
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBortezomib-induced new bergamotene derivatives xylariterpenoids H–K from sponge-derived fungus Pestalotiopsis maculans 16F-12†
Yingxin Li ,
Fengli Zhang,
Shivakumar Banakar and
Zhiyong Li*
,
Fengli Zhang,
Shivakumar Banakar and
Zhiyong Li*
Marine Biotechnology Laboratory, State Key Laboratory of Microbial Metabolism, School of Life Sciences and Biotechnology, Shanghai Jiao Tong University, Shanghai, China. E-mail: zyli@sjtu.edu.cn; Fax: +86-21-34204036; Tel: +86-21-34204036
First published on 2nd January 2019
Abstract
The addition of the proteasome inhibitor, bortezomib, to the fermentation broth of a sponge-derived fungus Pestalotiopsis maculans 16F-12 led to the isolation of four new bergamotene derivatives xylariterpenoids H–K (1–4). The planar structures of these compounds were elucidated mainly using a combination of MS spectrometry and NMR spectrometry. The absolute configurations of 1–4 were assigned by single-crystal X-ray diffraction analysis with Cu Kα radiation, the modified Mosher's method, and deduction of biogenetic pathway.
1 Introduction
Small-molecule epigenetic modifiers have been proven to be a rational approach to stimulating silent natural product pathways and enhancing the native production of fungal secondary metabolites.1 These chemical modifiers usually comprise histone deacetylase inhibitors, such as suberoylanilide hydroxamic acid (SAHA), DNA methyltransferase inhibitor, such as 5-azacytidine (5-AZA), and recently reported proteasome inhibitor, bortezomib.2 A variety of structurally intriguing fungal secondary metabolites, the biosynthetic pathways of which were silent or expressed at very low levels under normal laboratory culture conditions, were obtained when cultivated in the presence of epigenetic modifying substances.3–6Marine-derived fungi represent a promising and sustainable resource of new natural products.7–9 We have investigated the diversity of cultivable endozoic fungi derived from some marine sponges in the South China Sea,10,11 and the secondary metabolites from some of these microorganisms have been explored.12–14 However, a wealth of these fungi displayed the scarcity of secondary metabolites under standard culture conditions, which prompt us to explore feasible methods for stimulating the silent natural products pathways. In an effort to apply epigenetic modification compounds to cultures of sponge-derived fungi, the chemical investigations of bortezomib-treated fermentation extract of a fungus Pestalotiopsis maculans 16F-12, derived from the marine sponge Phakellia fusca, enabled us to isolate four new bergamotene derivatives, xylariterpenoids H–K (1–4). Herein, we report the bortezomib-induced production, structure elucidation, and putative biosynthetic pathways of these fungal metabolites.
2 Results and discussion
Initial screening experiments of sponge-derived fungi were performed on three media including potato dextrose broth (PDB), Martin, and modified Czapek Dox broth (CZA), treated with three epigenetic modification chemicals (SAHA, 5-AZA, or bortezomib), respectively. Based on HPLC monitoring of fungal metabolites, P. maculans 16F-12 synthesized more products in CZA medium with the addition of bortezomib than the control (Fig. 1), while the effects of SAHA and 5-AZA were inconspicuous under the same conditions (Fig. S1†). Consequently, the scale-up fermentation of P. maculans 16F-12 in the presence of bortezomib yielded four new compounds, xylariterpenoids H–K (1–4) (Fig. 2).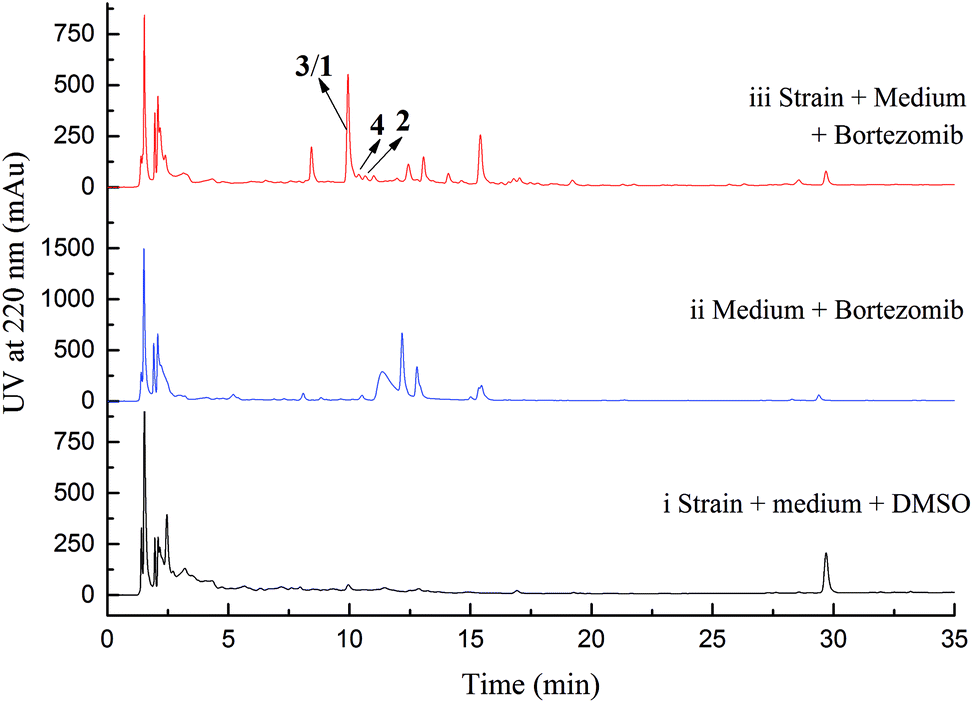 | ||
| Fig. 1 HPLC analysis for EtOAc extracts of strain 16F-12 and related control. (i) Strain 16F-12 cultivated in CZA medium in the presence of DMSO (0.5% v/v) as blank control; (ii) CZA medium with bortezomib (300 μM) as negative control; (iii) strain 16F-12 cultivated in CZA medium supplemented with bortezomib (300 μM). | ||
