
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInterspecies differences in stability kinetics and plasma esterases involved in hydrolytic activation of curcumin diethyl disuccinate, a prodrug of curcumin†
Pahweenvaj Ratnatilaka Na Bhuket ab,
Ponsiree Jithavechb,
Boonsri Ongpipattanakulcd and
Pornchai Rojsitthisak*be
ab,
Ponsiree Jithavechb,
Boonsri Ongpipattanakulcd and
Pornchai Rojsitthisak*be
aBiomedicinal Chemistry Program, Department of Biochemistry and Microbiology, Faculty of Pharmaceutical Sciences, Chulalongkorn University, Bangkok, Thailand
bNatural Products for Ageing and Chronic Diseases Research Unit, Chulalongkorn University, Bangkok, Thailand
cDepartment of Biochemistry and Microbiology, Faculty of Pharmaceutical Sciences, Chulalongkorn University, Bangkok, Thailand
dChulalongkorn University Drug and Health Products Innovation & Promotion Center, Faculty of Pharmaceutical Sciences, Chulalongkorn University, Bangkok, Thailand
eDepartment of Food and Pharmaceutical Chemistry, Faculty of Pharmaceutical Sciences, Chulalongkorn University, Bangkok, Thailand. E-mail: pornchai.r@chula.ac.th; Fax: +66-2-254-5195; Tel: +66-2-218-8310
First published on 6th February 2019
Abstract
The investigation of in vitro plasma metabolism of ester prodrugs is an important part of in vitro ADME assays during preclinical drug development. Here, we show that the in vitro metabolism including plasma stability and metabolizing enzymes of curcumin diethyl disuccinate (CDD), an ester prodrug of curcumin, in dog and human plasma are similar but markedly different from those in rat plasma. HPLC and nonlinear regression analyses indicated that the hydrolysis of CDD in plasma followed a consecutive pseudo-first order reaction. The rapid hydrolytic cleavage of CDD in rat, dog, and human plasma was accelerated by plasma esterases in the following order: rat ≫ human > dog. LC-Q-TOF/MS analysis showed that the cleavage of ester bonds of CDD is preferential at the phenolic ester. Monoethylsuccinyl curcumin is the only intermediate metabolite found in plasma metabolism of CDD in all tested species. Further investigation using different esterase inhibitors revealed that carboxylesterase is the major enzyme involved in the hydrolysis of CDD in rats while multiple plasma esterases play a role in dogs and humans. Thus, the difference in the hydrolysis rates and the metabolizing enzymes of CDD metabolism in rat, dog and human plasma observed here is of benefit to further in vivo studies and provides a rationale for designing ester prodrugs of CUR with esterase-specific bioactivation.
Introduction
Curcumin (CUR, Fig. 1A) is a yellow-orange pigment found in turmeric (Curcuma longa L.) which has been documented for its numerous biological activities including antioxidant,1 anti-inflammation2 and anticancer properties.3 CUR also demonstrated safety at high doses up to 8 g as shown in a phase II clinical trial for advanced pancreatic cancer treatment.4 However, the poor oral bioavailability of CUR due to rapid degradation in neutral to basic aqueous solutions5 and extensive metabolism6 hinders it from being developed as a therapeutic agent. Previously, we designed and synthesized an ester prodrug of CUR the so-called curcumin diethyl disuccinate (CDD, Fig. 1B) by acylating both phenolic hydroxyl groups of CUR with ethyl succinyl chloride in order to reduce the chemical degradation and phase II metabolism. CDD had better stability than CUR in phosphate buffer (pH 7.4)7 and exhibited good antinociceptive activities in central and peripheral pain models.8 In addition, a previous pharmacokinetic study of CDD in rats showed that CDD has superior tissue distribution in comparison with CUR.9 The data implied that CDD enhanced the oral bioavailability of CUR since both compounds were orally administered at the dose of 40 mg kg−1 while CDD has a molecular weight 1.7 times higher than CUR (MW 624 vs. 368, respectively). Therefore, CDD is a promising ester prodrug of CUR for further development as a drug candidate. | ||
| Fig. 1 Chemical structures of (A) curcumin (CUR), (B) curcumin diethyl disuccinate (CDD), (C) dimethylcurcumin (DMC, internal standard) and (D) monoethylsuccinyl curcumin (MSCUR). | ||
Several ester prodrugs have been developed to improve intestinal absorption and bioavailability of the parent drugs. In general, ester prodrugs undergo bioactivation by intestinal esterases after absorption across the gut. However, prodrug molecules may escape the bioconversion by intestinal esterases, enter the bloodstream as the intact prodrugs, and then are converted to active metabolites by plasma esterases. Esterases including acetylcholinesterase (AChE), butyrylcholinesterase (BChE), carboxylesterase (CES) and paraoxonase (PON, also known as arylesterase (ArE)) are known to be responsible for hydrolysis of ester prodrugs in plasma.10,11 For instance, PON1 has been shown to be the major enzyme responsible for the bioactivation of olmesartan medoxomil, an angiotensin receptor blocker, in human plasma.12
The distribution and activity of esterases in plasma vary between human and experimental animals. The high content and activity of CES are found in rat while this enzyme is not present in human and dog plasma.13,14 The activity of BChE is high in human and dog plasma but low in rat plasma.14,15 Due to interspecies differences in the distribution and activity of esterases, the plasma metabolism of ester-based drugs, in terms of rate and esterases involving the hydrolysis, may differ between animal species and human. For example, WS070117, a lipid-lowering drug candidate, was found to be rapidly hydrolyzed by CES and BChE in rat plasma but it was metabolized at the slower rates by BChE and PON in dog plasma and BChE in human plasma.16 Therefore, investigating the hydrolysis in the plasma of preclinical and clinical species is necessary for the development of ester prodrugs.
During the early stage of drug development, pharmacokinetic properties of a drug candidate are usually studied in rats. Ester prodrugs that are metabolized by CES tend to undergo extensive hydrolysis in this species because of the high abundance of CES in rat plasma. The results from a pharmacokinetic study in rats would lead to erroneous decisions for further investigation in human. For instance, we have previously performed a pharmacokinetic study of CDD in rats and showed that CDD was instantaneously converted to the active metabolite CUR after intravenous administration.17 We also found that either 10 mM bis(4-nitrophenyl) phosphate or 10 mM phenylmethanesulfonyl fluoride could inhibited ex vivo hydrolysis of CDD in rat plasma. This suggests that plasma CES may involve in the rapid clearance of CDD in rats. Due to the interspecies differences, the metabolism of CDD in human may be significantly different from rat. Therefore, the pharmacokinetic testing in alternative animal species, which has closer esterase expression and activity to human such as dog, is required to obtain more appropriate pharmacokinetic data for making go/no-go decision and appropriate allometric scaling of drug doses for human studies.
