
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGelation and luminescence of lanthanide hydrogels formed with deuterium oxide†
Yusuke Zamaa,
Kazushige Yanaib,
Juri Takeshitab,
Ayumi Ishiiacde,
Masamichi Yamanaka *be and
Miki Hasegawa*ae
*be and
Miki Hasegawa*ae
aCollege of Science and Engineering, Aoyama Gakuin University, 5-10-1 Fuchinobe, Chuo-ku, Sagamihara, Kanagawa 252-5258, Japan. E-mail: haesmiki@chem.aoyama.ac.jp; Fax: +81-42-759-6221
bDepartment of Chemistry, Faculty of Science, Shizuoka University, 836 Ohya, Suruga-ku, Shizuoka 422-8529, Japan. E-mail: yamanaka.masamichi@shizuoka.ac.jp
cJST, PRESTO, 4-1-8 Honcho, Kawaguchi, Saitama 332-0012, Japan
dGraduate School of Engineering, Toin University of Yokohama, Kurogane-cho, Aoba-ku, Yokohama, Kanagawa 225-8503, Japan
eMirai Molecular Material Design Institute, Aoyama Gakuin Universitiy, 5-10-1 Fuchinobe, Chuo-ku, Sagamihara, Kanagawa 252-5258, Japan
First published on 15th January 2019
Abstract
Gel formations and efficient lanthanide luminescence appeared in deuterium oxide (D2O) medium instead of light water (H2O), and their solvation possibilities by using luminescence lifetimes were discussed. The lanthanide ions in the hydrogel of 1 obtained by H2O (abbreviated as H2O-Ln1; Ln = Eu, Tb, and Gd) in our previous report act as the coupling part between neighbor molecules for the bundle structure. Here, D2O also acts as a medium to form the lanthanide-hydrogel of 1, and increases intensities of luminescence for Tb, because a soft crystalline state reducing resonance thermal relaxation is realized. The gel-formation and luminescence band positions of Ln1 in D2O corresponded to those in H2O. From the observation of luminescence lifetimes in H2O and D2O, the number of coordinating water molecules on Eu and Tb were estimated to be around 3 or 4 for both. The luminescence intensity of Eu1 did not increase even in D2O, due to a blue shift of the excited triplet state of 1, as compared to that in H2O.
Introduction
Deuterium oxide is a key matrix to induce ff-emission regarding trivalent lanthanide (abbreviated to Ln) complexes.1–5 For instance, Bruce et al. cordially explained the influence of the vibrational mode of a hydrogen/deuterium–oxygen for the enhancement or quenching of luminescence of Ln3+ such as Tb3+ and Eu3+ in their coordination compounds. The ff-emissions of Ln3+ are sensitized by the antenna effect by the coordination of π-electronic systems with high absorption coefficient. Lanthanide luminescent materials have recently attracted considerable attention, due to their brightness and sharp bands.1–4 For instance, trivalent Tb in a complex exhibits green emission, which originates from ff transitions that have no influence on any organic ligands because they exist independently in the core of the atom.5 Organic ligands act as photo-antennas to provide the excitation energy to the central metal. Additionally, the luminescence intensities of Tb complexes show temperature dependence, due to reverse energy transfer.6The combination of two hetero Ln ions such as Tb and Eu with organic components can accomplish colour mixing of luminescence, and induce inter-metal energy transfer from Tb to Eu by UV excitation of the ligand moiety.7–9 Such a stepwise energy transfer is also useful for developing luminescent sensors or devices. Recently, we reported mixed Tb/Eu systems in chain-structured complexes consisting of helical Ln complexes (abbreviated as LnL; Ln = Eu or Tb) linked by analogues of 1,4-benzenedicarboxylate (abbreviated as bdc).10 The initial compound LnL-bdc showed no inter-metal energy transfer between the energy donor (ligand) and acceptor (centre metal ion), whereas bdc substituted with NH2– or OH– induced inter-metal energy transfer in the chain complexes, and resulted in reddish-orange emission even with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of EuL and TbL, due to its amorphous structure.
:1 mixture of EuL and TbL, due to its amorphous structure.
It was reported that the intensity change of lanthanide luminescence is affected by O–H/O–D or C–H/C–D vibrational mode, due to harmonic resonance with the luminescent level in lanthanide ions.11,12 It is known that the quenching mechanism for trivalent Eu and Tb involves resonance of the third and fourth overtones of O–H vibrations (ca. 3400 cm−1) in H2O, respectively.13–15 We recently reported the effect of deuterium on the luminescent components of H/D-phenanthroline complexes with Eu, Tb, and Tm complexes, according to the appearance of polymorphism detected by the synchrotron X-ray powder diffraction (XRPD).13
A water-soluble lanthanide complex of helicate structure with π-electronic systems and two carboxyl groups, LnLCOOH also showed the change in luminescent intensity, when using D2O instead of H2O.16 LnLCOOH retains its molecular and spectral stability in a wide pH range of 2.6–9.7.
