
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructure–property relationship of nitramino oxetane polymers: a computational study on the effect of pendant chains
Yiding Ma a,
Yingzhe Liub,
Tao Yubc,
Weipeng Laib,
Zhongxue Ge*b and
Zhenyi Jiang*d
a,
Yingzhe Liub,
Tao Yubc,
Weipeng Laib,
Zhongxue Ge*b and
Zhenyi Jiang*d
aXi'an Modern Chemistry Research Institution, Xi'an 710065, China. E-mail: yiding@mail.ustc.edu.cn
bXi'an Modern Chemistry Research Institution, Xi'an 710065, China
cSchool of Chemistry and Chemical Engineering, Southeast University, Nanjing, China
dSchool of Physics Northwest University, Xi'an, China
First published on 22nd January 2019
Abstract
A comprehensive study of the effect of the structure of pendant chains on the energetic and mechanical properties of nitramino oxetane polymers has been conducted. Enthalpy of formation (EOF), density, glass transition temperature, and elastic moduli were calculated via quantum mechanics and molecular dynamic simulations. It is shown in this study that –CH2 groups are unfavorable for EOFs, densities, and elastic moduli of the polymers, whereas –NCH3NO2 groups are favorable for these parameters. The glass transition temperature (Tg) shows non-monotonic features with increasing –CH2 groups; it reaches a minimum value when the pendant chains consist of 1 or 2 –CH2 groups. Moreover, the location of the pendant chains can strongly affect Tg of the polymers. Our study suggests that the asymmetric structure, distantly located pendant chains and appropriate length of the pendant chains can effectively reduce Tg of the polymers with negligible compromise to other properties.
Introduction
Polymers are used widely in energetic materials such as solid propellants and plastic bonded explosives (PBX).1–6 They are eligible binders for the formation of a continuous phase and contribute to the integrity and plasticity of a material, which are important characteristics when considering the transport, processing and weaponization of energetic materials. Traditional polymeric binders, such as hydroxyl-terminated polybutadiene (HTPB), provide good mechanical properties in a wide range of temperatures.7–9 However, their non-energetic features limit the further improvement in the energy density of the propellants and explosives. For better performances of energetic materials, it has become important to design and screen polymers with good mechanical properties, compatibility with other ingredients, and high energy.10–12A feasible method of designing an energetic polymeric binder is to “decorate” the polymer chain with energetic groups, such as azido or nitramino groups, on the side chains. Some energetic polymeric binders have been synthesized and applied in recent years.13–15 Among these, nitramino oxetane polymers are potential candidates as next-generation energetic binders since the nitramine groups provide good amount of energy and good compatibility with widely used energetic additives in propellants and explosives, whereas the oxetane backbone provides flexibility and good mechanical properties to the polymer.
Due to safety concerns and limitations of experimental techniques, so far, only a few studies have been reported on nitramino oxetane polymers. Poly(3-methylnitramino methyl-3-methyl oxetane) (PMNAMMO) and poly(bis-(3-methyl nitramino-methyl)oxetane) (PBMNAMO) were first synthesized by Manser;15 the structural and decomposition features of PMNAMMO and PBMNAMO were measured.16,17 These polymers are considered to be suitable ingredients of energetic binders in rocket propellants,18 aluminized explosives,19 energetic elastomers,20 and gas-generating materials.21 In practical applications, researchers and engineers often pay attention to the enthalpy of formation (EOF),22 glass transition temperature (Tg),23 and mechanical properties24 of the polymeric binders. However, to date, only PMNAMMO and PBMNAMO have been synthesized and reported in experiments with theoretical values of their enthalpies of formation, whereas few tests are reported about their mechanical properties and glass transition temperatures. EOF represents the energy contained in the polymeric binder, Tg is the temperature range of applications of the polymeric binders, where they act as elastic rubbers rather than fragile solids, and the mechanical properties of the polymeric binders are significant in the application of energetic materials. For the application of nitramino oxetane polymers, these properties should be comprehensively investigated. Moreover, for effectively seeking new and applicable energetic binders, we require a systematic investigation of the structure–property relationship of nitramino oxetane polymers. It is known that the structure of pendant chains can remarkably affect the properties of the polymers. The length and the location of the pendant chains are closely related to the flexibility and entanglement of the polymers; also, the substituent group on the pendant chains can affect the polarity and other properties of the polymer. For example, PBMNAMO has an additional methylnitramino group compared to PMNAMMO; therefore, it is considered to be more energetic than PMNAMMO. Due to the symmetrical structure of PBMNAMO, it is assumed to be easy to crystallize and less flexible, which makes it a potential candidate for the “hard segment” of thermoplastic elastomers.25
Computer simulations have been testified to be effective and reliable in the prediction of properties of materials;26–38 means such as quantum mechanics26–28 and molecular dynamics29–38 allow high-flux designing and screening of new materials at lower costs compared to one-by-one experimentation. Dorofeeva27 calculated the enthalpy of formation of hexanitrohexaazaisowurtzitane (CL-20) using the quantum mechanics method; the results showed good consistency with those of the experiments. The mechanical and thermal properties of polymers can be predicted by molecular dynamic simulations.31 In this study, we combine quantum mechanics and molecular dynamic methods to study both the energetic and mechanical properties of nitramino oxetane polymers. A comprehensive investigation of the effect of pendant chains on the properties of the polymers has been conducted.
