
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControllable conversion of Prussian blue@yeast bio-template into 3D cage-like magnetic Fe3O4@N-doped carbon absorbent and its cohesive regeneration by persulfate activation†
Si Chenab,
Bo Bai *bcd,
Yunhua Heab,
Na Hucd,
Honglun Wangcd and
Yourui Suocd
*bcd,
Yunhua Heab,
Na Hucd,
Honglun Wangcd and
Yourui Suocd
aKey Laboratory of Subsurface Hydrology and Ecological Effects in Arid Region of the Ministry of Education, Chang'an University, No. 126 Yanta Road, Xi'an 710054, Shaanxi, China
bCollege of Environmental Science and Engineering, Chang'an University, Xi'an, 710054, P. R. China. E-mail: baibochina@163.com; Fax: +86 29 82339961; Tel: +86 29 82339052
cKey Laboratory of Tibetan Medicine Research, Northwest Institute of Plateau Biology, Chinese Academy of Sciences, Xining, 810008, China
dQinghai Provincial Key Laboratory of Tibetan Medicine Research, Xining, 810001, P. R. China
First published on 8th January 2019
Abstract
A multitude of heteroatom-doped carbon adsorbents have been explored to cope with ever-growing organic pollution. However, development of these advanced carbon materials with adequate activity and stability remains challenging. Herein, unique 3D cage-like magnetic N-doped Fe3O4@C adsorbents were rationally constructed by a one-step pyrolysis of Prussian blue@yeast (PB@yeast) bio-templates. By using yeast as an available biological support, the prepared Fe3O4@C hybrids were demonstrated to provide a sufficient number of Fe, N and C atoms for the novel cage-like microstructures, making them a new type of Fe, N co-doped carbon absorbents with a facile preparation procedure and remarkable adsorption behavior. Rhodamine B (RhB) removal indicated that the prepared N-doped Fe3O4@C adsorbents displayed high adsorption capabilities in a near-neutral solution, and Fe3O4@C (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.11) exhibited a maximum adsorption capability of 257.06 mg g−1. More importantly, spent N-doped Fe3O4@C absorbents, which could be recovered by magnetic separation and cohesive persulfate (PS) activated photo-Fenton regeneration, showed excellent adsorption reusability and high stability even after 5 cycles. Overall, this paper presents a simple method for fabrication of a 3D cage-like magnetic N-doped Fe3O4@C adsorbent, which provides a significant guidance for the study of Fe, N co-doped carbon adsorbents towards dye wastewater treatment.
:0.11) exhibited a maximum adsorption capability of 257.06 mg g−1. More importantly, spent N-doped Fe3O4@C absorbents, which could be recovered by magnetic separation and cohesive persulfate (PS) activated photo-Fenton regeneration, showed excellent adsorption reusability and high stability even after 5 cycles. Overall, this paper presents a simple method for fabrication of a 3D cage-like magnetic N-doped Fe3O4@C adsorbent, which provides a significant guidance for the study of Fe, N co-doped carbon adsorbents towards dye wastewater treatment.
1. Introduction
Prussian blue (PB) and its analogues (PBA), owing to their diverse morphologies and peculiar linkage of transition metals with N-containing organic ligands, have attracted considerable attention as templates and precursors for the development of heteroatoms-doped carbon materials.1 Previously, several metal/nitrogen (N) co-doped carbonaceous composites have been fabricated using PB or PBA as raw materials for various applications in wastewater treatment. For instance, Hu et al.2 reported the fabrication of FeCo nanocrystals with nitrogen-doped graphene shells, obtained by annealing Fe3[Co(CN)6]2, which acted as an efficient catalyst for 4-nitrophenol reduction. Lin and co-workers3 used Co3[Fe(CN)6] to prepare a magnetic carbon/cobalt/iron (MCCI) nanocomposite, which showed good catalytic activity during peroxymonosulfate degradation of Rhodamine B. Wen et al.4 synthetized porous magnetic N-doped Fe/Fe3C nanoparticles that were prepared from poly-dopamine coated Prussian blue microcubes with thermal treatment. Batch adsorptive experiments demonstrated that N-doped Fe/Fe3C@C-800 exhibited maximum sorption capacity for uranium(VI) removal. In brief, it is believed that chemical activity and stability of conventional carbon atoms could be enhanced by the incorporation of metal (Fe, Co or Ni) and N, hereby, making the metal/N-doped carbon composites very useful in wastewater treatment.5,6 In other words, co-doping of metal and nitrogen into carbon hosts has been recognized as an effective approach to enhance physical and chemical properties of conventional carbon materials.7,8 Nevertheless, despite the superiority of PB or PBA as starting materials, once they are used as the only precursors, shortage of C and N elements in the PB or PBA frameworks inevitably leads to the lack of sufficient N–C formation during the subsequent growth of carbonaceous layers. From previous experience,2,9 the deficiency of C and N in the final products not only limits their adjustable physicochemical properties, but also reduces their chemical stability. To overcome this dilemma, many N-containing carbon substrates have been utilized to modify PB-derived compounds, where C and N can be enriched, thus, obtaining improved metal/N–C hybrids with high activity, sustainable stability and better environmental tolerance. Unfortunately, to date, the vast majority of the above-mentioned carbonaceous supports are expensive and low in abundance, greatly constraining their practical application.10,11 In this regard, abundant and low cost carbonaceous supports are highly desirable, and there is still considerable room for development of advanced carbons with enriched nitrogen, transition metal and carbon sources.Yeast, commonly used for food fermentation, pharmaceuticals and biofuels, is readily available in nature and easy to artificially culture.12,13 The cell wall of yeast consists of approximately 90% polysaccharides, including glucan and mannan, and a small portion of protein, chitin and lipid, which offers an abundance of functional groups to the fine biostructure, such as –OH, –COOH, –NH2, –CONH2, etc.14,15 Benefiting from these properties, yeast has been used as a raw material for the preparation of functional carbon materials via thermal decomposition.16,17 Specifically, N-enrichment within the yeast cell (about 10.6 wt%) can be further converted to N-doped carbon and, thus, enable the yeast-based carbon to exhibit stronger chemical activity than traditional pristine carbon materials.18,19 Moreover, compared to the formation of some other N-doped carbon materials that introduce N into the carbon structure by using NH3, amines, urea or melamine, yeast can be directly transformed into N-functionalized carbon without any additions,20 making it a qualified green feedstock for the production of valuable N-doped carbon materials. From Shen and co-workers,19 a hierarchical microporous carbon material with high content of nitrogen (>5.3 wt%) was prepared from yeast, and it was proved to maintain excellent CO2 adsorption capacity. This finding strongly encouraged us to use yeast to prepare high value-added carbon materials and to explore their adsorption applications.
In this study, we proposed a synthesis strategy for a facile, economical, and scalable synthesis of 3D cage-like magnetic N-doped Fe3O4@C adsorbent with uniform iron-based composites embedded in N-enriched carbon microspheres. In particular, Prussian blue@yeast (PB@yeast) bio-template was previously synthesized using yeast and potassium ferricyanide as raw materials and then directly annealed at 550 °C for 3 h. By adjusting the content ratio of raw materials, several controllable hierarchical N-doped Fe3O4@C samples were rationally obtained. The prepared N-doped Fe3O4@C products were characterized by FE-SEM, EDS, XRD, FT-IR and Zeta potentiometry techniques. Based on the characterization results, the detailed mechanism for the formation of N-doped Fe3O4@C microcages was discussed. Afterwards, application of N-doped Fe3O4@C microcages in RhB removal was investigated in batch experiments. The adsorption isotherm was constructed, and kinetics and thermodynamics were explored to evaluate the absorption capabilities and to study the adsorption mechanism of N-doped Fe3O4@C adsorbent. The effects of pH, ionic strength and urea were also assessed to understand the possible interaction mechanism between RhB molecules and N-doped Fe3O4@C adsorbent in water. Furthermore, the reusability of the saturated N-doped Fe3O4@C adsorbent was achieved via magnetic separation and persulfate (PS, S2O82−) activated photo-Fenton regeneration. The results indicated that N-doped Fe3O4@C microcages with high adsorption activity and prolonged reusability would be promising candidates for practical applications in dye-containing wastewater treatment.
