
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectroless deposition of RuO2-based nanoparticles for energy conversion applications†
Jing-Mei Li *ab,
Chi-Chang Hu*a,
Tzu-Ho Wua and
Yung-Jung Hsub
*ab,
Chi-Chang Hu*a,
Tzu-Ho Wua and
Yung-Jung Hsub
aDepartment of Chemical Engineering, National Tsing Hua University, Hsinchu 30013, Taiwan. E-mail: cchu@che.nthu.edu.tw; maylinli_may@hotmail.com
bDepartment of Materials Science and Engineering, National Chiao Tung University, Hsinchu 30010, Taiwan
First published on 1st February 2019
Abstract
This study reports a delicate electroless approach for the deposition of RuO2·nH2O nanoparticles on the VOx·mH2O nanowires and this method can be extended to deposit RuO2·nH2O nanoparticles on various material surfaces. Electrochemical characterizations, including linear sweep voltammetry (LSV), electrochemical quartz crystal microbalance (QCM) analysis and rotating ring-disc electrode (RRDE) voltammetry, were carried out to investigate the growth mechanism. The deposition involves the catalytic reduction of dissolved oxygen by the V4+ species of VOx·mH2O, which drives the oxidation of RuCl3 to proceed with the growth of RuO2·nH2O. This core/shell VOx·mH2O/RuO2·nH2O shows a better catalytic activity of the oxygen reduction reaction (ORR) than RuO2·nH2O, which is ascribed to the pronounced dispersion of RuO2·nH2O. Such an electroless approach was applicable to the preparation of a RuO2-based nanoparticle suspension as well as the deposition of nanocrystalline RuO2·nH2O on other functional supports like TiO2 nanowires. The thus-obtained RuO2-decorated TiO2 nanorods exhibit significantly an enhanced photoactivity toward photoelectrochemical water oxidation. The versatility of the current electroless approach may facilitate the widespread deployment of nanocrystalline RuO2·nH2O in a variety of energy-related applications.
1. Introduction
With exceptional electrochemical and optoelectronic properties, ruthenium oxide (RuO2) has garnered considerable attention for numerous technological applications. For example, hydrous ruthenium oxide (RuO2·nH2O) is considered to be one of the most promising electrode materials for supercapacitors due to its capability of driving reversible multi-electron transfer redox reactions with over-lapped potentials.1–5 In electrocatalysis, RuO2 can function as an active component for chlorine generation from HCl,6 catalytic CO2 reduction7,8 as well as the efficient catalyst for the oxygen and hydrogen evolution reactions.9–11 Furthermore, promising potentials for photocatalytic water splitting,12–15 sodium-ion batteries,16 aerobic oxidation of alcohols,17 nitrous oxide (N2O) decomposition,18 photocatalytic CO2 reduction,19 carbon-free H2 production,20 and room-temperature CO oxidation21 have also been demonstrated by exploiting the intrinsic properties of RuO2. Recently the lithium–oxygen (Li–O2) battery has attracted tremendous attention and RuO2 plays a key role as bifunctional catalyst for the oxygen reduction reaction (ORR) and oxygen evolution reaction (OER) to reduce the overpotential of discharging/charging process.22–25 However, the high cost of ruthenium precursors has hindered the large-scale deployment of RuO2. Especially for the bulk, thick RuO2 films, the relatively low specific capacitance resulting from the long diffusion lengths of ions and electrons has limited their practical applications.7 Thanks to the appealing attributes derived from the size effect, nanocrystalline RuO2 with high specific surface area and enhanced kinetics of mass and charge transport can provide a promising solution to address the above issues.8 Consequently, countless efforts have been dedicated to the preparation of RuO2 nanostructures with miscellaneous structural forms, e.g., nanoparticles and nanotubes, whose capacitive performance can be accordingly enhanced.In view of the ease of handling and the feasibility of massive production, nanoparticles of RuO2 have proven their prowess in energy storage26,27 and energy conversion applications.11,28,29 To achieve homogeneous distribution and thereby satisfactory performance, depositing RuO2 nanoparticles on specific substrates with unique functionalities is desirable.17,25,30–34 For instance, carbon nanotubes were utilized as the template to grow highly dispersed RuO2 with the size down to 1.35 nm.17 With the striking structural uniformity, the resultant composites exhibited high activity and excellent selectivity toward catalytic aerobic oxidation. Moreover, RuO2 nanoparticles were deposited on TiO2 nanoflowers to form nanocomposites with three-dimensional porous architectures, which displayed high power capacitive characteristics relative to the un-supported RuO2.30,31 The superiority was ascribed to the porous nature from the nanoflower backbones of TiO2 and the connected electron pathways endowed by the well distributed RuO2. Till now, a vast body of the literature has documented various synthetic approaches for the deposition of RuO2 nanoparticles on functional substrates.33 Most of them however involved complicated preparation processes, such as prolonged reaction time,17,34 energy-consuming steps, for example, the need of post-annealing treatment at an elevated temperature,9,24,35 or the most concerned issue, i.e. the use of large amount of ruthenium precursors,17,22,23,34 which may further obstruct the practical utilization of nanocrystalline RuO2. Hence, the creation of a facile, cost-effective approach to obtain highly dispersive RuO2 with structural integrity is imperative more than ever before.
