
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePharmacokinetic profile and metabolite identification of bornyl caffeate and caffeic acid in rats by high performance liquid chromatography coupled with mass spectrometry†
Baimei Shi,
Lingjian Yang,
Tian Gao,
Cuicui Ma,
Qiannan Li,
Yefei Nan,
Shixiang Wang,
Chaoni Xiao,
Pu Jia* and
Xiaohui Zheng *
*
Key Laboratory of Resource Biology and Biotechnology in Western China, Ministry of Education/College of Life Science, Northwest University, 195# Mail Box, No. 229 Northern Taibai Road, Xi'an 710069, P. R. China. E-mail: Jiapu77@126.com; zhengxh@nwu.edu.cn; Fax: +86-29-88302686; Tel: +86-29-88302686
First published on 30th January 2019
Abstract
Bornyl caffeate was initially discovered as a bioactive compound in medicinal plants. Despite the promising pharmacological activities including anti-tumor and antibacterial activities, the pharmacokinetics of the compound remain open. This work developed a high performance liquid chromatography-tandem mass spectrometric method for the determination of bornyl caffeate and caffeic acid (major metabolite and a main unit of bornyl caffeate) in vivo. Successful application of the method included identification of its metabolites and investigation on the drug pharmacokinetics. A total of 30 compounds were identified as the metabolites of bornyl caffeate in rats. We attributed these metabolites to phase I metabolic routes of reduction, oxidation, hydrolysis and phase II metabolic reactions of glucuronidation, sulfation, O-methylation and glycine. Glucuronidation, sulfation, O-methylation and reduction were the main metabolic pathways of bornyl caffeate. The method presented a linear range of 1–4000 ng mL−1. The pharmacokinetic profile of bornyl caffeate was found to be a three compartment open model, while caffeic acid fitted to a two compartment open model when it was administered alone or served as the main metabolite of bornyl caffeate. The time to peak concentration (Tmax) and the maximum plasma concentration (Cmax) of bornyl caffeate were 0.53 h and 409.33 ng mL−1. Compared with original caffeic acid, the compound displayed an increased half-life of elimination (T1/2β), area under the concentration time curve from 0 to t (AUC0–t) and area under the concentration time curve from 0 to ∞ (AUC0–∞), a decreased half-life of absorption (T1/2α) and an identical Cmax. Taking together, we concluded that bornyl caffeate is able to rapidly initiate therapeutic effect and last for a relatively long time in rats; metabolic pathways of O-methylation and reduction is key to interpret the mechanism and toxicity of bornyl caffeate.
1. Introduction
Caffeic acid esters present a large group of bioactive compounds existed in medicinal plants and animals.1–3 Among them, bornyl caffeate (Fig. 1s(a)†) is originally isolated and identified from numerous plants such as Valeriana wallichii,12 Piper caninum (Piperaceae), Piper philippinum, Coreopsis mutica var. mutica and Verbesina turbacenina Kunth.4 The compound has exhibited a broad spectrum of activities including anti-inflammatory,5 antibacterial,6 anti-platelet7 and anti-tumor activity.4 More interestingly, bornyl caffeate possesses a good inhibitory activity towards human neutrophil elastase (HNE),8,9 HIV integrase,10 trypanosome cysteine protease11 and anti-leishmaniasis activity.12 These reports pave the way to further development of the compound to a new drug. This necessitates pharmacokinetic investigation of bornyl caffeate to assess the druggability of the compound at an early stage.In terms of pharmacokinetics and stability, a survey of literature has showed that there are a number of methods for the determination of caffeic acid esters.13–15 Among these assays, high performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS) is the most powerful and canonical technique to determine several caffeic acid esters such as 3,4-dihydroxyphenethyl caffeate,16 phenethyl caffeate17 and their metabolites in plasma. In most cases, these studies have focused on the stability of the compounds, but much less attention was paid to their pharmacokinetics. The main conclusion of the abovementioned reports is the hydrolyzation of caffeic acid esters to caffeic acid in vivo. Such conclusion has referenced us to take caffeic acid (CA, Fig. 1s(b)†) into account during the pharmacokinetic investigation of bornyl caffeate because it is a dominant metabolite of caffeic acid esters.
Pharmacokinetic study is the key to evaluate the bioactivities and monitor the dynamic processes of a drug in vivo.18–20 Several methodologies have applied in this case, including HPLC-MS, HPLC-UV and HPLC-DAD. As the most widespread assay, HPLC-MS/MS is advantageous due to high sensitivity, good selectivity and impressive accuracy.21,22 To our knowledge scope, few literatures have concerned about the establishment of such method to simultaneous investigate the pharmacokinetics of bornyl caffeate and CA, in particular, to identify theirs metabolite. To obtain better pharmacokinetic and metabolic information of bornyl caffeate and CA, we firstly identified 30 metabolites of the compound by HPLC-Q-TOF MS/MS. We further validated an HPLC-Ion-Trap-MS/MS method to determine the concentration of bornyl caffeate and CA in rats. By these results, we declared that bornyl caffeate has potential to become a drug candidate.
2. Experimental
2.1 Chemicals and reagents
Bornyl caffeate was home synthesized and identified by infrared spectrum, mass spectrometry and nuclear magnetic resonance spectroscopy. The purity was determined to be 99.0% by HPLC. Caffeic acid (purity > 99%, Fig. 1s(b)†) and internal standard (IS) phenethyl caffeate (purity > 98%, Fig. 1s(c)†) were purchased from Sigma-Aldrich Corp. (St. Louis, MO, USA). HPLC grade methanol was purchased from Fisher Scientific (Pittsburgh, PA, USA). HPLC grade formic acid was acquired from Kermel Chemical Reagent Co., Ltd. (Tianjin, China). Deionized water (>18 mΩ) was prepared by a Milli-Q water purification system (Bedford, MA, USA). Other reagents were of analytical grade unless specially stated.2.2 Animals
Sprague-Dawley rats (200–220 g) were acquired from the Experimental Animal Center of Xi'an Jiaotong University (Xi'an, China). The rats were raised in an air-conditioned breeding room (temperature: 20–23 °C; humidity: 60 ± 10%; 12 h light/dark cycle). In prior to further experiment, the animals were fed with standard laboratory food and acclimatized for a week. The rats were fasted for 12 h with free access to water before conducting the experiment. All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Northwest University and approved by the Animal Ethics Committee of Northwest University.2.3 Preparation of stock solutions, calibration standards and quality control samples
Primary stock solutions of bornyl caffeate, CA and the IS were prepared at a concentration of 1.6 mg mL−1 by dissolving the accurately weighed compounds in methanol. A series of standard working solutions of bornyl caffeate and CA were achieved at 0.01, 0.04, 0.20, 0.80, 2.00, 4.00, 8.00, 20.00, 40.00 μg mL−1 by diluting the stock solution with appropriate volume of methanol. The working solution of IS was prepared at 160 ng mL−1 by similar method.Reference standard solutions for calibration curves were prepared by spiking 10 μL of the appropriate working solution with 100 μL blank rat plasma. The final concentrations were 1.0, 4.0, 20.0, 80.0, 200.0, 400.0, 800.0, 2000.0, 4000.0 ng mL−1. Using the same method, we prepared quality control solutions at high, medium, low concentration (4, 200 and 3000 ng mL−1). All samples were stored at 4 °C and were kept away from light until further analysis.