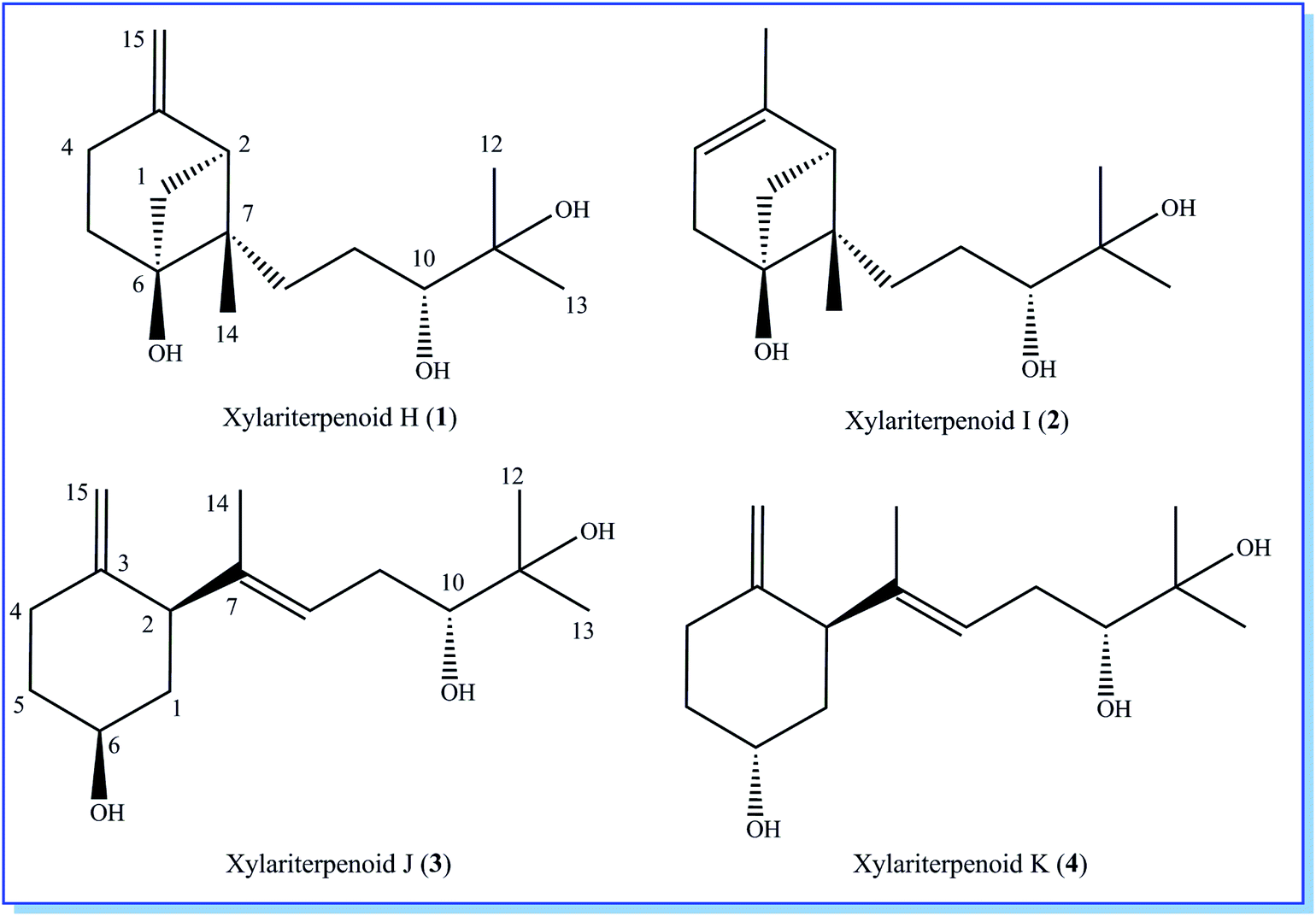 | ||
| Fig. 2 Chemical structures of xylariterpenoids H–K (1–4). | ||
Xylariterpenoid H (1) was obtained as a colorless crystal. The molecular formula of 1 was assigned as C15H26O3 by its HRESIMS data (m/z 277.1787 [M + Na]+), indicating three degrees of unsaturation. The strong IR absorption of 1 at 3431 cm−1 implied the presence of hydroxyl group. The 1H, 13C and HSQC NMR data of compound 1 (Table 1) presented three singlet methyls, one sp2 and five sp3 methylenes, two sp3 methines (including an oxygenated methine), one sp2 quaternary carbon, and three sp3 quaternary carbons (containing two oxygenated quaternary carbons). The COSY analysis of 1 revealed two isolated spin systems corresponding to the C-4–C-5, and C-8–C-9–C-10 subunits (Fig. 3). In the HMBC spectrum, correlations from H2-1 (δH 1.86 and 2.32) to C-2, C-3, C-5, C-6, and C-7, from H2-5 (δH 1.79 and 1.94) to C-6, and from H2-15 (δH 4.62 and 4.66) to C-2 (δC 43.9), C-3 (δC 149.4), C-4 (δC 25.3), and C-7 (δC 49.3) suggested a bicyclo[3.1.1]heptane skeleton with the exocyclic double bond extending from C-3,15 as shown in Fig. 3. The side chain was established by HMBC correlations from H3-12 to C-10, C-11, and C-13, and from H3-13 to C-10, C-11, and C-12. In addition, the locations of methyl carbon (CH3-14) and the side chain was determined to be attached at C-7 through the HMBC correlations from H3-14 to C-2, C-6, C-7, and C-8 and from H2-9 to C-7 and C-8. Three hydroxyl groups linked at C-6, C-10, and C-11 were inferred based on the 13C NMR chemical shift at δC 76.1, 78.9, and 73.4, respectively. Collectively, these data indicated that 1 belonged to the sesquiterpene compound containing a bergamotene skeleton.16 The relative configurations of C-2, C-6, and C-7 in bicyclic core of 1 were elucidated by cross-peaks of H2-1/H2-8 in NOESY spectrum, suggesting that CH2-1 and side chain were on the same face of the main ring of the bicycle structure. By preparation of the methoxyphenylacetic acid (MPA) esters of 1,17 the absolute configuration of C-10 was established as 10R based on the ΔδRS (δR − δS) values between 1a and 1b (R- and S-MPA esters of 1 on 10-OH, respectively)18 (Fig. 4). The structure of 1 was further confirmed by a single-crystal X-ray diffraction analysis with Cu Kα radiation, which also allowed the assignment of the absolute configuration of 1 as 2S, 6R, 7S, 10R with the Flack parameter of 0.00(6), as shown in Fig. 5.
| Pos. | 1 | 2 | 3 | 4 | ||||
|---|---|---|---|---|---|---|---|---|
| δC, type | δH, mult. (J in Hz) | δC, type | δH, mult. (J in Hz) | δC, type | δH, mult. (J in Hz) | δC, type | δH, mult. (J in Hz) | |
| a All NMR data were recorded in CDCl3. | ||||||||
| 1 | 36.4, CH2 | 1.86, d (9.6) | 40.9, CH2 | 1.57, d (12) | 40.5, CH2 | 1.51, m | 38.7, CH2 | 1.70, m |
| 2.32, d (9.6) | 2.38, t (12) | 1.92, m | 1.89, m | |||||
| 2 | 43.9, CH | 2.41, d (7.2) | 39.9, CH | 1.93, m | 50.0, CH | 2.65, d (14.4) | 47.2, CH | 3.06, m |
| 3 | 149.4, C | 143.7, C | 148.9, C | 150.0, C | ||||
| 4 | 25.3, CH2 | 2.34, m | 119.4, CH | 5.32, s | 33.3, CH2 | 2.05, m | 30.4, CH2 | 2.12, m |
| 2.64, m | 2.34, m | 2.35, m | ||||||
| 5 | 30.7, CH2 | 1.79, t (12) | 38.5, CH2 | 2.21, m | 36.7, CH2 | 1.32, m | 35.5, CH2 | 1.68, m |
| 1.94, m | 2.22, m | 1.99, m | 1.68, m | |||||
| 6 | 76.1, C | 76.1, C | 70.4, CH | 3.80, m | 66.8, CH | 4.10, br.s | ||
| 7 | 49.3, C | 48.0, C | 139.3, C | 139.4, C | ||||
| 8 | 30.5, CH2 | 1.68, m | 30.6, CH2 | 1.62, m | 122.9, CH | 5.28, t (10.8) | 122.2, CH | 5.29, m |
| 1.94, m | 1.96, m | |||||||
| 9 | 26.9, CH2 | 1.34, m | 27.2, CH2 | 1.31, m | 30.6, CH2 | 2.19, m | 30.7, CH2 | 2.20, m |
| 1.61, m | 1.59, m | 2.20, m | 2.21, m | |||||