Since dogs are unlikely to be used in the early phase of drug development, the in vitro plasma metabolism studies should be a reasonable alternative for evaluating and comparing the hydrolysis of ester prodrugs between preclinical species and human. Herein, we evaluated and compared the interspecies differences in the in vitro plasma metabolism of CDD in rat, dog, and human and found that the plasma metabolism of CDD in terms of plasma stability and plasma esterases involving the metabolism in dog and human are similar but obviously different from rat.
Experimental
Chemicals and reagents
CDD (purity > 98% by HPLC) and CUR (purity > 99% by HPLC) were synthesized using previously described method with some modifications.7 For the synthesis of dimethylcurcumin (DMC, Fig. 1C; purity > 99% by HPLC), 3,4-dimethoxybenzaldehyde was used instead of vanillin. Monoethylsuccinyl curcumin (MSCUR, Fig. 1D) was synthesized using the similar method for the synthesis of CDD with some modifications (see ESI†). All synthesized compounds were structurally confirmed by NMR. HPLC grade acetonitrile was purchased from Burdick & Jackson (Ulsan, Korea). Formic acid (98–100%) was obtained from Merck (Darmstadt, Germany). Sodium fluoride (NaF) was bought from BDH Merck (Poole, Dorset, UK). Bis(4-nitrophenyl) phosphate (BNPP) was procured from Tokyo Chemical Industry (Tokyo, Japan). Ethylenediaminetetraacetic acid disodium (Na2EDTA) was bought from Ajax Finechem (NSW, Australia). Phenylmethanesulfonyl fluoride (PMSF), 1,5-bis(4-allyldimethylammoniumphenyl)pentan-3-one dibromide (BW284c51), 4-(2-aminoethyl-)benzenesulphonylfloride hydrochloride (Pefabloc® SC), N-α-tosyl-L-lysine chloromethylketone hydrochloride (TLCK), ethyleneglycol-bis(2-aminoethylether)-tetraacetic acid (EGTA), tetraisopropyl pyrophosphoramide (iso-OMPA), 5,5′-dithiobis-2-nitrobenzoic acid (DTNB) and eserine hemisulfate salt were purchased from Sigma-Aldrich (MO, USA). Potassium dihydrogen phosphate was obtained from Scharlau (Sentmenat, Spain). Ultrapure water was prepared using an Elga Maxima 21F water purification system (Veolia Water Technologies, UK). Pooled blank rat plasma was obtained from the National Laboratory Animal Center (Mahidol University, Thailand). Pooled blank beagle dog and pooled normal human plasma were purchased from Innovative Research (MI, USA). Sodium heparin was used as an anticoagulant in the obtained blank plasma.Chromatographic conditions for HPLC analysis
The chromatographic conditions were modified from the previously published method.17 The chromatographic analysis was performed on an Agilent 1200 series liquid chromatographic system (Agilent, USA) consisting of degasser unit, quaternary pump and UV detector. The HPLC system and data processing were operated using ChemStation software. The separation of analytes was achieved by a Halo C18 column (4.6 × 50 mm, 2.7 μm). The injection volume was 10 μL and the temperature of the column oven was maintained at 35 °C. The autosampler temperature was set at 10 °C. The mobile phase comprising of acetonitrile–0.2% formic acid in water (70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30, v/v) was delivered in an isocratic mode at a flow rate of 0.5 mL min−1. The UV detection was set at 400 nm and the total run time was 5 min for each injection.
:30, v/v) was delivered in an isocratic mode at a flow rate of 0.5 mL min−1. The UV detection was set at 400 nm and the total run time was 5 min for each injection.
LC-Q-TOF/MS conditions for metabolite identification
The chromatographic separation was performed on a Dionex Ultimate 3000 UHPLC system (Thermo Scientific, USA). The separation of analytes was achieved on an Acquity UPLC® BEH C18 column (2.1 × 50 mm, 1.7 μm) with an isocratic mobile phase consisting of acetonitrile–0.2% formic acid in water (70:30 v/v) at a flow rate of 120 μL min−1. The injection volume was 20 μL and the column oven temperature was set at 35 °C. The sampling needle was washed with 50% methanol in water between each injection.
The mass spectrometric analysis was conducted in a micrOTOF-Q II mass spectrometer (Bruker Daltonics, Germany) using positive ion electrospray mode. The ESI mass spectra were recorded over the range of m/z 50–1000. Mass parameters were set as follows: end plate offset – 500 V; capillary voltage – 4.5 kV; nebulizer pressure 2.5 bar; drying gas flow rate 8.0 L min−1 and drying gas temperature 220 °C. For MS/MS acquisition, the mass parameters were the same as aforementioned except for the collision energy, isolation width and mass range which were set at 20 eV, Δm/z 10 and m/z 50–700, respectively. The calibration solution (sodium formate solution) was infused at the beginning of each run and all spectra were calibrated prior to spectral analysis. Bruker HyStar 3.2 and Data Analysis 4.0 software were used for system control, data acquisition and analysis.