2.4 Sample pretreatment
Frozen plasma samples were thawed to room temperature without any other treatments. An aliquot of 100 μL plasma samples were spiked with 10 μL aliquot of IS working solution. Following the inclusion of 300 μL methanol, we vortexed the result mixture for 2.0 min to precipitate protein and free the conjugated drugs. The protein was removed by centrifuging the suspension for 10 min with a speed of 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm and a temperature of 4 °C. After collection of the supernatant to a 1.5 mL Eppendorf tube, 200 μL methanol was applied to wash the precipitate protein. The supernatant was collected into the same tube and was dried under a stream of N2 at room temperature. The residue was dissolved by 100 μL 50% (v/v) methanol/water containing 0.1% formic acid. The result solution was filtered with 0.22 μm polytetrafluoroethylene membrane before HPLC-MS/MS analysis.
000 rpm and a temperature of 4 °C. After collection of the supernatant to a 1.5 mL Eppendorf tube, 200 μL methanol was applied to wash the precipitate protein. The supernatant was collected into the same tube and was dried under a stream of N2 at room temperature. The residue was dissolved by 100 μL 50% (v/v) methanol/water containing 0.1% formic acid. The result solution was filtered with 0.22 μm polytetrafluoroethylene membrane before HPLC-MS/MS analysis.
An aliquot of 600 μL urine was extracted by 1.8 mL methanol using the same method. Feces samples were dried naturally and then ground into crude powder 0.5 g of the powder was extracted with 4 mL methanol in an ultrasonic bath for 40 min, and the extracted solution was treated using the same method.
2.5 HPLC-MS/MS conditions
:1 before the small fraction was introduced into an Agilent Jet Stream dual spray electrospray ionization (ESI) interface. A reference standard solution of purine, hexakis(1H,1H,3H-tetrafluoropropoxy)phosphazine and ammonium trifluoroacetate was utilized to control the performance of ESI source by continuously introducing the solution into the interface. Under negative ion mode, the two solutions produced ions of m/z 112.9855 and 1033.9881. Such ion pattern was utilized for real-time internal mass calibration for pursuing the accuracy mass of the analyte ions. The MS/MS experiments were performed in target MS/MS mode with collision energies of 10, 20, 30, 40 eV for all the compounds. The MS data were acquisitioned at a rate of 1 spectrum per s in the range of m/z 50–500.Identification of the original compound and the metabolites in the real samples was achieved by comparisons of the experimental mass and MS/MS spectra with accurate mass data and spectra in the online opened databases including the massbank (http://www.massbank.jp/index.html), the human metabolite database (http://hmdb.ca) and the METLIN database (http://metlin.scripps.edu). The comparison of accurate mass, retention time and MS/MS spectra with authentic reference standards or the data from the literature was also performed for the identification of several metabolites.
The MS/MS data were collected using a rate of 2 spectra per s over a range of m/z 20 to m/z 500 and a medium isolation width (∼4 Da). As showed in Fig. 1, quantitation were performed using ion patterns of m/z 315.2 [M − H]− to m/z 161.0 [M − H–C10H18O]− for bornyl caffeate, m/z 179.0 [M − H]− to m/z 135.0 [M − H–CO2]− for CA and m/z 283.1 [M − H]− to m/z 135.0 [M − H–C9H9O2]− for IS.
 | ||
| Fig. 1 The MS2 spectra of bornyl caffeate (a), caffeic acid (b) and phenethyl caffeate ((c) IS). | ||
2.6 Method validation
Validation of the proposed method was examined in terms of selectivity, linearity, precision and accuracy, matrix and extraction recovery and stability according to FDA requirements of bioanalytical method validation. Selectivity was evaluated by comparing chromatograms of six different batches of blank plasma obtained from six rats with those corresponding to standard plasma samples spiked with bornyl caffeate, CA and IS.Calibration curves were plotted by the peak area ratios (y) of bornyl caffeate, CA to IS against the nominal concentration (x) of bornyl caffeate and CA. Linear regression using 1/x2 as weighting factor was utilized to assess the calibration curves. The lower limit of quantification (LLOQ) was defined as the lowest drug concentration that is determined with both accuracy (relative error, RE) and precision (relative standard deviation, RSD) within ±20%.
Precision and accuracy of the method was evaluated in six replicates at three QC levels on the same day and three analytical batches on three consecutive days. Intra- and inter-day precisions (relative standard deviation, RSD) were required to be below 15%. The accuracy (relative error, RE) of such cases was within ±15%.
The extraction recoveries of the analytes were determined at three QC levels by comparing the mean peak area of QC samples with those of the blank plasma spiked with neat solutions after extraction (n = 6). The recovery of IS was determined in a similar way at 160 ng mL−1. The matrix effect was measured by referring the post-extracted spiked sample to the unextracted sample, using the following equation:
| % matrix effect = (A − B)/B × 100 |
Stability of bornyl caffeate and CA in plasma samples were investigated under varieties of storing and processing conditions. In case of freeze–thaw stability, three QCs were stored at −20 °C for 24 h and thawed at room temperature. Subsequent refreezing of the samples was performed under the same conditions. After three freezing–thawing cycles, the concentrations of bornyl caffeate and CA were determined by the proposed HPLC-MS/MS method. Long-term stability was examined using the three QCs after the plasma samples were stored at −20 °C for 3 weeks. Short-term stability was investigated after the three QCs were kept at room temperature for 24 h. In addition, for post-preparative stability, the processed QCs were placed in the autosampler at 20 °C for 24 h. The samples was regarded as stable if the relative error was within ±15%.
2.7 Metabolite identification
In the case of metabolite identification, we randomly divided 18 male Sprague-Dawley rats into three groups (six rats per group) assigned as A for plasma collection, B for urine and feces collection, and C for collection of blank plasma, urine and feces. We suspended bornyl caffeate in 5% poloxamer to prepare 7.5 mg mL−1 of the drug suspension. Such suspension was administered to the rats through intragastric gavage with a dosage of 30 mg kg−1. Same dose of 5% poloxamer was given to the rats assigned as blank group. Following administration, blood samples (0.2 mL) were collected from the ophthalmic veins at 0.5, 1, 2 h and 4 h and placed into heparinized tubes. Urine and feces samples were collected from each rat in group B at 12 h pre-dose and 0–12 h and 12–24 h post-dose using separate metabolic cages.2.8 Pharmacokinetic study
To determine the pharmacokinetic profile of bornyl caffeate, we prepared bornyl caffeate at the concentration of 30 mg kg−1 using 5% poloxamer aqueous solution. According to the equimolar transformation from bornyl caffeate to caffeic acid, we prepared caffeic acid at 17 mg kg−1 using 5% poloxamer aqueous solution. 200 μL blood samples were collected from the ophthalmic veins into heparinized Eppendorf under anesthesia introduced by sevoflurane39 at predefined time points: 0, 0.08, 0.17, 0.25, 0.5, 1, 2, 4, 8, 12 h pre- and post-dosage. The blood samples were centrifuged at 12000 rpm for 10 min at 4 °C to collect plasma samples. The collected plasma was stored at −20 °C until further pretreatment.
2.9 Data analysis
Acquisition of the LC/MS data was achieved by Agilent Mass Hunter Qualitative Analysis software (version 4.0, Agilent Technologies). Using Molecular Feature Extractor (MFE), we mined the rough data to present the compounds in the sample by isotope peaks and adduct ions. The compounds were filtered when their ion intensities are larger than 5000 counts. Following normalization of the abundance, the peaks of diverse samples were aligned by a mass window of 10 ppm and a retention time window of 0.2 min. The differential metabolites exported from Agilent Mass Profiler Professional (MPP) software (version 12.0, Agilent Technologies) were confirmed by the Mass Hunter software with their ion peak area and symmetry for the pursuit of reducing the error caused by statistical analysis.Pharmacokinetic analysis of all data was processed by the DAS 3.0.0 software (DAS, T.C.M. Shanghai, China). The parameters including the area under curve (AUC), the maximum plasma concentration (Cmax) and the time to achieve maximum plasma concentration (Tmax), the half-life of absorption (T1/2α) were calculated to describe the pharmacokinetic properties of bornyl caffeate and CA. All results were expressed as arithmetic mean ± standard deviation (SD).