To overcome the quenching problem related to Tb or Eu in H2O, some investigators reported using a capsulated molecular structure to prohibit solvent molecules from reaching Ln ions.17,18 A Ln complex (Ln = Eu, Tb, Dy, and Sm) with a 2-hydroxyisophthalamide derivative with a multidentate ligand has water solubility, and shows a 60% luminescence quantum yield for Ln = Tb, even in water.17 Cluster-type Zn complexes with Ln [Zn8(ad)4(BPDC)6O·2Me2NH2, 8DMF, 11H2O] (ad = adeninate; BPDC = biphenyldicarboxylate; DMF = dimethylformamide) form the metal–organic frameworks and show Ln luminescence even in water.18
Supramolecular gels, which are formed by self-assembly of small molecules, have attracted interest in recent decades because of their wide applicability.19,20 Some luminescent lanthanide complexes in organogels and hydrogels have been developed and reported for applications such as paintable luminescent thermo sensors and color tunable LED fibers.21–28 We also recently succeeded in obtaining bright luminescent Tb complexes with tris-urea-based hydrogelator 1 in H2O.29 The gelator shown in Fig. 1 has multiple roles in hydrogel formation, by coordinating with cations such as protons, divalent calcium, trivalent Ln ions, and serving as a photo-antenna to transfer excitation energy to Ln ions. Compound 1 also shows Ln selectivity in luminescence; the hydrogel of the lanthanide complex with 1 obtained with H2O (abbreviated as H2O-Ln1; Ln = Eu, Tb, and Gd) shows different luminescence quantum yields based on the Ln; for H2O-Eu1 and H2O-Tb1, the quantum yields are <0.1% and 5.4%, respectively. It was recently revealed that the abovementioned lanthanide-including hydrogels of 1 act as media for organic fluorophores such as Rhodamine 6G,30 and the interaction between the gelator and fluorophore was clearly enhanced in the luminescence as well as its lifetime.
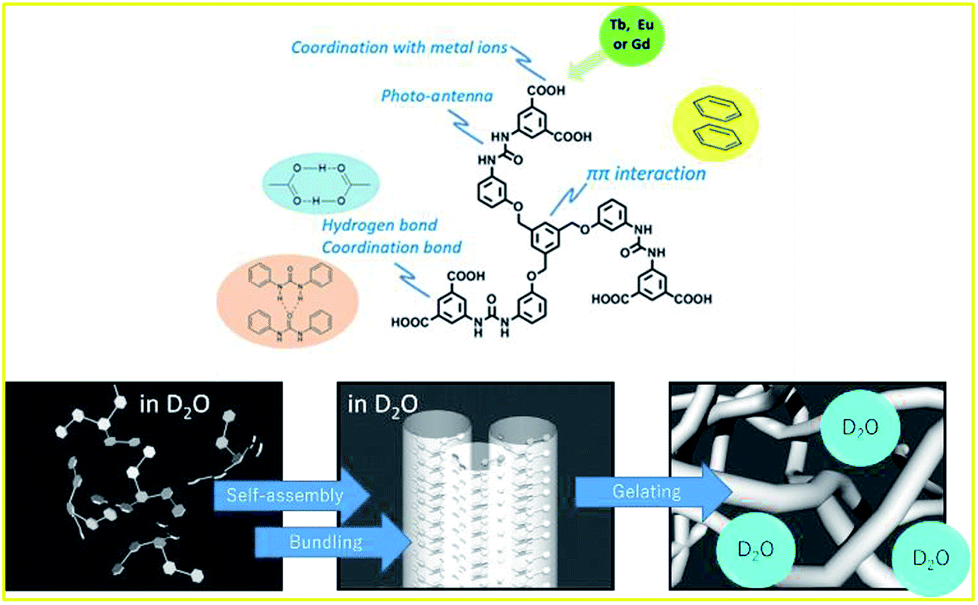 | ||
| Fig. 1 Role of substances of 1 in the metal gelation (top), and schematic representation of gel formation in D2O (bottom). | ||
Heavy water (D2O) has been considered to be nearly equivalent to light water (H2O), due to its physical properties in supramolecular hydrogels. Therefore, nuclear magnetic resonance (NMR) and small-angle neutron scattering (SANS) experiments have been performed in D2O as an alternative to H2O.31 The similarities and dissimilarities of D2O and H2O are still unclear, and more study is required to clarify them. Here, we report a heavy water effect in the luminescence of a series of D2O-Ln1 based on measurements of electronic absorption, excitation, and luminescence spectra. Furthermore, the inter-metal energy transfer between Eu and Tb was induced in D2O-Ln1. The luminescence lifetime and quantum yield were also measured to evaluate the luminescence efficiency in these hydrogels with Ln.