In this work, we developed a simple yet effective electroless method to deposit RuO2·nH2O nanoparticles on hydrated vanadium oxide (VOx·mH2O) nanowires. Here, VOx·mH2O nanowire arrays were employed as supports for RuO2·nH2O deposition. The nanowire backbones not only provided growth platform for homogeneous distribution of RuO2·nH2O deposits but also formed abundant structural interstices to create three-dimensional porosities, which is beneficial to the applications requiring high surface area and facile charge transport, such as electrochemical energy conversion, solar energy conversion, catalysis and sensing. The deposition was conducted at room temperature and completed within 1 h without the post-heat treatment. The resultant RuO2·nH2O crystallites had an average size of 1.9 nm and were homogeneously distributed on the VOx·mH2O nanowire surface with an ultrahigh density of 2.1 × 1013 particles per μm2. The results of linear sweep voltammetry (LSV), quartz crystal microbalance (QCM) analysis and rotating ring-disc electrode (RRDE) voltammetry suggested that the V4+ species of VOx·mH2O nanowires catalyzed the reduction of dissolved oxygen, which drove the oxidation of Ru3+ to deposit RuO2·nH2O. Such a delicate electroless deposition process can be extended for the deposition of nanocrystalline RuO2 on other oxides supports, for example TiO2 nanowires, which has significant implications in the development of multi-functional nanocomposites for a variety of energy-related applications ranging from photoelectrochemical energy conversion to electrochemical energy storage.
2. Materials and methods
2.1. Preparation of RuO2·nH2O-decorated VOx·mH2O nanowires
VOx·mH2O nanowires were first grown on graphite substrates (1 × 1 cm) using the anodic deposition method.36,37 The substrate deposited with VOx·mH2O nanowires was then immersed into a reaction solution (50 mL) containing 10 mM of RuCl3·xH2O and 10 mM of CH3COONa. Upon steady stirring at room temperature for 1 h, RuO2·nH2O nanoparticles were deposited on the VOx·mH2O nanowire surface. The substrate was then taken out, rinsed with de-ionized water and dried at room temperature. To study the growth mechanism, the reaction solution of V4+ species was used and prepared by adding 0.2 g of VOSO4 into 50 mL of deionized water. To prepare the reaction solution of V5+, a stoichiometric content of H2O2 was added along with 0.2 g of VOSO4 in the 50 mL deionized water, which led to the complete oxidation of V4+ to V5+. The homogeneous growth of RuO2 nanoparticle suspensions was conducted by employing the reaction solution containing 10 mM of RuCl3·xH2O, 10 mM of CH3COONa and a given amount of V4+.Rutile TiO2 nanorod arrays were grown on fluorine-doped tin oxide (FTO) glass substrates using a hydrothermal method reported in the literature.38,39 Concentrated HCl (15 mL) was diluted with deionized water (15 mL) and then mixed with titanium n-butoxide (Ti(OBu)4, 0.5 mL) in a 100 mL beaker. The resulting clear solution was transferred to a Teflon-lined stainless-steel autoclave (40 mL in volume), where a clean FTO glass substrate was submerged into the solution. The sealed autoclave was heated in an electric oven at 150 °C for 5 h and then slowly cooled to room temperature. A white TiO2 nanorod film was uniformly coated on the FTO glass substrate after cooling. The samples were thoroughly washed with deionized water and then annealed in air at 550 °C for 3 h to improve the crystallinity.
2.2. Characterizations
The morphology and dimensions of the samples were observed with a field-emission scanning electron microscope (FE-SEM, Hitachi S-4800) and a field-emission transmission electron microscope (FE-TEM, Philips, Tecnai F20 G2). The elemental analysis of the samples was conducted with energy dispersive X-ray spectrometer (EDS), an accessory of TEM. The electrochemical quartz crystal microbalance (EQCM) analysis was performed on a commercially available Au/Ti-sputtered quartz electrode with an electrochemical workstation (CH Instruments, CHI 4051a). The chemical environment information was acquired with an X-ray photoelectron spectrometer (XPS, ULVAC-PHI, Quantera SXM) using Al Kα radiation (hν = 1486.69 eV) under high vacuum conditions. Electrochemical characterizations were carried out using an electrochemical workstation (CH Instruments, CHI 633A). To study the oxygen reduction reaction (ORR) kinetics, the RRDE voltammetry was employed and the potential values were represented with respect to the reversible hydrogen electrode (RHE). The measurements were conducted at 900 rpm with a scan rate of 5 mV s−1. The photoelectrochemical measurements were performed and in 1.0 M NaOH under a three-electrode configuration where the Ru-based nanoparticles (NPs) decorated TiO2 nanorods, Pt foil, and Ag/AgCl serve as the working, counter, and reference electrodes, respectively. The simulated sunlight irradiation was produced using a solar simulator (LCS-100 Solar Simulator) with an AM 1.5 global filter with the light intensity of 100 mW cm−2.3. Results and discussion
3.1. Structural investigations
Fig. 1(A) shows the SEM image of VOx·mH2O nanowires obtained from the anodic deposition process. These nanowires were bundled, mutually interlaced to form porous urchin-like structures. By virtue of this highly porous structural feature, the VOx·mH2O nanowires have been demonstrating promising potentials as the cathode of asymmetric Li-ion supercapacitors.37 After reacted with RuCl3 in the electroless reaction, the nanowires became thicker in diameter and transformed into coral-like structures with rounded ends. As clearly observed in Fig. 1(B), the drastic morphology change indicates that certain deposits should have grown on the nanowire surface. | ||
| Fig. 1 SEM images of (A) pristine VOx·mH2O and (B) RuO2·nH2O-decorated VOx·mH2O nanowires. | ||
TEM observations were carried out to examine the microstructural details. As evident in Fig. 2(A), the nanowire surface was covered with a large quantity of nanoparticles. The deposited nanoparticles showed an average size of 1.9 nm and were uniformly distributed on the nanowire surface with an ultrahigh density of 2.1 × 1013 particles per μm2. The corresponding TEM-EDS elemental maps in Fig. 2(B) verify that the deposited particles were composed of Ru and O, suggesting the formation of Ru oxide-based nanoparticles on the surface of VOx·mH2O nanowires.