3. Results
3.1 Method validation
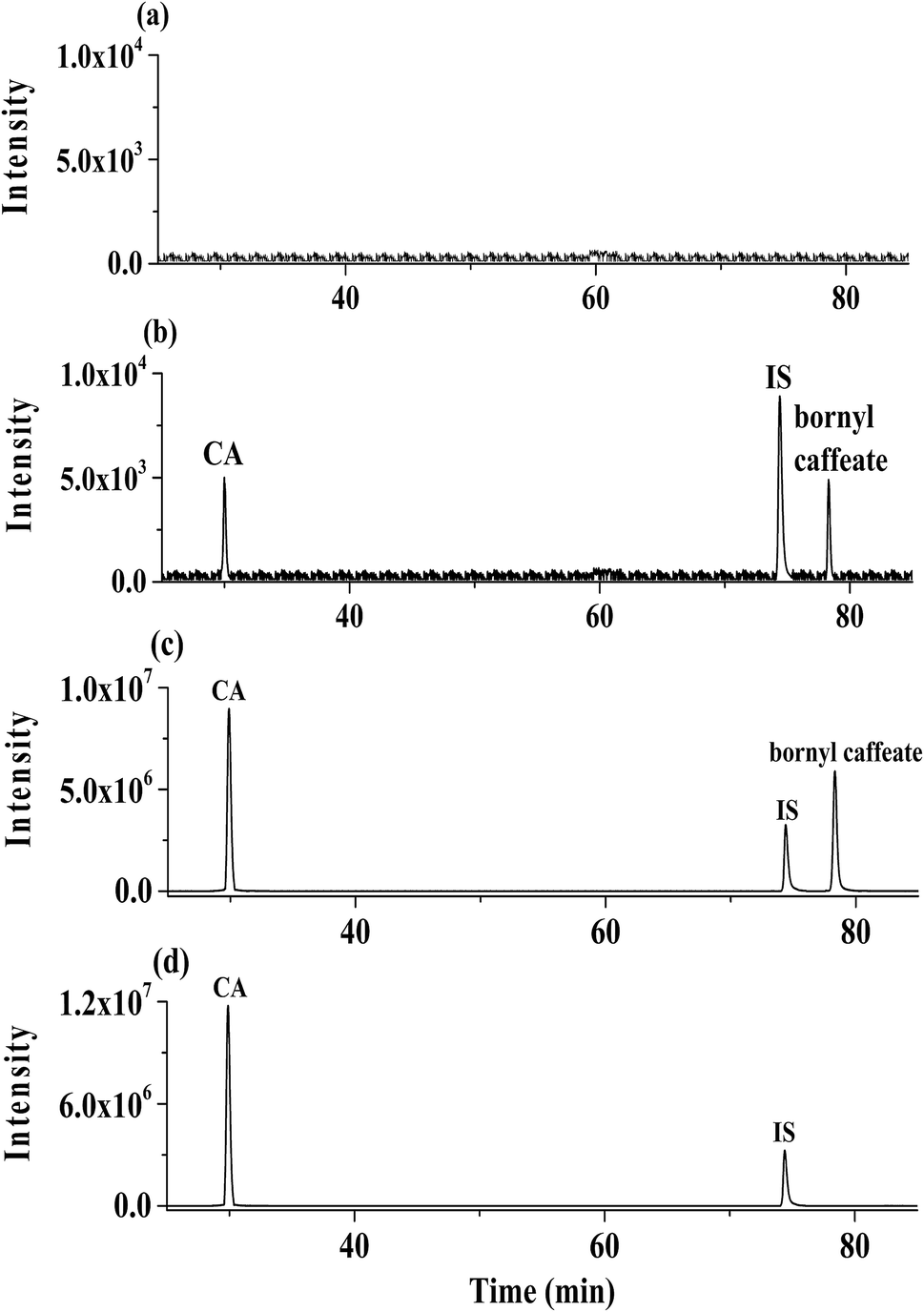 | ||
| Fig. 2 Representative extracted ion chromatograms of rat plasma samples: (a) blank plasma; (b) blank plasma spiked with bornyl caffeate (0.7 ng mL−1) and caffeic acid (0.3 ng mL−1) at LLOQ; (c) plasma collected at 0.53 h after a single oral administration of 30 mg kg−1 bornyl caffeate; (d) plasma collected at 0.25 h after a single oral administration of 17 mg kg−1 caffeic acid. | ||
| Compounds | Concentration added (ng mL−1) | Intra-day | Inter-day | ||||
|---|---|---|---|---|---|---|---|
| Concentration measured (ng mL−1) | (RSD%) | (RE%) | Concentration measured (ng mL−1) | (RSD%) | (RE%) | ||
| Bornyl caffeate | 4 | 3.94 ± 0.13 | 3.23 | 1.54 | 3.9 ± 0.28 | 7.07 | 2.42 |
| 200 | 195.72 ± 7.1 | 3.63 | 2.14 | 196.49 ± 9.93 | 5.05 | 1.75 | |
| 3000 | 2930.49 ± 103.04 | 3.51 | 2.32 | 2928.34 ± 199.48 | 6.81 | 2.39 | |
| CA | 4 | 3.90 ± 0.21 | 5.30 | 2.50 | 3.93 ± 0.20 | 5.20 | 1.71 |
| 200 | 194.84 ± 10.18 | 5.33 | 2.58 | 197.32 ± 7.71 | 3.91 | 1.34 | |
| 3000 | 2910.78 ± 149.34 | 5.13 | 2.97 | 3054.66 ± 119.08 | 3.90 | 1.82 | |
| Compound | Concentration added (ng mL−1) | Extraction recovery% | RSD% | Matrix effect | RSD% |
|---|---|---|---|---|---|
| Bornyl caffeate | 4 | 86.04 ± 3.77 | 4.37 | 10.89 ± 3.22 | 6.08 |
| 200 | 87.34 ± 3.49 | 3.99 | 5.87 ± 1.98 | 2.67 | |
| 3000 | 81.63 ± 3.13 | 3.84 | 7.22 ± 5.43 | 1.59 | |
| Caffeic acid | 4 | 92.25 ± 6.25 | 2.87 | 8.54 ± 2.73 | 2.73 |
| 200 | 95.41 ± 3.57 | 3.75 | 8.38 ± 3.39 | 3.20 | |
| 3000 | 98.83 ± 3.08 | 3.21 | 9.49 ± 3.05 | 2.53 | |
| IS | 160 | 89.62 ± 1.74 | 1.94 | 4.78 ± 2.09 | 1.37 |
| Compounds | Concentration added (ng mL−1) | Freeze–thaw | Short-term | Long-term | Autosampler stability | ||||
|---|---|---|---|---|---|---|---|---|---|
| Concentration measured (ng mL−1) | Stability (%) | Concentration measured (ng mL−1) | Stability (%) | Concentration measured (ng mL−1) | Stability (%) | Concentration measured (ng mL−1) | Stability (%) | ||
| Bornyl caffeate | 4 | 3.77 ± 0.14 | 94.29 | 3.97 ± 0.15 | 99.38 | 4.03 ± 0.18 | 100.79 | 3.97 ± 0.12 | 99.25 |
| 200 | 202.1 ± 6.48 | 101.05 | 196.41 ± 4.57 | 98.2 | 197.22 ± 6.86 | 98.61 | 193.54 ± 9.16 | 96.77 | |
| 3000 | 2835.08 ± 9.86 | 94.23 | 3009.81 ± 6.45 | 100.33 | 2990.99 ± 4.97 | 99.7 | 2966.96 ± 11.78 | 98.89 | |
| Caffeic acid | 4 | 3.77 ± 0.14 | 94.29 | 3.97 ± 0.15 | 99.38 | 4.03 ± 0.18 | 100.79 | 3.93 ± 0.16 | 98.25 |
| 200 | 202.1 ± 6.48 | 101.05 | 196.41 ± 4.57 | 98.2 | 197.22 ± 6.86 | 98.61 | 191.9 ± 6.19 | 95.95 | |
| 3000 | 2835.08 ± 9.86 | 94.23 | 3009.81 ± 6.45 | 100.33 | 2990.99 ± 4.97 | 99.7 | 2991.17 ± 12.48 | 99.7 | |
3.2 Metabolite identification
| Parameters | Bornyl caffeate | CA-metabolite | CA-ig |
|---|---|---|---|
| T1/2α1 (h) | 0.17 ± 0.04 | 0.24 ± 0.03 | 0.40 ± 0.09 |
| T1/2α2 (h) | 0.79 ± 0.13 | — | — |
| T1/2β (h) | 3.37 ± 0.24 | 3.56 ± 0.97 | 1.34 ± 0.22 |
| AUC(0–t) (μg L−1 h) | 1466.67 ± 67.76 | 1177.22 ± 77.37 | 786.84 ± 80.33 |
| AUC(0–∞) (μg L−1 h) | 1491.29 ± 66.42 | 1357.72 ± 76.85 | 820.77 ± 86.23 |
| Tmax (h) | 0.53 ± 0.28 | 0.37 ± 0.02 | 0.25 ± 0.01 |
| T1/2Ka (h) | 0.18 ± 0.09 | 0.14 ± 0.05 | 0.12 ± 0.03 |
| Cmax (ng mL−1) | 409.33 ± 92.94 | 414.42 ± 9.81 | 450.07 ± 13.8 |
| Metabolite | tR (min) | Elemental composition | Assigned identify | Calculated mass m/z | Measured mass m/z | Error (ppm) | Fragment ions | Metabolic pathway | Location |
|---|---|---|---|---|---|---|---|---|---|
| a Analysis was performed on plasma (p), urine (u) and feces (f) samples. Bornyl caffeate, bornyl 3-(3,4-dihydroxyphenyl)-2-acrylic acid. | |||||||||
| M0 | 78.3 | C19H24O4 | Caffeic acid bornyl ester | 315.1602 | 315.1602 | 0 | 179.0355, 161.0253, 134.0379 | Parent | p, u, f |
|
|||||||||
| Phase II metabolites of bornyl caffeate | |||||||||
| M1a | 73.7 | C25H32O10 | Bornyl 3-(3-O-glucuronyl-4-hydroxyphenyl)-2-acrylic acid | 491.1923 | 491.1923 | 1.49 | 315.1569, 179.0315, 175.0238, 161.0239, 134.0348, 113.0348, 85.0284 | Glucuronidation | p, u |
| M1b | 74.7 | C25H32O10 | Bornyl 3-(3-hydroxyphenyl-4-O-glucuronyl)-2-acrylic acid | 491.1923 | 491.1925 | 2.61 | 315.1587, 179.0305, 161.0219, 134.0339, 113.0364, 85.0287 | Glucuronidation | p, u |