In vitro metabolism of CDD in rat, dog, and human plasma
The in vitro metabolism of CDD in plasma was investigated in rat, dog, and human plasma. Blank plasma samples were pre-incubated at 37 °C for 10 min. The reaction was initiated by adding a stock solution of CDD (2% of total reaction volume) into the pre-incubated plasma to obtain a final concentration at 1.5 μM. The incubation temperature was maintained during the study. The reaction was quenched by mixing a 50 μL of the plasma sample at different time with a 250 μL of acetonitrile containing 0.6 μM of DMC (an internal standard). Subsequently, the samples were subjected to centrifugation at 4000 rpm at 5 °C for 5 min. Then, the supernatants were transferred to HPLC vials and subjected to HPLC analysis. Experiments were performed in triplicates. Recoveries of CDD, CUR and DMC have been previously reported.17 The recovery of MSCUR was assumed to be similar to CDD. Because concentrations of analytes are proportional to UV-vis absorbance, peak area ratios between the analytes and DMC were used in calculating kinetic parameters by fitting to the consecutive pseudo-first order kinetic model. The degradation of CDD in phosphate buffer pH 7.4 was assessed to determine the instability due to chemical degradation. Nonlinear least square analysis was performed by Microsoft Excel software, using equations described in the ESI.† 4-Nitrophenylacetate, a general substrate of esterase enzymes, was used to verify esterase activities in plasma of all tested species.Metabolite identification
CDD at 20 μM was incubated in rat, dog, and human plasma at 37 °C for 10 s, 15 min and 5 min, respectively. The reaction was stopped by transferring a 50 μL of each sample to a 250 μL of acetonitrile in separated tubes. The resulting mixtures were centrifuged at 4000 rpm at 5 °C for 5 min. Subsequently, the supernatants were diluted 10 times with 50% acetonitrile in water and transferred to HPLC vials for LC-Q-TOF/MS analysis.Investigation of esterase enzymes involved in the metabolism of CDD in plasma
Rat, dog, and human plasma were pre-incubated at 37 °C with various esterase inhibitors for 30 min. The final concentrations of each esterase inhibitor were based on reported literatures.16,18,19 To initiate the reaction, an appropriate volume of the CDD stock solution was spiked into plasma samples to obtain a final concentration of 1.5 μM. The temperature of reaction mixtures was maintained at 37 °C for 5 s, 15 min and 5 min for rat, dog, and human plasma, respectively. The plasma samples spiked with solvents of the esterase inhibitors were used as controls. The reaction was terminated by combining a 50 μL of each sample with a 250 μL of acetonitrile containing 0.6 μM of DMC. The % inhibition was used to indicate the type of plasma esterase involved in the hydrolysis of CDD which was calculated as follows:| % inhibition = [1 − (Dwith inhibitor/Dwithout inhibitor)] × 100 |
Results
In vitro hydrolysis of CDD in rat, dog, and human plasma
Representative chromatograms of CDD hydrolysis in rat, dog, and human plasma at different time intervals are shown in Fig. 2. The hydrolysis of CDD in plasma of all species gave the formation of two metabolites at the retention time of 1.36 and 2.05 min, which were designated as M1 and M2, respectively. The peak area of CDD gradually decreased while the peak of M2 simultaneously appeared in the chromatograms. During the course of CDD hydrolysis and emergence of M2, M1 appeared in the chromatograms which was expected to be converted from M2. These metabolites were subsequently identified using LC-Q-TOF/MS as described in the following section. A small peak at the retention time about 3.45 min (in front of the peak of CDD) was identified as another tautomer of CDD, which was similar to that of other curcuminoids.20,21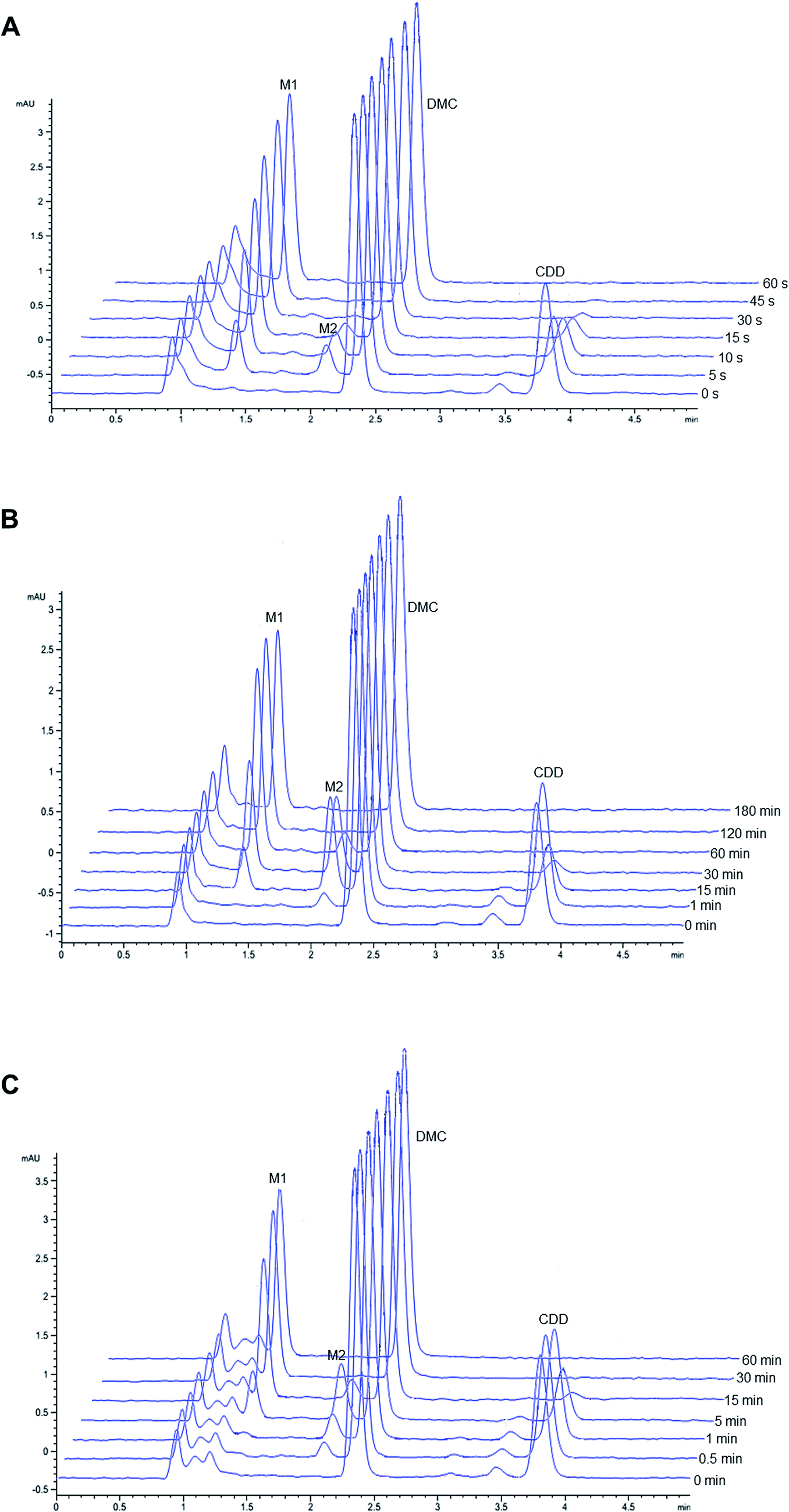 | ||
| Fig. 2 Representative chromatograms of CDD hydrolysis at 37 °C in (A) rat, (B) dog and (C) human plasma at different incubation times. | ||
The pseudo-first-order rate constants and half-lives of CDD hydrolysis in plasma are summarized in Table 1. CDD was hydrolyzed rapidly in plasma of all species. Degradation rate constants of CDD (k1) in plasma are far higher than in phosphate buffer pH 7.4 (5300, 60 and 100 times for rat, dog, and human plasma, respectively), suggesting the involvement of plasma esterases in the hydrolysis of CDD. The k1 values of CDD hydrolysis in plasma are in the following order: rat ≫ human > dog. The k1 value of CDD hydrolysis in human plasma is 50 times lower than rat but 1.6 times higher than dog. The conversion rate constants of M2 to M1 (k2) are also significantly different among the tested species. The k2 value in human is 68 times smaller than rat while it is 4.5 times higher than that in dog. The time courses for the metabolism of CDD in plasma of the three species are shown in Fig. 3.