 | ||
| Fig. 2 (A) TEM image and (B) TEM-EDS mapping results of RuO2·nH2O-decorated VOx·mH2O nanowires. Inset in (A) shows the enlarged image at the marked region. | ||
3.2. Deposition mechanism
To manipulate the present electroless process for depositing Ru oxide-based nanoparticles, it is essential to understand the growth mechanism behind. For electroless deposition of metal oxides in the higher oxidation state in comparison with their precursors (e.g., RuO2 deposited from RuCl3), oxidizing agents are required to drive the metal ion oxidation. Since no specific oxidant was added in the current synthetic process, the interactions among the reaction species, i.e., RuCl3 precursor, VOx·mH2O electrode, and dissolved oxygen molecules, might be responsible for the initiation of the Ru3+ oxidation. We first analyzed the oxidation kinetics of Ru3+ with electrochemical characterization methods and evaluated the possible involvement of VOx·mH2O electrode. Fig. 3(A) presents the LSV curves for two different electrodes, graphite substrates and VOx·mH2O nanowires, recorded in the electrolyte containing RuCl3. For graphite substrate, a prominent anodic peak at 1.15 V vs. Ag/AgCl was noticed, which was attributed to the oxidation of Ru3+ into Ru4+. The Ru3+ oxidation peak remained eminent for the VOx·mH2O nanowires electrode, exhibiting a higher current density with a less positive potential relative to the peak on the graphite substrate. This phenomenon suggests that VOx·mH2O nanowires may facilitate the electrochemical oxidation of Ru3+ by improving the reaction kinetics. Note that VOx·mH2O nanowires were composed of mixed V4+ and V5+ oxy-hydroxyl species,36,37 shown in Fig. S1.† To clarify the species responsible for facilitating the oxidation of Ru3+, we carried out the electroless reaction by using the reaction solution of V4+ or V5+ and compared the results with QCM. Fig. 3(B) displays the mass change of the QCM analyzer for the reaction solutions of V4+ and V5+. Also Fig. 3(B) includes the QCM result from the V4+ solution with N2 purging to clarify the possible role of the dissolved oxygen in the electroless deposition of RuO2. Several important features can be observed from Fig. 3(B). First, the electroless deposition was totally inhibited in the reaction solution of V5+, revealing that V4+ was the main species for facilitating the RuCl3 oxidation. Since the composition of VOx·mH2O electrode was unchanged during the electroless deposition process, V4+ was considered to be a catalyst for RuCl3 oxidation instead of an oxidant agent. Second, the deposition rate was hindered as the reaction solution of V4+ was purged with N2, suggesting that the dissolved oxygen is the oxidizing agent in the catalytic oxidation of RuCl3. Furthermore, when the RuCl3 solution without V4+ was purged with air for 1 h or exposed to the ambient air for 7 days, no precipitation could be gathered. Accordingly, we propose that the V4+ species of VOx·mH2O nanowires catalyze the reduction of dissolved oxygen to drive the oxidation of RuCl3 in order to proceed with the RuO2·nH2O deposition.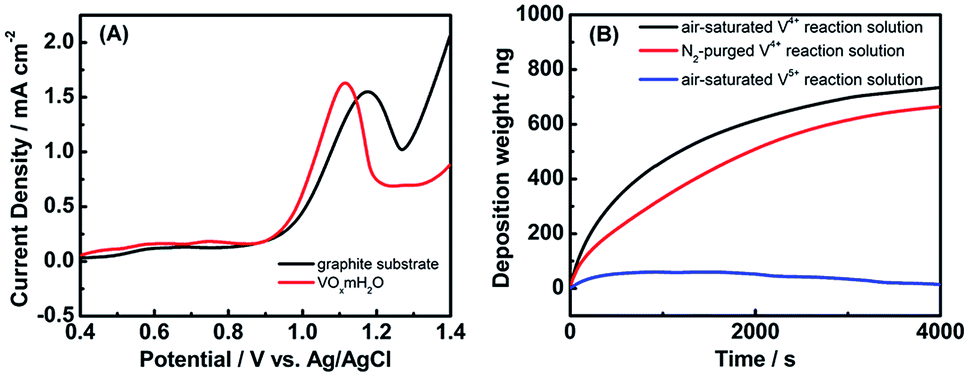 | ||