2. Methodology
2.1 Materials
Yeast powder was purchased from Angel Yeast Co., Ltd., China. Potassium ferricyanide (K4Fe(CN)6·3H2O) and Rhodamine B were acquired from Tianjin Kermel Chemical Reagent Co., Ltd., China. Polyvinylpyrrolidone (PVP, K-30) was supplied by Shanghai Lanji Co., Ltd., China. Persulfate (PS, Na2S2O8) was purchased from Aladdin Co., Ltd, China. Hydrochloric acid (HCl, 36.5%), sodium hydroxide (NaOH), and ethanol (C2H5OH, 95%) were purchased from Xi'an Chemical Agent Corp. All chemical reagents were used without further purification. Doubly deionized water was used for all experiments.2.2 Synthesis of 3D cage-like magnetic N-doped Fe3O4@C adsorbent
:0.11)). For comparison, several controllable N-doped Fe3O4@C samples (named Fe3O4@C (1:0.05) and Fe3O4@C (1:0.22)) also were prepared by adjusting the initial content ratio of yeast and K4Fe(CN)6·3H2O. That is, the K4Fe(CN)6·3H2O reagent was adjusted to 0.05 g or 0.22 g in the pre-prepared synthesis while other conditions remained unchanged.2.3 Characterization
The formation of N-doped Fe3O4@C hybrids was confirmed by field emission scanning electron microscopy (FE-SEM) on a HITACHI S4800 instrument. Elemental distribution of iron, nitrogen, carbon, and oxygen in as-prepared samples was evaluated by energy-dispersive spectroscopy (EDS) analysis (equipped with the FE-SEM). All of the samples were coated with Pt before fixing them on the FE-SEM equipment. Their composition was characterized by X-ray diffraction (XRD) on a Bruker D8 Advanced X-ray diffractometer (Cu Kα radiation λ = 0.15418 nm). Functional groups on N-doped Fe3O4@C were characterized using Fourier-transform infrared (FT-IR) spectra on a PerkinElmer Spectrum Two in the range 500–4000 cm−1. The ζ-potential value was recorded by Malvern ZEN3690. Surface chemical composition of N-doped Fe3O4@C was analyzed by using X-ray photoelectron spectroscopy (XPS) on PHI 5000 VersaProbe III. Surface area and pore volume were determined by specific surface area pore size analyzer (Builder SSA-6000).2.4 Batch experiments
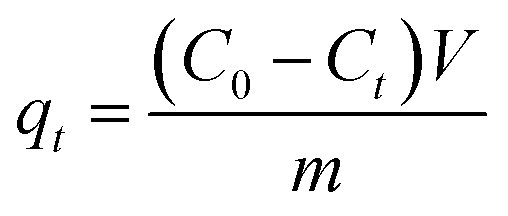 | (1) |
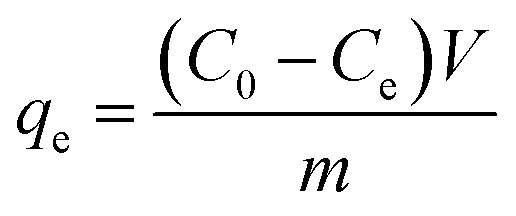 | (2) |
:0.11) adsorbent was saturated with 25 mg L−1 of RhB solution. The saturated adsorbent was then immersed into the 50 mL deionized water with mechanical agitation. Thirty mg of PS was added while xenon lamp was turned on (PLS-SXE300C, Beijing). The mixture was continuously stirred for 30 min to remove the adsorbed RhB molecules completely. Then, the regenerated adsorbent was magnetically separated from the solution by placing a magnet nearby and dried for more adsorption experiments.3. Results and discussion
3.1 Conversion of PB@yeast bio-template into 3D cage-like magnetic N-doped Fe3O4@C adsorbent
Scheme 1 shows the formation of 3D cage-like magnetic N-doped Fe3O4@C.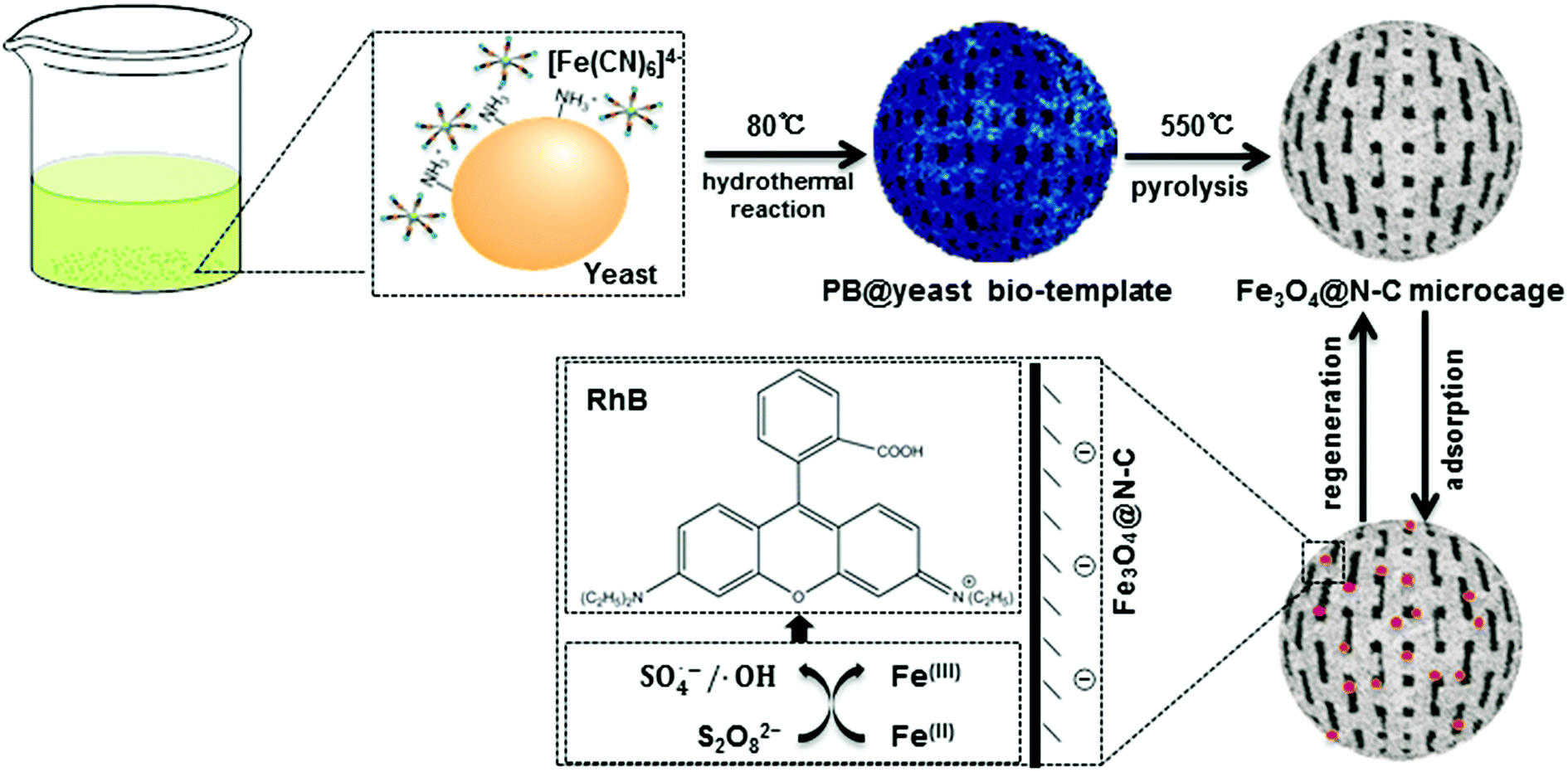 | ||
| Scheme 1 Schematic illustration of the formation of 3D cage-like N-doped Fe3O4@C adsorbent. | ||
According to previous reports,4,21 PB nanocubes could be easily synthesized by hydrothermal treatment at 80 °C using potassium ferricyanide (K4Fe(CN)6). The obtained PB product consisted of a face-centered-cubic crystal structure with Fe(II)–C–N–Fe(III) sequence, which was clarified as a mixed-valence iron(III) hexacyanoferrate(II) compound (Fe4[Fe(CN)6]3). Yeast is one type of elliptical unicellular fungus with sturdy cell walls. Owing to the polysaccharide layer constructed from coiled β-1,3-glucan in the cell wall, yeast can maintain the stability of the whole cell system even in relatively harsh environments.17 Previous studies also confirmed that when the initial pH value was below 3.0, the glucosamine unit (–NH2) of yeast cells could be easily converted into the protonated form (–NH3+), leaving the exposed surface of yeast cells positively charged in acidic solution.22 In current study, bare yeast and K4Fe(CN)6 were mixed together in 0.10 mol L−1 of HCl solution. The negatively charged [Fe(CN)6]4− could be easily attracted to the protonated –NH3+ of yeast by electrostatic force, allowing the two feedstocks to collect and form homogeneous Fe-containing yeast microspheres. Afterward, the mixture was transferred into a Teflon-lined stainless-steel autoclave for hydrothermal treatment. In the succeeding process, [Fe(CN)6]4− ions were in situ transformed into a well-defined PB crystals on the cell wall of the yeast microspheres. The color of the solution changed from light green to dark blue during the synthesis, confirming successful nucleation of PB shell on the surface of the yeast substrate (denoted here as PB@yeast). This way, the –NH2 units of the yeast could be continuously linked to PB nanoparticles by ionic interactions, which greatly contributed to the stability of PB@yeast microspheres.23,24 In addition, aggregation of PB nanoparticles was substantially avoided due to the tight and uniform adhesion of PB nanoparticles to the high-surface-area yeast substrate.
In view of the integrated features of PB@yeast, such as N/C enrichment of the novel microstructure and uniform Fe encapsulation into the framework, it can be accepted that the PB@yeast microspheres have acted as an ideal bio-template for the formation of advanced Fe, N co-doped carbonaceous materials. Hence, the PB@yeast microspheres were then calcined at 550 °C for 3 h. During the pyrolysis process, continuous decomposition of the PB shell and the yeast core occurred contemporaneously, resulting in a 3D hierarchical Fe3O4@N-doped carbon product (denoted as N-doped Fe3O4@C). In other words, as a result of constant decomposition of PB shell, Fe-containing nanocubes were produced while the yeast was carbonized, accompanied by certain N doping into the carbon layer to form N-doped carbon microspheres. From the above analysis, it can be proposed that the PB@yeast bio-template shows more promising potential for the formation of Fe, N-based carbon materials than PB nanocubes that act as a single sacrificial template/precursor.10,25 In particular, yeast cells plays a key bi-functional role in the formation of the final N-doped Fe3O4@C products. On one hand, the cell-wall polysaccharide network of yeast provides an excellent high-surface-area platform and enriched N-functional groups, which significantly facilitate the uniform distribution of Fe3O4 nanoparticles embedded in N-enriched carbonaceous microspheres. For this reason, agglomeration of Fe3O4 nanoparticles is avoided and a sufficient amount of N/C sources is added simultaneously. On the other hand, yeast cell consists of 2 kinds of components, namely, an amorphous matrix and a fibrillar network.26 During heat treatment, the former is gradually gasified into CO2, CO and H2,27 while the latter is burned to form a carbon material, making the yeast convert into a hollow carbonaceous microsphere with penetrable meso- and macropores.17,19 Based on this fact, a cage-like structured microsphere was thereby obtained by pyrolysis of the PB@yeast template. Similarly, decomposition of PB nanocubes also occurred, accompanied by the release of nitrogen-containing gases,28 resulting in regular Fe-based nanoboxes anchoring to the surface of N-enriched carbon microcage. From this point of view, the final prepared N-doped Fe3O4@C microcages, inheriting the attractive integrated features of PB@yeast bio-template, would show significantly improved activity and stability for adsorption applications.
To verify the successful formation of N-doped Fe3O4@C adsorbent, field-emission scanning electron microscope (FE-SEM) was employed to characterize the morphology and structure of the precursors and the final products.
Fig. 1 shows the FE-SEM images of the products.
