
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNephelauxetic effect of the hydride ligand in Sr2LiSiO4H as a host material for rare-earth-activated phosphors†
Tong Wu a,
Asako Ishikawaa,
Takashi Hondab,
Hiromu Tamatsukurib,
Kazutaka Ikedab,
Toshiya Otomob and
Satoru Matsuishi*a
a,
Asako Ishikawaa,
Takashi Hondab,
Hiromu Tamatsukurib,
Kazutaka Ikedab,
Toshiya Otomob and
Satoru Matsuishi*a
aMaterials Research Center for Element Strategy, Tokyo Institute of Technology, 4259 Nagatsuta-cho, Midori-ku, Yokohama 226-8503, Japan. E-mail: matsuishi@mces.titech.ac.jp
bInstitute of Materials Structure Science, High Energy Accelerator Research Organization, Tsukuba 305-0801, Japan
First published on 12th February 2019
Abstract
Strontium lithium orthosilicate hydride Sr2LiSiO4H was synthesized by the reaction of Sr2SiO4 with LiH at 700 °C in a H2 rich atmosphere. Rietveld refinement of the neutron powder diffraction pattern revealed that Sr2LiSiO4H is isostructural to Sr2LiSiO4F (space group P21/m) and its channel-like structure preferentially accommodates H− ions over F− ions. In addition, Sr2LiSiO4H is stable in air and its Eu2+-doped analog exhibits yellow photoluminescence with an emission band at 544 nm and a broad excitation band ranging from 250 to 450 nm. These bands were observed in the longer wavelength region when compared with those displayed by Sr2LiSiO4F:Eu2+. The red shift, which is induced by H− substitution, is consistent with the constrained density functional theory calculations, predicting the photo-excitation and emission energies of 4f–5d transitions. The present study reports the synthesis of stable oxyhydrides acting as phosphor hosts for rare earth ions. The phosphor hosts exhibit large nephelauxetic effects owing to the presence of H− ligands.
Introduction
The hydride ion H− is a monovalent anion composed of one proton and two electrons with no p orbitals in the outer shell.1 The unique atomic structure of H− strongly influences its chemical bonding.2 Owing to its multivalent property and rather low electronegativity, which is lower than that of oxygen, hydrogen can work both as an electron donor and acceptor in oxide-based materials. The unique properties of hydrogen offer promising applications for oxyhydrides, in which both oxide and hydride ions are presented, toward energy storage in semiconducting as well as superconducting materials.3–7 However, knowledge surrounding oxyhydrides with exclusive properties is limited, thereby generating much interest in pursuing further exploration of such oxyhydride materials.Over recent decades, lanthanide ion-doped compounds have been typically examined in phosphor-converted white light-emitting diodes. Examples of such compounds include YAG:Ce3+ and Eu2+-doped SiAlON phosphors on blue GaInN chips.8,9 Among them, Eu2+-doped host phosphor materials have received the most attention owing to its parity-allowed electronic dipole 4f → 5d transitions with comparable strong emitting intensities.10 Unlike the 4f electrons, the binding energy of the unshielded 5d electrons strongly depends on the crystalline environment, which can be described as the centroid shift and crystal field splitting of 5d levels that reflect on absorption energies.11 Therefore, to design phosphors with different emitting color requirements, oxides, nitrides, halides, and mixed ligand systems have been used as host materials.12 Compared to conventional host materials, hydride and hydride-based mixed anion compounds are expected to induce larger centroid shifts due to the expansion of electron cloud of the 5d electron. This so-called nephelauxetic effect is generally based on the covalent bonding between cation and anion ligand. Since hydrogen has a lower electronegativity (2.20 in Pauling scale) than those of oxygen (3.44), nitrogen (3.04), and fluorine (3.98),13 the stronger covalence bonding can be formed and then the large red shift of the excitation band is expected. For the past decades, hydrides and mixed anion hydrides containing Eu2+ dopant have been reported to exhibit green-red luminescence.10,14–20 However, their poor stability in air and moisture limits their application. In contrast, we have recently demonstrated that oxyhydride GdHO is stable in ambient air and can act as a phosphor host for Tb3+. Then, the green luminescence corresponding to 5D4 → 7FJ transition was observed upon excitation in near-UV light.21 Therefore, in the present study, we aim to evaluate stable oxyhydrides that can be used as phosphor hosts for Eu2+ with strong photoluminescence.
In the preliminary of this study, we investigated the conditions required to obtain oxyhydride-based phosphor from air-stable oxyhydrides. To avoid optical absorption in the visible region, which is typically due to the presence of the d electrons of transition metals,4,7,22 the host lattice of the phosphor should not contain transition-metal. Furthermore, hydride ions can be stably incorporated in transition-metal-free anion-encaging compounds with a cage and channel structure. Examples of such compounds include H-doped mayenite [Ca12Al14O32]2+(O1−x2−H2x−) and apatite [Ca10(PO4)6]2+(O1−x2−X2x−) (X = H, OH).3,6,23 Using this approach, we focused on strontium lithium orthosilicate fluoride Sr2LiSiO4F doped with Eu2+ and/or Ce3+. This material has shown potential as phosphor material in white light-emitting diodes with a broad emission range from blue to green.24–26 Due to the same valence and comparable ionic radii of H− and F−,27 we expected complete substitution of F− in Sr2LiSiO4F with H−. In the hydride analog Sr2LiSiO4H, only H− occupies the anion site in the channel space similarly to H-substituted apatite. In contrast, the anion sites in mayenite and apatite are partially occupied by both O2− and H−. The former arrangement is advantageous to suppressing optical absorption by electrons in anion vacancies generated by photo-dissociation of H− (O2− + H− → OH− + 2e−). Thus, Sr2LiSiO4H is expected to be stable in air and suitable as a host material for phosphor agents. As Eu2+ ion has an ionic radius similar to that of Sr2+ ion, Eu2+ ion can easily be incorporated into Sr sites. Because of aforementioned, Eu2+ is an appropriate dopant for investigating the influence of hydride substitution on the coordination structure and optical properties. In Sr2LiSiO4H:Eu2+, we expect a larger redshift in the Eu 5d energy levels, corresponding to smaller absorption energies, when compared with Sr2LiSiO4F:Eu2+.