Computational methods
Design of the energetic polymers
Polymers are made up of a long and stretching main chain, and the pendant chains grafted from it. In our study, the main chain structure of nitramine oxetane polymers was chosen to be the poly(n-oxapropylene) structure. At most two pendant chains, which consist of a few –CH2 groups and terminate with a hydrogen atom or –NCH3NO2 group, were grafted from the β-carbon of the oxygen atom. Each pendant chain contains 3 –CH2 groups at most. For clarity, each polymer is named by the length and the terminating groups of the pendant chains. For example, the polymer in Fig. 1 is named as “1N2H” since it has two pendant chains; one of them contains one –CH2 group and terminates with –NCH3NO2 group (for the letter “N”), and another has two –CH2 groups and terminates with –H atom (for the letter “H”). We designed 26 polymers in total, and the degree of polymerization of each polymer is 10. | ||
| Fig. 1 The naming of nitramino oxetanes; “N” and “H” represent the terminating groups of each pendant chain, and the number before them shows the length of the alkyl chains. | ||
Enthalpies of formation (EOFs) and heats of detonation
EOFs and heats of detonation are the parameters used to measure the energetic features of the polymers. EOFs are calculated by constructing the atomization reactions from the polymer to the constituent atoms and by investigating the enthalpy changes of the reactions. To reduce the cost of the calculation, we assume that the enthalpy contribution of every monomeric unit is a constant, so that we can obtain the EOF of every monomeric unit by linear fitting of EOFs of the oligomers22 (degree of polymerization is 2–6 in this study). Then, the heats of detonation are obtained by constructing the detonation reactions of the polymers and by calculating the enthalpy differences between the products and reactants. Here, the oligomers are optimized and their EOFs are calculated with the Density Functional Theory (DFT) method under the B3LYP/6-311G++(d,p) basis, and the products of the detonation reactions are assumed to be N2, C, CO2, and H2O.40,41Density, glass transition temperature (Tg), and mechanical properties
Density, glass transition temperature (Tg), and mechanical properties were predicted with molecular dynamics (MD) simulations using the Material Studio 8.0 (MS) software of Accelrys Inc. COMPASS force field39 was applied in the MD simulations, electrostatic interactions were treated with Ewald summation, and the cut-off distance of non-bonded interactions was set as 12.5 Å. Three-dimentional periodic boundary conditions were applied to the constructed cell. Berenden thermostat and barostat were applied to control the temperature and pressure of the systems, respectively. For each polymer, four independent initial configurations were created by putting 20 polymer chains randomly in the simulation box and setting the initial density of the system to 1.0 g cm−3. Then, 1000 ps simulation was carried out to make sure that the polymer systems reach thermal equilibrium, so that the locations of the polymers are independent of the initial configuration of the system; NVT ensemble was applied here and the temperature was 1000 K. The thermal equilibrated configurations were further applied to a series of NPT simulations, each of which lasted for 200 ps of simulation time. To obtain the configurations of the systems in ambient temperature and pressure, the temperature and pressure of the simulation were decreased step by step in the following order: 900 K–3 GPa, 800 K–300 MPa, 700 K–30 MPa, 600 K–3 MPa, 500 K–0.3 MPa, 400 K–0.1 MPa, 298 K–0.1 MPa. The density of each polymer was obtained from the average density of the four independent systems.According to Fox–Flory's free volume theory, there is a turning point around Tg where the specific volume of polymers decreases more rapidly with decreasing temperature. Thus, as shown in Fig. 2, we investigated the temperature dependence of the specific volume of the polymers in the range of 400–100 K, and Tg was determined by the changing point of the specific volume–temperature plot. To determine the cooling rate, we conducted simulations at the cooling rates of 15 K/200 ps and 10 K/200 ps; the results (Fig. 2) showed that the Tg values in these cooling rates are very close. Therefore, considering the accuracy and the cost of computations, we chose 15 K/200 ps as the cooling rate for the systems in our study. The uncertainty of Tg data in our study was about ±15 K (5%).
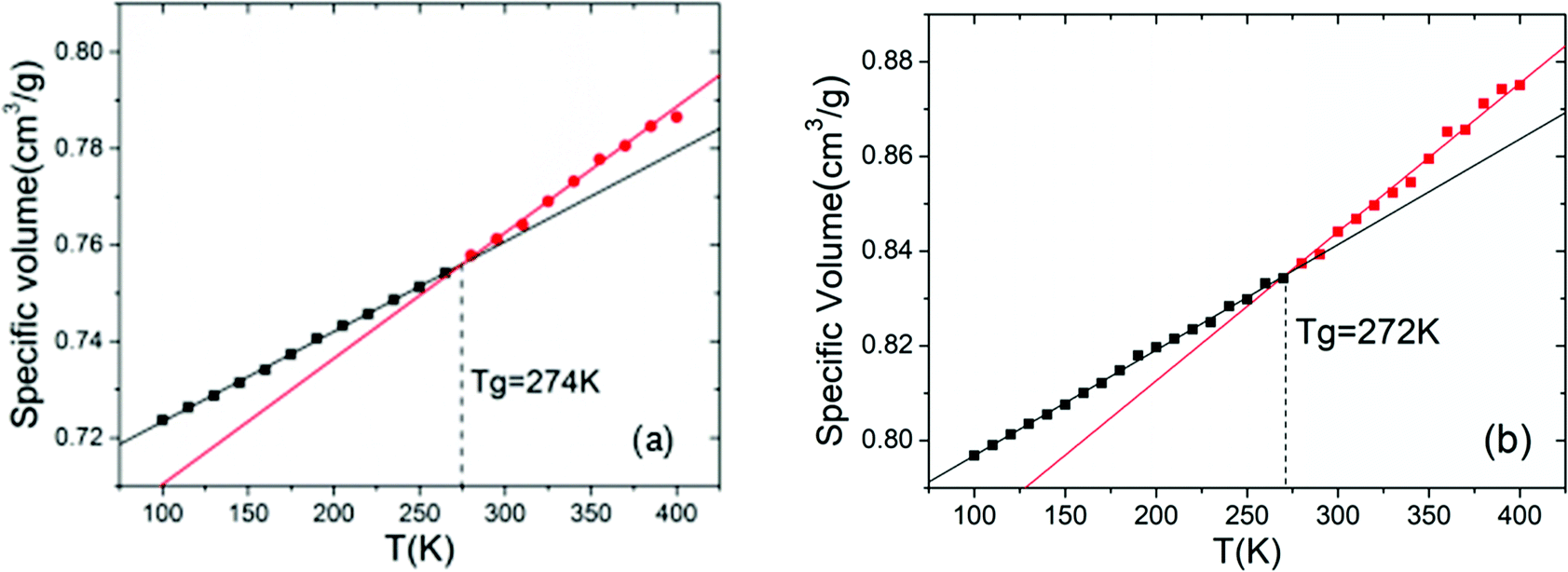 | ||
| Fig. 2 The calculation of Tg of PBMNAMO at the cooling rates of 15 K/200 ps (a) and 10 K/200 ps (b), respectively. The intersection points of the black and red lines, which are the linear fits of the black and red dots, mark Tg (274 K and 272 K, respectively). | ||
Mechanical properties are characterized by the elastic moduli and Poisson's ratio. The parameters are calculated on the basis of the Hooke's law (eqn (1) and (2)) by applying 0.5% deformation to the systems under 298 K and atmospheric pressure. The elastic moduli are calculated according to eqn (3) [Cij] is the elastic coefficient matrix containing 36 elements, and Lame coefficients (λ and μ) were introduced (C12 = λ, C11–C12 = 2μ).