Results and discussion
Hydrogel formation and structure of D2O-Ln1
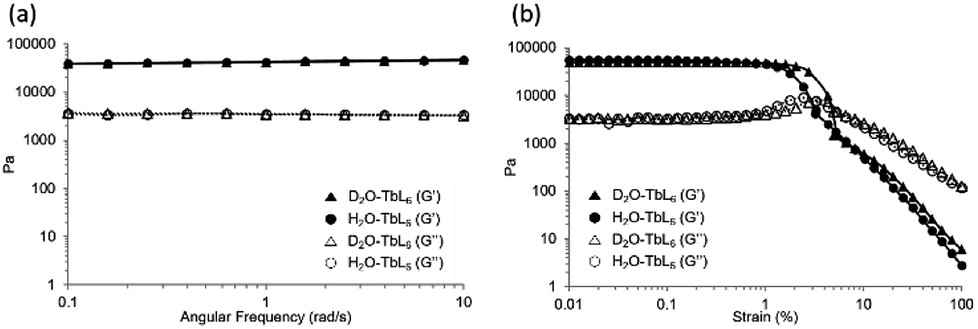 | ||
| Fig. 2 G′ and G′′ values for D2O-Tb1 (triangles) and H2O-Tb1 (circles) (a) with a frequency sweep; (b) with a strain sweep. | ||
:1 mixture of hydrogels containing Eu1 and Tb1 is shown in Fig. 3. The peaks for Eu and Tb appear at 1.15 and 1.25 keV, respectively. The component ratios of elements are C, 73.8%; N, 9.8%; O, 13.8%; Tb, 1.9%; and Eu, 1.7%. Tb and Eu are localized on the fiber in equal amounts, which is consistent with the results of a previous study.29
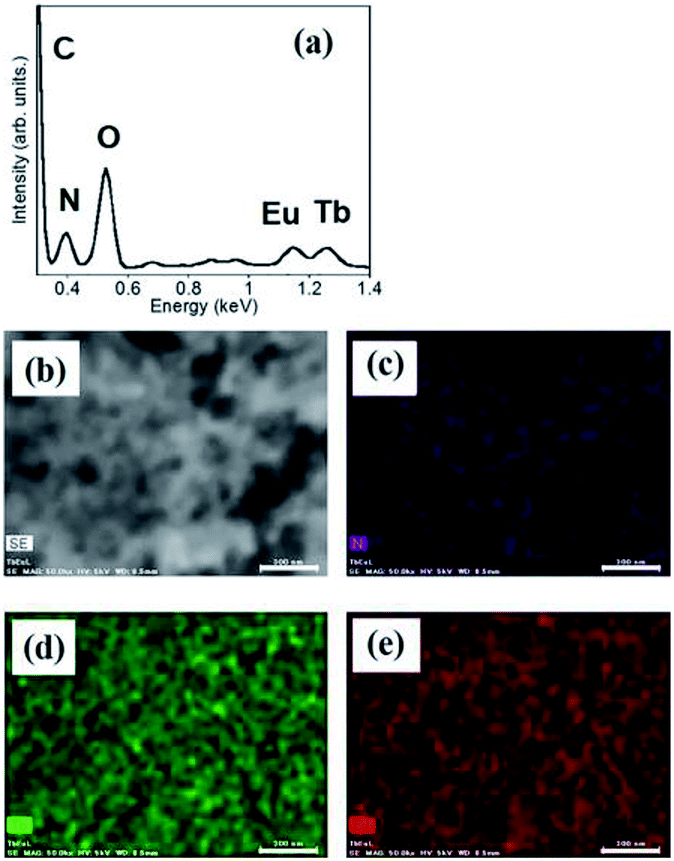 | ||
| Fig. 3 EDX spectrum (a), SEM image (b), and element maps ((c) N; (d) Tb and (e) Eu) of the xerogel obtained from hydrogels by mixing of Eu1 and Tb1. Scale bar shows 100 μm. | ||
Luminescence properties of D2O-Ln1
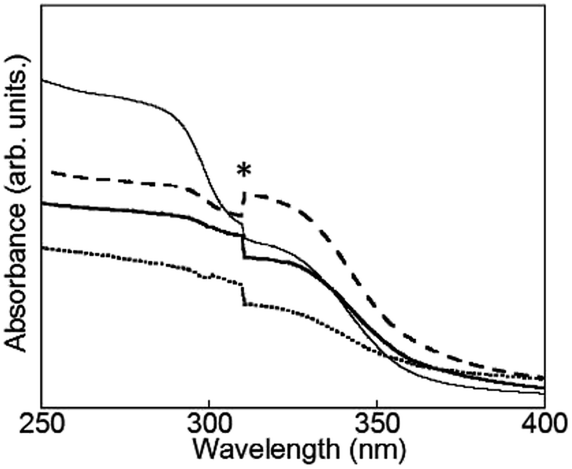 | ||
| Fig. 4 Electronic absorption spectra of D2O-Tb1 (solid line), D2O-Eu1 (dotted line), D2O-Gd1 (dashed line), and D2O-H1 (thin line). * Due to apparatus. | ||
Luminescence spectra of D2O-Tb1 and D2O-Eu1 are shown in Fig. 5. D2O-Tb1 shows sharp luminescence bands at 489, 544, 583, and 621 nm assigned to the 5D4 → 7F6, 5D4 → 7F5, 5D4 → 7F4, and 5D4 → 7F3 transitions, respectively. The excitation spectra monitored at these luminescence band positions correspond to those for the electronic absorption spectra (Fig. S1†). Luminescence and excitation bands for D2O-Tb1 reproduced those of H2O-Tb1, indicating that gelator 1 coordinates to the Ln ions while maintaining a suitable distance, to induce excitation energy transfer even in D2O-Tb1.