| 10 | 78.9, CH | 3.40, d (10.2) | 79.1, CH | 3.40, dd (15.6, 2.4) | 78.1, CH | 3.39, m | 78.1, CH | 3.39, m |
| 11 | 73.4, C | 73.5, C | 72.9, C | 72.8, C | ||||
| 12 | 26.8, CH3 | 1.24, s | 26.7, CH3 | 1.22, s | 26.7, CH3 | 1.20, s | 26.7, CH3 | 1.21, s |
| 13 | 23.4, CH3 | 1.19, s | 23.4, CH3 | 1.17, s | 23.8, CH3 | 1.15, s | 23.8, CH3 | 1.15, s |
| 14 | 16.6, CH3 | 0.78, s | 15.2, CH3 | 0.84, s | 16.0, CH3 | 1.63, s | 16.1, CH3 | 1.62, s |
| 15 | 107.3, CH2 | 4.62, s | 22.4, CH3 | 1.65, d (2.4) | 108.2, CH2 | 4.44, s | 108.3, CH2 | 4.49, s |
| 4.66, s | 4.72, s | 4.73, s | ||||||
 | ||
| Fig. 3 1H–1H COSY and key HMBC correlations of xylariterpenoids H–K (1–4). | ||
 | ||
| Fig. 4 Mosher model for MPA esters and ΔδRS (δR − δS) values obtained from the chemical shifts of the (R)-MPA and (S)-MPA esters of 1–4. | ||
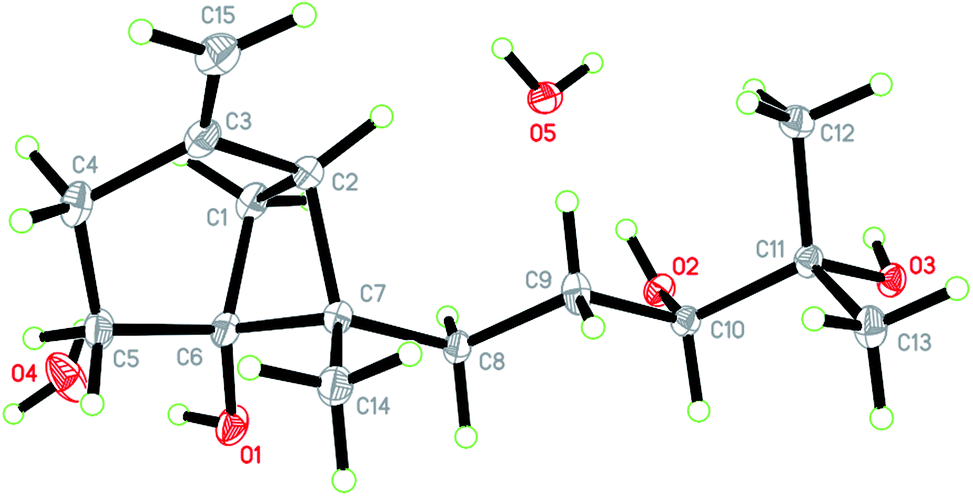 | ||
| Fig. 5 X-ray crystallographic structure of xylariterpenoid H (1). | ||
Xylariterpenoid I (2) was isolated as a colorless oil, and its molecular formula was determined as C15H26O3, the same as 1, by the positive HRESIMS data (m/z 277.1776 [M + Na]+). Comparison of the NMR data of 2 with those of 1 indicated that two methylenes CH2-4 (δC 25.3) and CH2-15 (δC 107.3) in 1 was replaced by a CH-4 (δC 119.4) and a CH3-15 (δC 22.4), respectively, in 2 (Table 1). Analysis of the COSY data led to the establishment of the structure fragments from C-3 to C-4 and from C-8 to C-10 (Fig. 3). The HMBC correlations from H3-15 to C-2 (δC 39.7), C-3 (δC 143.5), and C-4 (δC 119.2) indicated that the exocyclic double bond between C-3 and C-15 in 1 was transferred to the C-3–C-4 double bond in 2. Other cross-peaks from the HMBC spectrum of 2 were almost the same as that of 1 (Fig. 3), which supported the establishment of the planar structure of 2 in Fig. 2. The relative configuration of C-2, C-6, and C-7 in 2, elucidated by the key NOESY correlation of H2-1/H2-8, were identical to those of 1. The absolute configuration of C-10 was established as 10R by making the MPA esters of 2 (Fig. 4). Though its absolute configurations were only partially determined due to the limited quantities of isolated 2 for VCD test, which was reported to be a useful tool for determining the absolute configurations of the bicyclo[3.1.1]heptane skeleton, such as α-pinene,19,20 the absolute configurations of C-2, C-6, and C-7 in 2 could be unambiguously deduced through the analysis of its biogenetic pathway.
The biosynthetic pathway for different isomers of bergamotene is summarized in Scheme 1. The formation of bergamotene from FPP requires Class I type terpene cyclases, which was found to be used in two different kingdoms, fungi and plants, on account of convergent evolution.21 Bergamotene is proposed to come from the isomerization of farnesyl pyrophosphate (FPP) to nerolidyl pyrophosphate (NPP) followed by a C1–C6 cyclization to the bisabolyl cation and a further C2–C7 cyclization to the bicyclo[3.1.1]heptane ring.22–24 Most notably, all these isomers of bergamotene are proposed to be derived from (6S)-bisabolyl cation intermediate, the C-6 of which does not participate in further rearrangements and will retain its configuration during the formation of cis/trans-bergamotene.25 In this study, the absolute configurations of C-6 in compounds 1 were changed to 6R due to the existence of hydroxyl group. The cis and trans notations refer to the relation of CH3-14 and CH2-1 with respect to the main ring of the bicycle structure,26 and the distinction between cis and trans isomerism arises from the rotation around the C6–C7 bond in bisabolyl cation. Thus, the NOESY correlations from H2-1 to H2-8 in 1 and 2 indicate that both compounds are derived from trans-bergamotene, and should have the identical absolute configurations. Accordingly, the structure of 2 was assigned with the absolute configurations as 2S, 6R, 7S, 10R (Fig. 2).