| System | k1 (min−1) | k2 (min−1) | t1/2 (min) | tmax (min) |
|---|---|---|---|---|
| Rat plasma | 7.61 ± 0.50 | 15.0 ± 0.4 | 0.0914 ± 0.0062 | 0.0921 ± 0.0033 |
| Dog plasma | 8.97 ± 1.26 (×10−2) | 4.91 ± 0.60 (×10−2) | 7.82 ± 1.02 | 15.0 ± 1.6 |
| Human plasma | 0.147 ± 0.027 | 0.220 ± 0.007 | 4.85 ± 0.95 | 5.58 ± 0.57 |
| Phosphate buffer (pH 7.4) | 1.43 ± 0.30 (×10−3) | — | 499.1 ± 107.6 | — |
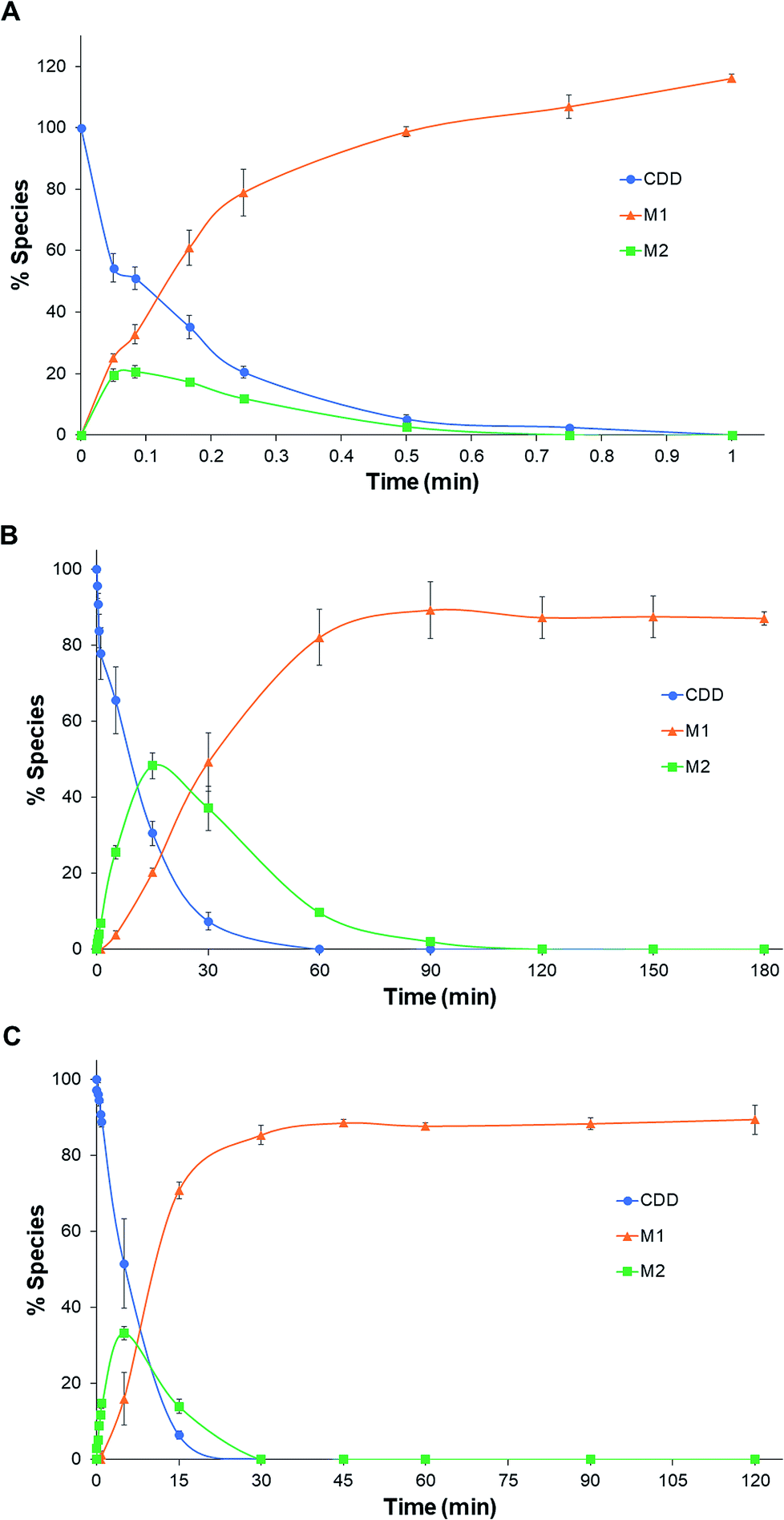 | ||
| Fig. 3 Time courses for the hydrolysis of CDD in rat (A), dog (B), and human (C) plasma at 37 °C. | ||
Identification of metabolites of CDD
The retention time and m/z of parent and product ions of CUR, MSCUR, CDD, M1, and M2 are summarized in Table 2 according to extracted-ion chromatograms (XIC) and mass spectra shown in Fig. S1–S12† (see ESI†). Under positive mode, MS spectra of CUR, MSCUR and CDD exhibited the ions at m/z 369.1332 [M + H]+, m/z 497.1805 [M′ + H]+ and m/z 625.2279 [M′′ + H]+, respectively. CDD and MSCUR also formed a sodium adduct with m/z 647.2081 [M′′ + Na]+and 519.1621 [M′ + Na]+, respectively. The MS2 spectrum of CUR shows several characteristic product ions including m/z 285 [M + H–C4H4O2]+, m/z 245 [M + H–C7H8O2]+ and m/z 177 [M + H–C11H12O3]+. For MSCUR, it loses an ethyl succinyl group resulting in the formation of CUR ion (m/z 369 [M′ + H–C6H8O3]+). In addition, it was fragmented to yield several product ions e.g. m/z 285, 245 and 177. In the case of CDD, the MS2 spectrum shows the loss of an ethyl succinyl group to form a peak at m/z 497 [M′′ + H–C6H8O3]+ as the major product ion. CDD also loses two ethyl succinyl groups yielding a product ion with m/z 369 [M′′ + H–2C6H8O3]+. The fragmentations of CUR, MSCUR, and CDD are proposed in Fig. S13.†| ID | Source | tR (min) | Predicted m/z | Measured m/z | Error (ppm) | Formula | Characteristic product ions (m/z) |
|---|---|---|---|---|---|---|---|
| a tR = retention time; RP = rat plasma; DP = dog plasma; HP = human plasma. | |||||||
| CDD | Standard | 3.77 | 625.2280 | 625.2279 | 0.0 | C33H37O12 | 497.1815, 369.1324, 245.0774, 177.0531 |
| CUR | Standard | 1.76 | 369.1333 | 369.1332 | 0.1 | C21H21O6 | 285.1107, 245.0747, 177.0522 |
| MSCUR | Standard | 2.39 | 497.1806 | 497.1805 | 0.3 | C27H29O9 | 369.1333, 285.1114, 245.0799, 177.0547 |
| M1 | RP | 1.77 | 369.1333 | 369.1337 | −1.1 | C21H21O6 | 285.1116, 245.0805, 177.0544 |
| DP | 1.76 | 369.1333 | 369.1332 | 0.2 | C21H21O6 | 285.1098, 245.0818, 177.0551 | |
| HP | 1.77 | 369.1333 | 369.1330 | 0.7 | C21H21O6 | 285.1111, 245.0792, 177.0567 | |
| M2 | RP | 2.41 | 497.1806 | 497.1796 | 1.9 | C27H29O9 | 369.1318, 285.1124, 245.0805, 177.0556 |