| σi = Cijεj, i, j = 1,…6 | (1) |
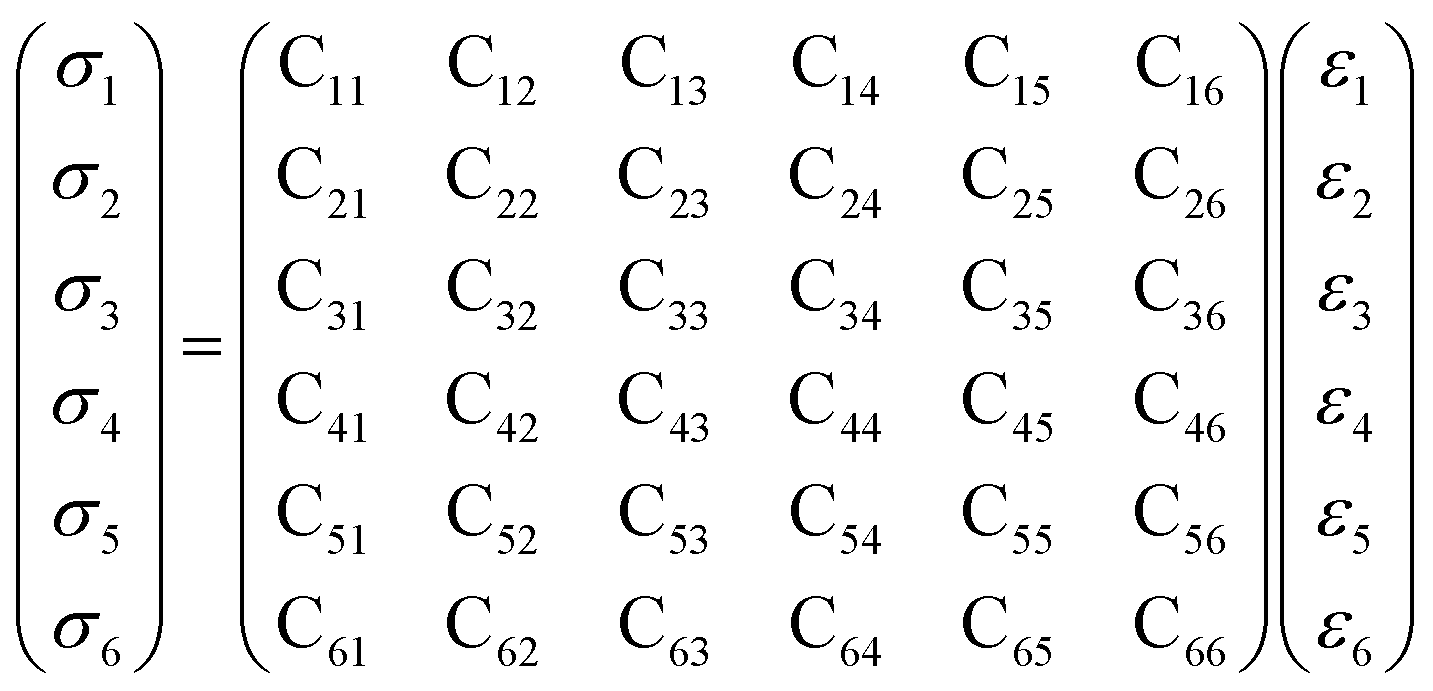 | (2) |
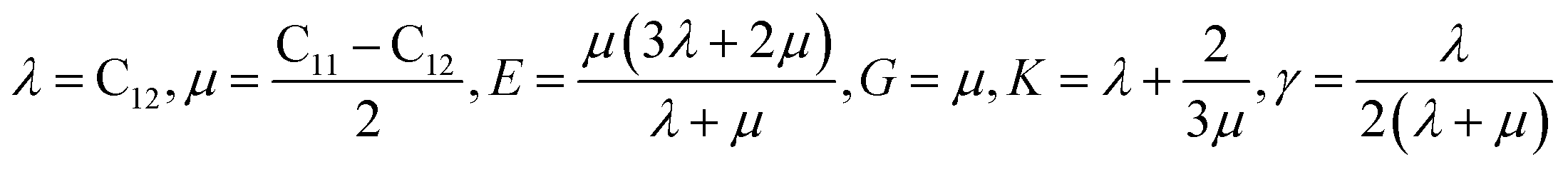 | (3) |
Results and discussions
One of the most important parameters of energetic polymeric binders is the glass transition temperature Tg, which is the key factor in deciding the temperature range of processing and application. It is the mark of a second-order phase transition of the polymers from glassy state to rubbery state. As shown in Fig. 3, polymers with more –NCH3NO2 groups exhibit slightly higher Tg because the polarity of the polymers increases with more –NCH3NO2 groups, and the mobility of the polymers decreases since the electrostatic interactions between molecules are stronger. Moreover, the existence of –NCH3NO2 group enhances the rotation barriers of the main chains of the polymers. These two factors are not beneficial to the flexibility of the polymers and result in higher Tg. This is also supported by the study by Lu et al.,42 which showed that strong intermolecular interactions and hydrogen bonding result in higher Tg.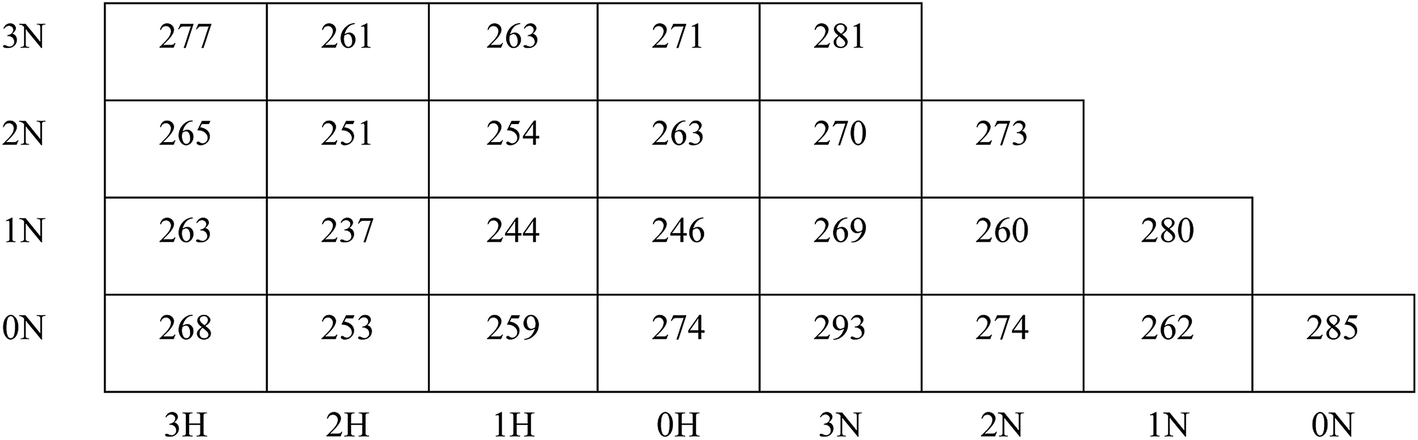 | ||
| Fig. 3 Glass transition temperature (in K) of the designed polymer. The data in the brackets are experimental values. The vertical axis represents the structure of one pendant chain and the horizontal axis represents the structure of another pendant chain. For example, information on the bottom-left is the density of polymer 0N3H. The uncertainty of the data of polymers is less than 5%. | ||
On the other hand, Tg of the polymers shows a non-monotonic feature when the pendant chains grow longer: Tg decreases at first, reaches a minimum value when the pendant chains contain 1–2 alkyl groups, and then increases with further increase in the length of the pendant chains. According to the free volume theory, the free volume of the system increases with the introduction of additional –CH2 groups, which benefits the flexibility of the polymers, and Tg tends to decrease. At the same time, the