In the present study, we report the synthesis of oxyhydride Sr2LiSiO4H by heating a mixture of LiH and Sr2SiO4 in H2 gas and its photoluminescence (PL) property activated by partial substitution of Eu2+ for Sr2+. Neutron powder diffraction (NPD) measurements of the deuterium-enriched sample demonstrated the formation of a crystalline phase isostructural to Sr2LiSiO4F and full occupation of the F sites by deuterium. Since the synthesized crystalline phase is stable in ambient air, all measurements were taken in ambient environment. The Eu-free Sr2LiSiO4H sample, with an optical bandgap larger than 5.2 eV, is light grey under daylight. With Eu incorporation, the sample is light yellow and emits intense yellow luminescence under near-UV light (375 nm). To investigate the increase in centroid shift of the Eu 5d level upon substitution of F− with H−, we performed constrained density functional theory (cDFT) calculations on the Eu2+-doped Sr2LiSiO4X systems (X = F, H).
Experimental section
Unlike most oxyhydrides that are synthesized by topochemical reaction of oxide precursors with a hydride ion source, polycrystalline Sr2−xEuxLiSiO4H was directly synthesized by reaction of Sr2−xEuxSiO4 with LiH at a high temperature. LiH is a highly thermally stable hydride, with a melting point of 689 °C and a decomposition temperature of >900 °C under ambient pressure condition. The stability of LiH is considered to be the key factor for the direct synthesis of the oxyhydride Sr2LiSiO4H. The precursor compounds were prepared by solid-state reaction of SrCO3, Eu2O3 and SiO2 under the reduction gas environment (95% Ar/5% H2) and hydrogenation of Li metal, respectively. The precursor compounds were then mixed and ground into fine powders with a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.05 and heated up to 700 °C in H2 gas at 0.9 MPa. For the powder X-ray diffraction (XRD) measurements and PL study, 2% Eu2+-doped sample i.e., Sr1.96Eu0.04LiSiO4H (Sr2LiSiO4H:Eu2+) was used. As for the NPD measurements, 100% deuterium-enriched sample (Sr2LiSiO4D) was synthesized using Sr2SiO4 and LiD. Detailed descriptions of the synthesis of the samples and characterization techniques including thermal desorption spectroscopy (TDS), XRD, NPD, diffuse reflectance spectroscopy, photoluminescence spectra (PL), magnetization measurements and DFT calculations are provided in the ESI.†
:1.05 and heated up to 700 °C in H2 gas at 0.9 MPa. For the powder X-ray diffraction (XRD) measurements and PL study, 2% Eu2+-doped sample i.e., Sr1.96Eu0.04LiSiO4H (Sr2LiSiO4H:Eu2+) was used. As for the NPD measurements, 100% deuterium-enriched sample (Sr2LiSiO4D) was synthesized using Sr2SiO4 and LiD. Detailed descriptions of the synthesis of the samples and characterization techniques including thermal desorption spectroscopy (TDS), XRD, NPD, diffuse reflectance spectroscopy, photoluminescence spectra (PL), magnetization measurements and DFT calculations are provided in the ESI.†
Result and discussion
The XRD patterns of Sr2LiSiO4H:Eu2+ measured with Cu Kα radiation, (Fig. S1(a) in ESI†) could be indexed to the space group P21/m, with lattice parameters a = 6.5854(2), b = 5.4206(2), and c = 6.9458(2) Å, and β = 112.568(2)°. These parameters are comparable with those of Sr2LiSiO4F (a = 6.5825(9), b = 5.4158(8), and c = 6.9266(6) Å, and β = 112.525(8)°), implying the formation of isostructural compound accommodating H− in place of F−. After exposing to ambient air for 16 h, no change was observed in the XRD pattern, indicating the air and moisture stability of Sr2LiSiO4H:Eu2+. Thus, subsequent measurements were taken in ambient environment. The refinement of the XRD pattern indicated the presence of small amount of Li2SrSiO4 (1.36 wt%), which could not be reduced even after the optimization of synthesis temperature and atmospheric condition. To determine the occupancy of the hydrogen site, NPD and thermal desorption spectroscopy (TDS) measurements were respectively performed for Sr2LiSiO4D and Sr2LiSiO4H samples. Fig. 1 shows the NPD pattern