| DP | 2.41 | 497.1806 | 497.1800 | 1.2 | C27H29O9 | 369.1323, 285.1125, 245.0807, 177.0556 | |
| HP | 2.41 | 497.1806 | 497.1801 | 1.1 | C27H29O9 | 369.1333, 285.1124, 245.0773, 177.0552 | |
In the extracted ion chromatograms, M1 was eluted at the retention time about 1.77 min similar to that of CUR standard. It showed the molecular ion ([M1 + H]+) at m/z 369.1337, 369.1332 and 369.1330 in rat, dog and human plasma, respectively. Its MS2 spectrum demonstrates several product ions including m/z 285, m/z 245 and m/z 177. The MS results indicate that M1 is CUR. The M2 metabolite appeared in the extracted mass chromatograms has the same retention time as MSCUR standard about 2.4 min. M2 in rat, dog and human plasma produced a molecular ion ([M2 + H]+) at m/z 497.1796, 497.1800 and 497.1801, respectively. Further fragmentation of M2 ion generated product ions at m/z 369, m/z 285, m/z 245 and m/z 177. The M2 metabolite was therefore identified as MSCUR.
Effect of esterase inhibitors on CDD hydrolysis
Plasma esterases involved in the ester hydrolysis of CDD were investigated using various types of esterase inhibitors. The effect of esterase inhibitors expressed as % inhibition is summarized in Table 3.| Inhibitors | Target | Concentration | % inhibitiona | ||
|---|---|---|---|---|---|
| Rat | Dog | Human | |||
| a % inhibition < 10% is considered as no inhibition. | |||||
| BNPP | CES | 0.1 mM | 83.0 ± 8.9 | 16.6 ± 8.2 | <10% |
| 1 mM | 97.0 ± 7.7 | 63.6 ± 1.8 | 42.2 ± 6.7 | ||
| BW284c51 | AChE | 10 μM | <10% | <10% | <10% |
| 100 μM | <10% | <10% | <10% | ||
| Iso-OMPA | BChE | 0.1 mM | 99.5 ± 11.2 | 16.9 ± 7.1 | <10% |
| 1 mM | 108.3 ± 5.2 | 17.6 ± 6.7 | <10% | ||
| Eserine | ChE | 10 μM | <10% | <10% | <10% |
| 100 μM | <10% | <10% | <10% | ||
| TLCK | Serine hydrolase | 0.1 mM | <10% | <10% | <10% |
| 1 mM | <10% | 17.0 ± 10.3 | <10% | ||
| PMSF | Serine hydrolase | 0.1 mM | <10% | <10% | <10% |
| 1 mM | 96.0 ± 9.2 | 29.6 ± 5.3 | <10% | ||
| Pefabloc® SC | Serine hydrolase | 0.1 mM | 19.4 ± 2.1 | <10% | <10% |
| 1 mM | 99.8 ± 7.9 | 25.8 ± 3.1 | <10% | ||
| DTNB | Arylesterase | 0.1 mM | <10% | <10% | <10% |
| 1 mM | <10% | 47.8 ± 0.83 | 52.0 ± 1.5 | ||
| EDTA | PON | 5 mM | <10% | <10% | <10% |
| 10 mM | <10% | <10% | <10% | ||
| EGTA | PON | 5 mM | <10% | 17.3 ± 6.1 | 23.2 ± 11.4 |
| 10 mM | <10% | 19.9 ± 8.0 | 26.5 ± 3.6 | ||
| NaF | Non-specific | 5 mM | <10% | <10% | <10% |
| 10 mM | 24.4 ± 7.1 | <10% | <10% | ||
In human plasma, BNPP showed the partial inhibition at 1 mM. DTNB exerted moderate inhibition at 1 mM. EGTA also partially inhibited the hydrolysis of CDD. BW284c51, eserine, TLCK, PMSF, Pefabloc® SC, iso-OMPA, EDTA and NaF had no inhibition in human plasma.