| M2a | 73.6 | C26H34O10 | Bornyl 3-(3-O-glucuronyl-4-O-methyl)-2-acrylic acid | 505.2079 | 505.2085 | −2.90 | 329.1806, 315.1627, 175.0258, 113.0264, 85.0313 | O-Methylation + glucuronidation | p, u |
| M2b | 74.7 | C26H34O10 | Bornyl 3-(3-O-methyl-4-O-glucuronyl)-2-acrylic acid | 505.2079 | 505.2086 | 0.69 | 329.1756, 315.1581, 175.0272, 113.0248, 85.0302 | O-Methylation + glucuronidation | p, u |
|
|||||||||
| Degradation product of bornyl caffeate | |||||||||
| M3 | 29.9 | C9H8O4 | Caffeic acid24–29 | 179.0350 | 179.0355 | 0.31 | 135.0461 | Hydrolysis | p, u, f |
|
|||||||||
| Phase I metabolites of caffeic acid | |||||||||
| M4 | 23.0 | C9H10O4 | Dihydrocaffeic acid27,28 | 181.0506 | 181.0508 | −2.51 | 163.0433, 136.9874 | Reduction | p, u, f |
| M5 | 35.5 | C9H10O3 | m-Hydrophenyl propionic acid30,31,33 | 165.0557 | 165.0551 | −2.23 | 147.0428, 121.0647, 106.0408, 77.0407 | Dehydroxylated + reduction | p, u, f |
| M6 | 39.2 | C9H8O3 | m-Coumaric acid32 | 163.0401 | 163.0401 | 119.0502 | Dehydroxylated | p, u, f | |
| M7 | 20.5 | C9H9 N O4 | m-Hydroxy hippuric acid32 | 194.0459 | 194.0458 | 0.56 | 150.0552, 93.0348 | Dehydroxylated + reduction + glycine | u, f |
| M8 | 42.9 | C7H6O2 | Benzoic acid34 | 121.0295 | 121.0292 | −1.96 | 77.0402 | -2C + dehydroxylated | u, f |
| M9a | 33.4 | C10H12O4 | Dihydroferulic acid (DHFA)28 | 195.0663 | 195.0661 | −8.48 | 178.0286, 151.0751, 136.0535, 119.9982 | Reduction | f |
| M9b | 37.6 | C10H12O4 | Dihydroisoferulic acid (DHiFA)28 | 195.0663 | 195.0663 | −8.13 | 178.0279, 151.0763, 136.0527, 119.9979 | Reduction | u, f |
| M10a | 28.2 | C10H12O7 S | Dihydroferulic-3-O-sulfate28 | 275.0230 | 275.0230 | −9.16 | 195.0668, 177.0563, 151.0771, 136.0535, 123.0456, 79.9588 | Reduction + sulfation | u, f |
| M10b | 29.9 | C10H12O7 S | Dihydroferulic-4-O-sulfate28 | 275.0231 | 275.0235 | 1.94 | 195.0670, 177.0571, 151.0785, 136.0547, 123.0462, 79.9590 | Reduction + sulfation | u, f |
| M11a | 24.5 | C16H20O10 | Dihydroferulic-3-O-glucuronide28 | 371.0984 | 371.0986 | 0.6 | 195.0662, 177.0554, 175.0251, 133.0661, 113.0256, 85.0308 | Reduction + glucuronidation | u |
| M11b | 27.4 | C16H20O10 | Dihydroferulic-4-O-glucuronide28 | 371.0984 | 371.0987 | 1.48 | 195.0709, 177.0588, 175.0274, 133.0682, 113.0268, 85.0325 | Reduction + glucuronidation | u |
| M12 | 22.2 | C8H8O4 | Vanillic acid34–36 | 167.0350 | 167.0352 | −6.12 | 152.0144, 123.0455, 108.0257, 77.0411 | -2C | u, f |
| M13 | 23.4 | C10H11NO5 | Vaniloyglycine34,37 | 224.0564 | 224.0558 | −9.84 | 180.0643, 165.0409, 123.0433, 100.0040, 74.0230 | -2C + glycine | u |
|
|||||||||
| Phase II metabolites of caffeic acid | |||||||||
| M14a | 31.71 | C10H10O4 | Ferulic acid (FA)25,26 | 193.0506 | 193.0501 | −1.71 | 178.0258, 149.0602, 134.0368 | O-Methylation | p, u |
| M14b | 42.5 | C10H10O4 | Isoferulic acid (IFA)25,26 | 193.0506 | 193.0505 | −0.67 | 178.0262, 149.0597, 134.0365 | O-Methylation | p, u, f |
| M15a | 26.8 | C9H8O7S | Caffeic acid 3-sulfate28 | 258.9918 | 258.9919 | −0.57 | 179.0345, 135.0443, 96.9598 | Sulfation | p, u, f |
| M15b | 29.9 | C9H8O7S | Caffeic acid 4-sulfate28 | 258.9918 | 258.9914 | 1.48 | 179.0341, 135.0446, 96.9584 | Sulfation | p, u, f |
| M16a | 22.8 | C15H16O10 | Caffeic acid 3-O-glucuronide28 | 355.0671 | 355.0665 | 0.56 | 311.0758, 179.0344, 175.0248, 135.0455, 113.0250, 85.0309 | Glucuronidation | p, u |
| M16b | 25.6 | C15H16O10 | Caffeic acid 4-O-glucuronide28 | 355.0671 | 355.0680 | −1.64 | 311.0799, 179.0368, 175.0264, 135.0441, 113.0258, 85.0290 | Glucuronidation | p, u |
| M17a | 33.2 | C16H18O10 | 3-(3-Methoxy-4-O-glucuronyl-phenyl)crylic acid24,29 | 369.0827 | 369.0825 | −0.61 | 193.0474, 178.0241, 149.0617, 134.0399, 113.0227, 85.0283 | O-Methylation + glucuronidation | p, u, f |
| M17b | 35.2 | C16H18O10 | 3-(3-O-Glucuronyl-4-methoxy-phenyl)crylic acid24,29 | 369.0827 | 369.0826 | 0.51 | 193.0490, 178.0267, 175.0233, 149.0678, 134.0367, 113.0267, 85.0295 | O-Methylation + glucuronidation | p, u, f |
| M18a | 31.7 | C10H10O7S | Ferulic-4-O-sulfate28 | 273.0074 | 273.0072 | 0.15 | 193.0501, 178.0266, 149.0603, 134.0371, 96.9602 | O-methylation + sulfation | p, u, f |
| M18b | 34.2 | C10H10O7S | Isoferulic-3-O-sulfate28 | 273.0074 | 273.0075 | 0.91 | 193.0508, 178.0265, 149.0405, 134.0369, 96.9608 | O-Methylation + sulfation | p, u, f |