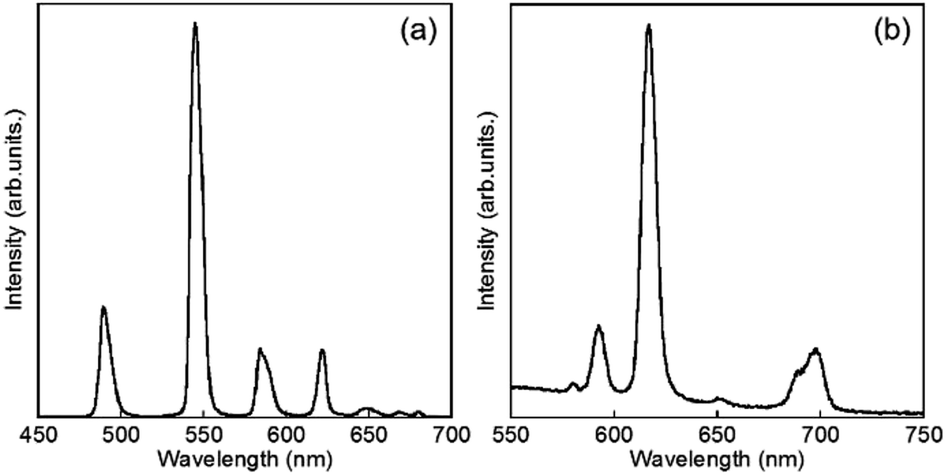 | ||
| Fig. 5 Luminescence spectra of D2O-Tb1 (a) and D2O-Eu1 (b). λex = 323 nm. | ||
To evaluate the luminescence of D2O-Tb1 quantitatively, luminescence quantum yields and lifetimes were measured and are summarized in Table 1. From the luminescence decay curves monitored at 545 nm for both D2O-Tb1 and H2O-Tb1 (Fig. 6), it is clear that the luminescence decay for Tb in D2O-Tb1 was much slower than that for H2O-Tb1. This indicates that the luminescence rate is influenced by the vibrational mode of the water medium. Based on the decomposition of the decay curves, D2O-Tb1 has at least three luminescent components, whereas the corresponding components in H2O-Tb1 were shorter than those in D2O-Tb1. Surprisingly, the longest luminescence lifetime was for D2O-Tb1. A typical value in terbium complexes in the solid state is ca. 1.5–1.7 ms. Thus, deuterium oxide accelerates Tb luminescence efficiently in the gel of D2O-Tb1, and this is known as the heavy water effect. The luminescence quantum yield for D2O-Tb1 is twice that for H2O-Tb1, which also supports the above conclusion. Thus, heavy water acts as an accelerator for Tb luminescence even in hydrogels.
| Quantum yields | Lifetimes/ms (amp); λex = 340 nmc | |||
|---|---|---|---|---|
| τ1 | τ2 | τ3 | ||
| a λobs = 450–700 nm.b λobs = 550–750 nm.c λmon = 545 and 616 nm for Tb and Eu, respectively. | ||||
| D2O-Tb1 | 10%a | 0.20(14%) | 0.83(41%) | 1.7(45%) |
| H2O-Tb1 | 5.4%a | 0.08(8%) | 0.37(37%) | 0.70(54%) |
| D2O-Eu1 | <0.1%b | 0.32(25%) | 1.06(75%) | — |
| H2O-Eu1 | <0.1%b | 0.10(23%) | 0.22(77%) | — |
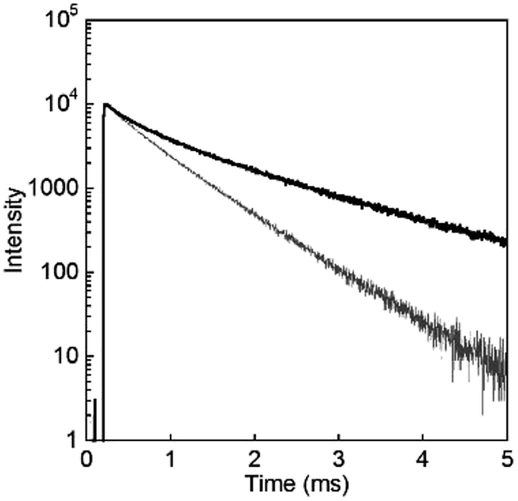 | ||
| Fig. 6 Luminescence decay profiles for D2O-Tb1 (solid line) and H2O-Tb1 (thin grey line). λmon = 545 nm and λex = 323 nm. | ||
D2O-Eu1 also exhibits luminescence bands at 580, 592, 616, 650, and 697 nm, which are assigned to the 5D0 → 7F0, 5D0 → 7F1, 5D0 → 7F2, 5D0 → 7F3, and 5D0 → 7F4 transitions, respectively. These band positions are mostly the same for H2O-Eu1 (Fig. S2†). The luminescence quantum yields for Eu in D2O-Eu1 and H2O-Eu1 are quite low (Table 1). The luminescence decay profile for D2O-Eu1 was divided into two components, whose lifetimes are longer than those of H2O-Eu1. This is in contrast to the two gel complexes with Tb, and may be also due to the vibrational mode of water as the medium.
