0.051 to 0.078 mmol gCat−1 min−1. In parallel, thermal regeneration of U-NCNT-1 at 350 °C, 550 °C, and 750 °C resulted in increased catalytic activities of 0.098, 0.12 and 0.11 mmol gCat−1 min−1, respectively. In contrast to CNT, the catalytic activities of regenerated NCNT were much lower than that of the fresh catalyst (0.18 mmol gCat−1 min−1). Characterization results from XPS and Boehm titration showed that both chemical reduction and thermal treatment of U-NCNT-1 substantially eliminated O-containing functionalities, particularly carboxyl groups. Concurrently, markedly increased basic sites were observed. As a result, the catalytic activities of the regenerated NCNT were partially restored. Notably, the N contents of NCNT-750, T-NCNT-350, and T-NCNT-550 were determined to be 2.56, 2.30 and 1.90 at%, respectively. Moreover, the N content in T-NCNT-750 was even below the determination level, reflecting increased N loss with regeneration temperature. Considering that pyridinic, pyrrolic and graphitic N species act as highly active sites for PMS activation, the much lower catalytic activities of the regenerated catalysts can thus be attributed to the N loss during the regeneration process.
4. Conclusions
In the present study, the reuse, deactivation and regeneration of CNT and NCNT catalysts in the catalytic activation of PMS were studied, and the variation of the surface functionalities of the catalysts was investigated. The results show that both CNT and NCNT are highly active catalysts in catalytic PMS activation for phenol degradation, while marked catalyst deactivation was observed on the used catalysts. The catalyst deactivation was predominantly due to the formation of O-containing functionalities and decreased PZC, which substantially decreases the active sites of the catalyst and suppresses access of PMS to the active sites due to electrostatic repulsion interactions. Consequently, markedly low catalytic activities are obtained on used CNT and NCNT. Additionally, phenol degradation is highly dependent on PMS concentration, following the Langmuir–Hinshelwood model; this indicates that the catalytic activation of adsorbed PMS is the rate-controlling step. Chemical reduction and thermal treatment of used CNT and NCNT can remove surface O-containing groups, restoring their catalytic activities. More effective regeneration can be achieved using thermal treatment compared to chemical reduction, and the regeneration effect of thermal treatment is strongly dependent on the treatment temperature. For CNT, the regenerated catalyst exhibits even higher catalytic activity than the fresh catalyst. For NCNT, however, thermal treatment regeneration leads to a marked loss of N-containing functionalities and even to complete N loss at 750 °C, giving rise to lower catalytic activity than that of the fresh catalyst. These results show that the surface functionalities of CNT and NCNT vary during catalyst reuse, deactivation and regeneration, which are strongly correlated to the catalytic activities of CNT and NCNT for PMS activation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial support from the National Natural Science Foundation of China (no. 21507056, 21407074 and 21577056) and the Natural Science Foundation of Jiangsu Province (BK20150568) is gratefully acknowledged.Notes and references
- S. K. Ling, S. B. Wang and Y. L. Peng, J. Hazard. Mater., 2010, 178, 385–389 CrossRef CAS PubMed.
- H. Lee, H. J. Lee, J. Jeong, J. Lee, N.-B. Park and C. Lee, Chem. Eng. J., 2015, 266, 28–33 CrossRef CAS.
- C. J. Liang and H. W. Su, Ind. Eng. Chem. Res., 2009, 48, 5558–5562 CrossRef CAS.
- J. M. Monteagudo, A. Durán, R. González and A. J. Expósito, Appl. Catal., B, 2015, 176–177, 120–129 CrossRef CAS.
- Z. H. Wang, L. Y. Sun, X. Y. Lou, F. Yang, M. Feng and J. S. Liu, J. Colloid Interface Sci., 2017, 507, 51–58 CrossRef CAS PubMed.
- Y. T. Lin, C. J. Liang and J. H. Chen, Chemosphere, 2011, 82, 1168–1172 CrossRef CAS PubMed.