of Sr2LiSiO4D collected on a neutron total scattering spectrometer (NOVA; beam-line BL21) at the Japan Proton Accelerator Research Complex (J-PARC), and Rietveld refinement was performed using the Z-Rietveld code.28 Except for the weak peaks originating from Sr2LiSiO4 (1.97(1) wt%) and SrO impurities (0.42(1) wt%), the major reflections could be indexed to the space group P21/m, with lattice parameters a = 6.5820(5), b = 5.4197(4), and c = 6.9475(5) Å, and β = 112.5628(2)°. Rietveld refinement of the NPD pattern was easily converged by using the Sr2LiSiO4F-type initial structure in which the fluorine atoms were fully replaced by deuterium atoms without noticeable vacancies. The hydrogen stoichiometry determined by TDS was H/Sr2LiSiO4 = 0.97, confirming complete occupation of fluoride sites in Sr2LiSiO4F by hydrogen without crystal structure transformation.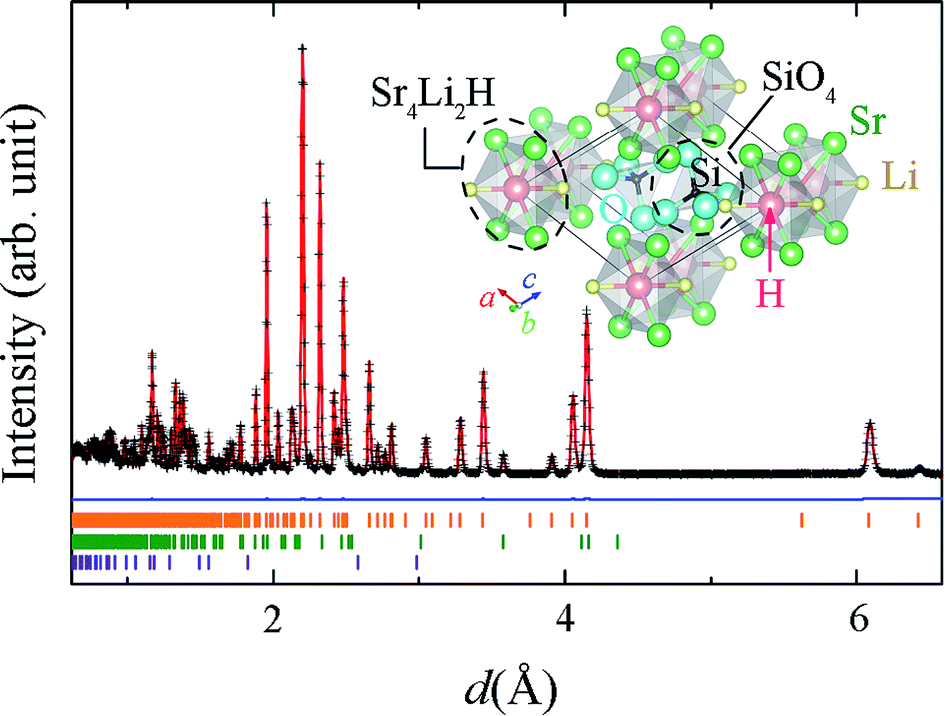 | ||
| Fig. 1 Time-of-flight NPD pattern of Sr2LiSiO4D and Rietveld fit (red line). The blue line represents the difference between the pattern and the fitted pattern. Orange, green, and purple markers represent the Bragg diffraction positions calculated for Sr2LiSiO4D, SrLi2SiO4, and SrO, respectively. Inset shows the structure model of Sr2LiSiO4H visualized by VESTA.43 | ||
The structure model of Sr2LiSiO4H along the b axis is shown in the inset of Fig. 1, and the interatomic distances in Sr2LiSiO4D (Sr2LiSiO4D data obtained from NPD) compared with those in Sr2LiSiO4F are listed in Table S6.† In this crystal, hydrogen or deuterium occupies the anion site that is octahedrally coordinated by four Sr and two Li atoms, forming a chain of face-sharing octahedra. The unit cell contains two types of 10-coordinated Sr sites, 4-coordinated Si atoms, and 6-coordinated H atoms. The Sr1 site is surrounded by one O1, four O2, and three O3 atoms and two H1 sites, whereas the Sr2 site is surrounded by three O1, four O2, and one O3 atoms, and two H1 sites. This monoclinic crystal structure is composed of face-shared (Sr4Li2H)4+ octahedra with isolated orthosilicate (SiO4)4– groups. All oxygen ions are shared at the vertices of the SiO4 tetrahedra. The H-centered octahedra provide one-dimensional (1D) chains of hydride ions with a H–H distance of 2.73 Å. Since the substitution of F− by H− does not cause any structural changes, hydride ion is highly stable in this 1D channel structure and its presence offers chemical stability to the corresponding oxyhydride material against air and moisture. Because of their similarity in ionic radius, Sr sites will be substituted upon introduction of divalent europium ions into the system.
To examine the effect of F− substitution by H−, DFT calculations were performed using the VASP code with projector-augmented plane-wave method and Perdew–Burke–Ernzerhof functional (PBE) functional.29–31 The calculated electronic band structure and density of state (DOS) are shown in Fig. 2. To emphasize the domination of hydride ions on the top of valence band, the energy axes were further adjusted by the O 2s orbital which located below Fermi level around 18–19 eV. The bandgaps of Sr2LiSiO4H and Sr2LiSiO4F estimated from the band structure, were 4.32 eV and 4.82 eV, respectively. However, according to the tendency that well-established in the literature, PBE calculations underestimate the actual bandgap energy.32,33 To obtain more realistic gap values, the calculations were performed using G0W0.34 Indeed, we obtained bandgap values of 6.29 eV and 6.87 eV for Sr2LiSiO4H and Sr2LiSiO4F, respectively. For Sr2LiSiO4F, the conduction band minima are primarily composed of Sr 3d orbitals, whereas the valence bands maxima are composed of O 2p orbitals. The 2p orbitals of F− ion have lower energies and overlap on the O 2p orbitals, thus have no effect on the bandgap of Sr2LiSiO4F. In contrast, the smaller bandgap of Sr2LiSiO4H is attributed to the hydride ions. Since its the valence band is composed of hydride 1s orbital which has a higher energy than the O 2p orbitals, a narrower bandgap was observed.