| Fig. 3 (A) LSV curves of RuCl3 oxidation for graphite substrates and VOx·mH2O nanowires recorded in the reaction solution. (B) QCM mass change on the electrode recorded from the reaction solutions containing V4+ and V5+, respectively. The result of performing N2 purging was also included for comparison. | ||
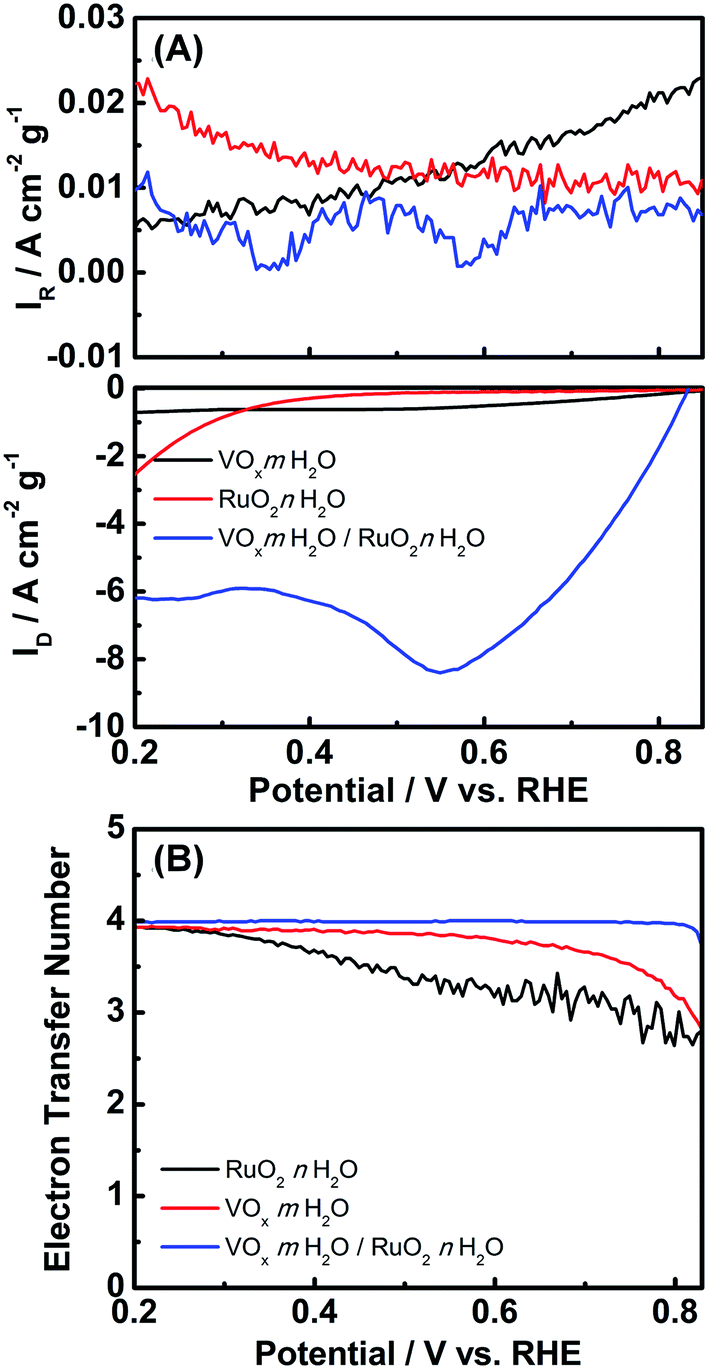
To manifest the above oxygen reduction contention, we investigated the electrochemical behavior of VOx·mH2O nanowires toward the ORR. It should be noted that the ORR in aqueous solutions occurs mainly through two pathways: the direct 4-electron reduction pathway from O2 to H2O, and the 2-electron reduction pathway from O2 to H2O2.40 For the 4-electron transfer pathway, the catalytic oxidation of Ru3+ was driven by the direct electron transfer from Ru3+ to the oxygen molecules adsorbed on the VOx·mH2O surface, which generated OH− ions to enable the growth of RuO2·nH2O. As to the two-electron transfer pathway, highly oxidative H2O2 could be produced in the vicinity of the VOx·mH2O surface, subsequently oxidizing Ru3+ to proceed with the RuO2·nH2O deposition.1,17 Interestingly, the concentration of proton at the surface of VOx·mH2O without potential bias was monitored after RuCl3 was added (in Fig. S2†). The result shows that the concentration of proton increased suddenly at first and reached a plateau, which indicates that the ORR and RuO2 deposition involve hydroxyl ions. Note that both 4-electron and 2-electron pathways of the ORR consume protons under the acidic circumstance to oxidize Ru(III) to form the Ru(IV) ions (i.e., decrease in proton concentration). Since every Ru(IV) ion will react with four OH− to form Ru(OH)4, which significantly increases the proton concentration, we cannot determinate the deposition mechanism from the sudden increase in the proton concentration. Accordingly, the RRDE method was employed to give a deep investigation. Fig. 4(A) shows the RRDE voltammograms on the GC electrodes deposited with VOx·mH2O in 0.5 M Na2SO4 electrolyte with pH = 3, a RuCl3-free electrolyte. When the applied potentials were swept from 0.8 to 0.2 V vs. RHE, the disk current density continuously increased. The apparent electron transfer number (n) can be determined from the equation, 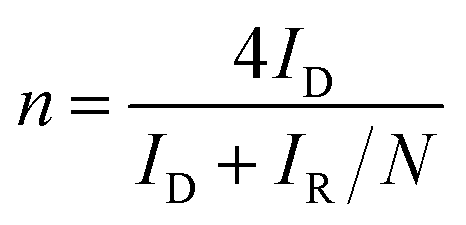 , where ID is the disk current, IR is the ring current, and N is the ring collection coefficient (0.38). As shown in Fig. 4(B), the apparent electron transfer number of ORR varied from 2.6 to 3.9 for the RuCl3-free electrolyte. In fact, the ORR on VOx·mH2O in the electrolyte containing RuCl3 was studied as well. However, the result was not shown here because of the enormous possibility of direct reduction of Ru3+ into Ru metal according to the ruthenium Pourbaix diagram,41 which may lead to misunderstanding. The RRDE results manifest that the ORR catalyzed by VOx·mH2O is shifted from the 2-electron transfer pathway to the 4-electron transfer pathway with the application of more negative potentials. This phenomenon suggests that without the potential bias, the ORR should obey the 2-electron transfer pathway, resulting in the phenomenon of increasing the proton concentration.