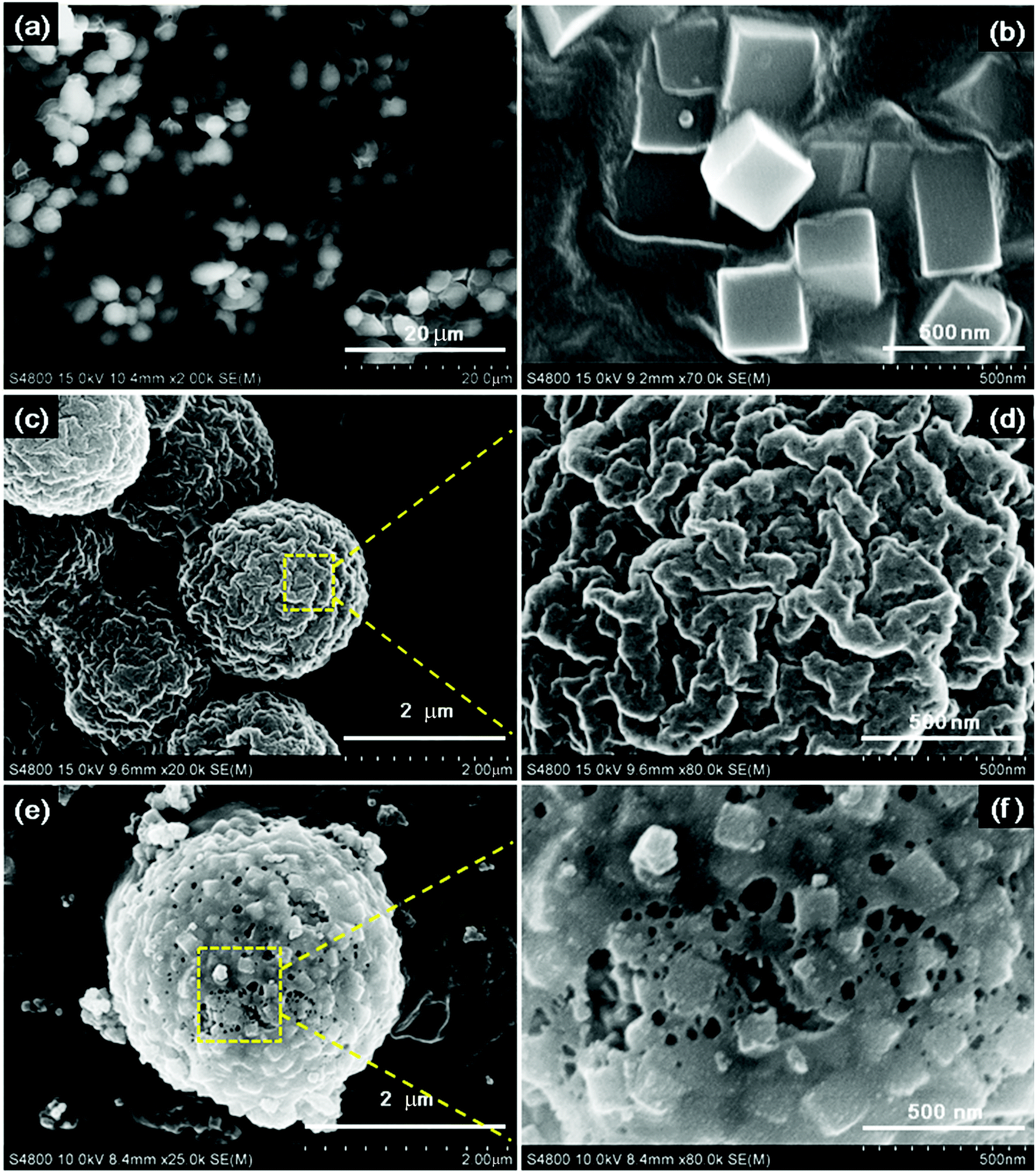 | ||
| Fig. 1 FE-SEM images of (a) naked yeast; (b) PB nanocubes; (c) PB@yeast (1:0.11) bio-template; (d) magnified details for PB@yeast (1:0.11) bio-template; (e) the sample Fe3O4@C (1:0.11); and (f) magnified details for Fe3O4@C (1:0.11). | ||
The uniform original yeast (Fig. 1a) with ellipsoidal shape and PB nanocubes (Fig. 1b) with an edge length of about 300 nm were obtained. Both showed a very smooth surface. In Fig. 1(c and d), the PB@yeast bio-template with a core/shell structure can be seen, which is slightly larger than naked yeast, and PB nanocubes are uniformly distributed on the surface of the microsphere. An enlarged image (Fig. 1d) illustrates that the surface of the microsphere became rough with several nanoparticles uniformly adhered to the exterior structure, and plenty of holes exist on the surface. This observation may be attributed to the relatively loosely assembled PB shell and evaporation of water in the structure of yeast during the hydrothermal reaction. In other words, some hydrolysis channels had emerged in the microsphere, which would conduce to the possible formation of cage-like microsphere after continued carbonization. After pyrolysis, as shown in Fig. 1(e and f), the N-doped Fe3O4@C (1:0.11) sample remained a core–shell structure, in which the closely arranged PB nanocubes in the outer shell were transformed into small individual and well-defined nanoboxes with sizes of approximately 150 nm, and the inner core maintained its own microsphere morphology. Close observation (Fig. 1f) revealed that the outer shell consisted of stacked assembled nanocubes, and the pores remained enlarged. This happened mainly because the individual hydrolysis channels of yeast kept enlarging to form penetrable meso- and macropores at high temperature.16 Also, the polycondensation and decomposition of PB contributed to the enlarged pores.29 Finally, 3D cage-like N-doped Fe3O4@C was obtained, which may be beneficial in sorption of target pollutants.
To understand the controllable conversion of PB@yeast bio-template, the parallel hierarchical Fe3O4@C samples were also formed by adjusting different mass ratios between yeast and K4Fe(CN)6 under the same conditions (donated Fe3O4@C (1:0.05) and Fe3O4@C (1:0.22)). The obtained FE-SEM images are shown in Fig. S1.† Compared to Fe3O4@C (1:0.11) in Fig. 1(e and f), the 3D hierarchical structures were also observed, which retained the microsphere shape roughly. Nevertheless, the Fe3O4@C (1:0.05) sample contained irregular nanoparticles wrapped around the microspheres with many holes on the surface, whereas the Fe3O4@C (1:0.22) sample displayed uniform nanoparticles from 20 to 40 nm that were completely coating the microsphere surface. Obviously, it appears that a greater number of PB nanocubes resulted in more compact microspheres, while a lower number led to the more sizable pores. These novel findings demonstrated that polycondensation and decomposition of yeast cells could significantly contribute to the formation of pores during pyrolysis. Additionally, the conditions of the yeast surface varied from different quantities of nanocube-assembled shells, resulting in different morphological parameters of the final product.
Further energy-dispersive spectroscopy (EDS) analysis of N-doped Fe3O4@C samples confirmed the observed distribution peaks of C, N, O and Fe elements (Fig. S2 and S3†). For comparison, the mass fractions of the given elements are list in Table 1.
| Sample | Weight percent (wt%) | Atomic percent (at%) | ||||||
|---|---|---|---|---|---|---|---|---|
| C | N | O | Fe | C | N | O | Fe | |
| Yeast-based sample | 64.94 | 10.90 | 11.99 | — | 76.91 | 11.07 | 10.66 | — |
| PB-based sample | 14.30 | — | — | 70.16 | 46.44 | — | — | 49.01 |
| Fe3O4@C (1:0.05) |
47.17 | 10.46 | 16.56 | 17.98 | 64.45 | 12.25 | 16.99 | 5.24 |
| Fe3O4@C (1:0.11) |
47.26 | 10.53 | 17.68 | 19.71 | 64.01 | 12.22 | 17.98 | 5.79 |
| Fe3O4@C (1:0.22) |
39.76 | 4.68 | 23.47 | 23.74 | 59.42 | 5.88 | 25.56 | 7.29 |
As shown in Table 1, the Fe element was observed in all 3 N-doped Fe3O4@C samples with 17.98, 19.71 and 23.74 wt%. The highest ratio of a sample (1:0.22) confirmed that the Fe source in prepared N-doped Fe3O4@C samples was provided entirely by the PB shell of the bio-template, and its quantity increased with the enhanced loading of PB nanocubes on the yeast core. Regarding C and N elements, their mass percent scaled up as the mass ratio of feedstocks increased from 1:0.05 to 1:0.11, but declined when the mass ratio reached to 1:0.22. This phenomenon may be caused by the fact that the surface of the core was completely enwrapped and difficult to detect by EDS analysis, which is consistent with the observations of the FE-SEM images. It is worth to note that the N distributed peak was not detected by EDS when using PB nanocubes as a single template, reminding few N composition in PB-derived carbonaceous compounds. As a result, it can be concluded that PB nanocubes provided a sufficient amount of Fe in order for the products and yeast to make a big enough contribution to the sources of C, N elements. EDS mapping images of the N-doped Fe3O4@C (1:0.11) sample (Fig. S3†) revealed that Fe, N and O atoms were evenly distributed on C layer with strong intensity. In particular, Fe atoms evenly covered the entire selected zone, demonstrating that Fe was uniformly enriched on the surface of the N-doped Fe3O4@C sample. A similar distribution was observed between the C and N element mapping images, confirming the uniform doping of N along the carbon matrix. So far, it can be concluded that the N-doped Fe3O4@C hybrid with enriched N, O, Fe elements, uniformly distributed on C substrate, would find application in organic wastewater treatment.
Further, XPS characterization was performed to gain more insight into the chemical compositions and the surface properties of the prepared N-doped Fe3O4@C samples (Fig. 2).
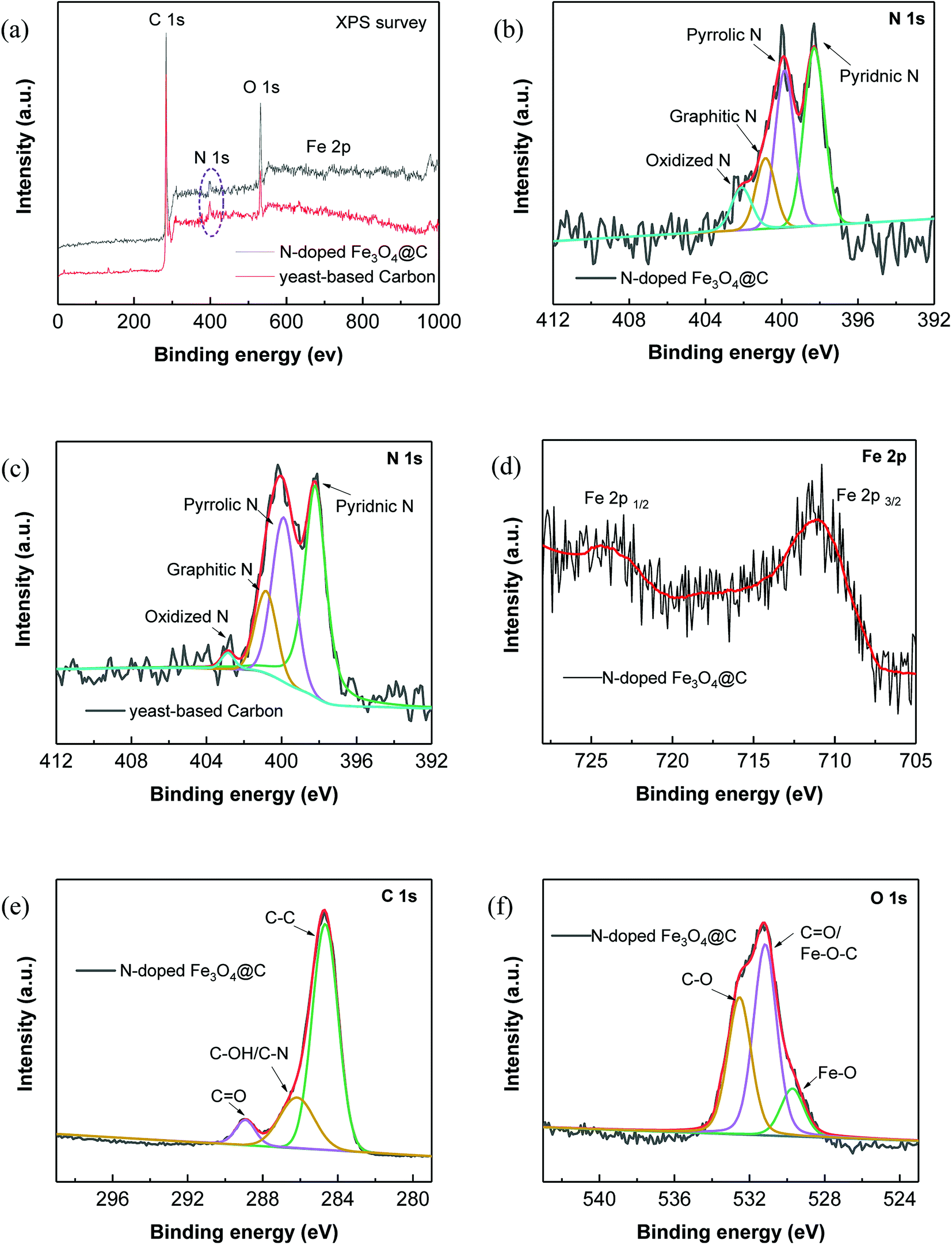 | ||