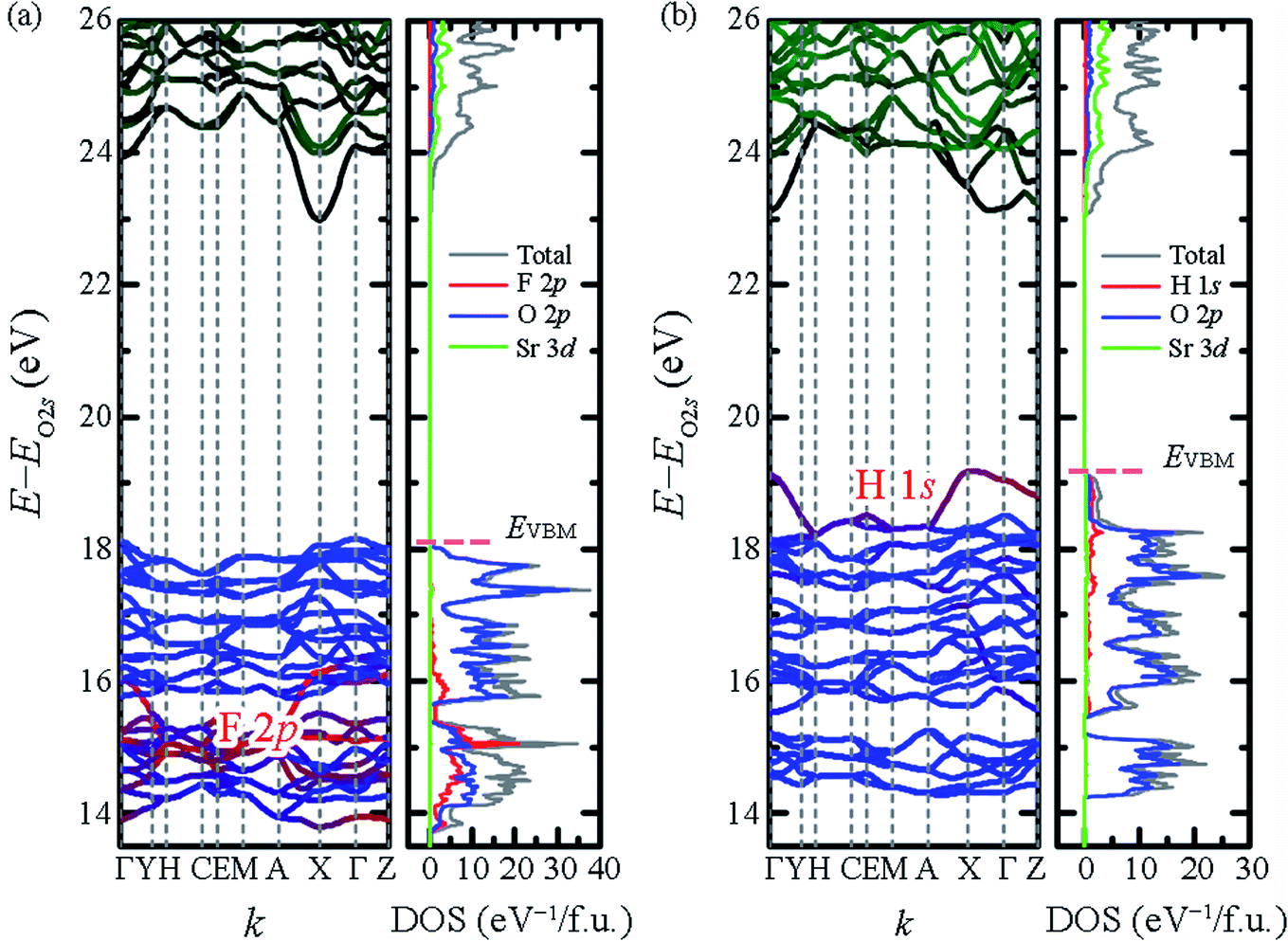 | ||
| Fig. 2 Band structures and DOS of (a) Sr2LiSiO4F and (b) Sr2LiSiO4H. The energy axes are adjusted by the O 2s levels located at −18 to −19 eV below each EVBM. | ||
The diffuse reflectance spectra of the Sr2LiSiO4H and Sr2LiSiO4F samples were recorded at room temperature within the wavelength range of 200–1500 nm. The optical absorption spectra transformed from the relative diffuse reflectance R using the Kubelka–Munk function F(R) = (1 − R)2/2R are shown in Fig. 3.35 For Sr2LiSiO4H, the strong optical absorption edge located around 250 nm could be associated with electron transitions from the valence band to the conduction band of the host lattice. This absorption edge was red-shifted relative to that of Sr2LiSiO4F (∼230 nm). This result further confirmed the narrower bandgap of Sr2LiSiO4H when compared with that of Sr2LiSiO4F. More intuitive bandgap (Eg) values can be calculated using the following equation:36 F(R)hv ∝ (hv − Eg)n/2, where hν is the photon energy and the value of n is dictated by the type of transition (n = 4 for indirect transition and n = 1 for direct transition). In the present study, (F(R)hν)2–hν plots were used to determine Eg, as shown in the inset of Fig. 3. This relationship gave a better linearity than the (F(R)hν)1/2–hν relationship (Fig. S2†). By extrapolating the former plot to 0, the direct bandgap of Sr2LiSiO4H was estimated to be ∼5.2 eV, whereas that of Sr2LiSiO4F was estimated to be larger i.e., ∼5.5 eV. These values are 1.1–1.4 eV smaller than those predicted by G0W0 calculations, indicating that the observed absorption below 6 eV is induced by bulk excitons or defects such as O2− and OH− in X− (X− = F−, H−) sites. Accordingly, Sr2LiSiO4H and Sr2LiSiO4F powder samples appear white-grey and white under daylight, respectively.