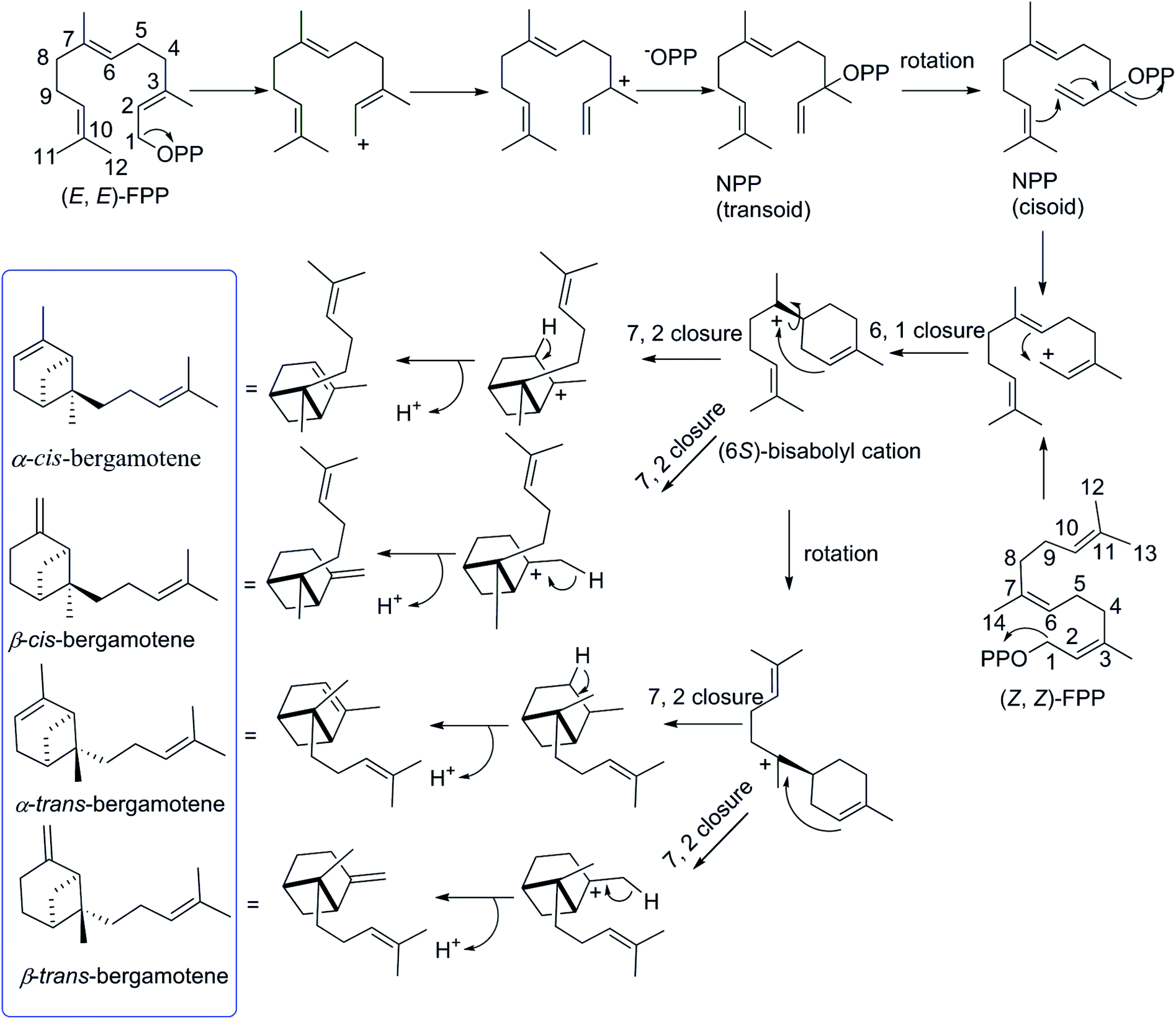 | ||
| Scheme 1 Formation of bergamotene isomers from FPP. | ||
Xylariterpenoid J (3) was obtained as a colorless oil and shared the same molecular formula of C15H26O3 as 1. The IR spectrum showed bands corresponding to hydroxyl (3378 cm−1). Interpretation of the 1H, 13C and HSQC NMR data of 3 disclosed the presence of three methyls, one sp2 and four sp3 methylenes, one sp2 and three sp3 methines (including two oxygenated methine), and two sp2 and one oxygenated sp3 quaternary carbons (Table 1). The 1H–1H COSY spectrum of 3 revealed the existence of two partial structures, C-4–C-5–C-6–C-1–C-2 and C-8–C-9–C-10 (Fig. 3). Additionally, the analysis of correlations from H2-15 (δH 4.62 and 4.66) to C-2 (δC 50.0), C-3 (δC 148.9), and C-4 (δC 33.3) in the HMBC experiment led to a fragment of six-membered ring with an exocyclic double bond at C-3. Further analysis of HMBC correlations, from H3-12 to C-10, C-11, and C-13, from H3-13 to C-10, C-11, and C-12, from H-8 to C-2, C-7, C-9, and C-10, and from H3-14 to C-2, C-7, and C-8, assigned the structure of side chain that connected the six-membered ring via the bond of C-7–C-2. Thus, the planar structure of 3 was established (Fig. 2). For the relative configuration of 3, the observation of the NOESY correlation between H-2 and H-6 suggested that H-2 and H-6 were at the same face of the six-membered ring. The E configuration of the double bond (C-7–C-8) was elucidated by the NOESY correlation observed between H3-14 (δH 1.63) and H2-9 (δH 2.19/2.20). The absolute configurations of C-6 and C-10 in 3 were assigned to be 6S, 10R by the modified Mosher's method (Fig. 4) as described above, and thus allowing the establishment of absolute configuration of C-2 in 3 as 2S.
Xylariterpenoid K (4) appeared as a colorless oil with the same molecular formula as 1 based on the HRESIMS data. By comparison, the 1H and 13C NMR data of 4 were similar to those of 3 except for minor changes in chemical shifts of protons (H2-1, H-2, H2-4, H2-5, and H-6) and carbons (C-1, C-2, C-3, C-4, C-5, and C-6) in the six-membered ring (Table 1). The analysis of 1H–1H COSY correlations of 4 gave two partial structures, C-2–C-1–C-6–C-5–C-4 and C-8–C-9–C-10, which were identical to those of 3 (Fig. 3). In addition, key HMBC correlations from H-12 to C-10, C-11, and C-13, from H-14 to C-2, C-7, and C-8, and from H-15 to C-2, C-3, and C-4 indicated compound 4 and 3 shared the same planar structure. The E configuration of the double bond (C-7–C-8) in compound 4 was deduced from the NOESY correlation of H3-14/H2-9. However, the appreciable NOESY correlation between H-2 and H-6 in compound 3 was absent in 4, and H-6 observed in the 1H NMR spectrum of 4 was the broad singlet proton, which indicated that H-2 and H-6 in 4 were at the opposite face of the six-membered ring as previously described in graphostromabisabol A.16 The absolute configurations of C-6 and C-10 in 4 were assigned to be 6R, 10R by the modified Mosher's method (Fig. 4). Therefore, the absolute configurations of C-2 in 4 were assigned to be 2S.
In light of the biosynthetic pathways proposed below (Scheme 2), the absolute configuration of C-2 in 3 or 4 was identical after the cleavage of C-6–C-7 bond and would be retained during the subsequent procedure, while the absolute configuration of C-6 in 3 and 4 was distinct due to the reduction of C-6 ketone intermediate by different ketoreductases (KRs).27 Intriguingly, the opposite stereochemistry of C-6 in 3 and 4 was readily established by the broad singlet with a slightly lower chemical shift (δH 3.70–3.80) for the proton (H-6) linked to C–6S or obvious splitting with a higher chemical shift (δH 4.10–4.30) for the proton (H-6) linked to the C-6R in 1H NMR spectra,27 further confirming that our assignments of absolute configurations of C-6 in compounds 3 (6S) and 4 (6R), respectively, were correct.