| Fig. 2 (a) Survey XPS spectrum of N-doped Fe3O4@C; High-resolution XPS spectra of N 1s in (b) N-doped Fe3O4@C and (c) yeast-based carbon; high-resolution XPS spectra of (d) C 1s; (e) Fe 2p and (f) O 1s in N-doped Fe3O4@C sample. | ||
The survey XPS spectrum in Fig. 2a reveals that the N-doped Fe3O4@C sample consisted of C, N, O and Fe elements, while no peaks of Fe element were observed in the yeast-based carbon. Notably, N element was clearly seen in the yeast-based carbon, clarifying the N-doping effect of yeast. To analyze the doped N element, the high-resolution XPS spectrum of N 1s was subsequently examined. As shown in Fig. 2b, there are 4 fitted peaks corresponding to pyridinic N (398.3 eV), pyrrolic N (399.8 eV), graphitic N (400.8 eV) and oxidized N (402.1 eV).30 The area percentages of the 4 dominating peaks were 41.0, 33.4, 15.2 and 10.4%, successively, corresponding to the content percentages of the 4 N species. Similarly, 49.9% of pyridinic N, 33.1% of pyrrolic N, 15.0% of graphitic N and 2.0% of oxidized N were obtained in the N 1s spectrum of yeast-based carbon (Fig. 2c), which further demonstrate the contribution of the yeast matrix to N-doping. High-resolution C 1s spectrum (Fig. 2e) shows 4 peaks located at 284.6, 286.2 and 288.9 eV, which were assigned to C–C/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, C–OH/C–N and OC groups, respectively.10,31 Fe 2p spectrum of the N-doped Fe3O4@C sample (Fig. 2d) shows 2 major characteristic peaks at 711.0 and 724.7 eV, which are assigned to Fe 2p 3/2 and Fe 2p 1/2 of Fe3O4.32 Additionally, the peak centered at 529.7 eV in the O 1s spectrum corresponds to the binding energy of lattice oxygen in Fe3O4,33 which also confirms the existence of Fe3O4 in the hybrid. A maximum peak at 531.2 eV in the O 1s spectrum corresponds to the carbonyl oxygen in CO and the Fe–O–C bonds in the hybrid.34,35
C, C–OH/C–N and OC groups, respectively.10,31 Fe 2p spectrum of the N-doped Fe3O4@C sample (Fig. 2d) shows 2 major characteristic peaks at 711.0 and 724.7 eV, which are assigned to Fe 2p 3/2 and Fe 2p 1/2 of Fe3O4.32 Additionally, the peak centered at 529.7 eV in the O 1s spectrum corresponds to the binding energy of lattice oxygen in Fe3O4,33 which also confirms the existence of Fe3O4 in the hybrid. A maximum peak at 531.2 eV in the O 1s spectrum corresponds to the carbonyl oxygen in CO and the Fe–O–C bonds in the hybrid.34,35
XRD patterns of the prepared N-doped Fe3O4@C are shown in Fig. 3a and the XRD patterns of PB@yeast bio-templates are shown in Fig. S4† to compare with N-doped Fe3O4@C data. As can be seen, 4 major peaks are located between 17.5° and 39.7, which confirm the presence of the face-centered cubic phase of PB nanoparticles.36 A broad peak around 2θ = 20°, assigned to the amorphous phase of yeast, also existed in the templates, verifying successful assembly of PB@yeast composites. After pyrolysis, the diffraction peaks at 30.2°, 35.5°, 43.3°, 53.7°, 57.1° and 62.8° in Fig. 3a correspond to (220), (311), (400), (422), (511), (440) planes of Fe3O4 (JCPDS 79-0419).37 A small and broad diffraction peak around 26.2° corresponds to the (002) plane of the graphitic carbon, endorsing that the iron compound catalyzed the graphitization of amorphous carbon somehow during the pyrolysis process.38 Different from previous report where PB was picked as a single precursor, the characteristic diffraction peaks of Fe3C were hard to detected, which may be attributed to the metastable state of iron carbides.39,40 Furthermore, the yeast matrix containing oxygen-enriched functional groups could support the formation of iron oxide. After reaction was complete, similar XRD patterns of the N-doped Fe3O4@C adsorbent were observed, indicating the chemical stability of N-doped Fe3O4@C adsorbent.
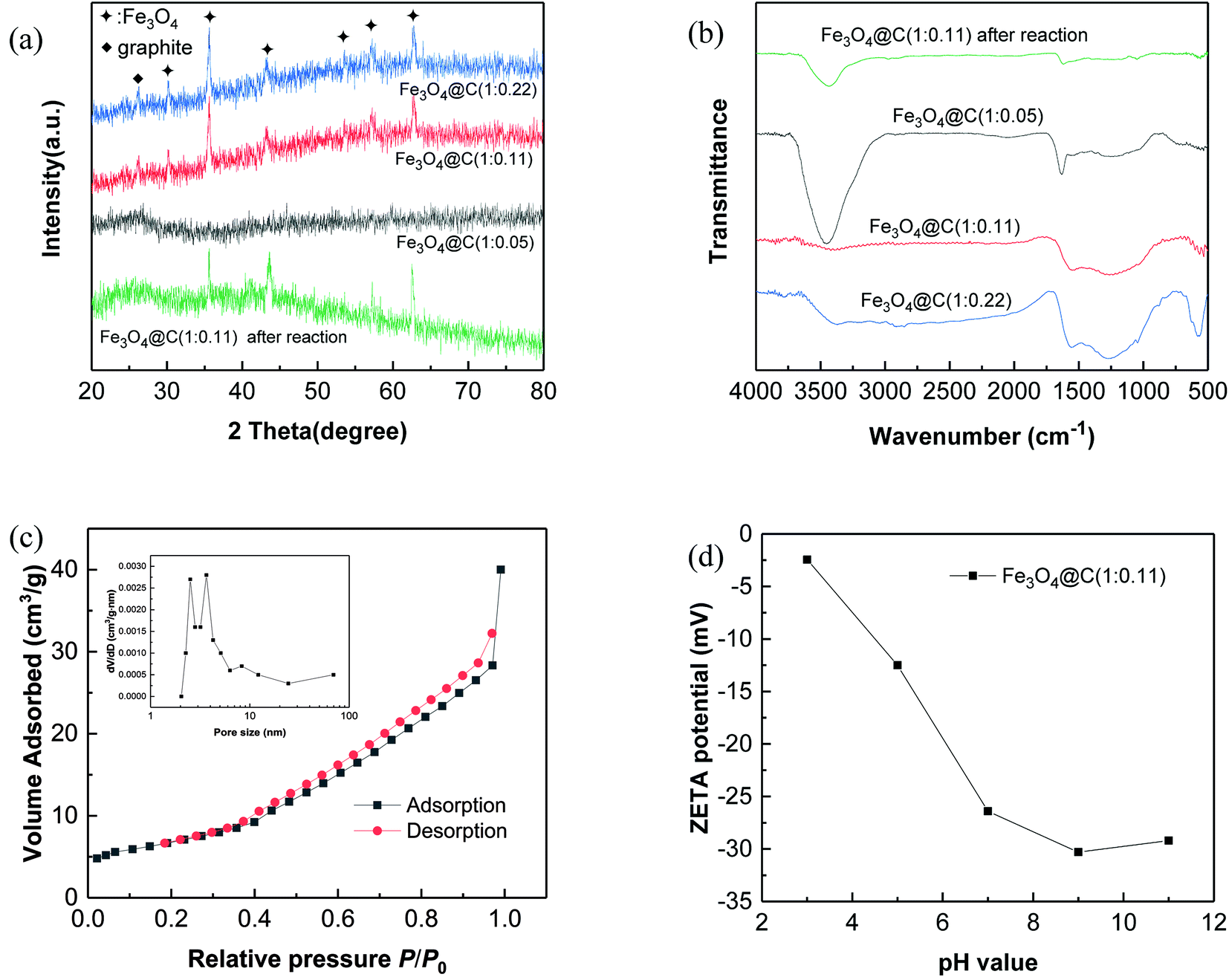 | ||
| Fig. 3 Characterization of N-doped Fe3O4@C: (a) XRD patterns; (b) FT-IR spectra; (c) N2 adsorption–desorption isotherm and pore size distribution curve (inset) and (d) zeta potential. | ||
Fig. 3b illustrates the FT-IR spectra of N-doped Fe3O4@C samples. A strong peak around 3300–3600 cm−1 corresponds to the adsorption band of O–H bending vibrations, indicating the existence of a large number of residual hydroxyl groups. The peaks at 1554 cm−1 and 880 cm−1 are likely assigned to N–H bending vibrations, and the peak at 1254 cm−1 corresponds to the C–N stretching vibration overlapping with the absorption bands of aromatic C–O.41 These characteristic absorption peaks indicate the presence of amide and hydroxyl groups in the prepared N-doped Fe3O4@C hybrids, all of which came from yeast substrates. In addition, the band at 579 cm−1 may correspond to the Fe–O stretching vibration of the Fe3O4@C surface.42 Combined with the results of XRD analysis, it can be concluded that Fe3O4 was indeed formed on the N-containing carbonaceous composites. Additionally, comparing these 3 products, similar XRD patterns and FT-IR spectra were obtained, indicating the universal production of N-doped Fe3O4@C hybrids.
The N2 adsorption–desorption isotherm for N-doped Fe3O4@C hybrid is given in Fig. 3c. According to BET analysis, a specific surface area of 31.26 m2 g−1 with meso- and macroporous features was obtained.43 In addition, the pore volume was found to be 0.0265 cm3 g−1, and the containing pores of N-doped Fe3O4@C ranged from a few nanometers to 69 nm (inset of Fig. 3c), which is essential for the adsorption performance of N-doped Fe3O4@C, since it may provide more contact area and retain a coherent channel for the transmission of target molecules. Additionally, the surface potential in Fig. 3d indicated that the surface of Fe3O4@C (1:0.11) was negatively charged over the entire pH range. Since the hydroxyl groups ionized to form –O− at high pH, the negative charge of the surface was getting stronger as the pH value increased. The potential reached −26.4 mV at pH 7.0, demonstrating good stability of the product at neutral conditions, which could be important for long-term applications.