size of the pendant chains increases and the rotation barriers of the main chains of the polymers rise due to the steric hindrance between the pendant chains and other segments of the polymers. When the pendant chains are short, the introduction of extra free volume is the dominant effect in the flexibility of the polymers. Thus Tg decreases with increasing length of the pendant chains; with longer pendant chains, the steric hindrance is dominant, the polymers become less flexible and therefore, Tg increases. This suggestion is also supported by the comparison of the isomers, for example, 3N0H, 2N1H, 1N2H and 0N3H. When the –CH2 groups are displaced in two shorter side chains rather than in a single pendant chain, the free spaces in the systems are almost the same, whereas the steric hindrance of the free rotation of the polymer main chain is lower; hence, lower Tg is obtained. Please note that the polymers of symmetrical structures, i.e., 0N0N, 1N1N, 2N2N, and 3N3N do not obey such a trend; they have significantly higher Tg than the polymers having more or less –CH2 groups and have higher Tg than their isomers. The symmetric polymers are easy to crystallize; the chains in the crystalline region are folded and regularly arranged so that the chain segments can only move freely at higher temperatures. This is consistent with the difference between PBAMO and PAMMO;43,44 PBAMO is easy to crystallize and PAMMO is a flexible polymer, and the Tg values of two polymers are 246 K and 238 K, respectively.
The mechanical properties were characterized by the Young's modulus (E), bulk modulus (K), shear modulus (G), and Poisson's ratio, which indicate the capacity of tensile elasticity, incompressibility, rigidity, and the lateral deformation of the materials, respectively. The results are displayed in Fig. 4, and the uncertainty of the data of polymer mechanical properties is less than 5%. The Poisson's ratios of all the designed polymers are about 0.3, indicating good plastic properties.