- S. N. Su, W. L. Guo, C. L. Yi, Y. Q. Leng and Z. M. Ma, Ultrason. Sonochem., 2012, 19, 469–474 CrossRef CAS PubMed.
- T. Zeng, X. L. Zhang, S. H. Wang, H. Y. Niu and Y. Q. Cai, Environ. Sci. Technol., 2015, 49, 2350–2357 CrossRef CAS PubMed.
- Y. S. Zhao, C. Sun, J. Q. Sun and R. Zhou, Sep. Purif. Technol., 2015, 142, 182–188 CrossRef CAS.
- D. D. Tang, G. K. Zhang and S. Guo, J. Colloid Interface Sci., 2015, 454, 44–51 CrossRef CAS PubMed.
- Y. J. Yao, H. Chen, C. Lian, F. Y. Wei, D. W. Zhang, G. D. Wu, B. J. Chen and S. B. Wang, J. Hazard. Mater., 2016, 314, 129–139 CrossRef CAS PubMed.
- S. Indrawirawan, H. Q. Sun, X. G. Duan and S. B. Wang, Appl. Catal., B, 2015, 179, 352–362 CrossRef CAS.
- X. G. Duan, C. Su, L. Zhou, H. Q. Sun, A. Suvorova, T. Odedairo, Z. H. Zhu, Z. P. Shao and S. B. Wang, Appl. Catal., B, 2016, 194, 7–15 CrossRef CAS.
- X. G. Duan, Z. M. Ao, D. G. Li, H. Q. Sun, L. Zhou, A. Suvorova, M. Saunders, G. X. Wang and S. B. Wang, Carbon, 2016, 103, 404–411 CrossRef CAS.
- X. G. Duan, K. O'Donnell, H. Q. Sun, Y. X. Wang and S. B. Wang, Small, 2015, 11, 3036–3044 CrossRef CAS PubMed.
- X. G. Duan, H. Q. Sun, Y. X. Wang, J. Kang and S. B. Wang, ACS Catal., 2015, 5, 553–559 CrossRef CAS.
- X. G. Duan, H. Q. Sun, J. Kang, Y. X. Wang, S. Indrawirawan and S. B. Wang, ACS Catal., 2015, 5, 4629–4636 CrossRef CAS.
- H. Q. Sun, S. Z. Liu, G. L. Zhou, H. M. Ang, M. O. Tadé and S. B. Wang, ACS Appl. Mater. Interfaces, 2012, 4, 5466–5471 CrossRef CAS PubMed.
- Y. X. Wang, Z. M. Ao, H. Q. Sun, X. G. Duan and S. B. Wang, Appl. Catal., B, 2016, 198, 295–302 CrossRef CAS.
- H. Q. Sun, C. K. Kwan, A. Suvorova, H. M. Ang, M. O. Tadé and S. B. Wang, Appl. Catal., B, 2014, 154–155, 134–141 CrossRef CAS.
- X. G. Duan, Z. M. Ao, H. Q. Sun, S. Indrawirawan, Y. X. Wang, J. Kang, F. L. Liang, Z. H. Zhu and S. B. Wang, ACS Appl. Mater. Interfaces, 2015, 7, 4169–4178 CrossRef CAS PubMed.
- C. Wang, J. Kang, H. Q. Sun, H. M. Ang, M. O. Tadé and S. B. Wang, Carbon, 2016, 102, 279–287 CrossRef CAS.
- C. Lu and H. Chiu, Chem. Eng. Sci., 2016, 61, 1138–1145 CrossRef.
- J. Vakros, C. Kordulis and A. Lycourghiotis, Chem. Commun., 2002, 1980–1981 RSC.
- K. Bourikas, J. Vakros, C. Kordulis and A. Lycourghiotis, J. Phys. Chem. B, 2003, 107, 9441–9451 CrossRef CAS.