The number of coordinating water molecules on Ln ions in hydrogels can be estimated using the experimental Horrocks' equation for the lifetime:32–35
| qLn = x(1/τH2O − 1/τD2O) | (1) |
| τ = (a1τ12 + a2τ22)/(a1τ1 + a2τ2) | (2) |
To clarify the energy relaxation process for the water vibrational mode, ligand-centred emissions such as fluorescence and phosphorescence were observed. Fig. 7 shows fluorescence spectra of D2O-H1 and phosphorescence spectra of D2O-Gd1. The compounds D2O-H1 and D2O-Tb1 also formed stable hydrogels similar to D2O-Tb1. The use of Gd ion instead of a luminescent Ln ion is the conventional means for the determination of the singlet and triplet states as the energy donor in the π-electron system.37 In our previous report, the fluorescence and phosphorescence bands for 1 using the hydrogel H2O-Gd1 were at 363 and 465 nm, respectively.29 The corresponding bands appear at 363 (27500 cm−1) and 477 (21000 cm−1) nm, respectively, in the present compounds. The 477 band acts as an energy donor to Tb or Eu in hydrogels. The phosphorescence band position for D2O-Gd1 is lower than that for H2O-Gd1 by 12 nm, and may affect the energy transfer efficiency of D2O-Eu1 which has acceptor bands around 394 or 416 nm.38 Additionally, H2O-Tb1 and D2O-Tb1 show broad luminescence bands in laser Raman spectra at 637 (15700 cm−1) and 625 (16000 cm−1) nm, respectively (Fig. 8). These bands do not correspond to the fluorescence and phosphorescence in Fig. 6, suggesting that the luminescence bands are caused by strong intermolecular interactions by the formation of supramolecular hydrogels. It is noteworthy that the band position for self-assemblies of the gelator covers the Eu luminescence bands. In other words, the O–D vibrational mode intensified the luminescence of Tb in D2O-Tb1. In contrast, the weakened luminescence for Eu in D2O-Eu1 was caused by the O–D vibration in deuterium oxide and by superimposition of the emission of self-assembly in the gelator and luminescence of Eu.
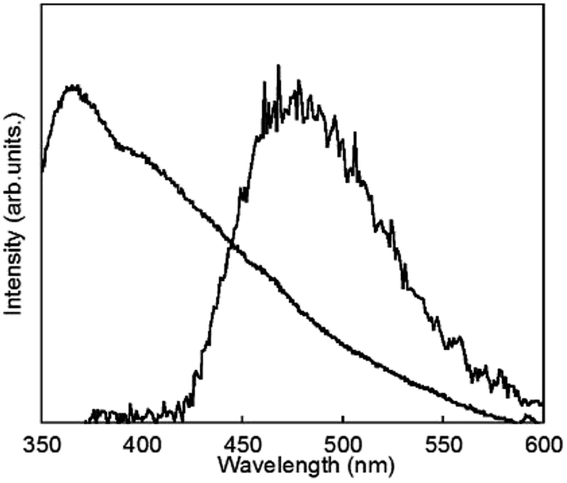 | ||
| Fig. 7 Fluorescence spectrum of D2O-H1 (shorter wavelength band and phosphorescence spectrum at 77 K) and of D2O-Gd1 (longer wavelength band) localized on 1 (λex = 323 nm). | ||
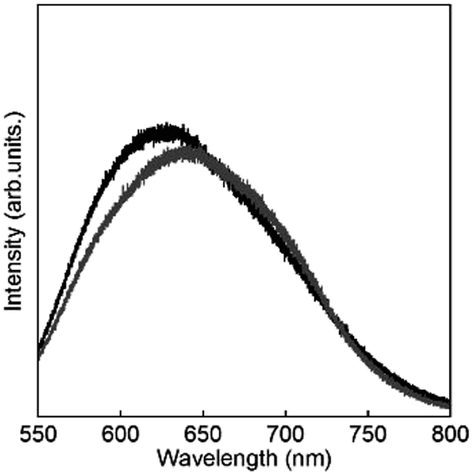 | ||
| Fig. 8 Luminescence spectra of D2O-Tb1 (black line) and H2O-Tb1 (grey line) observed by Raman spectroscopy (λex = 532 nm). | ||
:1 mixed hydrogels of Eu and Tb with 1 in H2O and D2O, abbreviated as H2O-Tb/Eu1 and D2O-Tb/Eu1, respectively. From the EDX measurement as described above, nitrogen in 1 was also found at the similar position with the metal ions. This supports luminescence effects such as inter-metal interactions between Tb and Eu in the mixed hydrogel of Eu1 and Tb1.Fig. 9 shows luminescence spectra of hydrogels containing Tb and Eu ions in a 1:1 ratio, which are abbreviated as H2O-Tb/Eu1 and D2O-Tb/Eu1 for light and heavy water, respectively. Bands at 544, 545, 584, 585, 620, 650(sh), and 679(sh) nm, assigned to the overlap of ff transitions of Tb3+ and Eu3+, are observed for H2O-Tb/Eu1. The corresponding bands for D2O-Tb/Eu1 appear at 544, 584, 590, 615, 649, and 696 nm, respectively.