In rat plasma, BNPP and iso-OMPA effectively inhibited the hydrolytic cleavage of CDD even at 0.1 mM (>80% inhibition). PMSF and Pefabloc® SC also showed significant inhibition (>90% inhibition) at concentrations of 1 mM. NaF at 10 mM partially prevented CDD hydrolysis. However, BW284c51, eserine, TLCK, EDTA, as well as DTNB did not prevent the hydrolytic cleavage of CDD.
BNPP, TLCK, PMSF and Pefabloc® SC slightly inhibited the hydrolysis of CDD in dog plasma. DTNB showed moderate inhibition at 1 mM. The inhibitory actions of iso-OMPA and EGTA were fewer than those of the aforementioned esterase inhibitors. BW284c51, eserine, EDTA and NaF showed no inhibition against CDD hydrolysis.
Discussion
The hydrolysis of an ester prodrug to its active metabolite in biological matrices is desirable for further development but one should consider the interspecies differences when conducting in vivo studies. For ester prodrugs intended to be administered intravenously, plasma is the first and important matrix in the bioconversion of ester prodrugs to their respective metabolites. Our in vitro plasma stability experiment clearly showed that CDD underwent rapid hydrolysis in rat, dog, and human at different rates. The in vitro half-lives of CDD in dog and human plasma are comparable but markedly differ from that in rat. These results suggest that the plasma clearance of CDD in dog is closer to human than rat. This can be explained by the interspecies differences in esterase activity. It has been reported that the relative esterase activity in plasma is as following order: rat > human > dog.16,22 However, the esterase expression and activity in dogs are similar to humans than rats. Our findings are in agreement with other ester-containing compounds such as clevidipine, oseltamivir, and WS070117.13,16,23 We could not establish the k2 value in phosphate buffer (pH 7.4) due to the absence of MSCUR during HPLC analysis. We hypothesized that MSCUR was rapidly degraded. Additional degradation kinetic experiment was performed for MSCUR in phosphate buffer (pH 7.4) and showed that the degradation rate constant (k) of MSCUR was about 70 times higher than that of CDD (unpublished data). Though we could not establish the k2 value in phosphate buffer (pH 7.4), the obtained k2 values from plasma of the studied species suggested the involvement of plasma esterases in the conversion of MSCUR to CUR. Interestingly, the k2 value in human plasma is slightly higher than the k1 but vice versa in dog, indicating that CDD and MSCUR are hydrolyzed by plasma esterases at the comparable rate in both species. In contrast, the k2 in rat plasma is extremely high and is two times higher than the k1 value. These results imply that, in rat, plasma esterases may preferential hydrolyze MSCUR. Therefore, the interspecies differences in the rate of conversion of CDD in plasma should be taken into consideration when selecting animal species for future in vivo studies.CDD contains four ester bonds; there are five possible metabolites that can be generated by plasma esterases. However, the LC-Q-TOF/MS analysis showed only two hydrolytic metabolites similar to those found by HPLC. In addition, metabolites with disrupted chromophore were not detected since esterases are the major drug-metabolizing enzymes in plasma.10,13,24 We subsequently performed mass fragmentation to obtain mass fragmental patterns of standards for comparing with those of metabolites. MS and MS2 analyses confirm that M1 metabolite is CUR because this metabolite shares the same m/z and product ions with those of CUR standard (Fig. S4 and S7–S9†) and as reported in the literature.25 This evidence indicates that M1 is CUR. For M2, it was identified as MSCUR because it showed the same m/z to the standard MSCUR and the major product ion of CDD (m/z 497, Fig. S5A and S6B† vs. Fig. S10A–12A†). The MS2 spectra of M2 from plasma of all tested species (Fig. S10B–S12B†) showed the similar fragmentation profiles to that of standard MSCUR (Fig. S5B†). Interestingly, MSCUR is the only intermediate metabolite formed during plasma metabolism. It could be due to the fact that the phenolic group of CDD exerts stronger electron-withdrawing effect than the ethoxy group. This makes the carbonyl carbon adjacent to the phenolic group more electrophilic and susceptible towards nucleophilic attack by nucleophilic groups within the active site of esterases than that of the ethoxy group. In addition, the phenolate anion is a more stable leaving group than an ethoxide anion due to the lower pKa value. Therefore, the cleavage of CDD would be preferred to take place at the phenolate ester of CDD.
The use of various esterase inhibitors is one of the approaches employed to determine plasma esterases involving the ester hydrolysis of xenobiotics.16,18,26,27 Our results showed that BW284c51 did not show inhibition in all species, indicating that AChE did not involve in the hydrolytic cleavage of CDD. In human plasma, the moderate inhibition by DTNB and EGTA suggests that PON/ArE may involve in the hydrolysis of CDD. These inhibitors also partially inhibited CDD hydrolysis in dog plasma. These results imply that PON/ArE play a role in the plasma metabolism of CDD. In addition to PON/ArE, BChE also participated in the hydrolytic cleavage of CDD in dog plasma as suggested by the partial inhibition of iso-OMPA, PMSF and Pefabloc® SC. It should be noted that BNPP showed the inhibition against CDD hydrolysis in dog and human plasma, implying that CES may involve in CDD hydrolysis. However, several studies reported that this enzyme is absent in dog and human plasma.13,14 The prevention of CDD hydrolysis in dog and human plasma by BNPP could be due to the inhibition on serum albumin. Serum albumin has been demonstrated for its capability to hydrolyze several ester-containing molecules e.g. organophosphate pesticides, aspirin and ester prodrugs.28–32 Because BNPP is an organophosphate, it may react with Tyr411 residue similar to other organophosphate compounds.32 Similarly, the reduction in CDD hydrolysis by DTNB could be derived from the inhibition on serum albumin in addition to ArE. Serum albumin has been reported for the thioesterase activity.33 This thioesterase activity is attributed to the reactive Cys34 residue which can undergo a thiol-disulfide exchange with thiol-containing molecules including disulfiram,34 2,2′-dithiodipyridine35 and DTNB.36,37 Serum albumin and ArE might play a vital role in the hydrolysis of CDD in dog and human plasma. In rat, CDD was hydrolyzed by plasma CES as supported by the inhibition of BNPP, PMSF and Pefabloc® SC. It was surprised that iso-OMPA, a BChE inhibitor, also inhibited the hydrolysis of CDD in rat plasma at the similar extent to BNPP. This observation is consistent with the previous study showing that iso-OMPA could inhibit plasma CES in rats which is required for the detoxification of soman, a toxic organophosphate ester.38 Also, eserine, a cholinesterase inhibitor, did not inhibit the hydrolytic cleavage of CDD. Hence, BChE did not participate in the metabolism of CDD in rat plasma. TLCK, another serine hydrolase inhibitor, weakly prevented the hydrolytic cleavage of CDD which may be due to the different acylation site (His residue) in the catalytic triad of the enzymes.39 PON/ArE did not participate in the hydrolysis of CDD in rat plasma, as demonstrated by no inhibition in the presence of DTNB, EDTA and EGTA. Because MSCUR contains a phenolate ester as found in CDD, we postulate that MSCUR can be hydrolyzed to CUR by esterases similar to ester hydrolysis of CDD. Therefore, the esterase inhibitors that can inhibit the ester hydrolysis of CDD may also affect the conversion of MSCUR to CUR. The use of esterase inhibitors allows the appropriate selection of purified or recombinant enzymes that might involve in the hydrolysis of CDD for further mechanistic investigation. The overall results suggest that dogs would be a better animal model, compared to rats, in the investigation of pharmacokinetic properties of CDD for allometric scaling of drug doses for human studies. The proposed metabolism map of CDD in the plasma of the three studied species is shown in Fig. 4.