3.2 Adsorption performance of N-doped Fe3O4@C in RhB removal
RhB, a typical cationic dye, has attracted a great deal of attention because of its industrial relevance and inflexible nature.44,45 Herein, the adsorption properties of N-doped Fe3O4@C samples were evaluated by removing RhB from water. Fig. 4a shows the adsorption performance for RhB adsorption onto as-prepared N-doped Fe3O4@C adsorbents. Experimental data showed that the uptake capacities of N-doped Fe3O4@C increased gradually with the increase of initial concentration, which was attributed to the increased driving force from the concentration gradient that readily accelerated the diffusion of the RhB molecules.46 In addition, the adsorption capacity of Fe3O4@C (1:0.11) was higher than that of Fe3O4@C (1:0.05) or Fe3O4@C (1:0.22). From reported studies,47,48 the presence of N could increase the electron density of N-containing carbon surface, thus, enhancing the interaction between dye molecules and absorbents. In this study, the N content of Fe3O4@C (1:0.11) was higher than that of Fe3O4@C (1:0.05), which further evidenced that the increased N-doped content could play a significant role in the improvement of adsorption performance. Also, good dispersion of Fe3O4 provided a strong coarse surface, which would be beneficial to the RhB molecules adsorbing on the N-doped Fe3O4@C. In addition, the unique 3D cage-like structure made an indispensable contribution to the high adsorption. However, the experimental result of (1:0.22) was not as good as that of Fe3O4@C (1:0.11), which probably was due to the external shell tightness, blocking plenty of targeted RhB molecules diffusing from the aqueous solution to the N-doped carbon surface. In brief, all the synthesized N-doped Fe3O4@C adsorbents had the ability to adsorb RhB in solution, and Fe3O4@C (1:0.11) performed better within 25–200 mg L−1 of the initial RhB concentration. This was mainly attributed the prominent comprehensive characteristics of Fe3O4@C (1:0.11) such as a regular 3D cage-like structure, N-modified synergy, and the presence of well-dispersed Fe3O4 nanoparticles on the carbon matrix. To explore the adsorption process, data were fitted by frequently-used Langmuir and Freundlich models. The former Langmuir model can be presented as:
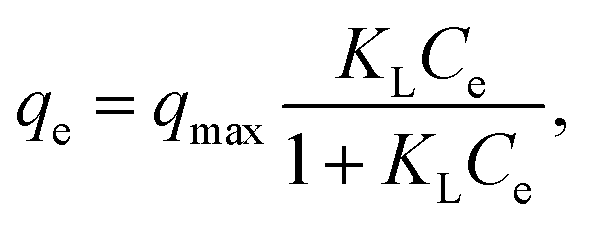 | (3) |
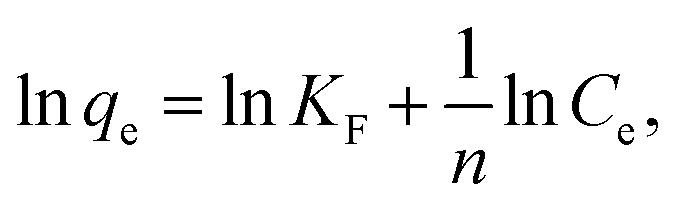 | (4) |
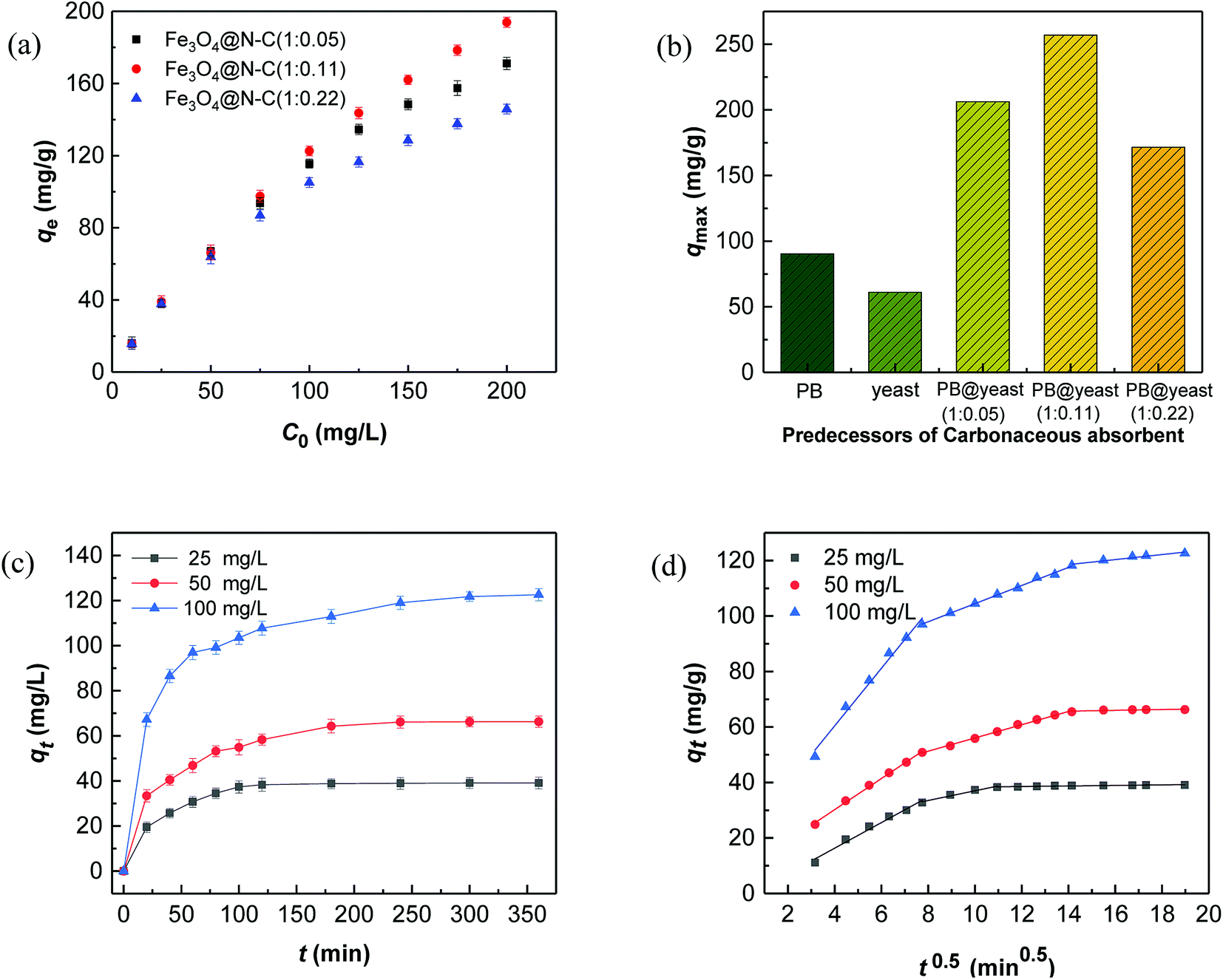 | ||
| Fig. 4 RhB adsorption performance of (a) Fe3O4@C adsorbents and (b) different predecessor-derived adsorbents. (c) Adsorption kinetics curves of Fe3O4@C (1:0.11). (d) Intra-particle diffusion model. | ||
The dimensionless separation factor (RL) was supplemented to mark the favorability of adsorption model, which is defined as:49,50
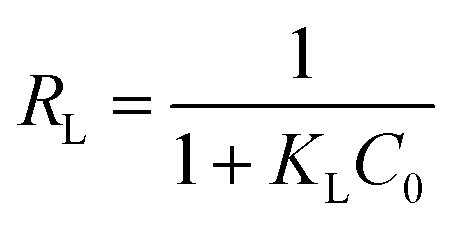 | (5) |
The curve fitting of isotherm models using different adsorbents is presented in Fig. S5(a and b),† and the corresponding parameters are listed in Table 2.
| Sample | Langmuir model | Freundlich model | ||||
|---|---|---|---|---|---|---|
| qmax (mg g−1) | KL (L g−1) | R2 | 1/n | KF (L g−1) | R2 | |
| Fe3O4@C (1:0.05) |
206.19 | 0.0439 | 0.9961 | 0.4019 | 9.0635 | 0.9824 |
| Fe3O4@C (1:0.11) |
257.06 | 0.0343 | 0.9961 | 0.4874 | 8.5676 | 0.9733 |
| Fe3O4@C (1:0.22) |
171.53 | 0.0442 | 0.9955 | 0.3575 | 9.0228 | 0.9873 |
The results illustrated that the adsorption of RhB on the 3 N-doped Fe3O4@C adsorbents were well fitted by both the Langmuir model (R2 = 0.9961, 0.9961, 0.9955) and the Freundlich model (R2 = 0.9824, 0.9733, 0.9873). From the calculated data, the 1/n values for 3 kinds of N-doped Fe3O4@C adsorbents were below 1 and the RL values were in the range of 0.10–0.37, suggesting that Langmuir model performed better than the Freundlich model. The Fe3O4@C (1:0.11) sample achieved the highest adsorption capacity (257.06 mg g−1), higher than those of Fe3O4@C (1:0.05) (206.19 mg g−1) and Fe3O4@C (1:0.22) (171.53 mg g−1). Still, the adsorption capacities of all prepared N-doped Fe3O4@C samples were much higher than those of either PB-based or yeast-based carbonaceous adsorbents under the same conditions (Fig. 4b, 90.34, 61.03 mg g−1). These data further support our prediction with regards to the N-doped Fe3O4@C adsorbents showing outstanding adsorption abilities for RhB removal because of the comprehensive conditions, discussed earlier. Compared with previous literatures results (see in the Table S1†), the maximum adsorption capacity for RhB by N-doped Fe3O4@C adsorbents was relatively higher than that of other carbonaceous adsorbents. These results demonstrated that the N-doped Fe3O4@C material derived from PB@yeast bio-templates can be used as a high-efficiency adsorbent for RhB removal in aqueous solution.
3.3 Adsorption kinetics
Fig. 4c shows the data for the selected initial concentrations of RhB adsorption by Fe3O4@C (1:0.11) at different contact times. It was found that fast adsorption occurred at the first 60 min because of abundant adsorption sites on the Fe3O4@C surface, indicating that strong binding forces exist between the adsorbent and adsorbate. For understanding the kinetic adsorption mechanism, 3 kinetic models, namely, pseudo-first-order model (eqn (6)), pseudo-second-order model (eqn (7)), and an intra-particle diffusion model (eqn (8)) were applied to fit the experimental data.51–53
These models can be written as:
|
ln(qe − qt) = lnqe − k1t
| (6) |
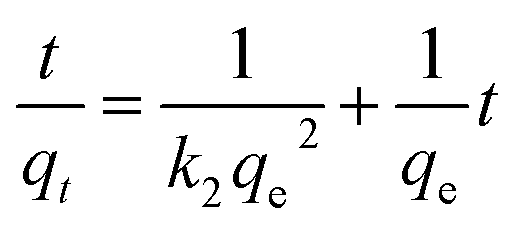 | (7) |
| qt = kidt0.5 + C | (8) |
The corresponding plots are shown in Fig. S5(c, d)† and 4d, and the related kinetic parameters are listed in Table S2.† It was revealed in Fig. S5(c and d)† that a better linear curve was generated by t/qt versus t for pseudo-second-order model than ln(qe − qt) versus t for pseudo-first-order model, and the higher correlation coefficient (R2 = 0.9977, 0.9989, 0.9993) was obtained compared to that of the pseudo-first-order model (R2 = 0.9643, 0.9515, 0.9747). In addition, the theoretical qe,cal values, calculated using the pseudo-second-order model, were very close to the qe,exp values. These results suggest that adsorption followed the pseudo-second-order kinetic model well, demonstrating that physicochemical interactions, such as valence forces between adsorbent and adsorbate, are responsible for the removal of RhB from solution.54,55
Ordinarily, the overall adsorption by carbonaceous adsorbent may occur in several stages, such as external surface adsorption, intra-particle diffusion and interior surface adsorption.56 It has been affirmed that hierarchical pores exist in the unique 3D cage-like structured N-doped Fe3O4@C adsorbent, which is sufficient for the fast diffusion of RhB molecules. As shown in Fig. 4d, the fitting curves displayed multi-linearity in general but did not pass through the origin, signifying that intra-particle diffusion played an important role in the adsorption process, even though it was not the only rate-controlling step in this mode. From Table S2,† the rate constant of the first stage was greatest out of all of 3 sets of data. It is reasonable that the surface adsorption through boundary layer diffusion usually occurs in the first step, which is significantly affected by the resistance of mass transfer around the adsorbate. Apparently, it was prominent in the early stage of adsorption because of the diffusion driven force of high initial RhB concentration, followed by the gradual adsorption stage dominated by the intra-particle diffusion, and the final equilibrium stage with low RhB concentration in solution.57,58 The experimental results of RhB diffusion revealed that both film diffusion and intra-particle diffusion were functioned during the adsorption process. In view of the unique morphology of the N-doped Fe3O4@C adsorbent, a cage-like structure with numerous visible pores can greatly promote the transport of guest molecules into the pores to reduce the diffusion resistance, thereby, leading to high adsorption of RhB.