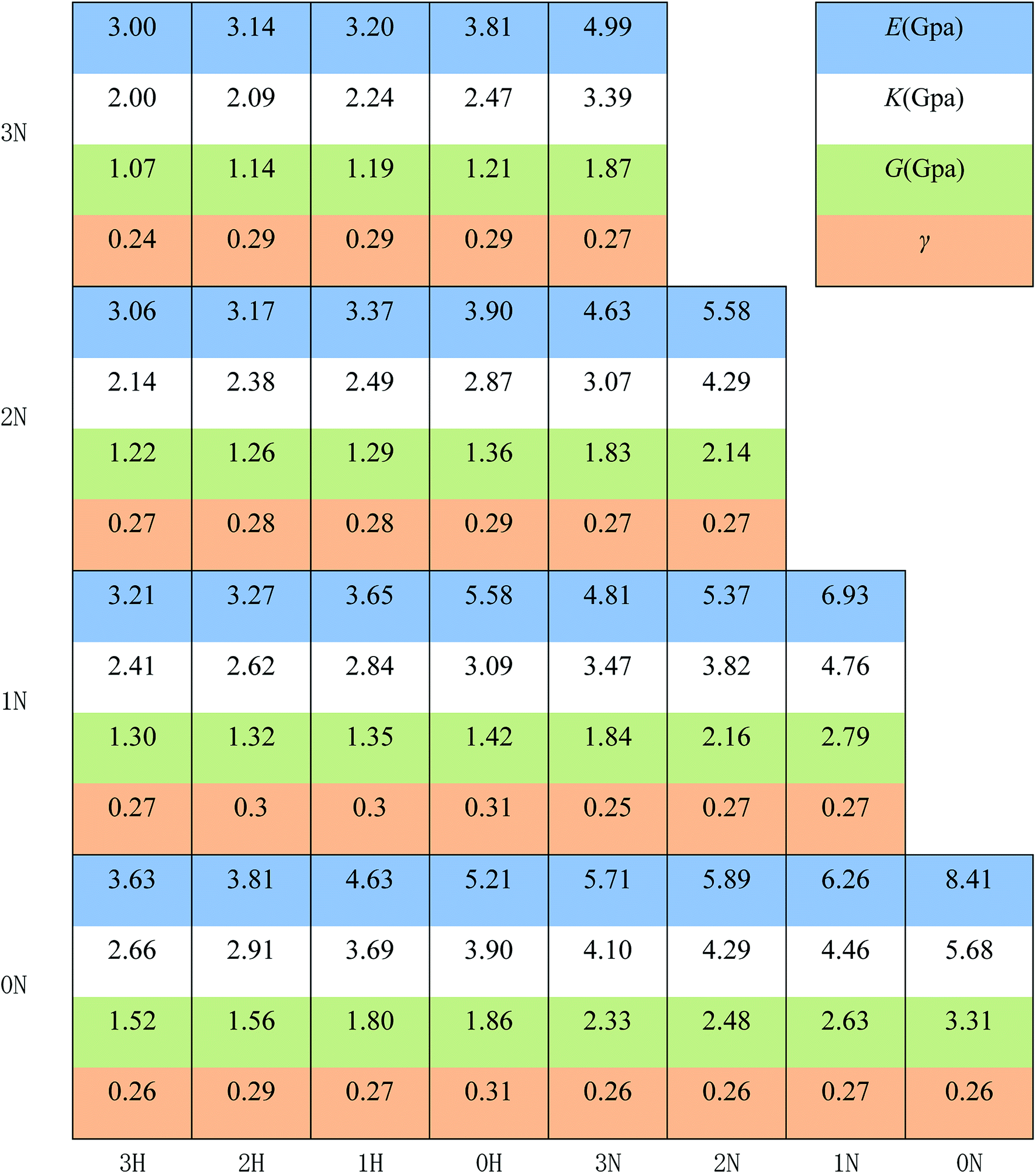 | ||
| Fig. 4 Young's modulus (E, in blue cells), bulk modulus (K, in green cells), shear modulus (G, in orange cells) and Poisson's ratio (γ, in white cells). The vertical axis represents the structure of one pendant chain and the horizontal axis represents the structure of another pendant chain. For example, information on the bottom-left is the density of polymer 0N3H. The uncertainty of the data of polymer mechanical properties is less than 5%. | ||
It is shown that E, K, and G decrease monotonously with increasing number of –CH2 groups. The rubbery features of polymeric binders originate from their long and entangled main chains. When the pendant chains grow longer, the fraction of the main chains becomes lower and the entanglement of the chains becomes weaker with additional free volume, thus lowering the moduli.
Fig. 4 also shows that polymers with –NCH3NO2 groups exhibit significantly higher moduli. The polarity of –NCH3NO2 groups may account for this result since the polymers are polarized by more –NCH3NO2 groups and the electrostatic interactions between the molecules are stronger, thus making the materials more resistant to elastic deformations. Combining the result in Fig. 4 and the data of Tg, we can see that PBMNAMO has higher elastic moduli, higher Tg and tendency to crystallize, indicating that PBMNAMO is a suitable choice for the “hard segment” of thermoplastic elastomers. On the other hand, PMNAMMO has better flexibility and is suitable for the “soft segment” of thermoplastic elastomers. In practice, PBMNAMO and PMNAMMO are used as described above.20
As is well-known, the density of the energetic binder is an important parameter since it is closely related to the density and solid content of solid propellants and explosives; thus, high energy density can be achieved by applying the binder with high density. The uncertainty of the calculated densities is about 7%. As shown in Table 1, irrespective of termination with nitramine groups, when the number of alkyl groups increases, the densities of the polymers decrease. When the pendant chains grow longer, additional degrees of freedom are introduced into the system and the free volume between polymer chains increases; therefore, the densities of the polymers decrease due to volume exclusion effects. Interestingly, the densities of the isomers, such as 3N0H, 2N1H, 1N2H, and 0N3H, are almost the same considering the uncertainty of the calculation, suggesting that the densities of the polymers are more sensitive to the number of alkyl groups rather than their location. Moreover, the densities of the polymers containing more nitramine groups are higher because the intermolecular interactions increase with the number of nitramine groups due to increasing polarity.