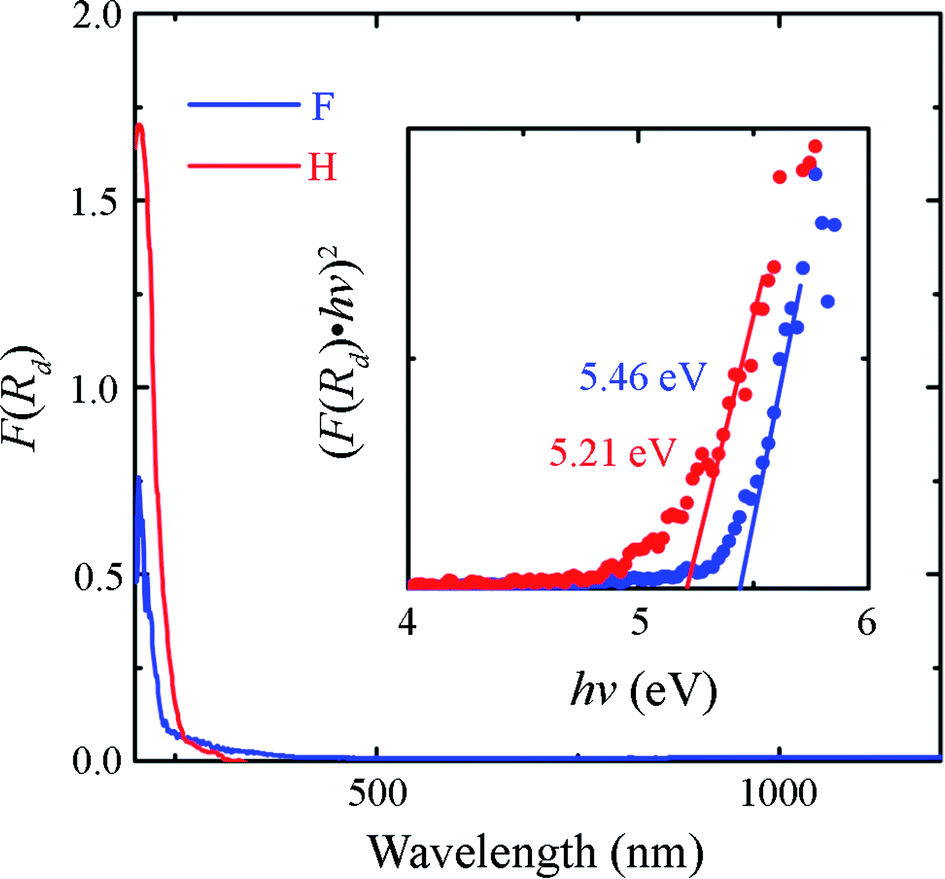 | ||
| Fig. 3 Diffuse reflectance spectra of Sr2LiSiO4F and Sr2LiSiO4H. Inset shows (F(R)hv)2–hv plots to estimate band gap energies. The (F(R)hν)2 data of Sr2LiSiO4F were multiplied by 10 so that both sets of data could be observed within the same coordinate axis range. | ||
The photoluminescence excitation (PLE) and PL spectra of Sr2LiSiO4H:Eu2+ recorded at room temperature are depicted in Fig. 4. As a reference, the spectra of Sr1.96Eu0.04LiSiO4F (Sr2LiSiO4F:Eu2+) were also recorded under the same conditions. Under an excitation light of 293 nm, Sr2LiSiO4H:Eu2+ displayed a single emission peak centered at 544 nm with a full width at half maximum (FWHM) of 0.42 eV, whereas Sr2LiSiO4F:Eu2+ displayed an emission peak at 506 nm with a FWHM of 0.7 eV under an excitation light of 324 nm. The absence of sharp emission lines characteristic of 4f–4f transitions in Eu3+ confirmed a complete reduction from Eu3+ to Eu2+ for both samples. This result is consistent with the magnetization data indicating that the most of Eu atom form divalent states with 8S7/2 configuration (see Fig. S3†). The broad emission band was attributed to the parity-allowed electronic dipole transitions in Eu2+ from 4f65d to 4f7 configuration. The band could be deconvoluted into two Gaussian peaks, which could be assigned to Eu occupying two different Sr sites. Sr2LiSiO4H:Eu2+ possesses an emission band redshifted from that of Sr2LiSiO4F:Eu2+ and emits intense yellow luminescence under illumination by near-UV (375 nm) LED as shown in the inset of Fig. 4.
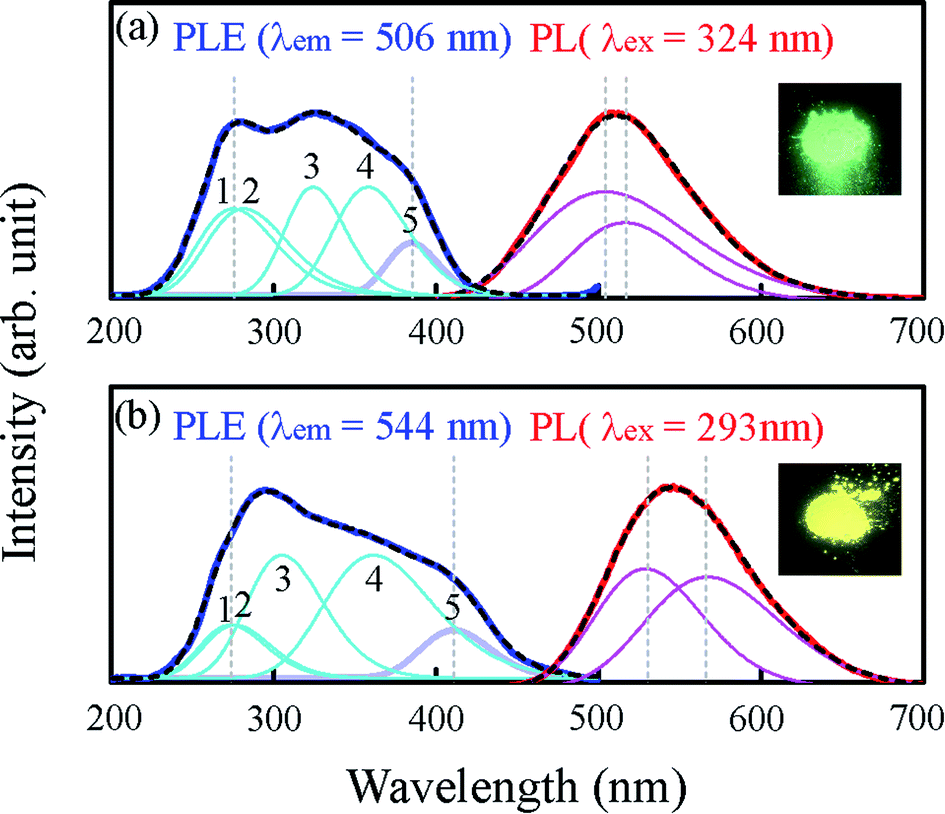 | ||
| Fig. 4 Photoluminescence properties of (a) Sr2LiSiO4F:Eu2+ and (b) Sr2LiSiO4H:Eu2+. PL and PLE spectra and corresponding photographs of the materials under illumination with near-UV light of 375 nm. Sr2LiSiO4F:Eu2+ was excited at 324 nm and monitored at 506 nm. Sr2LiSiO4H Eu2+ was excited at 293 nm and monitored at 544 nm. Both PLE and PL spectra were deconvoluted by Gaussian fitting. | ||
In each PLE spectrum recorded, there was only one broad absorption band consisting of at least five or more overlapping Gaussian peaks. Each broad band could be ascribed to the crystal field splitting of Eu 5d levels of 4f7 → 4f65d1. For Sr2LiSiO4H:Eu2+, the broad band was observed within the wavelength region of 250–450 nm, whereas in Sr2LiSiO4F:Eu2+, the broad band was observed within the higher photon energy region of 230–410 nm. An energy level scheme was established accordingly, with each peak within the broad excitation band corresponding to the Eu 5d levels and the position of the lowest-excitation peak (as marked “5”) was considered as the Eu lowest 5d energy level. In more details, the absolute position of the lowest 5d level depends on the redshift which typically consists of centroid shift and crystal field splitting. Centroid shift is mainly influenced by nephelauxetic effects, whereas crystal field splitting is dependent on the site symmetry.37 The lowest-excitation peak positions for Sr2LiSiO4F:Eu2+ and Sr2LiSiO4H:Eu2+ were 3.22 eV and 3.02 eV, indicating the downshift of Eu lowest 5d energy level and further confirming the high likelihood that the replacement of fluoride with hydride leads to the red shift of the absorption band due to large nephelauxetic effects exerted by H− ligands.