3.4 Adsorption thermodynamics
The temperature-dependent adsorption capacities for RhB on Fe3O4@C (1:0.11) adsorbent at 10, 25, 40, and 55 °C are shown in Fig. 5, and the detailed analysis is presented in Table S3.†
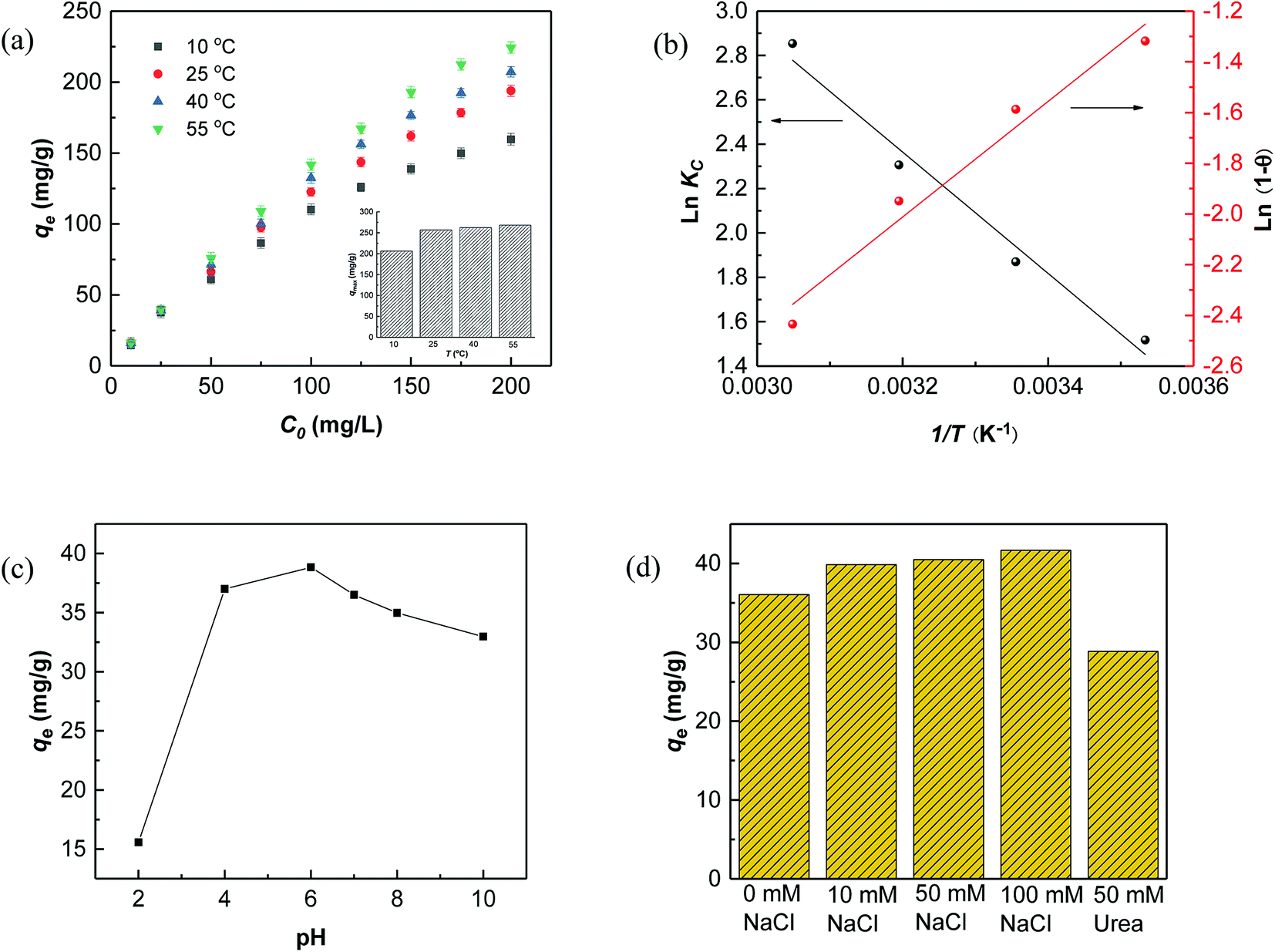 | ||
| Fig. 5 (a) Adsorption capacity at 10, 25, 40, and 55 °C; (b) the linear plots of lnKC vs. 1/T and ln(1 − θ) vs. 1/T for RhB adsorption on Fe3O4@C (1:0.11) at 10, 25, 40, and 55 °C. (c) Effect of pH; (d) effects of ionic strength and urea. | ||
Thermodynamic parameters were determined from the distribution coefficient (Kc) defined by eqn (9):
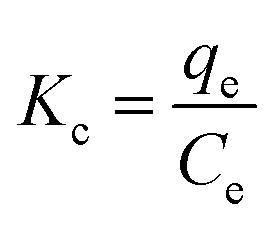 | (9) |
The corresponding changes in standard free energy (ΔGo), standard enthalpy (ΔHo) and standard entropy (ΔSo) for RhB can be defined by the following eqn (10)–(12):59
| ΔGo = ΔHo − ΔSoT | (10) |
|
ΔGo = −RTlnKC
| (11) |
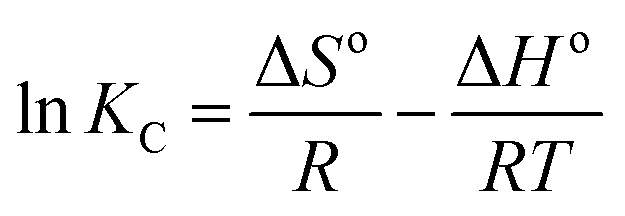 | (12) |
KC versus 1/T, and thermodynamic parameters are summarized in Table 3.
:0.11)
| T (°C) | ΔGo (kJ mol−1) | ΔSo (J mol−1 K−1) | ΔHo (kJ mol−1) | Ea (kJ mol−1) | S* |
|---|---|---|---|---|---|
| 10 | −3.42 | 92.40 | 22.73 | 18.95 | 0.0395 |
| 25 | −4.81 | ||||
| 40 | −6.19 | ||||
| 55 | −7.58 |
The results in Fig. 5a showed that the adsorption capacities increased explicitly from 206.61 mg L−1 to 268.10 mg L−1 as the temperature rose from 10 °C to 55 °C, indicating that higher temperature would be favorable for RhB adsorption by Fe3O4@C (1:0.11) adsorbent. The increased temperature phenomenon might occur due to the improved mobility of the large RhB molecules, which could speed up the transfer of the dye from aqueous solution to the Fe3O4@C surface. As seen in Table 3, positive values of ΔHo and ΔSo indicate an endothermic adsorption process and an increased randomness at the solid–liquid interface during RhB adsorption on Fe3O4@C (1:0.11) product, which is similar to previously reported results.60 The negative value of ΔGo, ranging from −3.43 to −7.58 kJ mol−1, revealed that RhB adsorption was spontaneous and feasible. Moreover, the values decreased with the increase in temperature, revealing a more effective removal of RhB with a degree of spontaneity at higher temperatures. From Ma and co-workers,61 the Gibb's free energy, ΔGo, changes within the range of −20 and 0 kJ mol−1 and indicates a physical adsorption process. To further affirm the physical domination adsorption mechanism for RhB removal on N-doped Fe3O4@C adsorbent, a modified Arrhenius equation (eqn (13)) was used to estimate the activation energy (Ea) and sticking probability (S*):62
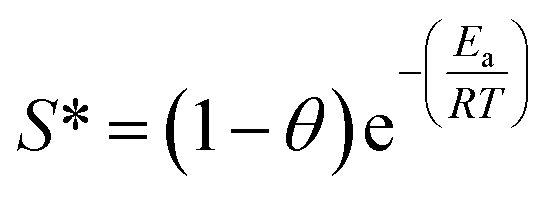 | (13) |
The surface coverage (θ) could be calculated using eqn (14):
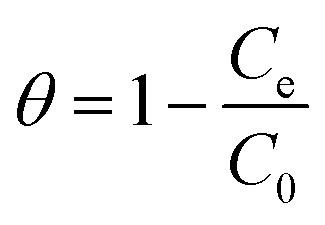 | (14) |
A plot of ln(1 − θ) versus 1/T is displayed in Fig. 4b and the corresponding values of Ea and S* are listed in Table 3. The results showed that the value of sticking probability (S*) was between 0 and 1, indicating favorable sticking between the adsorbent and adsorbate.63 These data in combination with ΔGo values ranging from −3.43 to −7.58 kJ mol−1 in the temperature range from 10 °C to 55 °C indicate that adsorption of RhB on Fe3O4@C (1:0.11) product matched physisorption predominant mechanism well, which is preferential for separation and reusability of the adsorbent.
3.5 Mechanism of RhB adsorption
Based on the above results, the description of a possible interaction mechanism between RhB molecules and Fe3O4@C (1:0.11) in a wide pH range of 2.0–10.0 is displayed in Fig. 5c. It shows that the equilibrium adsorption capacity dramatically increased from pH 2.0 to 6.0 but decreased with further increasing pH values. This phenomenon could be interpreted by the surface electrical properties of Fe3O4@C (1:0.11) adsorbent and the relative ionization. As shown in Fig. 2d, the point of pHpzc of Fe3O4@C (1:0.11) was remarkably lowered to pH 3.0. Therefore, the Fe3O4@C (1:0.11) was positively charged at pH 2.0, which would electrostatically repel cationic RhB molecules. As the pH value increased to 6.0, the RhB adsorption behavior was promoted by increasing electronegativity of the Fe3O4@C (1:0.11), which was ascribed to the enhanced electrostatic attraction and decreased competitive interactions with H+. For pH values higher than 6.0, a few RhB molecules initially existed in zwitterion form (RhB±) and then formed dimers, which could block the small pores to some degree.64,65 Meanwhile, the free Fe(II)/Fe(III), stemming from iron corrosion, may bind OH− and further block the pores on the surface of Fe3O4@C (1:0.11) under alkaline conditions, thereby, leading to decreased adsorption amounts.
The effects of ionic strength and urea on RhB adsorption by Fe3O4@C (1:0.11) at pH 7.0 were also assessed. The results, depicted in Fig. 5d, showed that adsorption improved slightly with increased ionic strength, which might be attributed to the electrostatic screening effect of NaCl that weakened the electrostatic repulsion between RhB molecules at pH 7.0.66 In addition, the salting-out effect synchronously functioned with increasing ionic strength, which made it easier for RhB molecules to spread throughout the Fe3O4@C/water interface, driven by hydrophobic interactions, thus, increasing the adsorption amounts of RhB on the Fe3O4@C surface.67 Urea is widely accepted as a hydrogen bond inhibitor since it can preferentially form hydrogen bonds between itself and the carbon/water interface. Here, the absorption of RhB decreased a little after adding urea (Fig. 5d), which was due to the competing adsorption between urea and RhB molecules at the Fe3O4@C (1:0.11)/water interface. This result confirmed that hydrogen bonding also played a role in the adsorption process. In summary, the mechanism of RhB adsorption on Fe3O4@C (1:0.11) may combine electrostatic, hydrophobic and hydrogen bonding interactions.