| Polymer | ρ (g cm−3) | ΔHf (kJ mol−1) | Q (kJ mol−1) |
|---|---|---|---|
| 0N0H | 1.27 | −90.5 | 634.9 |
| 0N1H | 1.22 | −115.1 | 610.1 |
| 0N2H | 1.19 | −120.6 | 603.9 |
| 0N3H | 1.15 | −134.0 | 591.5 |
| 1N0H | 1.22 | −132.0 | 593.3 |
| 1N1H | 1.18 | −147.1 | 578.4 |
| 1N2H | 1.15 | −152.7 | 572.4 |
| 1N3H | 1.12 | −165.5 | 559.6 |
| 2N0H | 1.18 | −146.3 | 581.0 |
| 2N1H | 1.14 | −157.0 | 568.2 |
| 2N2H | 1.12 | −164.4 | 560.5 |
| 2N3H | 1.09 | −173.6 | 551.5 |
| 3N0H | 1.14 | −159.2 | 566.4 |
| 3N1H | 1.12 | −174.3 | 553.4 |
| 3N2H | 1.09 | −178.1 | 545.3 |
| 3N3H | 1.07 | −190.4 | 535.1 |
| 0N0N | 1.66 | −9.1 | 1200.1 |
| 0N1N | 1.37 | −52.8 | 1156.4 |
| 0N2N | 1.30 | −70.6 | 1138.6 |
| 0N3N | 1.27 | −87.8 | 1121.4 |
| 1N1N | 1.31 | −94.6 | 1114.3 |
| 1N2N | 1.26 | −115.6 | 1093.6 |
| 1N3N | 1.23 | −128.1 | 1081.2 |
| 2N2N | 1.22 | −120.7 | 1088.5 |
| 2N3N | 1.19 | −141.3 | 1068 |
| 3N3N | 1.16 | −154.1 | 1055 |
The energetic features distinguish the energetic binders from traditional binders, providing the propellants and explosives with higher energy density and better performances. Here, we chose EOF and heat of detonation as representative parameters of the energetic features. As shown in Table 1, EOFs of the polymers decrease monotonously when the number of –CH2 groups increase, and the heats of detonation of the polymers decrease with additional –CH2 groups accordingly, indicating that the –CH2 group is unfavorable for the energy of the polymers. On the other hand, the –NCH3NO2 group positively contributes about 40 kJ mol−1 to EOFs of the polymers, which does not seem significant; however, it also contributes about 540 kJ mol−1 to the heat of detonation of the polymers, which indicates that the –NCH3NO2 groups are the main source of energy of the polymers.
The location of the pendant chains is another important factor affecting the properties of polymeric binders; they occupy a large space in the system and can be decisive in the flexibility of the polymers. We designed isomers of PBMNAMO by altering the location of two pendant chains (P11–P13, Fig. 5), and we investigated their energetic and mechanical properties, as shown in Table 2. As can be easily understood, the free volume and the energy contributions induced by the pendant chains are almost the same in all the isomers; thus, the densities and EOFs are not significantly affected, as is shown by the data in Table 2. The flexibility of the polymers can be strongly affected due to the difference in the rotation barriers of the main chains of the isomers: the pendant chains in P13 are most distantly located from each other, and the rotation of the main chain is negligibly affected by them; the pendant chains of PBMNAMO and P11 are grafted from the α- and β-carbons of the oxygen atom, respectively. When the main chains of the polymers rotate, the two pendant chains make no contact with each other, but the rotation barriers rise because of the large occupied space of the α- and β-carbons. Hence, PBMNAMO and P11 have similar and higher Tg values. The rotation energies of α- and β-carbon bonds of the isomers are shown in Fig. 6; the energy barriers are 26.5 kJ mol−1, 83.7 kJ mol−1, 19.9 kJ mol−1, and 47.4 kJ mol−1 for P11, P12, P13, and PBMNAMO, respectively. The pendant chains of P13 strongly hinder the rotation of the bonds of α- and β-carbons due to volume exclusion effects, and Tg of P13 is significantly higher than that of other isomers. On the other hand, the elastic moduli of more flexible polymers are lower. We can see that the Tg values of the polymers can be lowered by 14 K by modifying the sites where the pendant chains are located, with negligible compromise of the mechanical properties while maintaining the energy density of the binder.
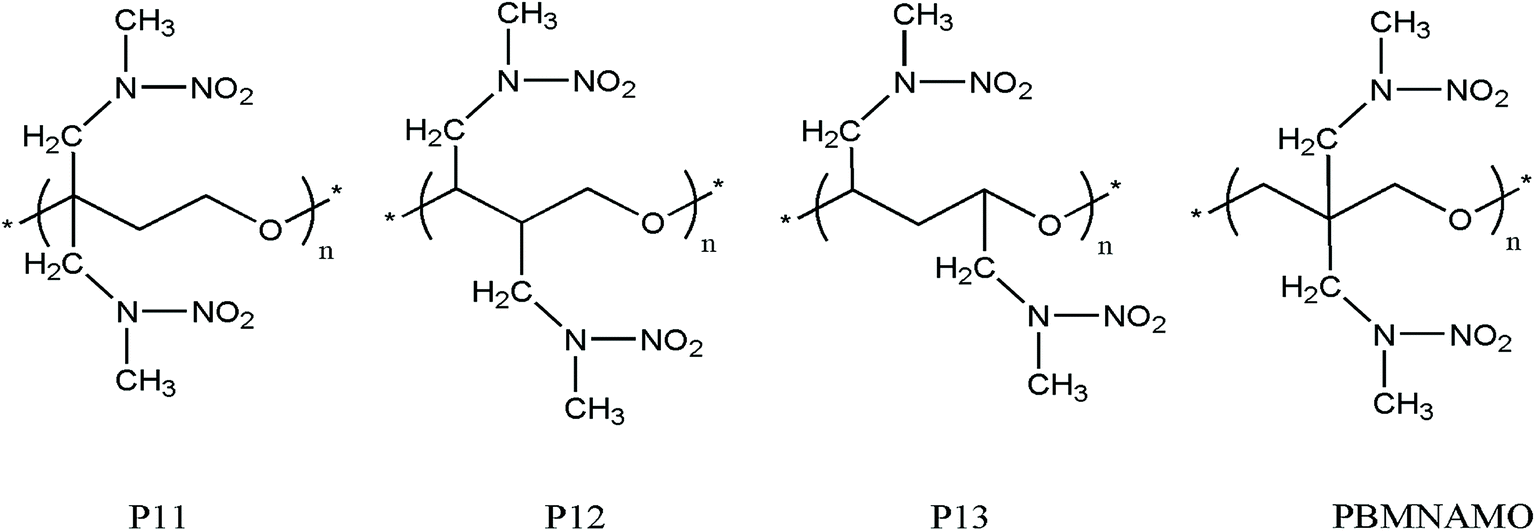 | ||