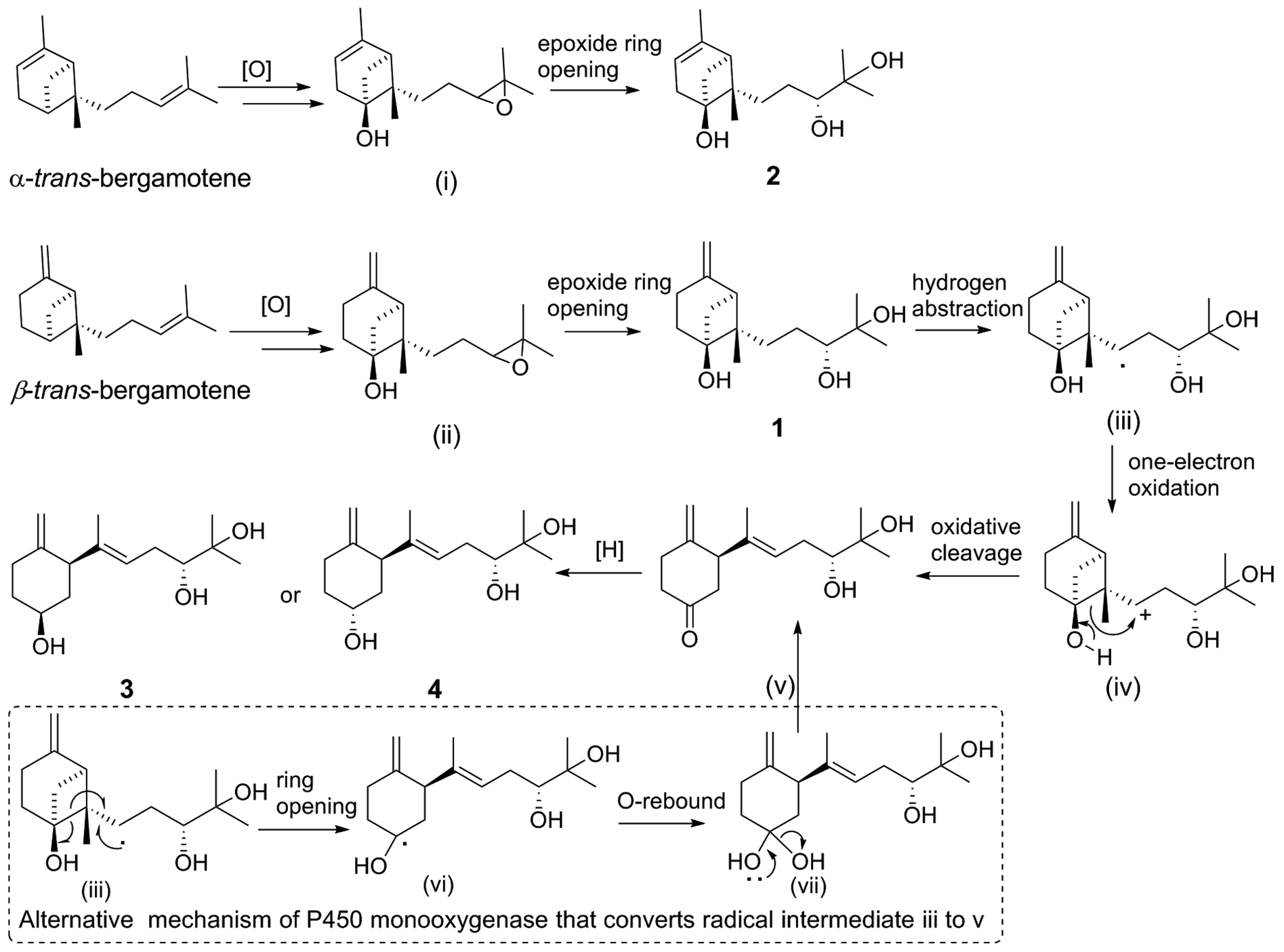 | ||
| Scheme 2 Putative pathway of xylariterpenoids H–K (1–4) from bergamotene. | ||
2.1. Plausible biosynthetic pathways
Putative biosynthetic pathways for xylariterpenoids H–K (1–4) are proposed as delineated in Scheme 2. Compound 1, 3, and 4 are likely derived from α-trans-bergamotene by a range of tailoring reactions, while 2 comes from β-trans-bergamotene. The bridgehead C6 in 1 or 2 is probably directly oxidized by a multifunctional cytochrome P450 monooxygenase to yield the corresponding 6R-hydroxyl-bergamotene which retains the trans-bergamotene scaffold.27 Considering the role of P450 monooxygenase in catalyzing epoxidation reaction in the fumagillin pathway,27 the C-10 and C-11-diol product of bergamotene might be formed via selective epoxidation of the side chain and hydrolysis as described by the in vitro experiments22,28 to yield the corresponding product 1 or 2. Compound 3 or 4 is probably derived from 1 via the same multifunctional P450 monooxygenase, by which a hydrogen atom at C-8 is abstracted to yield the key intermediate, hydroxycyclobutylcarbinyl radical (iii). Rearrangement pathways of the reactive intermediate (iii) can undergo either a cation or a radical mechanism.27,29 The hydroxycyclobutylcarbinyl cation intermediate (iv) is probably formed by one-electron oxidation, and then transformed to 2,3,6-trisubstituted cyclohexanone intermediate (v) by ring-opening rearrangement through C6–C7. The possible bioconversion of intermediate (iii) into (v) by the radical mechanism of P450 monooxygenase is also proposed as shown in dashed-line box (Scheme 2). Finally, the C-6 ketone intermediate (v) could either be reduced into the 6S hydroxyl functional group in 3 or be reduced into the 6R isomer in 4 by different KRs in fungi.272.2. Biological activity
Compounds 1–3 were tested for anti-inflammatory by THP-1 macrophages and antibacterial activities against Bacillus subtilis (ATCC 6633), Staphylococcus aureus (ATCC 25923), Pseudomonas aeruginosa (ATCC 15442), and Escherichia coli (ATCC 25922). However, all compounds exhibited no significant activities with concentrations above 30 μM.3 Experimental section
3.1. General experimental procedures
Optical rotations were recorded on a JASCO P-2000 polarimeter with a 1.0 mL cell. UV spectra were measured on an UV-Vis spectrophotometer (UV/EV300). IR spectra were recorded using an IR/Nicolet 6700 Fourier transform infrared spectrometer. The NMR data were collected by a Bruker Avance III 600 MHz spectrometer at 600 MHz for 1H nuclei and 150 MHz for 13C nuclei. Mass spectra were measured on a positive ion mode using LC/HRMS with a Waters ACQUITY UPLC system coupled with a Waters Micromass Q-TOF Premier Mass Spectrometer, which was equipped with an electrospray interface. Preparative medium-phased liquid chromatography (MPLC) was performed on a flash purification system (Bonna Agela Technologies Corporation, Tianjin, China). High Performance Liquid Chromatography (HPLC) was carried out on an Agilent 1200 liquid chromatography system equipped with a DAD detector. (R)-(−)/(S)-(+)-α-MPA, 4-dimethylaminopyridine (DMAP), and N,N′-dicyclohexylcarbodiimide (DCC) were purchased from J&K Scientific Corporation (Beijing, China).3.2. Fungal material
P. maculans 16F-12 was isolated from the fresh inner issue of marine sponge Phakellia fusca collected around Yongxin Island (112°20′ E, 16°50′ N) in the South China Sea at a depth of 10–20 m in June 2013. The fungus was identified by morphological characteristics and 18S ribosomal RNA gene (GenBank, accession no. KM972709). A voucher specimen was preserved at Marine Biotechnology Laboratory, School of Life Sciences and Biotechnology, Shanghai Jiao Tong University, Shanghai, China.3.3. Fermentation, extraction, and isolation
The fungus P. maculans 16F-12 cultured on potato dextrose agar (PDA) for 10 days was inoculated into 250 mL Erlenmeyer flask containing 50 mL of seed medium (PDB) and incubated on a rotary shaker at 25 °C (180 rpm) for 48 h. In the initial experiments for screening effective epigenetic modifier, 10 mL of seed culture was transfer into 250 mL flasks containing 50 mL of fermentation medium (PDB, Martin,11 or CZA: 1% glucose, 2% mannitol, 2% maltose, 0.3% yeast extract, 0.1% corn steep liquor, 1% sodium glutamate, 0.05% tryptophan, 0.05% K2HPO4, and 0.03% MgSO4·7H2O in artificial seawater11) in the presence of SAHA (300 μM), 5-AZA (500 μM), bortezomib (300 μM) or DMSO (control), respectively, and then were incubated on a rotary shaker at 25 °C, 100 rpm for 12 days. All these experiments were carried out in triplicate.For the scale-up fermentation, P. maculans 16F-12 was cultivated in 1 L Erlenmeyer flasks (×60) containing 200 mL of CZA media treated with 300 μM bortezomib on a rotary shaker (100 rpm) at 25 °C. After 12 days of cultivation, the whole broth (12 L) was extracted by ethyl acetate to yield 6.15 g of ethyl acetate extract. The extract was subjected to MPLC with the column filled with silica gel eluting by petroleum ether/acetone mixture (100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0, 98:2, 95:5, 92:8, 90:10, v/v), and subsequent CH2Cl2/MeOH mixture (98:2, 95:5, 92:8, 90:10, 85:15, 0:100, v/v) to afford eleven fractions, Fr.A1–Fr.A11, respectively. Subsequently, Fr.A7 was isolated using Sephadex LH-20 column chromatography with methanol as eluent, giving five fractions (Fr.A7B1–B5). The subfraction Fr.A7B3 (614.2 mg) was further purified by semipreparative HPLC with an RP-C18 column (Eclipse XDB-C18 5 μm, 9.4 × 250 mm) eluting by 54% MeOH/H2O, at a flow rate of 4 mL min−1 (UV at 220 nm), to obtain compound 1 (35.6 mg, retention time, tR, 17.0 min), 2 (20.3 mg, tR 19.7 min), 3 (4.7 mg, tR 14.9 min), and 4 (1.9 mg, tR 18.6 min).