Subsequently, we theoretically investigated the Eu2+ excitation and emission energies for Sr2LiSiO4H:Eu2+ and Sr2LiSiO4F:Eu2+ using cDFT calculations with the configurational coordinate diagram. To introduce Eu into the host lattices, 2 × 2 × 1 supercells were applied with one of the Sr atoms replaced by Eu, namely Sr31Eu1Li16Si16O64H16 and Sr31Eu1Li16Si16O64F16. Here, we considered two scenarios of Eu substituting into Sr1 or Sr2 site, respectively. According to the configurational coordinate diagram, excitation and emission processes occur among four electronic states that are ground state A0, excited state without structural relaxation 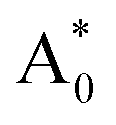 , excited state after relaxation A*, and ground state with the relaxed structure A. For the excitation process, one electron located at Eu 4f level was excited to the 5d level by absorbing a photon with energy larger than the energy difference between the A0 and
, excited state after relaxation A*, and ground state with the relaxed structure A. For the excitation process, one electron located at Eu 4f level was excited to the 5d level by absorbing a photon with energy larger than the energy difference between the A0 and 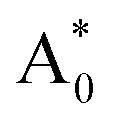 states. Lattice relaxation corresponding to non-radiative processes from
states. Lattice relaxation corresponding to non-radiative processes from 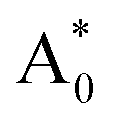 to A*, proceeded owing to imbalance of the electronic configuration in Eu2+. The excited electron in the 5d level then returned to the 4f level by emission of a photon resulting in transition from an A* to an A state. On the basis of the total energy of each state, we calculated the absorption energy
to A*, proceeded owing to imbalance of the electronic configuration in Eu2+. The excited electron in the 5d level then returned to the 4f level by emission of a photon resulting in transition from an A* to an A state. On the basis of the total energy of each state, we calculated the absorption energy 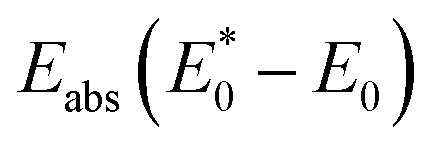 , emission energy Eem(E* − E), and the Stokes shift ΔS
, emission energy Eem(E* − E), and the Stokes shift ΔS 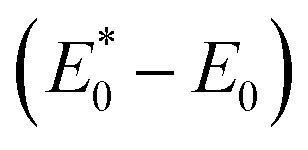 , which are listed in Table 1. The supercell structure was first optimized to obtain the A0 state by standard DFT calculation using the +U method with U = 6 eV for both Sr2LiSiO4H and Sr2LiSiO4F to localize the Eu 4f bands within the bandgap. In the ground state, narrow Eu 4f bands occupied by seven spin-up electrons, were located above the valence band maximum (VBM) of the host lattice. The
, which are listed in Table 1. The supercell structure was first optimized to obtain the A0 state by standard DFT calculation using the +U method with U = 6 eV for both Sr2LiSiO4H and Sr2LiSiO4F to localize the Eu 4f bands within the bandgap. In the ground state, narrow Eu 4f bands occupied by seven spin-up electrons, were located above the valence band maximum (VBM) of the host lattice. The 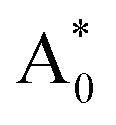 state was calculated by cDFT method which constrains the electronic occupancy of the highest-occupied band to be 0 and that of the lowest unoccupied band to be 1. Thus, two new bands were formed by the core-hole interaction. One is unoccupied 4f bands which shifts above the valence band and the other is occupied 5d band which shifts below the conduction band. The cDFT method was continuously utilized to maintain electronic configuration during the second geometry optimization while the energy of the Eu 5d band was reduced. For the A state, without constraining the electron occupancy, the excited electron automatically returned to the Eu 4f band with the same equilibrium configuration coordinates of the A* state. The band structures in each electronic state were assessed further for both Sr2LiSiO4F and Sr2LiSiO4H (Fig. S3†). Noteworthy, the cDFT calculations overestimate the absorption/emission energy relative to the experimental results. In fact, the cDFT method cannot be used to accurately demonstrate each splitting band position but only the tendency of energy transformations. However, the smaller absorption energy (Eabs) which indicates the downshifting of the Eu lowest 5d energy level in Sr2LiSiO4H from the calculated data well-agreed with experimental results. Compared with Sr2LiSiO4F, Sr2LiSiO4H showed lower absorption and emission energies irrespective of the Sr site substitution of Eu. This result is consistent with the observed red shifts in both absorption band and emission band induced by the substation of F− by H−.