| M19a | 20.2 | C12H13NO5 | Feruloylglycine34,38 | 250.0721 | 250.0721 | −10.88 | 206.0827, 191.0542, 177.0510, 163.0657, 149.0588, 134.0348, 100.0026, 79.9566 | O-Methylation + glycine | u |
| M19b | 28.2 | C12H13NO5 | Isoferuloyglycine34,38 | 250.0721 | 250.0726 | −11.91 | 206.0815, 191.0540, 177.0534, 163.0610, 149.0602, 134.0383, 100.0039, 79.9607 | O-Methylation + glycine | u |
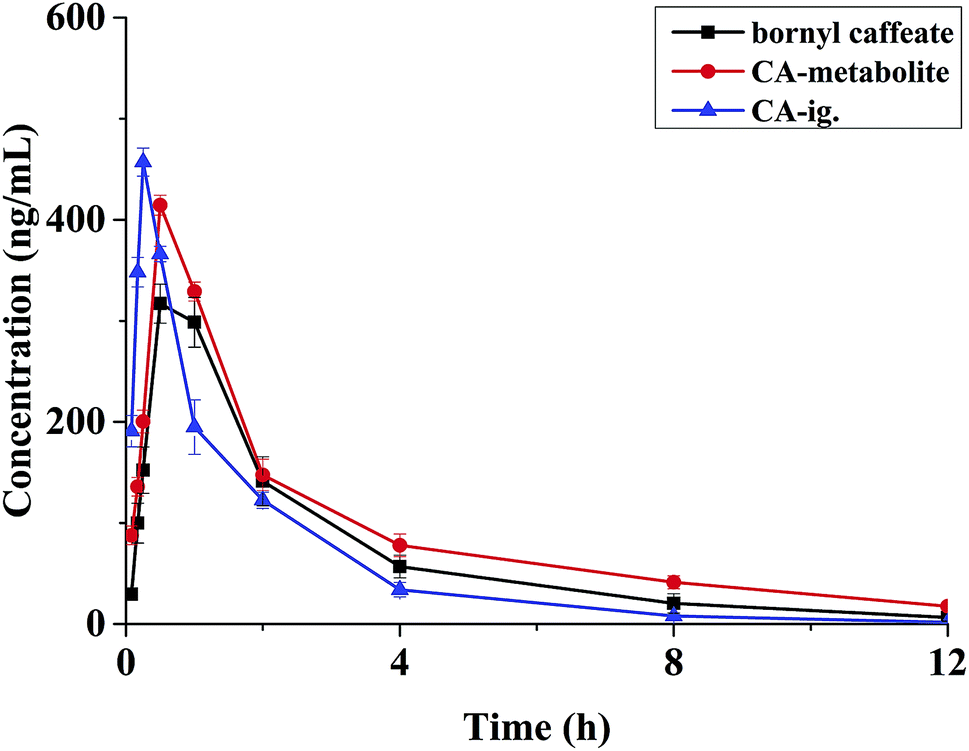 | ||
| Fig. 3 Mean plasma concentration–time profile of bornyl caffeate, CA-metabolite and CA-ig (n = 6). | ||
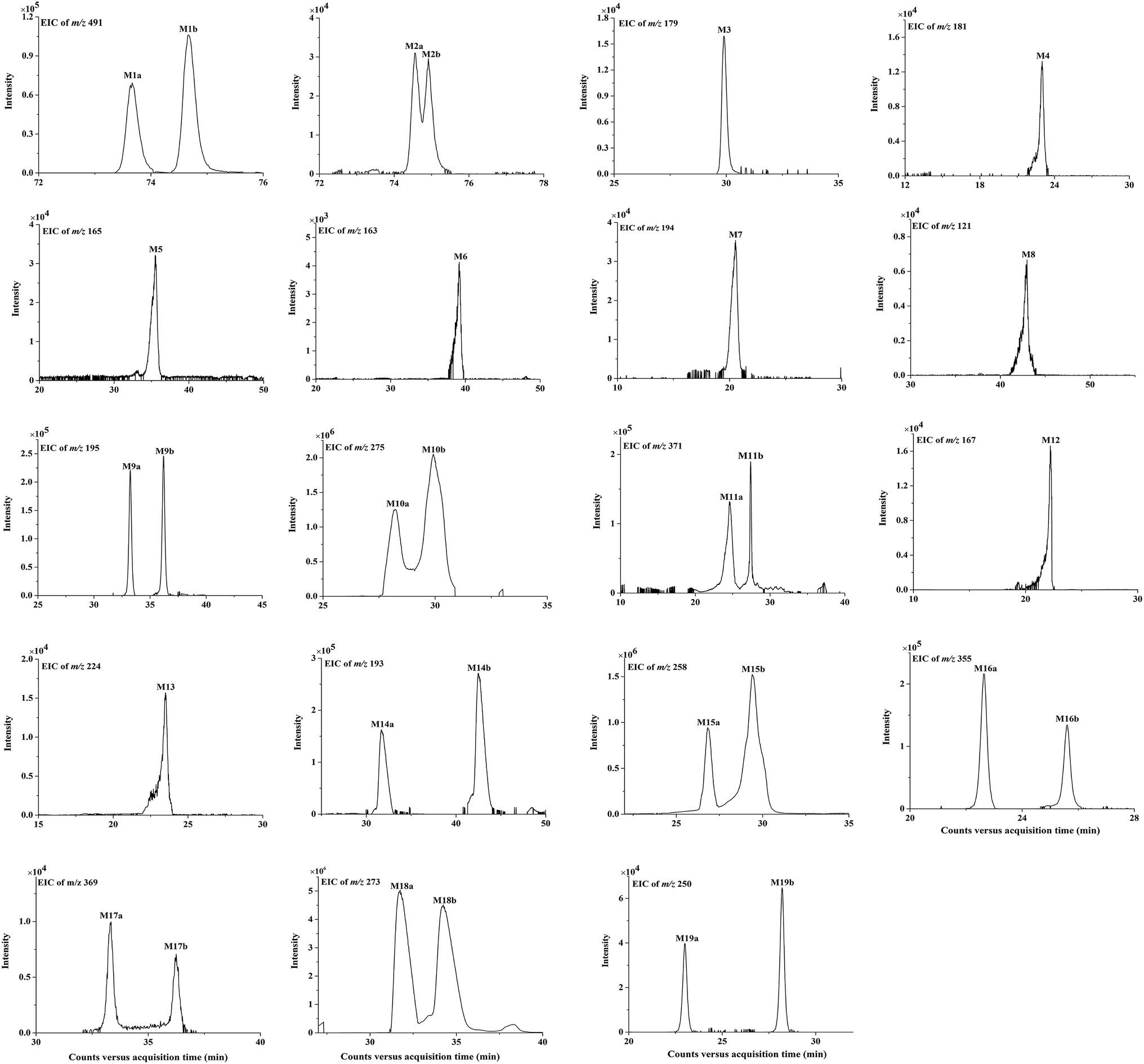 | ||
| Fig. 4 The extracted ion chromatograms (EICs) of in vivo metabolites M1 to M19 of bornyl caffeate in rat. | ||
 | ||
| Fig. 5 Proposed metabolic pathway and metabolites of bornyl caffeate. | ||
3.3 Pharmacokinetic study
A survey of literature has showed that caffeic acid esters can be easily hydrolyzed to CA in vivo.16,17 As a kind of caffeic acid ester, bornyl caffeate is composed of CA and borneol. We simultaneously examined the pharmacokinetics of bornyl caffeate and CA after administration of a single dose of bornyl caffeate. We investigated the pharmacokinetics of CA after single dose administration of the drug to elucidate the transformation from bornyl caffeate to CA in vivo. The mean concentration–time curves were presented in Fig. 3. The calculated pharmacokinetic parameters were listed in Table 4.Bornyl caffeate presented a pharmacokinetic profile of a three-compartment open model. The Cmax of bornyl caffeate was 409.33 ± 92.94 ng mL−1, with a Tmax of 0.53 ± 0.28 h after oral administration. The half-life for distribution (T1/2α1 and T1/2α2)and elimination (T1/2β) were 0.17 ± 0.04, 0.79 ± 0.13 and 3.37 ± 0.24 h, respectively. These results demonstrated that the drug has the properties of fast absorption from gastrointestinal tract into blood circulatory system, and has the capacity to act quickly. The AUC0–∞ and AUC0–t were determined to be 1491.29 ± 66.42 and 1466.67 ± 67.76 μg L−1 h. The ratio of the AUC value was below 120%, providing a proof of rationale time-points of blood collection for the pursuit of pharmacokinetic investigation.
4. Discussion
4.1 Method development
To obtain an efficient and sensitive qualitative analysis method for analyzing bornyl caffeate and caffeic acid, the conditions of mobile phase and MS were optimized. We compared the different kinds of mobile phase, such as formic acid and ammonium formate. When the mobile phase was formic acid, the peak intensities of analytes were 2.3 times more intense than those of the ammonium formate. Therefore, we selected the formic acid as the mobile phase. Subsequently, different percentages of formic acid (0.05, 0.1, 0.2%, v/v) were added to mobile phase to compare the sensitivity of the analytes. When the percentage of formic acid was 0.1% (v/v), the peak intensities of analytes were 1.1 and 1.2 times more intense than those in the mobile phase containing 0.05% and 0.2% formic acid, respectively. Ultimately, a solvent system consisting of 0.1% formic acid in water and methanol was selected for gradient elution. Besides that, we compared different fragmentor voltages (115 V, 125 V, 135 V, 145 V, 155 V) to optimum the sensitivity of the analytes. When the fragmentor voltage was 135 V, the peak area of analytes were 3.8, 3.6, 3.6, and 3.2 times intense than those whose fragmentor voltages were 115 V, 125 V, 145 V, 155 V.4.2 Metabolite identification
Conjugation with glucuronidation, sulfation, O-methylation and glycine were the predominant phase II metabolic reactions. A total of 16 phase II metabolites were identified in plasma, urine and feces after dosing bornyl caffeate. Besides of M1 and M2, the other phase II metabolites were formed from M3. The phase II metabolic pathways of M3 included O-methylation (M14a, M14b), sulfation (M15a, M15b), glucuronidation (M16a, M16b), O-methylation + glucuronidation (M17a, M17b), O-methylation + sulfation (M18a, M18b) and O-methylation + glycine (M19a, M19b). Although the metabolites conjugation reaction with glycine was not demonstrable in plasma, these metabolites served as major components in urine. This result agreed well with previous research.38 The other phase II metabolites produced by conjugation reactions with glucuronidation, sulfation and O-methylation were detectable in plasma. Phase I and phase II biotransformation