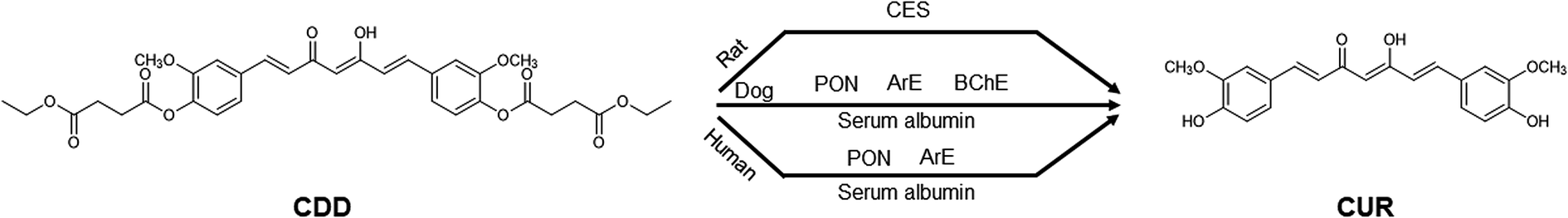 | ||
| Fig. 4 Summarized esterases involved in the hydrolysis of CDD in rat, dog, and human plasma. | ||
Conclusion
In summary, CDD undergoes rapid metabolism in plasma of rat, dog, and human. The marked interspecies differences in the rate of conversion could be observed. The hydrolysis process of CDD in plasma of all tested species is mediated by plasma esterases. The use of different esterase inhibitors provides the information of plasma esterases responsible for hydrolysis of CDD. Considering the rate of hydrolysis and plasma esterases involved in the hydrolysis of CDD, dog was found to be closer to human than rat. The variation in the metabolism of CDD in plasma of different animal species could be derived from the difference in types of esterases present as well as the structural differences of the same enzymes. These findings indicate that the future in vivo pharmacokinetic study of CDD for allometric scaling of drug doses for further clinical studies should be investigated in dog rather than rat because of the closer plasma metabolism between dog and human.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the scholarship from the Graduate School, Chulalongkorn University to Commemorate the 72nd Anniversary of His Majesty King Bhumibala Aduladeja, the 90th Anniversary Chulalongkorn University Fund (Ratchadaphiseksomphot Endowment Fund, Grant No. GCUGR1125602053M), the Overseas Research Experience Scholarship from the Graduate School and Faculty of Pharmaceutical sciences, Chulalongkorn University to P. Ratnatilaka Na Bhuket, and Ratchadaphiseksomphot Endowment Fund of Chulalongkorn University (Grant No. CU-GR_60_07_33_01) to P. Rojsitthisak. The authors would like to show gratitude to the Research Instrument Center of Faculty of Pharmaceutical Sciences at Chulalongkorn University, Chulalongkorn University Drug and Health Products Innovation Promotion Center (CU-D-HIP), the Medical Research Laboratory of Faculty of Medicine at Chulalongkorn University, the Department of Chemistry and Biochemistry at University of South Carolina and Pharma Nueva Co., Ltd for providing research facilities.References
- T. Ak and İ. Gülçin, Chem.-Biol. Interact., 2008, 174, 27–37 CrossRef CAS PubMed.
- W. Buadonpri, W. Wichitnithad, P. Rojsitthisak and P. Towiwat, J. Health Res., 2009, 23, 11–16 CAS.
- J. Nautiyal, S. S. Kanwar, Y. Yu and A. P. N. Majumdar, J. Health Res., 2011, 6, 7 CAS.
- N. Dhillon, B. B. Aggarwal, R. A. Newman, R. A. Wolff, A. B. Kunnumakkara, J. L. Abbruzzese, C. S. Ng, V. Badmaev and R. Kurzrock, Clin. Cancer Res., 2008, 14, 4491–4499 CrossRef CAS PubMed.
- Y.-J. Wang, M.-H. Pan, A.-L. Cheng, L.-I. Lin, Y.-S. Ho, C.-Y. Hsieh and J.-K. Lin, J. Pharm. Biomed. Anal., 1997, 15, 1867–1876 CrossRef CAS PubMed.
- P. Ratnatilaka Na Bhuket, A. El-Magboub, I. S. Haworth and P. Rojsitthisak, Eur. J. Drug Metab. Pharmacokinet., 2017, 42, 341–353 CrossRef CAS PubMed.
- W. Wichitnithad, U. Nimmannit, S. Wacharasindhu and P. Rojsitthisak, Molecules, 2011, 16, 1888–1900 CrossRef CAS PubMed.
- J. Wongsrisakul, W. Wichitnithad, P. Rojsitthisak and P. Towiwat, J. Health Res., 2010, 24, 175–180 CAS.