3.6 Regeneration and reusability
Regeneration of adsorbents using Fenton reaction is considered a promising alternative approach compared to traditional physical regeneration, since it can continuously generate active free radicals to attack dye molecules, leaving the organic contaminants degraded on-site. In particular, as a great advantage of PS-involved Fenton processes, the resulting sulfate radicals (SO4˙−) show higher oxidation potential and longer half-life than hydroxyl radicals (·OH), produced in the Fe/H2O2 system.3,68 In addition, introduction of a common transition metal and its oxides could significantly facilitate the generation of SO4˙−, thereby, effectively improving the degradation efficiency of organic pollutants.69,70 In this regard, it is reasonable to believe that the use of PS-activated Fenton reaction would offer a great potential for the regeneration of iron-modified adsorbents.Considering the integrated features of the prepared N-doped Fe3O4@C sample, a natural extension of this study allowed us to consider employing magnetic separation and PS activated Fenton regeneration to reuse the spent N-doped Fe3O4@C adsorbent. In the regeneration experiments, the quick response of Fe3O4@C (1:0.11) to a magnet, placed near the vessel (inset of Fig. 6b), stated clearly that it can be easily recycled from the solution in external magnetic field. The following PS activated Fenton regeneration was performed under visible light, and the regenerated Fe3O4@C (1:0.11) was reused for RhB adsorption. The reused adsorption performance is shown in Fig. 6.
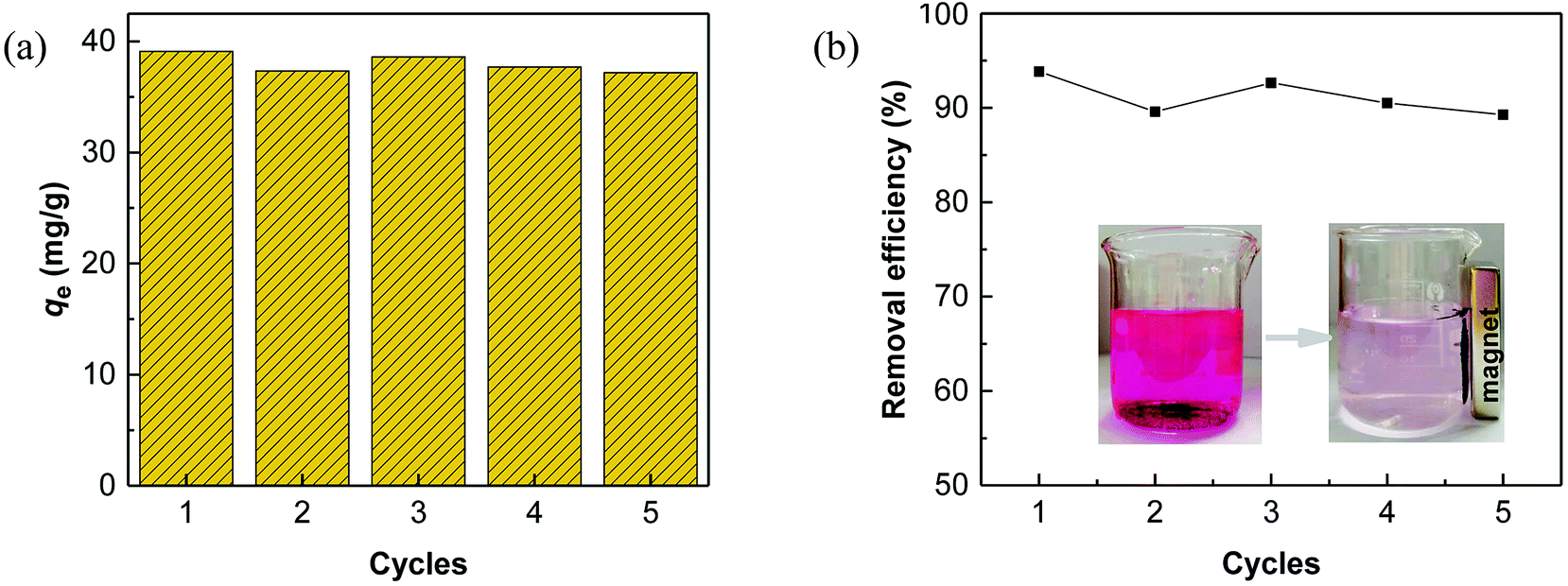 | ||
| Fig. 6 (a) Adsorption capacity and (b) adsorption efficiency of Fe3O4@C (1:0.11) for RhB removal in five consecutive adsorption–reduction cycles. Inset: images of RhB solution and the magnetic separation after adsorption. | ||
It revealed that the adsorption capacity toward RhB remained stable, and the removal efficiency of the regenerated Fe3O4@C (1:0.11) still reached 89.2%, even after 5 adsorption–regeneration cycles, indicating the high reusability obtained by Fenton regeneration. According to Gao and co-workers,71 the excited RhB* molecule under visible light, combined with Fe(II) formed on the surface of Fe–K, could activate PS to generate free radicals for RhB degradation. The probable processes could be described by eqn (15)–(20):
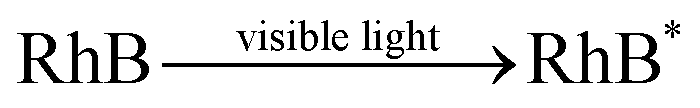 | (15) |
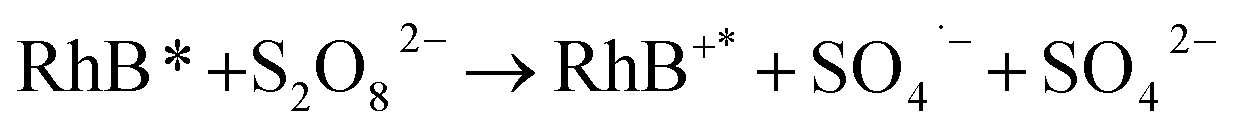 | (16) |
RhB* + ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Fe(III) → Fe(II) + RhB+* Fe(III) → Fe(II) + RhB+*
| (17) |
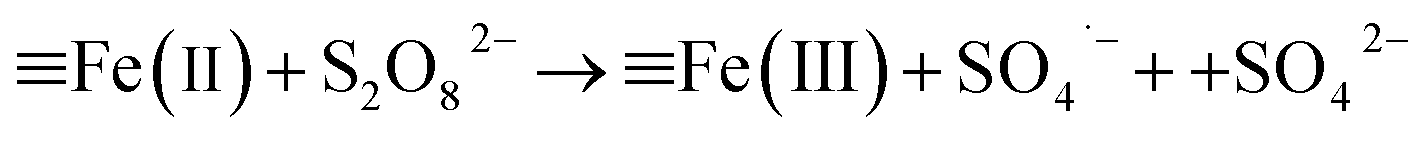 | (18) |
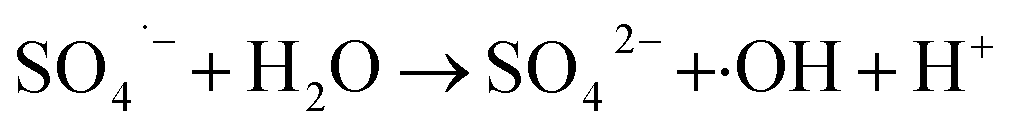 | (19) |
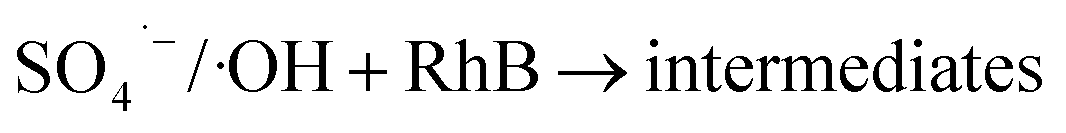 | (20) |
In term of the N-doped Fe3O4@C, the solution during the Fenton-like regeneration remained colorless, and the pH value decreased approximately from 6.8 to 4.7 (not shown), indicating that the RhB molecules, adsorbed on the surface of N-doped Fe3O4@C, were degraded directly without desorption to the solution and some acid intermediates were formed after photo-Fenton reaction. These results are essentially in agreement with the published studies.72,73 Based on previous results, it can be inferred that both Fe3O4 and N-doped graphitic carbon played significant role in the regeneration.74 The enriched N increased the content of π-electron rich sites on the surface of the carbon matrix, which facilitated the degradation of RhB in the photo-Fenton reaction and also ensured high stability of the product.41 In summary, Fe3O4@C (1:0.11) displayed high reusability, achieved by magnetic separation and PS activated photo-Fenton regeneration, and can be considered a valuable and effective material for RhB removal in aqueous solutions.
4. Conclusion
In this study, a facile and economical PB@yeast template-directed conversion was developed to prepare 3D cage-like magnetic N-doped Fe3O4@C adsorbent, using yeast cells and potassium ferricyanide as raw materials. Introduction of yeast cells not only enriches the N, C sources but also provides a unique cage-like microstructure, which endows the resultant products with significantly improved absorption performance. RhB was employed to study the adsorption behavior and the possible adsorption mechanism of N-doped Fe3O4@C for comparison with a batch system. The experimental results verified that the maximum adsorption capacity of 257.06 mg g−1 was achieved for Fe3O4@C (1:0.11), which was much better than that of a PB-derived carbon material. Further, the spent Fe3O4@C adsorbent was easily recovered by an applied magnetic field and regenerated by PS activated photo-Fenton reaction, which could be sustainably repeated 5 times without a significant drop in activity, demonstrating good reusability and stability of the adsorbent. In short, a novel easily constructed and practical N-doped Fe3O4@C microcage is a promising candidate for the dye-containing wastewater treatment.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Innovation platform for the development and construction of special project of Key Laboratory of Tibetan Medicine Research of Qinghai Province (No. 2017-ZJ-Y11) and Qinghai Provincial Science Foundation (2017-SF-A8).References
- L. Zhang, H. B. Wu, R. Xu and X. W. Lou, CrystEngComm, 2013, 15, 9332–9335 RSC.
- L. Hu, R. Zhang, L. Wei, F. Zhang and Q. Chen, Nanoscale, 2015, 7, 450–454 RSC.
- K. A. Lin and B. J. Chen, Chemosphere, 2017, 166, 146–156 CrossRef CAS PubMed.
- T. Wen, X. Wang, J. Wang, Z. Chen, J. Li, J. Hu, T. Hayat, A. Alsaedi, B. Grambow and X. Wang, Inorg. Chem. Front., 2016, 3, 1227–1235 RSC.
- Á. Sánchez-Sánchez, F. Suárez-García, A. Martínez-Alonso and J. M. D. Tascón, J. Colloid Interface Sci., 2015, 450, 91–100 CrossRef PubMed.
- H. Li, N. An, G. Liu, J. Li, N. Liu, M. Jia, W. Zhang and X. Yuan, J. Colloid Interface Sci., 2016, 466, 343–351 CrossRef CAS PubMed.
- L. Dai, Y. Xue, L. Qu, H. Choi and J. Baek, Chem. Rev., 2015, 115, 4823–4892 CrossRef CAS PubMed.