of bornyl caffeate mainly involved in reduction, oxidation, hydrolysis, glucuronidation, sulfation, O-methylation and glycine. In these reactions, the polarity of the metabolites proved to be enhanced, which was promising for their elimination from the body.A total of 30 metabolites were identified in this research. Among them, 17 were detected in plasma, 29 in urine, 19 in feces. The urine presented 10 metabolites more than that in feces. Among them, 4 metabolites of bornyl caffeate (M1a, M1b, M2a, M2b) and 4 metabolites of caffeic acid (M7, M13, M19a, M19b) were only detectable in urine. Different from the monoglucuronidation product of caffeic acid (M16a, Ma16b) in urine, both glucuronidated and methylated metabolite (M17a, M17b) presented in feces. Taking together, we concluded that the renal route of excretion was the main excretion pathway for bornyl caffeate and its metabolites. The conjugated products of bornyl caffeate with glucuronidation, sulfation and O-methylation were detected in plasma, urine and feces. They were believed to be catalyzed by glucuronosyltransferase, methyltransferase and sulfotransferase. Caffeic acid (CA, M3), hydrolyzed from bornyl caffeate by hydrolytic enzymes, was recognized in plasma, urine and feces. A. N. Booth et al. had demonstrated that some metabolites of caffeic acid conjugated with glycine in vitro, and then he verified that these metabolites can be detected in urine when it was administrated to humans and rats.33 In this research, the glycine of bornyl caffeate (M7, M13, M19a, M19b) were mainly detected in urine. This finding was in accordance with the previous studies.34,37,38 The dehydroxylated metabolites (M5, M6, M7, M8) were catalyzed by dehydroxylated enzymes.41 M4 and M9a, M9b may be produced by CA and M14a, M14b under the action of hydrogenation enzyme and reductase.42 M8 was detected in urine and feces, which was formed by dehydroxylation step and then beta-oxidation from CA.34 M12 was generated by the beta-oxidation from M14a. It generated M13 by direct conjugation with glycine.34
4.3 Pharmacokinetic study
After administration of bornyl caffeate, CA (the main metabolite) presented a pharmacokinetic profile of a two-compartment open model. This finding was in accordance with the results of CA administrated alone the declaration in previous reports.40 By comparing the mean concentration–time curves of bornyl caffeate and its metabolite (CA), we concluded that bornyl caffeate is easy to form CA through hydrolyzation in vivo. Compared with CA administration alone, the metabolite (CA) exhibited clear changes of pharmacokinetic parameters when equimolar bornyl caffeate was administrated. The parameters of T1/2β, AUC0–∞, AUC0–t and Tmax increased significantly, while T1/2α decreased obviously. The elimination of T1/2α and the growth of T1/2β indicated that bornyl caffeate enable sits main metabolite (CA) to act quickly and last longer in vivo. The increase of AUC values and the identical Cmax suggested that bornyl caffeate is capable of improving CA absorption but without increase the toxic dose. Taking together, we concluded that bornyl caffeate has potential to become a lead compound.5. Conclusion
In this work, a sensitive, selective and reliable HPLC-Ion-Trap-MS/MS method was developed and validated for the quantitation of bornyl caffeate and CA in rats. Successful application of the method was confirmed by pharmacokinetic study of these substances. Bornyl caffeate proved to act quickly and last a long therapeutic effects. The metabolites of bornyl caffeate were tentatively identified in rat plasma, urine and feces samples by HPLC-Q-TOF/MS. To our knowledge, this is the first study to establish the metabolism profile of the substance by HPLC-Q-TOF/MS. These results are helpful and lay a solid foundation for investigating the metabolism and the pharmacological effect of bornyl caffeate. The biotransformation of the substance will provide scientific basis for elucidating the pharmacological activity and toxicity of bornyl caffeate in vivo.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the grants from the National Natural Science Foundation of China [No. 81603259], the Project of Key Research and Development Plan of Shaanxi [No. 2017ZDCXL-SF-01-02-01, 2018SF-293], program for Changjiang Scholars and Innovative Research in University [IRT_15R55], the Education Department of Shaanxi Province [No. 09JS086].References