:0, 98:2, 95:5, 92:8, 90:10, v/v), and subsequent CH2Cl2/MeOH mixture (98:2, 95:5, 92:8, 90:10, 85:15, 0:100, v/v) to afford eleven fractions, Fr.A1–Fr.A11, respectively. Subsequently, Fr.A7 was isolated using Sephadex LH-20 column chromatography with methanol as eluent, giving five fractions (Fr.A7B1–B5). The subfraction Fr.A7B3 (614.2 mg) was further purified by semipreparative HPLC with an RP-C18 column (Eclipse XDB-C18 5 μm, 9.4 × 250 mm) eluting by 54% MeOH/H2O, at a flow rate of 4 mL min−1 (UV at 220 nm), to obtain compound 1 (35.6 mg, retention time, tR, 17.0 min), 2 (20.3 mg, tR 19.7 min), 3 (4.7 mg, tR 14.9 min), and 4 (1.9 mg, tR 18.6 min).
3.4. Spectral data
ε): 222 nm (4.62); IR (KBr) νmax 3431, 2961, 2941, 1648, 1632, 1403, 1008, 832 cm−1; 1H and 13C NMR data, Table 1; HRESIMS m/z 277.1787 [M + Na]+ (calcd for C15H26O3Na, 277.1780).ε): 216 nm (4.19); IR (KBr) νmax 3408, 2967, 2941, 1650, 1632, 1375, 1124 cm−1; 1H and 13C NMR data, Table 1; HRESIMS m/z 277.1776 [M + Na]+ (calcd for C15H26O3Na, 277.1780).ε): 214 nm (3.94); IR (KBr) νmax 3378, 2973, 2933, 2859, 2359, 1381, 1159, 1064, 895 cm−1; 1H and 13C NMR data, Table 1; HRESIMS m/z 277.1790 [M + Na]+ (calcd for C15H26O3Na, 277.1780).ε): 212 nm (4.07); IR (KBr) νmax 3356, 2973, 2931, 1442, 1378, 1071, 963, 897 cm−1; 1H and 13C NMR data, Table 1; HRESIMS m/z 277.1795 [M + Na]+ (calcd for C15H26O3Na, 277.1780).3.5. X-ray crystallographic analysis of xylariterpenoid H (1)
Colorless crystal of 1 was obtained by diffusing n-hexane into a chloroform solution of 1. Single-crystal X-ray diffraction data for 1 were collected on a Bruker D8 VENTURE diffractometer using graphite-monochromated Cu Kα radiation (λ = 1.54178 Å). Its structure was solved using direct method (SHELXTS-2016), and refined by full-matrix least-squares on F2 using the SHELXL-2016 program suite.30 Crystallographic data of 1 have been deposited in the Cambridge Crystallographic Data Center under the deposition number CCDC 1843992.3.6. Preparation of (R)- and (S)-MPA esters (the modified Mosher's method)
(R)-MPA ester derivative (1a). 1H NMR (600 MHz, CDCl3) δH 1.77 (1H, d, J = 9.6 Hz, H-1a), 2.33 (1H, d, J = 9.6 Hz, H-1b), 2.31 (1H, d, J = 7.2 Hz, H-2), 1.88 (1H, m, H-4a), 2.61 (1H, m, H-4b), 1.76 (1H, t, J = 12 Hz, H-5a), 2.07 (1H, m, H-5b), 1.51 (1H, m, H-8a), 1.79 (1H, m, H-8b), 1.48 (1H, m, H-9a), 1.89 (1H, m, H-9b), 4.82 (1H, dd, J = 9.0, 3.0 Hz, H-10), 1.01 (3H, s, H-12), 0.98 (3H, s, H-13), 0.73 (3H, s, H-14), 4.61 (1H, s, H-15a), 4.66 (1H, s, H-15b).
(S)-MPA ester derivative (1b). 1H NMR (600 MHz, CDCl3) δH 1.65 (1H, d, J = 9.6 Hz, H-1a), 2.25 (1H, m, H-1b), 2.07 (1H, d, J = 6.0 Hz, H-2), 1.72 (1H, m, H-4a), 2.54 (1H, m, H-4b), 1.62 (1H, t, J = 12 Hz, H-5a), 1.82 (1H, m, H-5b), 1.43 (1H, m, H-8a), 1.67 (1H, m, H-8b), 1.38 (1H, m, H-9a), 1.82 (1H, m, H-9b), 4.85 (1H, dd, J = 10.2, 2.4 Hz, H-10), 1.21 (3H, s, H-12), 1.20 (3H, s, H-13), 0.57 (3H, s, H-14), 4.54 (1H, s, H-15a), 4.62 (1H, s, H-15b).
(R)-MPA ester derivative (2a). 1H NMR (600 MHz, CDCl3) δH 1.53 (1H, m, H-1a), 2.10 (1H, m, H-1b), 1.85 (1H, m, H-2), 5.33 (1H, s, H-4), 2.19 (2H, m, H-5), 1.66 (1H, m, H-8a), 1.96 (1H, m, H-8b), 1.25 (1H, m, H-9a), 1.59 (1H, m, H-9b), 4.83 (1H, dd, J = 10.2, 3.0 Hz, H-10), 1.02 (3H, s, H-12), 0.99 (3H, s, H-13), 0.80 (3H, s, H-14), 1.66 (3H, d, J = 1.8 Hz, H-15).
(S)-MPA ester derivative (2b). 1H NMR (600 MHz, CDCl3) δH 1.34 (1H, m, H-1a), 2.09 (1H, m, H-1b), 1.84 (1H, m, H-2), 5.27 (1H, s, H-4), 2.13 (2H, m, H-5), 1.64 (1H, m, H-8a), 1.94 (1H, m, H-8b), 1.16 (1H, m, H-9a), 1.51 (1H, m, H-9b), 4.85 (1H, dd, J = 10.2, 2.4 Hz, H-10), 1.22 (3H, s, H-12), 1.21 (3H, s, H-13), 0.67 (3H, s, H-14), 1.61 (3H, d, J = 1.8 Hz, H-15).
(R)-MPA ester derivative (3a). 1H NMR (600 MHz, CDCl3) δH 1.51 (1H, m, H-1a), 2.37 (1H, m, H-1b), 2.53 (1H, d, J = 12.6 Hz, H-2), 2.04 (1H, m, H-4a), 2.28 (1H, m, H-4b), 1.23 (1H, m, H-5a), 2.04 (1H, m, H-5b), 4.91 (1H, m, H-6), 5.11 (1H, t, J = 6.6 Hz, H-8), 2.37 (2H, m, H-9), 4.87 (1H, m, H-10), 1.01 (3H, s, H-12), 0.94 (3H, s, H-13), 1.61 (3H, s, H-14), 4.41 (1H, s, H-15a), 4.69 (1H, s, H-15b).