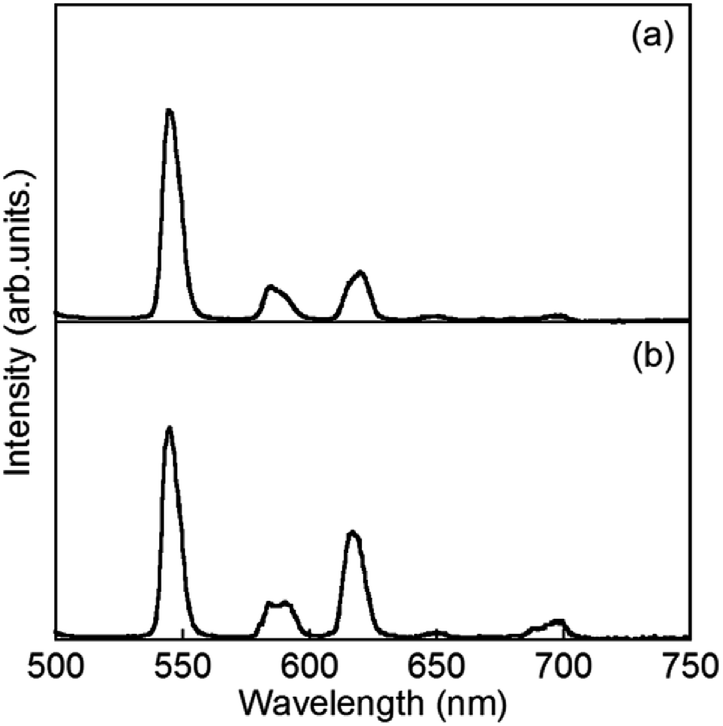 | ||
| Fig. 9 Luminescence spectra of H2O-Tb/Eu1 and D2O-Tb/Eu1 (λex = 323 nm). | ||
The luminescence decay profile of H2O-Tb/Eu1 was monitored at both the emission band positions of Tb and Eu (Fig. S3† and Table 2). The decay curve of H2O-Tb/Eu1 monitored at 545 nm for Tb was divided into three luminescent components, and that at 616 nm for Eu into two. In the luminescence decay profile monitored at 616 nm for H2O-Tb/Eu1, the luminescence rise curve for Eu was obtained and resulted in the time as 0.15 ms because the time range corresponds to the decay time of 0.19 ms for Tb. These results are consistent with Tb3+ ions transferring the excitation energy to Eu3+ in the mixed metal hydrogels. It also demonstrated that Tb and Eu would exist sufficiently close together, so as to promote metal-to-metal energy transfer in gels.39 Similar metal-to-metal energy transfer was also observed in the xerogel obtained from hydrogels with both Tb and Eu. It should note to worth that the number of luminescent decay component of Eu in D2O-Tb/Eu1 is one; however, the rise in luminescence lifetime of 0.29 ms is higher than that in H2O-Tb/Eu1. It suggests that the metal-to-metal energy transfer from Tb to Eu in the gel with D2O is more efficient than that with H2O.
| Quantum yielda | Lifetimes/ms (amp.) | |
|---|---|---|
| a λobs = 450–700 nm.b λmon = 545 nm for Tb.c λmon = 616 nm for Eu.d Minus values in lifetimes represent the rise times of luminescence. | ||
| H2O-Tb/Eu1 | <0.1% | 0.53 (40%), 0.19 (30%), 0.018 (30%)b |
| 0.51 (24%), 0.20 (76%), −0.15c | ||
| D2O-Tb/Eu1 | 2% | 1.20 (35%), 0.41 (33%), 0.034 (32%)b |
| 1.40 (100%), −0.29c | ||
| Xerogel of Tb/Eu1 | <0.1% | 0.47 (36%), 0.14 (28%), 0.026 (36%)b |
| 0.50 (22%), 0.23 (78%), −0.081c | ||
The total luminescence quantum yield of Tb/Eu1 was observed in the Tb and Eu emission region (500–750 nm). The quantum yield of D2O-Tb/Eu1 was 2%, which is higher than that of H2O-Tb/Eu1 and xerogel (<0.1%). These trends support the above discussion.
The xerogel obtained from hydrogels of Tb/Eu1 showed the luminescence spectra and quantitative properties similar to those of the initial compound. It is also consistent that heavy water in this hydrogel system plays a role in enhancing lanthanide luminescence and inter-metal energy transfer. The increase in the φ value for the xerogel at 77 K means the increase in forwarded energy transfer from 1 to the Tb ion, because of the existence of thermal equilibrium between the donor and accepter levels in the Tb complexes.7,9,40
The energy diagram for Tb/Eu1 systems is shown in Fig. 10. The excited triplet state of 1 acts as an energy donor to the terbium with less contribution of backward energy transfer.29 However, the energy difference between 1 and the Eu ion is not suitable for efficient energy transfer. The mixing of Tb and Eu in the hydrogel formed with D2O accomplished acceleration of Eu emission by Tb, through metal–metal energy transfer in D2O-Tb/Eu1.