| Fig. 5 The structures of isomers of PBMNAMO. | ||
| Polymer | ρ (g cm−3) | ΔHf (kJ mol−1) | Tg (K) | E (GPa) | K (GPa) | G (GPa) | γ |
|---|---|---|---|---|---|---|---|
| PBMNAMO | 1.29 | −94.6 | 274 | 4.01 | 2.98 | 1.56 | 0.28 |
| P11 | 1.28 | −92.2 | 274 | 3.91 | 2.86 | 1.48 | 0.28 |
| P12 | 1.29 | −96.3 | 307 | 4.47 | 3.17 | 1.72 | 0.27 |
| P13 | 1.28 | −95.5 | 260 | 2.75 | 2.67 | 1.28 | 0.32 |
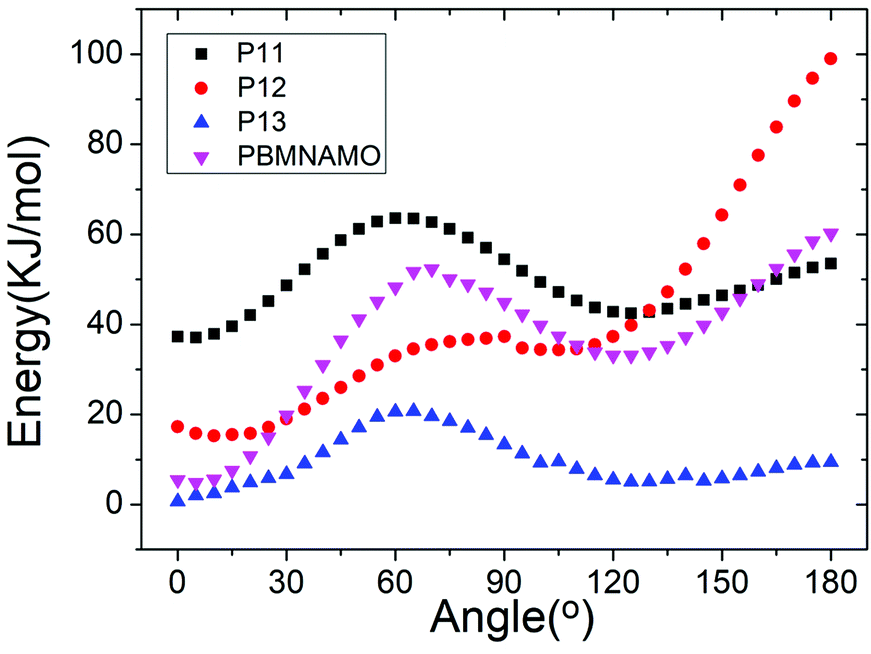 | ||
| Fig. 6 The rotation energy of α- and β-carbon bonds of the isomers. Data are shifted for clarity. | ||
Conclusion
To study the structure–property relationships and to provide guidance for designing and screening of nitramino oxetane polymers, polymers derived from poly(n-oxapropylene) main chain and pendant chains containing –NCH3NO2 group were designed. Their EOFs, glass transition temperatures, and mechanical properties were predicted by molecular dynamics simulations and quantum mechanics calculations. We found the following conclusions:(1) Tg of the polymers against the number of –CH2 groups showed a non-monotonic trend, having a minimum value when the length of the pendant chains contained 1–2 –CH2 groups and the –CH2 groups were equally displaced in two pendant chains.
(2) The introduction of –CH2 groups is not favorable to the densities, EOFs, and elastic moduli of the polymers, whereas the –NCH3NO2 group is the main contributor to EOFs and increases the densities and elastic moduli of the polymers.
(3) Tg of polymers can be significantly lowered by modifying the grafting sites of the pendant chains while maintaining energy density.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the Natural Science Foundation of Shaanxi Province under Grants (No. 2018JQ2076) and National Natural Science Foundation of China under Grants (No. 51572219, 11447030).References
- R. R. Soman, N. T. Agawane and A. Hazarika, et al., Synthesis, characterization and thermal degradation of Glycidyl Azide Polymer-Tetrahydrofuran (GAP-THF) copolymer, Int. J. Polym. Mater., 2003, 20, 423–433 CAS.
- U. R. Nair, S. N. Asthana, A. S. Rao and B. R. Gandhe, Advances in high energy materials (review paper), Def. Sci. J., 2010, 60(2), 137 CrossRef CAS.
- J. -J. Jutier, A. De Gunzbourg and R. E. Prud'Homme, Synthesis and characterization of poly (3,3 bis (azidomethyl) oxetane-co-ε-caprolactone)s, J. Polym. Sci., Part A: Polym. Chem., 1999, 37(7), 1027–1039 CrossRef CAS.
- H. S. Dong and F. F. Zhou, High energy explosives and correlative physical properties, Beijing, Science Press, 1989 Search PubMed.
- R. L. Simpson, P. A. Urtiew, D. L. Ornellas, G. L. Moody, K. J. Scribner and D. M. Hoffman, CL-20 performance exceeds that of HMX and its sensitivity is moderate, Propellants, Explos., Pyrotech., 1997, 22(5), 249–255 CrossRef CAS.
- A. H. Ghee, and P. Sreekumar, Energetic polymers, Wiley-VCH, Weinheim, Germany, 2012, ISBN 978-3-527-33155-0 Search PubMed.
- Y. K. Sinha, B. T. N. Sridhar and M. Santhosh, Thermal decomposition study of HTPB solid fuel in the presence of activated charcoal and paraffin, J. Therm. Anal. Calorim., 2015, 119, 557–565 CrossRef CAS.
- K. Subramanian, Hydroxyl-terminated poly(azidomethylethyleneoxide-b-butadiene-b-azidomethyl ethylene oxide)—synthesis, characterization and its potential as a propellant binder, Eur. Polym. J., 1999, 35(8), 1403–1411 CrossRef CAS.
- U. R. Nair, et al., Advances in high energy materials (review paper), Def. Sci. J., 2010, 60(2), 137 CrossRef CAS.
- U. R. Nair, et al., Advances in high energy materials (review paper), Def. Sci. J., 2010, 60(2), 137 CrossRef CAS.
- Y. Wu, Y. Luo and Z. Ge, Properties and Application of a Novel Type of Glycidyl Azide Polymer (GAP)-Modified Nitrocellulose Powders, Propellants, Explos., Pyrotech., 2015, 40(1), 67–73 CrossRef CAS.