, where ID is the disk current, IR is the ring current, and N is the ring collection coefficient (0.38). As shown in Fig. 4(B), the apparent electron transfer number of ORR varied from 2.6 to 3.9 for the RuCl3-free electrolyte. In fact, the ORR on VOx·mH2O in the electrolyte containing RuCl3 was studied as well. However, the result was not shown here because of the enormous possibility of direct reduction of Ru3+ into Ru metal according to the ruthenium Pourbaix diagram,41 which may lead to misunderstanding. The RRDE results manifest that the ORR catalyzed by VOx·mH2O is shifted from the 2-electron transfer pathway to the 4-electron transfer pathway with the application of more negative potentials. This phenomenon suggests that without the potential bias, the ORR should obey the 2-electron transfer pathway, resulting in the phenomenon of increasing the proton concentration.
 | ||
| Fig. 4 (A) The RRDE voltammograms and (B) the mean electron transfer number (n) of the ORR obtained from the GC disk electrode deposited with (1) VOx·mH2O/RuO2·nH2O, (2) VOx·mH2O, and (3) RuO2·nH2O in O2-saturated 0.5 M Na2SO4 with pH = 3 at 5 mV s−1 and a rotating speed of 900 rpm. | ||
3.3. Applications of the catalytic redox deposition of RuO2·nH2O/VOx·mH2O
Fig. 4(A) shows the RRDE voltammograms on the GC electrodes deposited with RuO2·nH2O/VOx·mH2O and RuO2·nH2O in 0.5 M Na2SO4 electrolyte with pH = 3. The onset potentials of the ORR on RuO2·nH2O/VOx·mH2O and RuO2·nH2O are about 0.83 and 0.34 V, respectively. Note here that the onset potential of the ORR was defined as the potential where the current density of oxygen reduction reached 0.5 A cm−2 g−1. Clearly, the usage of VOx·mH2O nanowires as the catalyst backbone promotes the onset potential, probably due to a higher electrolyte-accessible surface area in comparison with RuO2·nH2O. Also note that the catalytic activity of RuO2·nH2O is extremely low at potentials positive than 0.4 V while a sharp increase in the disc current density was found in the less positive potential region. This phenomenon may be assigned to that the Ru(III)/Ru(IV) redox couple is the main active species catalyzing the ORR.42–45 In Fig. 4(B), the mean electron transfer number, n, of RuO2·nH2O monotonously increases from ca. 2.6 to 3.9 with the negative shift in the electrode potential in Fig. 4(B). Surprisingly, the mean electron transfer number of the ORR on RuO2·nH2O/VOx·mH2O was approximately constant and very close to 4. This result reveals that RuO2·nH2O/VOx·mH2O is an excellent catalyst for the ORR, leading to the four-electron transfer pathway. From all the above results and discussion, the RuO2·nH2O/VOx·mH2O composite is a promising electrocatalyst of the ORR for the applications of H2O2 generation and fuel cells in weak acid media containing different supporting electrolytes.3.4. RuO2 suspensions and deposition on TiO2 nanowires
To realize the growth mechanism, we further performed the electroless reaction in a reaction solution simply containing V4+ species and the RuCl3 precursor. The synthesis was conducted under the ambient atmosphere for 6 h, which produced abundant nanoparticle suspensions in the bulk solution. The morphology and composition of resultant RuO2 precipitates were characterized with SEM and XPS analyses. The SEM image in Fig. 5(A) shows that the precipitates are particles of 100–200 nm in size. From a detailed examination, these particles are considered to be aggregates of tiny nanocrystals with sizes of 50–80 nm. The corresponding XPS spectrum in Fig. 5(B) suggests the diverse chemical environment of ruthenium species within the precipitates. The deconvolution of Ru 3p peak yields three chemical states, RuO2, RuO2·nH2O, and Ru(OH)3, with the percentage determined to be 37.8, 40.6, and 21.6 at%, respectively. This illustration highlights the versatility of our electroless approach which not only deposits RuO2·nH2O nanoparticles on the VOx·mH2O nanowires but also produces RuO2-based nanoparticle suspensions in the bulk solution.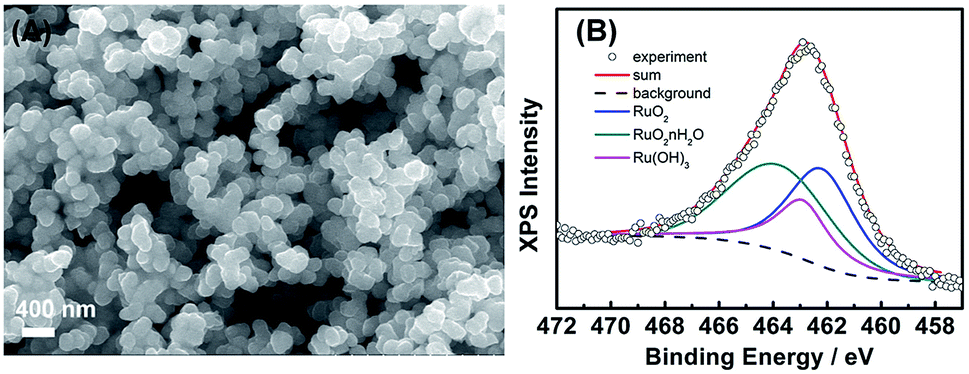 | ||