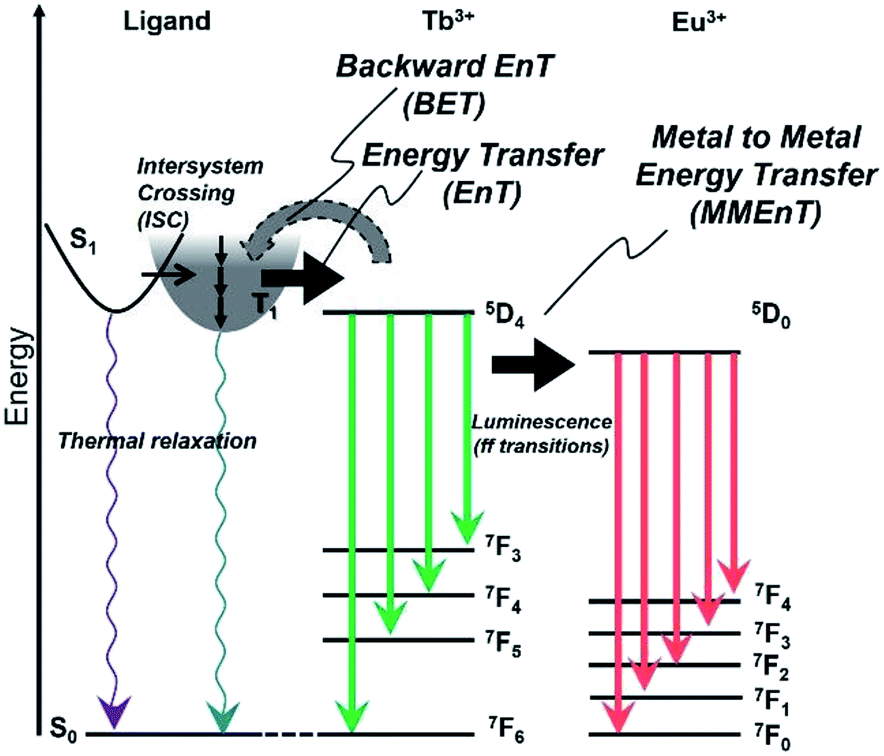 | ||
| Fig. 10 Schematic representation of energy diagram for 1 and Tb-sensitizing of the Eu luminescence in hydrogels. | ||
Experimental
Sample preparation
The gelator 1 and the gel containing Ln ions and D2O were prepared as given in the previous paper using H2O.29,30 Lanthanide-containing hydrogels were obtained from light water and D2O (Isotech Inc.), and lanthanide nitrates were used as the sources of metal ions (Ln = Eu, Tb and Gd). The 1:1 mixed hydrogels using Eu and Tb were also obtained by the mixing of europium nitrate and terbium nitrate equivalently. To obtain a metal-free hydrogel, hydrochloride and D2O were used.
Apparatus
Rheological measurements were carried out on a DHR-2 rheometer (TA Instruments), using a flat plate geometry (8 mm). Electronic absorption and luminescence spectra were recorded using a Shimadzu 3600S UV-VIS-NIR spectrophotometer and a Horiba Jovin-Yvon Fluorolog 3-22 spectrofluorometer, respectively. Luminescence quantum yields and lifetimes were measured by a Hamamatsu 9920-02 absolute photoluminescence quantum yield spectrometer and Quantaurus tau fluorescence lifetime spectrometer, respectively. SEM images were obtained using an Ultra-55 scanning electron microscope (Carl Zeiss AG) equipped with a secondary in-lens electron detector, together with a QUANTAX detector (Bruker Corporation) for EDX. The micro Raman spectra were obtained using Renishow via a Reflex spectrometer equipped with a 532 nm diode laser, calibrated to the 520.5 cm−1 line of silicon.Conclusion
Three types of lanthanide ions aggregate to form hydrogels with 1, even in heavy water. The viscoelastic property of the hydrogel D2O-Tb1 was identical to that of H2O-Tb1, whereas deuterium oxide in the gel D2O-Tb1 enhanced the efficiency of ff emission of Tb ion. There was no influence on the luminescence of D2O-Eu1 by heavy water. The fluorescence and phosphorescence bands localized on 1 were observed using D2O-H1 (metal free; prepared by using hydrochloride) and D2O-Gd1, respectively. Raman spectra supported the weak luminescence of Eu in this system. Finally, Eu emission was enhanced in the mixed hydrogel D2O-Tb/Eu1 through inter-metal energy transfer. These results will provide new aspects for designing hydrogelators in heavy water.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors acknowledge Grants-in-Aid for Scientific Research on Innovative Areas: KAKENHI “Soft Crystals (Area Number: 2903)”, No. 17H06374 for M. H., “Fusion Materials (Area Number: 2206)”, No. 25107730 for M. H., and No. 25107713 for M. Y. from Japan Society for the Promotion of Science, and Challenging Exploratory Research Centre Project for Private University and a matching fund subsidy from MEXT (2013–2017 for M. H.). M. H. also thanks Izumi Science and Technology Foundation, and Aoyama Gakuin University Soken Project for their support. EDX and SEM images measurements were supported at Centre for Instrumental Analysis, College of Science and Engineering, Aoyama Gakuin University.References
- J. L. Kropp and M. W. Windsor, J. Phys. Chem., 1963, 39, 2769 CrossRef CAS.
- J. I. Bruce, M. P. Lowe and D. Parker, in The Chemistry of Contrast Agents in Medical Magnetic Resonance Imaging, ed. A. E. Merbach and E. Tóth, John Wiley and Sons, 2001, ch 11, p. 437 Search PubMed.
- J. C. Bünzli and C. Piguet, Chem. Soc. Rev., 2005, 34, 1048 RSC.
- S. V. Eliseeva and J. C. Bünzli, Chem. Soc. Rev., 2010, 39, 189 RSC.
- J. C. Bünzli, Chem. Rev., 2010, 110, 2729 CrossRef PubMed.
- L. Prodi, M. Maestri, R. Ziessel and V. Balzani, Inorg. Chem., 1991, 30, 3798 CrossRef CAS.
- M. Latva, H. Takalo, V. Mukkala, C. Matachescu, J. C. Rodriguez-Ubis and J. Kankare, J. Lumin., 1997, 75, 149 CrossRef CAS.
- K. Miyata, Y. Konno, T. Nakanishi, M. Kobayashi, M. Kato, K. Fushimi and Y. Hasegawa, Angew. Chem., Int. Ed., 2013, 52, 6413 CrossRef CAS PubMed.
- M. Hatanaka, Y. Hirai, Y. Kitagawa, T. Nakanishi, Y. Hasegawa and K. Morokuma, Chem. Sci., 2017, 8, 423 RSC.