- H. Wang, T. Maiyalagan and X. Wang, ACS Catal., 2012, 2, 781–794 CrossRef CAS.
- J. D. Wigginscamacho and K. J. Stevenson, J. Phys. Chem. C, 2015, 113, 19082–19090 CrossRef.
- Y. Hou, T. Huang, Z. Wen, S. Mao, S. Cui and J. Chen, Adv. Energy Mater., 2014, 4, 1220–1225 Search PubMed.
- C. Zhang, J. Liu, Y. Ye, Z. Aslam, R. Brydson and C. Liang, ACS Appl. Mater. Interfaces, 2018, 10, 2423–2429 CrossRef CAS PubMed.
- Y. Wang, Z. Gong, X. Yang, H. Shen, Q. Wang, J. Wang and Z. K. Zhao, Process Biochem., 2015, 50, 1097–1102 CrossRef CAS.
- X. Zhang, W. He, Y. Yue, R. Wang, J. Shen, S. Liu, J. Ma, M. Lia and F. Xua, J. Mater. Chem., 2012, 22, 19948–19956 RSC.
- X. Yang, G. Jin, Z. Gong, H. Shen, Y. Song, F. Bai and Z. K. Zhao, Bioresour. Technol., 2014, 158, 383–387 CrossRef CAS PubMed.
- P. Magnelli, J. F. Cipollo and C. Abeijon, Anal. Biochem., 2002, 301, 136–150 CrossRef CAS PubMed.
- D. Z. Ni, L. Wang, Y. H. Sun, Z. R. Guan, S. Yang and K. B. Zhou, Angew. Chem., 2010, 122, 4319–4323 CrossRef.
- Z. Guan, L. Liu, L. He and S. Yang, J. Hazard. Mater., 2011, 196, 270–277 CrossRef CAS PubMed.
- X. Zhu, Y. Yan, W. Wan, Y. Wang, Y. Wu, X. He and Z. Lü, Mater. Lett., 2018, 215, 71–74 CrossRef CAS.
- W. Shen, Y. He, S. Zhang, J. Li and W. Fan, ChemSusChem, 2012, 5, 1274–1279 CrossRef CAS PubMed.
- S. Gao, X. Wei, H. Fan, L. Li, K. Geng and J. Wang, Nano Energy, 2015, 13, 518–526 CrossRef CAS.
- L. Zhang, H. B. Wu, S. Madhavi, H. H. Hng and X. W. Lou, J. Am. Chem. Soc., 2012, 134, 17388–17391 CrossRef CAS PubMed.
- R. Song, B. Bai and D. Jing, J. Chem. Technol. Biotechnol., 2014, 90, 930–938 CrossRef.
- L. Zhang, H. B. Wu, S. Madhavi, H. H. Hng and X. W. Lou, J. Am. Chem. Soc., 2012, 134, 17388 CrossRef CAS PubMed.
- S. C. Jang, S. M. Kang, Y. Haldorai, K. Giribabu, G. W. Lee, Y. C. Lee, M. S. Hyun, Y. K. Han, C. Roh and Y. S. Huh, Sci. Rep., 2016, 6, 38384 CrossRef CAS PubMed.
- X. Xu, R. Cao, S. Jeong and J. Cho, Nano Lett., 2012, 12, 4988–4991 CrossRef CAS PubMed.
- O. Necas, Bacteriol. Rev., 1971, 35, 149–170 CAS.
- M. A. Lillo-Ródenas, J. Juan-Juan, D. Cazorla-Amorós and A. Linares-Solano, Carbon, 2004, 42, 1371–1375 CrossRef.
- Z. Lin, G. Waller, Y. Liu, M. Liu and C. P. Wong, Adv. Energy Mater., 2012, 2, 884–888 CrossRef CAS.
- J. Liu, Y. Zheng, Z. Hong, K. Cai, F. Zhao and H. Han, Sci. Adv., 2016, 2, e1600858 CrossRef PubMed.
- Y. Liu, N. Zhang, C. Yu, L. Jiao and J. Chen, Nano Lett., 2016, 16, 3321–3328 CrossRef CAS PubMed.
- Z. H. Sheng, L. Shao, J. J. Chen, W. J. Bao, F. B. Wang and X. H. Xia, ACS Nano, 2011, 5, 4350–4358 CrossRef CAS PubMed.
- Y. Xue, H. Chen, D. Yu, S. Wang, M. Yardeni, Q. Dai, M. Guo, Y. Liu, F. Lu, J. Qu and L. Dai, Chem. Commun., 2011, 47, 11689–11691 RSC.
- Z. Geng, Y. Lin, X. Yu, Q. Shen, L. Ma, Z. Li, N. Pan and X. Wang, J. Mater. Chem., 2012, 22, 3527–3535 RSC.
- N. A. Zubir, C. Yacou, J. Motuzas, X. Zhang and J. C. Diniz da Costa, Sci. Rep., 2014, 4, 4954 Search PubMed.
- J. Zhou, H. Song, L. Ma and X. Chen, RSC Adv., 2011, 1, 782–791 RSC.
- Y. Lu, L. Wang, J. Cheng and J. B. Goodenough, Chem. Commun., 2012, 48, 6544–6546 RSC.
- J. Yan, M. Lei, L. Zhu, M. N. Anjum, J. Zou and H. J. Tang, J. Hazard. Mater., 2011, 186, 1398–1404 CrossRef CAS PubMed.
- D. Deng, L. Yu, X. Chen, G. Wang, L. Jin, X. Pan, J. Deng, G. Sun and X. Bao, Angew. Chem., Int. Ed. Engl., 2013, 52, 371–375 CrossRef CAS PubMed.
- B. K. Barman and K. K. Nanda, Green Chem., 2016, 18, 427–432 RSC.
- H. Yang, J. Xiao, L. Su, T. Feng, Q. Lv and X. Zhang, Chem. Commun., 2017, 53, 3882–3885 RSC.
- Y. F. Shen, S. L. Suib, M. Deeba and G. S. Koermer, J. Catal., 1994, 146, 483–490 CrossRef CAS.
- G. Zhao, J. J. Feng, Q. L. Zhang, S. P. Li and H. Y. Chen, J. Mater. Chem., 2005, 17, 3154–3159 CrossRef CAS.
- Z. S. Wu, S. Yang, Y. Sun, K. Parvez, X. Feng and K. Müllen, J. Am. Chem. Soc., 2012, 134, 9082–9085 CrossRef CAS PubMed.
- D. Wan, W. Li, G. Wang, K. Chen, L. Lu and Q. Hu, Appl. Surf. Sci., 2015, 349, 988–996 CrossRef CAS.
- P. P. Gan and S. F. Y. Li, Chem. Eng. J., 2013, 229, 351–363 CrossRef CAS.
- A. S. Özcan and A. Özcan, J. Colloid Interface Sci., 2004, 276, 39–46 CrossRef PubMed.
- A. Dhaouadi and N. Adhoum, Appl. Catal., B, 2010, 97, 227–235 CrossRef CAS.
- C. S. D. Rodrigues, O. S. G. P. Soares, M. T. Pinho, M. F. R. Pereira and L. M. Madeira, Appl. Catal., B, 2017, 219, 109–122 CrossRef CAS.
- S. Kaur, T. P. Singh Walia and I. Kansal, J. Surf. Sci. Technol., 2008, 24, 179–193 Search PubMed.
- F. Haghseresht and G. Q. Lu, Energy Fuels, 1998, 12, 1100–1107 CrossRef CAS.
- S. Lagergren, K. Sven. Vetenskapsakad. Handl., 1898, 24, 1–39 Search PubMed.
- W. J. Weber and J. C. Morris, J. Sanit. Eng. Div., Am. Soc. Civ. Eng., 1963, 89, 31–60 Search PubMed.
- F. C. Wu, R. L. Tseng and R. S. Juang, Chem. Eng. J., 2009, 153, 1–8 CrossRef CAS.
- Y. S. Ho and G. Mckay, Process Biochem., 1999, 34, 451–465 CrossRef CAS.
- G. Bayramoglu, B. Altintas and M. Y. Arica, Chem. Eng. J., 2009, 152, 339–346 CrossRef CAS.
- F. Hayeeye, M. Sattar, W. Chinpa and O. Sirichote, Colloids Surf., A, 2017, 513, 259–266 CrossRef CAS.
- Y. Li, Q. Y. Yue and B. Y. Gao, J. Hazard. Mater., 2010, 178, 455–461 CrossRef CAS PubMed.
- S. Guo and G. Zhang, RSC Adv., 2016, 6, 2537–2545 RSC.
- D. Duranoğlu, A. W. Trochimczuk and U. Beker, Chem. Eng. J., 2004, 99, 193–202 Search PubMed.
- G. Vijayakumar, R. Tamilarasan and M. Dharmendirakumar, J. Mater. Environ. Sci., 2012, 3, 157–170 CAS.
- J. Ma, F. Yu, L. Zhou, M. X. Yang, J. S. Luan, Y. H. Tang, H. B. Fan, Z. W. Yuan and J. H. Chen, ACS Appl. Mater. Interfaces, 2012, 4, 5749–5760 CrossRef CAS.
- A. Aziz, M. S. Ouali, E. H. Elandaloussi, L. C. De Menorval and M. Lindheimer, J. Hazard. Mater., 2009, 163, 441–447 CrossRef CAS PubMed.
- M. Ghaedi, F. Karimi, B. Barazesh, R. Sahraei and A. Daneshfar, J. Ind. Eng. Chem., 2013, 19, 756–763 CrossRef CAS.
- K. Shakir, A. F. Elkafrawy, H. F. Ghoneimy, S. G. E. Beheir and M. Refaat, Water Res., 2010, 44, 1449–1461 CrossRef CAS PubMed.
- G. Crini, Bioresour. Technol., 2006, 97, 1061–1085 CrossRef CAS PubMed.
- M. A. FontechaCámara, M. V. LópezRamón, M. A. ÁlvarezMerino and C. MorenoCastilla, Langmuir, 2007, 23, 1242–1247 CrossRef PubMed.
- M. Campinas and M. J. Rosa, J. Colloid Interface Sci., 2006, 299, 520–529 CrossRef CAS PubMed.
- P. Hu and M. Long, Appl. Catal., B, 2016, 181, 103–117 CrossRef CAS.
- H. Li, J. Wan, Y. Ma and Y. Wang, Chem. Eng. J., 2016, 301, 315–324 CrossRef CAS.
- Q. Yang, H. Choi, S. R. Al-Abed and D. D. Dionysiou, Appl. Catal., B, 2015, 88, 462–469 CrossRef.
- Y. Gao, Z. Zhang, S. Li, J. Liu, L. Yao, Y. Li and H. Zhang, Appl. Catal., B, 2016, 185, 22–30 CrossRef CAS.
- L. Guz, G. Curutchet, R. M. Torres Sánchez and R. Candal, J. Environ. Chem. Eng., 2014, 2, 2344–2351 CrossRef CAS.
- Y. Gao, Y. Wang and H. Zhang, Appl. Catal., B, 2015, 178, 29–36 CrossRef CAS.
- X. K. Kong, Z. Y. Sun, M. Chen, C. L. Chen and Q. W. Chen, Energy Environ. Sci., 2013, 6, 3260–3266 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra08886a |
| This journal is © The Royal Society of Chemistry 2019 |