state was calculated by cDFT method which constrains the electronic occupancy of the highest-occupied band to be 0 and that of the lowest unoccupied band to be 1. Thus, two new bands were formed by the core-hole interaction. One is unoccupied 4f bands which shifts above the valence band and the other is occupied 5d band which shifts below the conduction band. The cDFT method was continuously utilized to maintain electronic configuration during the second geometry optimization while the energy of the Eu 5d band was reduced. For the A state, without constraining the electron occupancy, the excited electron automatically returned to the Eu 4f band with the same equilibrium configuration coordinates of the A* state. The band structures in each electronic state were assessed further for both Sr2LiSiO4F and Sr2LiSiO4H (Fig. S3†). Noteworthy, the cDFT calculations overestimate the absorption/emission energy relative to the experimental results. In fact, the cDFT method cannot be used to accurately demonstrate each splitting band position but only the tendency of energy transformations. However, the smaller absorption energy (Eabs) which indicates the downshifting of the Eu lowest 5d energy level in Sr2LiSiO4H from the calculated data well-agreed with experimental results. Compared with Sr2LiSiO4F, Sr2LiSiO4H showed lower absorption and emission energies irrespective of the Sr site substitution of Eu. This result is consistent with the observed red shifts in both absorption band and emission band induced by the substation of F− by H−.
| Sr2LiSiO4F | Sr2LiSiO4H | |||
|---|---|---|---|---|
| Exp. (eV) | Cal. (eV) | Exp. (eV) | Cal. (eV) | |
| a For the calculations, standard DFT+U and cDFT were used. | ||||
| Eabs | 3.22 | 4.49(Eu1) | 3.02 | 4.12(Eu1) |
| 4.78(Eu2) | 4.20(Eu2) | |||
| Eem | 2.40 | 3.16(Eu1) | 2.18 | 2.97(Eu1) |
| 2.46 | 3.44(Eu2) | 2.34 | 3.40(Eu2) | |
| ΔS | 0.82 | 1.34(Eu1) | 0.84 | 1.15(Eu1) |
| 0.76 | 1.33(Eu2) | 0.68 | 0.81(Eu2) | |
Herein, we mainly discuss the absorption energies difference between Eu 5d levels in Sr2LiSiO4H:Eu2+ and Sr2LiSiO4F:Eu2+. According to the semiempirical model reported by Dorenbos,38–41 both centroid shift and crystal field splitting can separately cause downshifting of the lowest Eu 5d level. In particular, crystal field splitting εcfs can be described as εcfs = β/Rav2, where β is a measure of the shape and size of the Eu-central polyhedron and Rav is the average bonding length of the Eu site coordination.41 As shown in Tables S7 and S8,† the bond lengths and bond angles of the coordinate structures around Eu2+ in 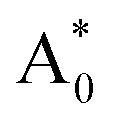 states were almost same in Sr2LiSiO4H and Sr2LiSiO4F, indicating that the degree of crystal field splitting for excitation energy was comparable in these two structures. This similarity is due to the identical charge and similar ionic radii of F− and H−. Therefore, the downshifting of the Eu 5d level corresponding to the redshift of PLE band in Sr2LiSiO4H should be attributed to the difference in the degree of centroid shift. As stated in introduction section, hydrogen has a lower electronegativity than fluoride and can give the strong nephelauxetic effect inducing large centroid shift which presents as the right shift of absorption band redshift in Sr2LiSiO4H:Eu2+. The present findings illustrate the potential of hydride ion substitution in oxyfluoride phosphor in inducing a red shift of the absorption bands. In a very recent study by Gehlhaar et al.42 on the synthesis and photoluminescence property of Sr2LiSiO4H:Eu2+, the authors also observed red shifts of the PL band induced by substitution of F− with H−. However, our research demonstrates that the red shifting of absorption band due to the strong nephelauxetic effect giving by hydride ligands in hydride analogue of oxyfluoride compounds are possibly predicted by cDFT calculation.
states were almost same in Sr2LiSiO4H and Sr2LiSiO4F, indicating that the degree of crystal field splitting for excitation energy was comparable in these two structures. This similarity is due to the identical charge and similar ionic radii of F− and H−. Therefore, the downshifting of the Eu 5d level corresponding to the redshift of PLE band in Sr2LiSiO4H should be attributed to the difference in the degree of centroid shift. As stated in introduction section, hydrogen has a lower electronegativity than fluoride and can give the strong nephelauxetic effect inducing large centroid shift which presents as the right shift of absorption band redshift in Sr2LiSiO4H:Eu2+. The present findings illustrate the potential of hydride ion substitution in oxyfluoride phosphor in inducing a red shift of the absorption bands. In a very recent study by Gehlhaar et al.42 on the synthesis and photoluminescence property of Sr2LiSiO4H:Eu2+, the authors also observed red shifts of the PL band induced by substitution of F− with H−. However, our research demonstrates that the red shifting of absorption band due to the strong nephelauxetic effect giving by hydride ligands in hydride analogue of oxyfluoride compounds are possibly predicted by cDFT calculation.
Conclusions
Sr2LiSiO4H was synthesized by high-temperature reaction of oxide and hydride precursors in H2 gas at 0.9 MPa. The synthesized compound was stable in air and the Eu2+-doped analog displayed strong yellow photoluminescence upon excitation by near-UV light. The cDFT calculations successfully simulated the lowering of the Eu 5d level upon H− substitution, and the calculated red shifts of the PL and PLE energies were consistent with the experimentally observed red shifts. In this oxyhydride, the mixed ligand sphere containing hydride gives the nephelauxetic effect stronger than that in oxyfluoride analogue, which influences on the centroid shift of the Eu 5d levels to lower energy region. The present findings demonstrate that the hydride ligand can be used to design novel phosphor materials and modify PL properties of conventional oxide-based materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the element strategy initiative of MEXT Japan and JSPS KAKENHI (Grant Numbers 16K05934 and 16H06441). The neutron experiments at the Materials and Life Science Experimental Facility of the J-PARC were performed under a user program (Proposal No. 2014S06).References
- N. H. Nickel, R. K. Willardson and E. R. Weber, Hydrogen in semiconductors II, 1999, vol. 61 Search PubMed.
- H. Kageyama, K. Hayashi, K. Maeda, J. P. Attfield, Z. Hiroi, J. M. Rondinelli and K. R. Poeppelmeier, Nat. Commun., 2018, 1, 772 CrossRef PubMed.