- Y. Otake and R. G. Jenkins, Carbon, 1993, 31, 109–121 CrossRef CAS.
- J. A. Menéndez, J. Phillips, B. Xia and L. R. Radovic, Langmuir, 1996, 12, 4404–4410 CrossRef.
- Y. X. Wang, Y. B. Xie, H. Q. Sun, J. D. Xiao, H. B. Cao and S. B. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 9710–9720 CrossRef CAS PubMed.
- U. Zielke, K. J. Huttinger and W. P. Hoffman, Carbon, 1996, 34, 983–998 CrossRef CAS.
- Q. L. Zhuang, T. Kyotany and A. Tomita, Carbon, 1994, 32, 539–540 CrossRef CAS.
- J. L. Figueiredo, M. F. R. Pereira, M. M. A. Freitas and J. J. M. Órfão, Carbon, 1999, 37, 1379–1389 CrossRef CAS.
- R. Arrigo, M. Haevecker, S. Wrabetz, R. Blume, M. Lerch, J. McGregor, E. P. J. Parrott, J. A. Zeitler and L. F. Gladden, J. Am. Chem. Soc., 2010, 132, 9616–9630 CrossRef CAS PubMed.
- J. B. Chen, L. M. Zhang, T. Y. Huang, W. W. Li, Y. Wang and Z. M. Wang, J. Hazard. Mater., 2016, 320, 571–580 CrossRef CAS PubMed.
- H. Q. Sun, Y. X. Wang, S. Z. Liu, L. Ge, L. Wang, Z. H. Zhu and S. B. Wang, Chem. Commun., 2013, 49, 9914–9916 RSC.
- H. Chen and K. C. Carroll, Environ. Pollut., 2016, 215, 96–102 CrossRef CAS PubMed.
- H. Lee, H.-I. Kim, S. Weon, W. Choi, Y. S. Hwang, J. Seo, C. Lee and J.-H. Kim, Environ. Sci. Technol., 2016, 50, 10134–10142 CrossRef CAS PubMed.
- H. Zhang, J. Joseph, M. Gurney, D. Becker and B. Kalyanaraman, J. Biol. Chem., 2002, 277, 1013–1020 CrossRef CAS PubMed.
- M. Y. Wei, L. Gao, J. Li, J. Fang, W. X. Cai, X. X. Li and A. H. Xu, J. Hazard. Mater., 2016, 316, 60–68 CrossRef CAS PubMed.
- R. A. Floyd and L. M. Soong, Biochem. Biophys. Res. Commun., 1977, 74, 79–84 CrossRef CAS PubMed.
- J. Y. Yao, X. L. Zeng and Z. Y. Wang, Chem. Eng. J., 2017, 330, 345–354 CrossRef CAS.
- X. X. Pan, J. Chen, N. N. Wu, Y. M. Qi, X. X. Xu, J. L. Ge, X. H. Wang, C. G. Li, R. J. Qu, V. K. Sharma and Z. Y. Wang, Water Res., 2018, 143, 176–187 CrossRef CAS PubMed.
- X. Cheng, H. G. Guo, Y. L. Zhang, X. Wu and Y. Liu, Water Res., 2017, 113, 80–88 CrossRef CAS PubMed.
- S. D. Yan, W. H. Xiong, S. Y. Xing, Y. Q. Shao, R. Guo and H. Zhang, Sci. Total Environ., 2017, 599–600, 1181–1190 CrossRef CAS PubMed.
- H. Chen, Z. Y. Xu, H. Q. Wan, J. Z. Zheng, D. Q. Yin and S. R. Zheng, Appl. Catal., B, 2010, 96, 307–313 CrossRef CAS.
- Z. M. de Pedro, J. A. Casas, L. M. Gomez-Sainero and J. J. Rodriguez, Appl. Catal., B, 2010, 98, 79–85 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra07696k |
| This journal is © The Royal Society of Chemistry 2019 |