- K. Bangphumi, C. Kittiviriyakul, P. Towiwat, P. Rojsitthisak and P. Khemawoot, Eur. J. Drug Metab. Pharmacokinet., 2016, 41, 777–785 CrossRef CAS PubMed.
- B. M. Liederer and R. T. Borchardt, J. Pharm. Sci., 2006, 95, 1177–1195 CrossRef CAS PubMed.
- Y.-h. Yang, H. Aloysius, D. Inoyama, Y. Chen and L.-q. Hu, Acta Pharm. Sin. B, 2011, 1, 143–159 CrossRef CAS.
- T. Ishizuka, I. Fujimori, A. Nishida, H. Sakurai, Y. Yoshigae, K. Nakahara, A. Kurihara, T. Ikeda and T. Izumi, Drug Metab. Dispos., 2012, 40, 374 CrossRef CAS PubMed.
- F. G. Bahar, K. Ohura, T. Ogihara and T. Imai, J. Pharm. Sci., 2012, 101, 3979–3988 CrossRef CAS PubMed.
- L. M. Berry, L. Wollenberg and Z. Zhao, Drug Metab. Lett., 2009, 3, 70–77 CrossRef CAS PubMed.
- E. V. Rudakova, N. P. Boltneva and G. F. Makhaeva, Bull. Exp. Biol. Med., 2011, 152, 73–75 CrossRef CAS PubMed.
- Y. Liu, J. He, Z. Abliz and H. Zhu, Xenobiotica, 2011, 41, 549–560 CrossRef CAS PubMed.
- P. Ratnatilaka Na Bhuket, N. Niwattisaiwong, P. Limpikirati, P. Khemawoot, P. Towiwat, B. Ongpipattanakul and P. Rojsitthisak, J. Chromatogr. B, 2016, 1033, 301–310 CrossRef PubMed.
- K. Tsujikawa, K. Kuwayama, H. Miyaguchi, T. Kanamori, Y. T. Iwata and H. Inoue, Xenobiotica, 2009, 39, 391–398 CrossRef CAS PubMed.
- M. Koitka, J. Höchel, H. Gieschen and H.-H. Borchert, J. Pharm. Biomed. Anal., 2010, 51, 664–678 CrossRef CAS PubMed.
- S.-i. Kawano, Y. Inohana, Y. Hashi and J.-M. Lin, Chin. Chem. Lett., 2013, 24, 685–687 CrossRef CAS.
- U. V. R. Vijaya Saradhi, Y. Ling, J. Wang, M. Chiu, E. B. Schwartz, J. R. Fuchs, K. K. Chan and Z. Liu, J. Chromatogr. B, 2010, 878, 3045–3051 CrossRef CAS PubMed.
- T. Minagawa, Y. Kohno, T. Suwa and A. Tsuji, Biochem. Pharmacol., 1995, 49, 1361–1365 CrossRef CAS PubMed.
- H. Ericsson, B. Tholander and C. G. Regårdh, Eur. J. Pharm. Sci., 1999, 8, 29–37 CrossRef CAS PubMed.
- B. Li, M. Sedlacek, I. Manoharan, R. Boopathy, E. G. Duysen, P. Masson and O. Lockridge, Biochem. Pharmacol., 2005, 70, 1673–1684 CrossRef CAS PubMed.
- H. Jiang, Á. Somogyi, N. E. Jacobsen, B. N. Timmermann and D. R. Gang, Rapid Commun. Mass Spectrom., 2006, 20, 1001–1012 CrossRef CAS PubMed.
- T. Hioki, T. Fukami, M. Nakajima and T. Yokoi, Drug Metab. Dispos., 2011, 39, 1345–1352 CrossRef CAS PubMed.
- G. Eichenbaum, J. Skibbe, A. Parkinson, M. D. Johnson, D. Baumgardner, B. Ogilvie, E. Usuki, F. Tonelli, J. Holsapple and A. Schmitt-Hoffmann, J. Pharm. Sci., 2012, 101, 1242–1252 CrossRef CAS PubMed.
- E. Walker John, FEBS Lett., 1976, 66, 173–175 CrossRef.
- M. A. Sogorb, S. García-Argüelles, V. Carrera and E. Vilanova, Chem. Res. Toxicol., 2008, 21, 1524–1529 Search PubMed.
- A. Salvi, P.-A. Carrupt, J. M. Mayer and B. Testa, Drug Metab. Dispos., 1997, 25, 395–398 CAS.
- S.-F. Ma, M. Anraku, Y. Iwao, K. Yamasaki, U. Kragh-Hansen, N. Yamaotsu, S. Hirono, T. Ikeda and M. Otagiri, Drug Metab. Dispos., 2005, 33, 1911–1919 CrossRef CAS PubMed.
- O. Lockridge, W. Xue, A. Gaydess, H. Grigoryan, S.-J. Ding, L. M. Schopfer, S. H. Hinrichs and P. Masson, J. Biol. Chem., 2008, 283, 22582–22590 CrossRef CAS PubMed.
- U. Kragh-Hansen, Biochim. Biophys. Acta, Gen. Subj., 2013, 1830, 5535–5544 CrossRef CAS PubMed.
- R. P. Agarwal, M. Phillips, R. A. McPherson and P. Hensley, Biochem. Pharmacol., 1986, 35, 3341–3347 CrossRef CAS PubMed.
- P. A. Overgaard and J. Jørgen, Eur. J. Biochem., 1980, 106, 291–295 Search PubMed.
- J. Stewart Alan, A. Blindauer Claudia, S. Berezenko, D. Sleep, D. Tooth and J. Sadler Peter, The FEBS Journal, 2004, 272, 353–362 CrossRef PubMed.
- M. J. Torres, L. Turell, H. Botti, L. Antmann, S. Carballal, G. Ferrer-Sueta, R. Radi and B. Alvarez, Arch. Biochem. Biophys., 2012, 521, 102–110 CrossRef CAS PubMed.
- Z. Grubič, D. Sket and M. Brzin, Arch. Toxicol., 1988, 62, 398–399 CrossRef.
- J. C. Powers, J. L. Asgian, Ö. D. Ekici and K. E. James, Chem. Rev., 2002, 102, 4639–4750 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra08594c |
| This journal is © The Royal Society of Chemistry 2019 |