- S. Matsuishi, K. Hayashi, M. Hirano and H. Hosono, J. Am. Chem. Soc., 2005, 127, 12454–12455 CrossRef CAS PubMed.
- G. Kobayashi, Y. Hinuma, S. Matsuoka, A. Watanabe, M. Iqbal, M. Hirayama, M. Yonemura, T. Kamiyama, I. Tanaka and R. Kanno, Science, 2016, 351, 1314–1317 CrossRef CAS PubMed.
- M. C. Verbraeken, C. Cheung, E. Suard and J. T. S. Irvine, Nat. Mater., 2015, 14, 95–100 CrossRef CAS PubMed.
- K. Hayashi, S. Matsuishi, T. Kamiya, M. Hirano and H. Hosono, Nature, 2002, 419, 462–465 CrossRef CAS PubMed.
- T. Hanna, Y. Muraba, S. Matsuishi, N. Igawa, K. Kodama, S. I. Shamoto and H. Hosono, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 024521 CrossRef.
- K. Bando, K. Sakano, Y. Noguchi and Y. Shimizu, J. Light Visual Environ., 1998, 22, 2–5 CrossRef.
- S. Ye, F. Xiao, Y. X. Pan, Y. Y. Ma and Q. Y. Zhang, Mater. Sci. Eng., R, 2010, 71, 1–34 CrossRef.
- N. Kunkel, H. Kohlmann, A. Sayede and M. Springborg, Inorg. Chem., 2011, 50, 5873–5875 CrossRef CAS PubMed.
- P. Dorenbos, J. Lumin., 2003, 104, 239–260 CrossRef CAS.
- H. Daicho, Y. Shinomiya, K. Enomoto, A. Nakano, H. Sawa, S. Matsuishi and H. Hosono, Chem. Commun., 2018, 54, 884–887 RSC.
- A. L. Allred, J. Inorg. Nucl. Chem., 1961, 17, 215–221 CrossRef CAS.
- N. Kunkel and H. Kohlmann, J. Phys. Chem. C, 2016, 120, 10506–10511 CrossRef CAS.
- N. Kunkel, A. Meijerink and H. Kohlmann, Phys. Chem. Chem. Phys., 2014, 16, 4807–4813 RSC.
- N. Kunkel, A. D. Sontakke, S. Kohaut, B. Viana and P. Dorenbos, J. Phys. Chem. C, 2016, 120, 29414–29422 CrossRef CAS.
- N. Kunkel, A. Meijerink and H. Kohlmann, Inorg. Chem., 2014, 53, 4800–4802 CrossRef CAS PubMed.
- G. Lefevre, A. Herfurth, H. Kohlmann, A. Sayede, T. Wylezich, S. Welinski, P. D. Vaz, S. F. Parker, J. Franc, P. Goldner and N. Kunkel, J. Phys. Chem. C, 2018, 122, 10501–10509 CrossRef CAS.
- N. Kunkel, D. Rudolph, A. Meijerink, S. Rommel, R. Weihrich, H. Kohlmann and T. Schleid, Z. Anorg. Allg. Chem., 2015, 641, 1220–1224 CrossRef CAS.
- D. Rudolph, D. Enseling, T. Jüstel and T. Schleid, Z. Anorg. Allg. Chem., 2017, 643, 1525–1530 CrossRef CAS.
- J. Ueda, S. Matsuishi, T. Tokunaga and S. Tanabe, J. Mater. Chem. C, 2018, 6, 7541–7548 RSC.
- T. Hanna, S. Matusishi, K. Kodama, T. Otomo, S. I. Shamoto and H. Hosono, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 020401 CrossRef.
- K. Hayashi, P. V. Sushko, Y. Hashimoto, A. L. Shluger and H. Hosono, Nat. Commun., 2014, 5, 1–8 CAS.
- X. Zhang and J. S. Kim, Appl. Phys. A: Mater. Sci. Process., 2009, 97, 549–552 CrossRef CAS.
- A. Akella and D. A. Keszler, Chem. Mater., 1995, 7, 1299–1302 CrossRef CAS.
- V. Sivakumar and U. V. Varadaraju, J. Electrochem. Soc., 2009, 156, J179–J184 CrossRef CAS.
- C. E. Messer, J. Solid State Chem., 1970, 2, 144–155 CrossRef CAS.
- R. Oishi, M. Yonemura, Y. Nishimaki, S. Torii, A. Hoshikawa, T. Ishigaki, T. Morishima, K. Mori and T. Kamiyama, Nucl. Instrum. Methods Phys. Res., Sect. A, 2009, 600, 94–96 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B, 1994, 50, 17953–17979 CrossRef.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. Klime, D. R. Bowler and A. Michaelides, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 195131 CrossRef.
- Z. Wu and R. E. Cohen, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 235116 CrossRef.
- M. Van Schilfgaarde, T. Kotani and S. Faleev, Phys. Rev. Lett., 2006, 96, 226402 CrossRef CAS PubMed.
- P. Kubelka, J. Opt. Soc. Am., 1948, 38, 448–457 CrossRef CAS PubMed.
- J. Tauc, R. Grigorovici and A. Vancu, Phys. Status Solidi, 1966, 15, 627–637 CrossRef CAS.
- W. M. Yen, S. Shigeo and H. Yamamoto, Phosphor Handbook, 2012 Search PubMed.
- P. Dorenbos, Phys. Rev. B, 2000, 62, 15640–15649 CrossRef CAS.
- P. Dorenbos, Phys. Rev. B, 2000, 62, 15650–15659 CrossRef CAS.
- P. Dorenbos, Phys. Rev. B, 2002, 65, 235110 CrossRef.
- P. Dorenbos, Phys. Rev. B, 2001, 64, 125117 CrossRef.
- F. Gehlhaar, R. Finger, N. Zapp, M. Bertmer and H. Kohlmann, Inorg. Chem., 2018, 57, 11851–11854 CrossRef CAS PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra08344d |
| This journal is © The Royal Society of Chemistry 2019 |