- C. Y. Rao, D. Desai, B. Simi, N. Kulkarni, S. Aniin and B. S. Redd, Inhibitory effect of caffeic acid esters on azoxymethane-induced biochemical changes and aberrant crypt foci formation in rat colon, Cancer Res., 1993, 53, 4182–4188 CAS.
- J. Wu, C. Omene, J. Karkoszka, M. Bosland, J. Eckard, C. B. Klein and K. Frenkel, Caffeic acid phenethyl ester (CAPE), derived from a honeybee product propolis, exhibits a diversity of anti-tumor effects in pre-clinical models of human breast cancer, Cancer Lett., 2011, 308, 43–53 CrossRef CAS PubMed.
- Z. Orban, N. Mitsiades, T. R. Burke Jr, M. Tsokos and G. P. Chrousos, Caffeic acid phenethyl ester induces leukocyte apoptosis, modulates nuclear factor-kappa B and suppresses acute inflammation, NeuroImmunoModulation, 2000, 7, 99–105 CrossRef CAS PubMed.
- C. B. Yang, W. J. Pei, J. Zhao, Y. Y. Cheng, X. H. Zheng and J. H. Rong, Bornyl caffeate induces apoptosis in human breast cancer MCF-7 cells via the ROS- and JNK-mediated pathways, Acta Pharmacol. Sin., 2013, 1–11 Search PubMed.
- E. Maldonado, M. T. R. Apan and A. L. Pérez-Castorena, Anti-inflammatory activity of phenyl propanoids from Coreopsis mutica var. mutica, Planta Med., 1998, 64, 660–661 CrossRef CAS PubMed.
- W. N. Setzer, M. C. Setzer, R. B. Bates, P. Nakkiew, B. R. Jackes, L. Q. Chen, M. B. McFerrin and E. J. Meehan, Antibacterial hydroxycinnamic esters from piper caninum from paluma, north queensland, australia. The crystal and molecular structure of (+)-bornyl coumarate, Planta Med., 1999, 64, 747–749 CrossRef PubMed.
- Y. C. Chen, C. H. Liao and I. S. Chen, Lignans, an amide and anti-platelet activities from Piper philippinum, Phytochemistry, 2007, 68, 2101–2111 CrossRef CAS PubMed.
- T. Steinbrecher, D. A. Case and A. Labahn, A multistep approach to structure-based drug design: studying ligand binding at the human neutrophil elastase, J. Med. Chem., 2006, 49, 1837–1844 CrossRef CAS PubMed.
- T. Steinbrecher, A. Hrenn, K. L. Dormann, I. Merfort and A. Labahn, Bornyl (3, 4, 5-trihydroxy)-cinnamate-An optimized human neutrophil elastase inhibitor designed by free energy calculations, Bioorg. Med. Chem., 2008, 16, 2385–2390 CrossRef CAS PubMed.
- C. N. Xia, H. B. Li, F. Liu and W. X. Hu, Synthesis of trans-caffeate analogues and their bioactivities against HIV-1 integrase and cancer cell lines, Bioorg. Med. Chem. Lett., 2008, 18, 6553–6557 CrossRef CAS.
- I. V. Ogungbe, R. A. Crouch, W. A. Haber and W. N. Setzer, Phytochemical investigation of Verbesina turbacensis Kunth: trypanosome cysteine protease inhibition by (-)-bornyl esters, Nat. Prod. Commun., 2010, 5, 1161–1166 CAS.
- J. Glaser, M. Schultheis, S. Hazra, B. Hazra, H. Moll, U. Schurigt and U. Holzgrabe, Anti-leishmanial lead structures from nature: analysis of structure-activity relationships of a compound library derived from caffeic acid bornyl ester, Molecules, 2014, 19, 1394–1410 CrossRef PubMed.
- N. Celli, L. K. Dragani, S. Murzilli, T. Pagliani and A. Poggi, In vitro and in vivo stability of caffeic acid phenethyl ester, a bioactive compound of Propolis, J. Agric. Food Chem., 2007, 55, 3398–3407 CrossRef CAS PubMed.
- X. Y. Wang, J. H. Pang, R. A. Newman, S. M. Kerwin, P. D. Bowman and S. Stavchansky, Quantitative determination of fluorinated caffeic acid phenethyl ester derivative from rat blood plasma by liquid chromatography-electrospray ionization tandem mass spectrometry, J. Chromatogr. B, 2008, 867, 138–143 CrossRef CAS PubMed.
- X. Y. Wang, P. D. Bowman, S. M. Kerwin and S. Stavchansky, Stability of caffeic acid phenethyl ester and its fluorinated derivative in rat plasma, Biomed. Chromatogr., 2007, 21, 343–350 CrossRef CAS PubMed.
- X. Guo, L. Shen, Y. H. Tong, J. Zhang, G. Wu, Q. He, S. Yu, X. W. Ye, L. B. Zou, Z. Z. Zhang and X. Y. Lian, Antitumor activity of caffeic acid 3,4-dihydroxyphenethyl ester and its pharmacokinetic and metabolic properties, Phytomedicine, 2013, 20, 904–912 CrossRef CAS PubMed.
- C. Tang and O. S. Sojinu, Simultaneous determination of caffeic acid phenethyl ester and its metabolite caffeic acid in dog plasma using liquid chromatography tandem mass spectrometry, Talanta, 2012, 94, 232–239 CrossRef CAS PubMed.