| Fig. 5 (A) SEM images and (B) XPS Ru 3p core-level spectra of RuO2-based nanoparticle suspensions. | ||
The feasibility in obtaining the highly dispersive and well suspending nanocrystalline RuO2 is particularly appealing for stimulating the practical applications. As a demonstration, TiO2 nanowires array was utilized as a frame in the electroless deposition of nanocrystalline RuO2. Here the high-resolution TEM image was employed to inspect the dispersion of RuO2 on the surface of TiO2 nanowires and Fig. 6 shows that TiO2 nanowire was covered with a 2 nm thin layer of nanocrystalline RuO2 to form a core/shell nanostructure. The lattice fringes of TiO2 frame with the interplanar spacing of 0.32 nm and 0.29 nm were observed, consistent with the d-spacing of (110) and (001) planes of rutile TiO2.38 On the other hand, the lattice fringes of RuO2 nanoparticles with the interplanar spacing of 0.221 nm and 0.225 nm were obtained, corresponding to the d-spacing of (111) and (200) planes of rutile RuO2, respectively.
 | ||
| Fig. 6 A high-resolution TEM image of a RuO2-decorated TiO2 nanowire. | ||
The capacitive behavior of TiO2 nanowires and TiO2/RuO2 core/shell structure was examined. For the comparison purpose, the electroless deposition of nanocrystalline RuO2 on an FTO was conducted as well. From a comparison of CV curves of the core/shell TiO2/RuO2 and RuO2-decorated FTO in Fig. 7(A), well-dispersed RuO2 nanocrystallites promotes the utilization of active species, leading to higher specific capacitance of the core/shell TiO2/RuO2 in comparison with the RuO2-decorated FTO electrode. Since RuO2 has been widely recognized as an efficient oxygen evolution co-catalyst in photo-electrochemical water splitting,46,47 the decoration of RuO2 nanocrystallites onto TiO2 may significantly enhance the photocatalytic activity of TiO2 for water oxidation. Here, the photoelectrochemical measurements were conducted to evaluate the potential of the RuO2-decorated TiO2 nanowire for water splitting. Fig. 7(B) compares the LSV curves of pristine TiO2 and RuO2-decorated TiO2 photoelectrodes for water oxidation. Evidently, RuO2-decorated TiO2 displays larger photocurrent densities in the entire potential widow than pristine TiO2. This phenomenon was ascribed to the improved water oxidation kinetics on the decorated RuO2.
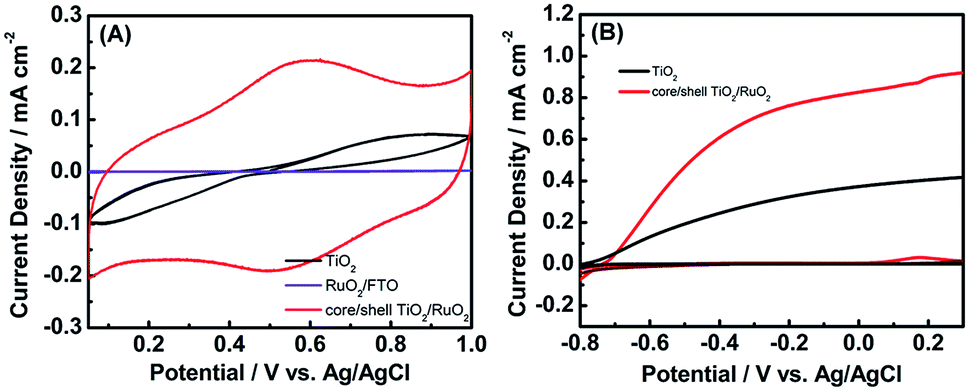 | ||
| Fig. 7 (A) CV curves of pristine TiO2 and RuO2-decorated TiO2 for supercapacitors. (B) LSV curves of pristine TiO2 and RuO2-decorated TiO2 photoanodes for water oxidation. | ||
To extend the application of this electroless deposition, NiCl2 was used as the precursor for Ni-based oxyhydroxide deposition and a precipitate was observed from the mixed solution of VOSO4 and NiCl2. In comparison with the precursor NiCl2, the precipitate displayed different Ni 2p spectrum shown in Fig. S3.† Ni 2p3/2 was further deconvoluted into two peaks cantered at 856.5 eV and 857.6 eV shown in Fig. S4,† which are corresponding to Ni(OH)2 and NiOOH.48 Again, this result demonstrates the oxidation of NiCl2 to form nickel oxyhydroxide and nickel hydroxide. Based on the above preliminary test, this V4+-catalyzed electroless deposition presents an enormous potential in transition metal oxide deposition.