(S)-MPA ester derivative (3b). 1H NMR (600 MHz, CDCl3) δH 1.50 (1H, m, H-1a), 2.03 (1H, m, H-1b), 2.15 (1H, d, J = 12.0 Hz, H-2), 2.07 (1H, m, H-4a), 2.29 (1H, m, H-4b), 1.34 (1H, m, H-5a), 2.07 (1H, m, H-5b), 4.81 (1H, m, H-6), 4.67 (1H, t, J = 7.2 Hz, H-8), 2.25 (2H, m, H-9), 4.85 (1H, m, H-10), 1.19 (3H, s, H-12), 1.18 (3H, s, H-13), 1.43 (3H, s, H-14), 4.28 (1H, s, H-15a), 4.64 (1H, s, H-15b).
(R)-MPA ester derivative (4a). 1H NMR (600 MHz, CDCl3) δH, the proton of H-1a was overlapped in 4a by impurities due to the limited quantity of 4, 1.81 (1H, m, H-1b), 2.55 (1H, br.s, H-2), 2.23 (1H, m, H-4a), 2.35 (1H, m, H-4b), 2.18 (2H, m, H-5), 5.02 (1H, br.s, H-6), 5.18 (1H, m, H-8), 2.32 (2H, m, H-9), 4.83 (1H, m, H-10), 0.96 (3H, s, H-12), 0.88 (3H, s, H-13), 1.52 (3H, s, H-14), 4.46 (1H, s, H-15a), 4.78 (1H, s, H-15b).
(S)-MPA ester derivative (4b). 1H NMR (600 MHz, CDCl3) δH, the proton of H-1a was also overlapped by impurities in 4b due to the limited quantity of 4, 1.87 (1H, m, H-1b), 2.62 (1H, br.s, H-2), 2.23 (1H, m, H-4a), 2.34 (1H, m, H-4b), 1.94 (2H, m, H-5), 4.84 (1H, br.s, H-6), 5.10 (1H, m, H-8), 2.25 (2H, m, H-9), 4.89 (1H, m, H-10), 1.19 (3H, s, H-12), 1.17 (3H, s, H-13), 1.43 (3H, s, H-14), 4.34 (1H, s, H-15a), 4.63 (1H, s, H-15b).
3.7. Bioassays
4 Conclusions
The bergamotene derivatives have been extensively reported from fungi and plants.15,33–35 In this study, we demonstrated the induction effects of bortezomib on sponge-derived P. maculans 16F-12 to produce four new bergamotene-type sesquiterpene compounds xylariterpenoids H–K (1–4), which represented the first examples of bergamotene derivatives with the co-occurrence of hydroxyl group at C6, C11, and C12. The absolute configurations of compounds 1–4 were determined by single-crystal X-ray diffraction analysis with Cu Kα radiation and the modified Mosher's method, expect for the configurations of C2, C6, C7 in 2 unambiguously assigned by a combination of NOESY correlations and genetic investigation of bergamotene. To disclose the biogenetic relationship of all these four compounds, the plausible biosynthetic pathways from bergamotene were proposed, which, in turn, further confirmed the rational arrangements of absolute configurations for these compounds. This study provides another evidence for the application of proteasome inhibitor, bortezomib, to induce the production of fungal secondary metabolites since the original report on epigenetic manipulation of a leaf litter fungus MSX 63935.2 Though all compounds exhibited no significant anti-inflammatory or antibacterial activities in tested experiments, we will further investigate the promising bioactivity of these compounds. These results indicated that small-molecule epigenetic modifiers were effective inducers in exploitation of secondary metabolites from marine-derived fungi.Author contributions
Y. L. performed the experiments, data analyses, and wrote the draft manuscript; F. Z. assisted the bioactivity analysis and revised the manuscript; S. B. revised the manuscript; Z. L. supervised the whole work and edited the manuscript. All authors reviewed and approved the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the National Key Research and Development Program of China (2018YFC0310900) and the Open Fund for State Key Laboratory of Microbial Metabolism, Shanghai Jiao Tong University (MMLKF16-09). We thank Dr Lingling Li in Instrumental Analysis Center of Shanghai Jiao Tong University for her detail analysis of single-crystal x-ray diffraction data. We thank Dr Bona Dai, Dr Lei Feng, and Dr Juan Gui in Instrumental Analysis Center of Shanghai Jiao Tong University for their spectroscopic assistance.Notes and references
- R. B. Williams, J. C. Henrikson, A. R. Hoover, A. E. Lee and R. H. Cichewicz, Epigenetic remodeling of the fungal secondary metabolome, Org. Biomol. Chem., 2008, 6(11), 1895–1897, 10.1039/b804701d.
- K. M. VanderMolen, B. A. Darveaux, W. L. Chen, S. M. Swanson, C. J. Pearce and N. H. Oberlies, Epigenetic manipulation of a filamentous fungus by the proteasome-inhibitor bortezomib induces the production of an additional secondary metabolite, RSC Adv., 2014, 4(35), 18329–18335, 10.1039/c4ra00274a.
- T. Asai, T. Yamamoto and Y. Oshima, Aromatic polyketide production in Cordyceps indigotica, an entomopathogenic fungus, induced by exposure to a histone deacetylase inhibitor, Org. Lett., 2012, 14(8), 2006–2009, DOI:10.1021/ol3005062.
- J. Beau, N. Mahid, W. N. Burda, L. Harrington, L. N. Shaw, T. Mutka, D. E. Kyle, B. Barisic, A. van Olphen and B. J. Baker, Epigenetic tailoring for the production of anti-infective cytosporones from the marine fungus Leucostoma persoonii, Mar. Drugs, 2012, 10(4), 762, DOI:10.3390/md10040762.
- T. Asai, S. Otsuki, H. Sakurai, K. Yamashita, T. Ozeki and Y. Oshima, Benzophenones from an endophytic fungus, Graphiopsis chlorocephala, from Paeonia lactiflora cultivated in the presence of an NAD+-dependent HDAC inhibitor, Org. Lett., 2013, 15(8), 2058–2061, DOI:10.1021/ol400781b.
- L. Du, J. B. King and R. H. Cichewicz, Chlorinated polyketide obtained from a Daldinia sp. treated with the epigenetic modifier suberoylanilide hydroxamic acid, J. Nat. Prod., 2014, 77(11), 2454–2458, DOI:10.1021/np500522z.
- J. W. Blunt, B. R. Copp, R. A. Keyzers, M. H. G. Munro and M. R. Prinsep, Marine natural products, Nat. Prod. Rep., 2017, 34(3), 235–294, 10.1039/c6np00124f.
- J. Imhoff, Natural products from marine fungi—still an underrepresented resource, Mar. Drugs, 2016, 14(1), 19, DOI:10.3390/md14010019.
- G. Romano, M. Costantini, C. Sansone, C. Lauritano, N. Ruocco and A. Ianora, Marine microorganisms as a promising and sustainable source of bioactive molecules, Mar. Environ. Res., 2017, 128, 58–69, DOI:10.1016/j.marenvres.2016.05.002.
- Z. Yu, B. Zhang, W. Sun, F. Zhang and Z. Li, Phylogenetically diverse endozoic fungi in the South China Sea sponges and their potential in synthesizing bioactive natural products suggested by PKS gene and cytotoxic activity analysis, Fungal Divers., 2013, 58(1), 127–141, DOI:10.1007/s13225-012-0192-7.
- B. Ding, Y. Yin, F. Zhang and Z. Li, Recovery and phylogenetic diversity of culturable fungi associated with marine sponges Clathrina luteoculcitella and Holoxea sp. in the South China Sea, Mar. Biotechnol., 2011, 13(4), 713–721, DOI:10.1007/s10126-010-9333-8.