- S. Sato, A. Ishii, C. Yamada, J. Kim, C. H. Song, A. Fujiwara, M. Takata and M. Hasegawa, Polym. J., 2015, 47, 195 CrossRef CAS.
- A. Beeby, I. M. Clarkson, R. S. Dickins, S. Faulkner, D. Parker, L. Royle, A. S. de Sousa, J. A. G. Williams and M. Woods, J. Chem. Soc., Perkin Trans. 2, 1999, 3, 493 RSC.
- R. S. Dickins, D. Parker, A. S. de Sousa and J. A. G. Williams, Chem. Commun., 1996, 6, 697 RSC.
- S. Ogata, A. Ishii, C. L. Lu, T. Kondo, N. Yajima and M. Hasegawa, J. Photochem. Photobiol., A, 2017, 334, 55 CrossRef CAS.
- W. D. Horrocks and D. R. Sudnick, Acc. Chem. Res., 1981, 14, 384 CrossRef CAS.
- G. Stein and E. Würzberg, J. Chem. Phys., 1975, 62, 208 CrossRef CAS.
- S. Ogata, T. Shimizu, T. Ishibashi, Y. Ishiyone, M. Hanami, M. Ito, A. I. S. Kawaguchi, K. Sugimoto and M. Hasegawa, New J. Chem., 2017, 41, 6385 RSC.
- S. Petoud, S. M. Cohen, J. C. Bünzli and K. N. Raymond, J. Am. Chem. Soc., 2003, 125, 13324 CrossRef CAS.
- J. An, C. M. Shade, D. A. Chengelis-Czegan, S. Petoud and N. L. Rosi, J. Am. Chem. Soc., 2011, 133, 1220 CrossRef CAS PubMed.
- A. R. Hirst, B. Escuder, J. F. Miravet and D. K. Smith, Angew. Chem., Int. Ed., 2008, 47, 8002 CrossRef CAS.
- X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115, 13165 CrossRef CAS PubMed.
- K. Nishiyama, Y. Watanabe, K. Watanabe and T. Harada, Chem. Lett., 2012, 41, 1697 CrossRef CAS.
- H. Kim and J. Y. Chang, RSC Adv., 2013, 3, 1774 RSC.
- O. Kotova, R. Daly, C. M. G. dos Santos, M. Boese, P. E. Kruger, J. J. Boland and T. Gunnlaugsson, Angew. Chem., Int. Ed., 2012, 51, 7208 CrossRef CAS PubMed.
- H. Wang, X. Li, F. Fang and Y. Yang, Dalton Trans., 2010, 39, 7294 RSC.
- S. Banerjee, R. Kandanelli, S. Bhowmik and U. Maitra, Soft Matter, 2011, 7, 8207 RSC.
- M. Li, Y. Wang, Y. Chen and S. A. Zhang, Photochem. Photobiol. Sci., 2014, 13, 1025 RSC.
- T. Wang, P. Li and H. Li, ACS Appl. Mater. Interfaces, 2014, 6, 12915 CrossRef CAS PubMed.
- G. M. Peters, L. P. Skala and J. T. Davis, J. Am. Chem. Soc., 2016, 138, 134 CrossRef CAS PubMed.
- M. Yamanaka, K. Yanai, Y. Zama, J. Tsuchiyagaito, M. Yoshida, A. Ishii and M. Hasegawa, Chem.–Asian J., 2015, 10, 1299 CrossRef CAS PubMed.
- J. Takeshita, Y. Hasegawa, K. Yanai, A. Yamamoto, A. Ishii, M. Hasegawa and M. Yamanaka, Chem.–Asian J., 2017, 12, 2029 CrossRef CAS PubMed.
- Z. Li, L. E. Z. Buerkle, M. R. Orseno, K. A. Streletzky, S. Seifert, M. A. Jamieson and S. J. Rowan, Langmuir, 2009, 26, 10093 CrossRef PubMed.
- D. W. Horrocks and D. R. A. Sudnick, J. Am. Chem. Soc., 1979, 101, 334 CrossRef.
- R. M. Supkowski and W. D. Horrocks, Inorg. Chim. Acta, 2002, 340, 44 CrossRef CAS.
- M. Li and P. R. Selvin, J. Am. Chem. Soc., 1995, 117, 8132 CrossRef CAS.
- M. Kawa and J. M. Fréchet, Chem. Mater., 1998, 10, 286 CrossRef CAS.
- A. Sillen and Y. Engelborghs, J. Photochem. Photobiol., A, 1998, 67, 475 CAS.
- M. Hasegawa, H. Ohtsu, D. Kodama, T. Kasai, S. Sakurai, A. Ishii and K. Suzuki, New J. Chem., 2014, 38, 1225 RSC.
- K. Binnemans, Coord. Chem. Rev., 2015, 295, 1 CrossRef CAS.
- P. R. Matthes, C. J. Höller, M. Mai, J. Heck, S. J. Sedlmaier, S. Schmiechen, C. Feldmann, W. Schnick and K. Müller-Buschbaum, J. Mater. Chem., 2012, 22, 10179 RSC.
- S. Katagiri, Y. Hasegawa, Y. Wada and S. Yanagida, Chem. Lett., 2011, 33, 1438 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Excitation and luminescence spectra and luminescence decay profiles. See DOI: 10.1039/c8ra08504h |
| This journal is © The Royal Society of Chemistry 2019 |