4. Conclusions
An effective electroless approach has been developed for the deposition of nanocrystalline RuO2 onto various functional supports such as VOx·mH2O and TiO2 nanowires as well as the preparation of RuO2-based nanoparticle suspensions in the bulk solution. The success of this synthetic approach relied on the presence of V4+ species which catalyzed the oxygen reduction reaction to drive RuCl3 oxidation for RuO2 deposition. The versatility of the present electroless approach shall foster the advanced applications of nanocrystalline RuO2 in a wide array of fields, which include electrochemical cells, room-temperature CO oxidation, and photocatalytic water splitting.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Ministry of Science and Technology (MOST) of Taiwan under grants MOST 105-2221-E-007-127-MY3, 106-2221-E-007-089-MY3, and 106-2923-E-007-005.References
- K.-H. Chang and C.-C. Hu, J. Electrochem. Soc., 2004, 151, A958–A964 CrossRef CAS.
- C.-C. Hu, K.-H. Chang, M.-C. Lin and Y.-T. Wu, Nano Lett., 2006, 6, 2690–2695 CrossRef CAS PubMed.
- C.-C. Hu, M.-J. Liu and K.-H. Chang, J. Power Sources, 2007, 163, 1126–1131 CrossRef CAS.
- I. L. Chen, Y.-C. Wei, T.-Y. Chen, C.-C. Hu and T.-L. Lin, J. Power Sources, 2014, 268, 430–438 CrossRef CAS.
- I. L. Chen, T.-Y. Chen, Y.-C. Wei, C.-C. Hu and T.-L. Lin, Nanoscale, 2014, 6, 2861–2871 RSC.
- D. Teschner, R. Farra, L. Yao, R. Schlögl, H. Soerijanto, R. Schomäcker, T. Schmidt, L. Szentmiklósi, A. P. Amrute, C. Mondelli, J. Pérez-Ramírez, G. Novell-Leruth and N. López, J. Catal., 2012, 285, 273–284 CrossRef CAS.
- M. Karamad, H. A. Hansen, J. Rossmeisl and J. K. Nørskov, ACS Catal., 2015, 5, 4075–4081 CrossRef CAS.
- J. Qu, X. Zhang, Y. Wang and C. Xie, Electrochim. Acta, 2005, 50, 3576–3580 CrossRef CAS.
- L. M. Torres-Martínez, R. Gómez, O. Vázquez-Cuchillo, I. Juárez-Ramírez, A. Cruz-López and F. J. Alejandre-Sandoval, Catal. Commun., 2010, 12, 268–272 CrossRef.
- Y. Lee, J. Suntivich, K. J. May, E. E. Perry and Y. Shao-Horn, J. Phys. Chem. Lett., 2012, 3, 399–404 CrossRef CAS PubMed.
- C. Gómez-Solís, J. C. Ballesteros, L. M. Torres-Martínez and I. Juárez-Ramírez, Fuel, 2016, 166, 36–41 CrossRef.
- K. Ikarashi, J. Sato, H. Kobayashi, N. Saito, H. Nishiyama and Y. Inoue, J. Phys. Chem. B, 2002, 106, 9048–9053 CrossRef CAS.
- S. D. Tilley, M. Schreier, J. Azevedo, M. Stefik and M. Graetzel, Adv. Funct. Mater., 2014, 24, 303–311 CrossRef CAS.
- J. Park, J. W. Lee, B. U. Ye, S. H. Chun, S. H. Joo, H. Park, H. Lee, H. Y. Jeong, M. H. Kim and J. M. Baik, Sci. Rep., 2015, 5, 11933 CrossRef PubMed.
- M. T. Uddin, O. Babot, L. Thomas, C. Olivier, M. Redaelli, M. D'Arienzo, F. Morazzoni, W. Jaegermann, N. Rockstroh, H. Junge and T. Toupance, J. Phys. Chem. C, 2015, 119, 7006–7015 CrossRef CAS.
- M. Peng, B. Li, H. Yan, D. Zhang, X. Wang, D. Xia and G. Guo, Angew. Chem., Int. Ed., 2015, 54, 6452–6456 CrossRef CAS PubMed.
- X. Fu, H. Yu, F. Peng, H. Wang and Y. Qian, Appl. Catal., A, 2007, 321, 190–197 CrossRef CAS.
- Q. Lin, Y. Huang, Y. Wang, L. Li, X. Y. Liu, F. Lv, A. Wang, W.-C. Li and T. Zhang, J. Mater. Chem. A, 2014, 2, 5178–5181 RSC.
- Q. Liu, Y. Zhou, J. Kou, X. Chen, Z. Tian, J. Gao, S. Yan and Z. Zou, J. Am. Chem. Soc., 2010, 132, 14385–14387 CrossRef CAS PubMed.
- K. Nagaoka, T. Eboshi, Y. Takeishi, R. Tasaki, K. Honda, K. Imamura and K. Sato, Sci. Adv., 2017, 3, e1602747 CrossRef PubMed.