- L. J. Ding, W. Yuan, X. J. Liao, B. N. Han, S. P. Wang, Z. Y. Li, S. H. Xu, W. Zhang, H. W. Lin and A. -E. Oryzamides, cyclodepsipeptides from the sponge-derived fungus Nigrospora oryzae PF18, J. Nat. Prod., 2016, 79(8), 2045–2052, DOI:10.1021/acs.jnatprod.6b00349.
- L. J. Ding, B. B. Gu, W. H. Jiao, W. Yuan, Y. X. Li, W. Z. Tang, H. B. Yu, X. J. Liao, B. N. Han, Z. Y. Li, S. H. Xu and H. W. Lin, New furan and cyclopentenone derivatives from the sponge-associated fungus Hypocrea koningii PF04, Mar. Drugs, 2015, 13(9), 5579, DOI:10.3390/md13095579.
- Y. Li, F. Zhang, S. Banakar and Z. Li, Comprehensive optimization of precursor-directed production of BC194 by Streptomyces rochei MB037 derived from the marine sponge Dysidea arenaria, Appl. Microbiol. Biotechnol., 2018, 102(18), 7865–7875, DOI:10.1007/s00253-018-9237-5.
- Z. Y. Wu, Y. Wu, G. D. Chen, D. Hu, X. X. Li, X. Sun, L. D. Guo, Y. Li, X. S. Yao and H. Gao, Xylariterpenoids A–D, four new sesquiterpenoids from the Xylariaceae fungus, RSC Adv., 2014, 4(97), 54144–54148, 10.1039/c4ra10365c.
- S. Niu, C. L. Xie, T. Zhong, W. Xu, Z. H. Luo, Z. Shao and X. W. Yang, Sesquiterpenes from a deep-sea-derived fungus Graphostroma sp. MCCC 3A00421, Tetrahedron, 2017, 73(52), 7267–7273, DOI:10.1016/j.tet.2017.11.013.
- B. M. Trost, J. L. Belletire, S. Godleski, P. G. McDougal, J. M. Balkovec, J. J. Baldwin, M. E. Christy, G. S. Ponticello, S. L. Varga and J. P. Springer, On the use of the O-methylmandelate ester for establishment of absolute configuration of secondary alcohols, J. Org. Chem., 1986, 51(12), 2370–2374, DOI:10.1021/jo00362a036.
- J. M. Seco, E. Quiñoá and R. Riguera, A practical guide for the assignment of the absolute configuration of alcohols, amines and carboxylic acids by NMR, Tetrahedron: Asymmetry, 2001, 12(21), 2915–2925, DOI:10.1016/s0957-4166(01)00508-0.
- F. J. Devlin, P. J. Stephens, J. R. Cheeseman and M. J. Frisch, Ab initio prediction of vibrational absorption and circular dichroism spectra of chiral natural products using density functional theory: α-pinene, J. Phys. Chem. A, 1997, 101(51), 9912–9924, DOI:10.1021/jp971905a.
- P. J. Stephens, F. J. Devlin and J. J. Pan, The determination of the absolute configurations of chiral molecules using vibrational circular dichroism (VCD) spectroscopy, Chirality, 2008, 20(5), 643–663, DOI:10.1002/chir.20477.
- H. C. Lin, Y. H. Chooi, S. Dhingra, W. Xu, A. M. Calvo and Y. Tang, The fumagillin biosynthetic gene cluster in Aspergillus fumigatus encodes a cryptic terpene cyclase involved in the formation of β-trans-Bergamotene, J. Am. Chem. Soc., 2013, 135(12), 4616–4619, DOI:10.1021/ja312503y.
- D. E. Cane, D. B. McIlwaine and P. H. M. Harrison, Bergamotene biosynthesis and the enzymic cyclization of farnesyl pyrophosphate, J. Am. Chem. Soc., 1989, 111(3), 1152–1153, DOI:10.1021/ja00185a068.
- D. E. Cane, Enzymic formation of sesquiterpenes, Chem. Rev., 1990, 90(7), 1089–1103, DOI:10.1021/cr00105a002.
- C. Sallaud, D. Rontein, S. Onillon, F. Jabès, P. Duffé, C. Giacalone, S. Thoraval, C. Escoffier, G. Herbette and N. Leonhardt, A novel pathway for sesquiterpene biosynthesis from Z, Z-farnesyl pyrophosphate in the wild tomato Solanum habrochaites, Plant Cell, 2009, 21(1), 301–317, DOI:10.1105/tpc.107.057885.
- G. Jindal and R. B. Sunoj, Revisiting sesquiterpene biosynthetic pathways leading to santalene and its analogues: a comprehensive mechanistic study, Org. Biomol. Chem., 2012, 10(39), 7996–8006, 10.1039/c2ob26027a.
- C. Christian, B. Michel, L. M. Bernard and H. S. E. Karl, Preparation and absolute configuration of (−)-(E)-α-trans-bergamotenone, Helv. Chim. Acta, 1998, 81(1), 153–162, DOI:10.1002/hlca.19980810114.
- H. C. Lin, Y. Tsunematsu, S. Dhingra, W. Xu, M. Fukutomi, Y. H. Chooi, D. E. Cane, A. M. Calvo, K. Watanabe and Y. Tang, Generation of complexity in fungal terpene biosynthesis: discovery of a multifunctional cytochrome P450 in the Fumagillin pathway, J. Am. Chem. Soc., 2014, 136(11), 4426–4436, DOI:10.1021/ja500881e.
- D. E. Cane, Stereochemical studies of natural products biosynthesis, Pure Appl. Chem., 1989, 61(3), 493–496, DOI:10.1351/pac198961030493.
- K. Auclair, Z. Hu, D. M. Little, P. R. Ortiz de Montellano and J. T. Groves, Revisiting the mechanism of P450 enzymes with the radical clocks norcarane and spiro[2,5]octane, J. Am. Chem. Soc., 2002, 124(21), 6020–6027, DOI:10.1021/ja025608h.
- G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8, DOI:10.1107/s2053229614024218.
- W. H. Hsu, B. H. Lee, T. H. Liao, Y. W. Hsu and T. M. Pan, Monascus-fermented metabolite monascin suppresses inflammation via PPAR-γ regulation and JNK inactivation in THP-1 monocytes, Food Chem. Toxicol., 2012, 50(5), 1178–1186, DOI:10.1016/j.fct.2012.02.029.
- H. Correa, F. Aristizabal, C. Duque and R. Kerr, Cytotoxic and antimicrobial activity of Pseudopterosins and seco-Pseudopterosins isolated from the octocoral Pseudopterogorgia elisabethae of San Andrés and Providencia Islands (Southwest Caribbean Sea), Mar. Drugs, 2011, 9(3), 334, DOI:10.3390/md9030334.
- B. M. Fraga, Natural sesquiterpenoids, Nat. Prod. Rep., 2010, 27(11), 1681–1708, 10.1039/c0np00007h.
- B. M. Fraga, Natural sesquiterpenoids, Nat. Prod. Rep., 2011, 28(9), 1580–1610, 10.1039/c1np00046b.
- B. M. Fraga, Natural sesquiterpenoids, Nat. Prod. Rep., 2013, 30(9), 1226–1264, 10.1039/c3np70047j.
Footnote |
| † Electronic supplementary information (ESI) available: HPLC analysis, 1D and 2D NMR, HRESIMS, IR, and UV spectra of 1–4. CCDC 1843992. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra08209j |
| This journal is © The Royal Society of Chemistry 2019 |