- M. Rowland, C. Peck and G. Tucker, Physiologically-based pharmacokinetics in drug development and regulatory science, Annu. Rev. Pharmacol. Toxicol., 2011, 51, 45–73 CrossRef CAS PubMed.
- J. H. Lin and A. Y. H. Lu, Role of pharmacokinetics and metabolism in drug discovery and development, Pharmacol. Rev., 1997, 49, 403–449 CAS.
- S. A. Roberts, Drug metabolism and pharmacokinetics in drug discovery, Curr. Opin. Drug Discovery Dev., 2003, 6, 66–80 CAS.
- D. Patil, M. Gautam, S. Mishra, S. Karupothula, S. Gairola, S. Jadhav, S. Pawar and B. Patwardhan, Determination of with a ferin A and with anolide A in mice plasma using high-performance liquid chromatography-tandem mass spectrometry: application to pharmacokinetics after oral administration of withania somnifera aqueous extract, J. Pharm. Biomed. Anal., 2013, 80, 203–212 CrossRef CAS PubMed.
- D. Du, B. Gao, G. Xin, A. Sunb, B. Z. Huang, R. Z. Zhang, H. Xing, Q. M. Chen, Y. He and W. Huang, Determination of deltonin in rat plasma by using HPLC–MS/MS and the application of this method in pharmacokinetic studies, J. Chromatogr. B, 2013, 931, 1–5 CrossRef CAS PubMed.
- N. Celli, B. Mariani, L. K. Dragani, S. Murzilli, C. Rossi and D. Rotilio, Development and validation of a liquid chromatographic–tandem mass spectrometric method for the determination of caffeic acid phenethyl ester in rat plasma and urine, J. Chromatogr. B, 2004, 810, 129–136 CrossRef CAS.
- Z. C. Zhang, M. Xu, S. F. Sun, X. Qiao, B. R. Wang, J. Han and D. A. Guo, Metabolic analysis of four phenolic acids in rat by liquid chromatography–tandem mass spectrometry, J. Chromatogr. B, 2008, 871, 7–14 CrossRef CAS PubMed.
- M. B. Hossain, D. K. Rai, N. P. Brunton, A. B. M. Diana and C. Barry-Ryan, Characterization of phenolic composition in Lamiaceae spices by LC-ESI-MS/MS, J. Agric. Food Chem., 2010, 58, 10576–10581 CrossRef CAS PubMed.
- F. Pellati, G. Orlandini, D. Pinetti and S. Benvenuti, HPLC-DAD and HPLC-ESI-MS/MS methods for metabolite profiling of propolis extracts, J. Pharm. Biomed. Anal., 2011, 55, 934–948 CrossRef CAS PubMed.
- F. Tomas-Barberan, R. García-Villalba, A. Quartieri, S. Raimondi, A. Amaretti, A. Leonardi and M. Rossi, In vitro transformation of chlorogenic acid by human gut microbiota, Mol. Nutr. Food Res., 2014, 58, 1122–1131 CrossRef CAS PubMed.
- C. Marmet, L. Actis-Goretta, M. Renouf and F. Giuffrida, Quantification of phenolic acids and their methylates, glucuronides, sulfates and lactones metabolites in human plasma by LC–MS/MS after oral ingestion of soluble coffee, J. Pharm. Biomed. Anal., 2014, 88, 617–625 CrossRef CAS PubMed.
- S. Y. Cao, Z. Zhang, Y. Y. Ye, L. J. Chen, Y. Li, X. Y. Yu, Y. Yang, L. Wang, Z. Li and L. Li, Metabolic transformation evidence of caffeic acid derivatives in male rats after the oral administration of functional food by UPLC coupled with a hybrid quadrupole-orbitrap mass spectrometer, RSC Adv., 2015, 5, 16960 RSC.
- M. M. Appeldoorn, J. P. Vincken, A. M. Aura, P. C. H. Hollman and H. Gruppen, Procyanidin dimers are metabolized by human microbiota with 2-(3,4-Dihydroxyphenyl)acetic acid and 5-(3,4-Dihydroxyphenyl)-γ-valerolactone as the major metabolites, J. Agric. Food Chem., 2009, 57, 1084–1092 CrossRef CAS PubMed.
- M. P. Gonthier, C. Remesy, A. Scalbert, V. Cheynier, J. M. Souquet, K. Poutanen and A. M. Aura, Microbial metabolism of caffeic acid and its esters chlorogenic and caftaric acids by human faecal microbiota in vitro, Biomed. Pharmacother., 2006, 60, 536–540 CrossRef CAS PubMed.
- M. P. Gonthier, J. L. Donovan, O. Texier, C. Felgines, C. Remesy and A. Scalbert, Metabolism of dietary of procyanidins in rats, Free Radical Biol. Med., 2003, 35, 837–844 CrossRef CAS PubMed.
- A. N. Booth, O. H. Emerson, F. T. Jones and F. Deeds, Urinary metabolites of caffeic and chlorogenic acids, J. Biol. Chem., 1957, 229, 51–59 CAS.
- J. Pekkinen, N. N. Rosa, O. I. Savolainen, P. H. Keski-Rahkonen, K. Mykkänen Poutanen, V. Micard and K. Hanhineva, Disintegration of wheat aleurone structure has an impact on the bioavailability of phenolic compounds and other phytochemicals as evidenced by altered urinary metabolite profile of diet-induced obese mice, Nutr. Metab., 2014, 11, 1–15 CrossRef PubMed.
- G. Jiří, N. Ondřej and S. Miroslav, Rapid analysis of phenolic acids in beverages by UPLC–MS/MS, Food Chem., 2008, 111, 789–794 CrossRef.
- N. B. Fang, S. G. Yu and R. L. Prior, LC/MS/MS characterization of phenolic constituents in dried plums, J. Agric. Food Chem., 2002, 50, 3579–3585 CrossRef CAS.
- R. Llorach-Asunción, O. Jauregui, M. Urpi-Sarda and C. Andres-Lacueva, Methodological aspects for metabolome visualization and characterization A metabolomic evaluation of the 24 h evolution of human urine after cocoa powder consumption, J. Pharm. Biomed. Anal., 2010, 51, 373–381 CrossRef PubMed.
- A. Stalmach, W. Mullen, D. Barron, K. Uchida, T. Yokota, C. Cavin, H. K. Steiling, G. Williamson and A. Crozier, Metabolite profiling of hydroxycinnamate derivatives in plasma and urine after the ingestion of coffee by humans: identification of biomarkers of coffee consumption, Drug Metab. Dispos., 2009, 37, 1749–1758 CrossRef CAS PubMed.
- O. Dale and B. R. Brown Jr, Clinical pharmacokinetics of the inhalational anaesthetics, Clin. Pharmacokinet., 1987, 3, 145–167 CrossRef PubMed.
- S. J. Wang, J. Zeng, B. K. Yang and Y. M. Zhong, Bioavailability of caffeic acid in rats and its absorption properties in the CaCO-2 cell model, Pharm. Biol., 2014, 9, 1150–1157 CrossRef PubMed.
- D. Liu, X. H. Zheng, Y. T. Tang, J. Zi, Y. F. Nan, S. X. Wang, C. N. Xiao, J. L. Zhu and C. Chen, Metabolism of tanshinolborneol ester in rat and human liver microsomes, Drug Metab. Dispos., 2010b, 38, 1464–1470 Search PubMed.
- M. Y. Moridani, H. Scobie and P. J. O. Brien, Metabolism of caffeic acid by isolated rat hepatocytes and subcellular fractions, Toxicol. Lett., 2002, 133, 141–151 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra07972b |
| This journal is © The Royal Society of Chemistry 2019 |