- Y. Jiao, H. Jiang and F. Chen, ACS Catal., 2014, 4, 2249–2257 CrossRef CAS.
- E. Yilmaz, C. Yogi, K. Yamanaka, T. Ohta and H. R. Byon, Nano Lett., 2013, 13, 4679–4684 CrossRef CAS PubMed.
- Z. Jian, P. Liu, F. Li, P. He, X. Guo, M. Chen and H. Zhou, Angew. Chem., Int. Ed., 2014, 53, 442–446 CrossRef CAS PubMed.
- X. Guo, P. Liu, J. Han, Y. Ito, A. Hirata, T. Fujita and M. Chen, Adv. Mater., 2015, 27, 6137–6143 CrossRef CAS PubMed.
- K. R. Yoon, G. Y. Lee, J.-W. Jung, N.-H. Kim, S. O. Kim and I.-D. Kim, Nano Lett., 2016, 16, 2076–2083 CrossRef CAS PubMed.
- X. Chuan, C. Wei, W. Xianbin, M. N. Hedhili, W. Nini and H. N. Alshareef, Adv. Energy Mater., 2015, 5, 1401805 CrossRef.
- H. Ma, D. Kong, Y. Xu, X. Xie, Y. Tao, Z. Xiao, W. Lv, H. D. Jang, J. Huang and Q.-H. Yang, Small, 2017, 13, 1701026 CrossRef PubMed.
- F. Li, D.-M. Tang, T. Zhang, K. Liao, P. He, D. Golberg, A. Yamada and H. Zhou, Adv. Energy Mater., 2015, 5, 1500294 CrossRef.
- X. Paquez, G. Amiard, G. de Combarieu, C. Boissière and D. Grosso, Chem. Mater., 2015, 27, 2711–2717 CrossRef CAS.
- C.-C. Hu, H.-Y. Guo, K.-H. Chang and C.-C. Huang, Electrochem. Commun., 2009, 11, 1631–1634 CrossRef CAS.
- C.-C. Hu, Y.-L. Yang and T.-C. Lee, Electrochem. Solid-State Lett., 2010, 13, A173–A176 CrossRef CAS.
- A. P. Seitsonen and H. Over, J. Phys. Chem. C, 2010, 114, 22624–22629 CrossRef CAS.
- Y. S. Min, E. J. Bae, K. S. Jeong, Y. J. Cho, J. H. Lee, W. B. Choi and G. S. Park, Adv. Mater., 2003, 15, 1019–1022 CrossRef CAS.
- H. Yu, Y. Wu, F. Peng, Y. Zhang, H. Wang and J. Yang, Catal. Lett., 2012, 142, 100–107 CrossRef CAS.
- D. A. Neamen, Semiconductor Physics And Devices, McGraw-Hill, 2003 Search PubMed.
- J.-M. Li, K.-H. Chang and C.-C. Hu, Electrochim. Acta, 2010, 55, 8600–8605 CrossRef CAS.
- J.-M. Li, K.-H. Chang and C.-C. Hu, Electrochem. Commun., 2010, 12, 1800–1803 CrossRef CAS.
- G. Wang, H. Wang, Y. Ling, Y. Tang, X. Yang, R. C. Fitzmorris, C. Wang, J. Z. Zhang and Y. Li, Nano Lett., 2011, 11, 3026–3033 CrossRef CAS PubMed.
- Y.-C. Pu, G. Wang, K.-D. Chang, Y. Ling, Y.-K. Lin, B. C. Fitzmorris, C.-M. Liu, X. Lu, Y. Tong, J. Z. Zhang, Y.-J. Hsu and Y. Li, Nano Lett., 2013, 13, 3817–3823 CrossRef CAS PubMed.
- R. Zhou, Y. Zheng, M. Jaroniec and S.-Z. Qiao, ACS Catal., 2016, 6, 4720–4728 CrossRef CAS.
- Y. Sugawara, A. P. Yadav, A. Nishikata and T. Tsuru, J. Electrochem. Soc., 2008, 155, B897–B902 CrossRef CAS.
- F. Schlottig, J. Schreckenbach and G. Marx, Fresenius. J. Anal. Chem., 1999, 363, 209–211 CrossRef CAS.
- J. W. Long, R. M. Stroud, K. E. Swider-Lyons and D. R. Rolison, J. Phys. Chem. B, 2000, 104, 9772–9776 CrossRef CAS.
- A. Yamada and J. B. Goodenough, J. Electrochem. Soc., 1998, 145, 737–743 CrossRef CAS.
- J.-M. Zen, A. S. Kumar and J.-C. Chen, J. Mol. Catal. A: Chem., 2001, 165, 177–188 CrossRef CAS.
- R. Srivastava and P. Strasser, ECS Trans., 2009, 25, 565–571 CAS.
- H. Suzuki, S. Nitta, O. Tomita, M. Higashi and R. Abe, ACS Catal., 2017, 7, 4336–4343 CrossRef CAS.
- N. Watanabe, T. Arakawa, Y. Sasaki, T. Yamashita and I. Koiwa, J. Electrochem. Soc., 2012, 159, A1949–A1953 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The concentration of proton at the surface of VOx· mH2O versus time, XPS spectra of VOx, precipitate from solution contained VOSO4 and NiCl2, and NiCl2. See DOI: 10.1039/c8ra07810f |
| This journal is © The Royal Society of Chemistry 2019 |