- J. Xiao, et al., A molecular dynamics study of interface interactions and mechanical properties of HMX-based PBXs with PEG and HTPB, J. Mol. Struct.: THEOCHEM, 2008, 851, 242–248 CrossRef CAS.
- L. Liao, H. Wei, J. Li, X. Fan, Y. Zheng, Y. Ji, X. Fu, Y. Zhang and F. Liu, Compatibility of PNIMMO with some energetic materials, J. Therm. Anal. Calorim., 2012, 109, 1571 CrossRef CAS.
- M. Guo, Z. Ma, L. He, W. He and Y. Wang, Effect of varied proportion of GAP-ETPE/NC as binder on thermal decomposition behaviors, stability and mechanical properties of nitramine propellants, J. Therm. Anal. Calorim., 2017, 130, 909 CrossRef CAS.
- G. E. Manser and R. W. Fletcher, Nitramine oxetanes and polyethers formed therefrom, US Pat. no. 4707540, U.S. Patent and Trademark Office, Washington, DC, 1987 Search PubMed.
- M. Farber, S. P. Harris and R. D. Srivastava, Mass Spectrometric Investigation of the Thermal Decomposition of Several Propellant and Explosive Ingredients, Space Sciences Inc, Monrovia CA, 1986 Search PubMed.
- J. J. Kaufman, Quantum Chemical Investigations of the Mechanism of Cationic Polymerization and Theoretical Prediction of Crystal Densities and Decomposition Pathways of Energetic Molecules (No. TR-9), Johns Hopkins Univ, Baltimore MD, 1988 Search PubMed.
- C. J. Campbell, Energetic oxetane propellants, US Pat. no. 6217682, 2001 Search PubMed.
- P. P. Vadhe, R. B. Pawar, R. K. Sinha, S. N. Asthana and A. S. Rao, Cast aluminized explosives, Combust., Explos. Shock Waves, 2008, 44(4), 461–477 CrossRef.
- A. J. Sanderson and W. W. Edwards, Method for the synthesis of energetic thermoplastic elastomers in non-halogenated solvents, US Pat. no. 6997997, 2006 Search PubMed.
- H. R. Blomquist, Gas generating material for vehicle occupant protection device, US Pat. no. 6802533, 2004 Search PubMed.
- M. A. S. Khan, A. Dey, J. Athar and A. K. Sikder, Calculation of enthalpies of formation and band gaps of polymeric binders, RSC Adv., 2014, 4(62), 32840–32846 RSC.
- R. Sarangapani, S. T. Reddy and A. K. Sikder, Molecular dynamics simulations to calculate glass transition temperature and elastic constants of novel polyethers, J. Mol. Graphics Modell., 2015, 57, 114–121 CrossRef CAS PubMed.
- W. Yao, J. Dai and H. Tan, Study on improving mechanical property by couple technology, Acta Armamentarii, 1994, 2, 11–15 Search PubMed.
- A. J. Sanderson, W. Edwards, L. F. Cannizzo and R. B. Wardle, Synthesis of energetic thermoplastic elastomers containing both polyoxirane and polyoxetane blocks, US Pat. no. 7101955, 2006 Search PubMed.
- L. Qiu, H. Xiao, W. Zhu, J. Xiao and W. Zhu, Ab initio and molecular dynamics studies of crystalline TNAD (trans-1,4,5,8-tetranitro-1,4,5,8-tetraazadecalin), J. Phys. Chem. B, 2006, 110(22), 10651–10661 CrossRef CAS PubMed.
- O. V. Dorofeeva and M. A. Suntsova, Enthalpy of formation of CL-20, Comput. Theor. Chem., 2015, 1057, 54–59 CrossRef CAS.
- R. Sarangapani, V. D. Ghule and A. K. Sikder, Computational screening of oxetane monomers for novel hydroxy terminated polyethers, J. Mol. Model., 2014, 20(6), 2253 CrossRef PubMed.
- D. Mark Hoffman and L. E. Caley, Polymer blends as high explosive binders, Polym. Eng. Sci., 1986, 26(21), 1489–1499 CrossRef.
- Y. Lu, et al., Theoretical simulations on the glass transition temperatures and mechanical properties of modified glycidyl azide polymer, Comput. Mater. Sci., 2017, 139, 132–139 CrossRef CAS.
- Q. Yang, X. Chen, Z. He, F. Lan and H. Liu, The glass transition temperature measurements of polyethylene: determined by using molecular dynamic method, RSC Adv., 2016, 6, 12053–12060 RSC.
- C. Li and A. Strachan, Molecular dynamics predictions of thermal and mechanical properties of thermoset polymer EPON862/DETDA, Polymer, 2011, 52, 2920–2928 CrossRef CAS.
- J. S. Bermejo and C. M. Ugarte, Chemical crosslinking of PVA and prediction of material properties by means of fully atomistic MD simulations, Macromol. Theory Simul., 2009, 18(4–5), 259–267 CrossRef CAS.
- D. N. Theodorou and U. W. Suter, Atomistic modeling of mechanical properties of polymeric glasses, Macromolecules, 1986, 19(1), 139–154 CrossRef CAS.
- U. W. Suter and B. E. Eichinger, Estimating elastic constants by averaging over simulated structures, Polymer, 2002, 43(2), 575–582 CrossRef CAS.
- C. Li and A. Strachan, Molecular dynamics predictions of thermal and mechanical properties of thermoset polymer EPON862/DETDA, Polymer, 2011, 52(13), 2920–2928 CrossRef CAS.
- S. Yang and J. Qu, Computing thermomechanical properties of crosslinked epoxy by molecular dynamic simulations, Polymer, 2012, 53(21), 4806–4817 CrossRef CAS.
- N. Berrahou, A. Mokaddem, B. Doumi, S. Hiadsi, N. Beldjoudi and A. Boutaous, Investigation by molecular dynamics simulation of the glass transition temperature and elastic properties of amorphous polymers PMMA, PMAAM and PMMA-co-PMAAM copolymers, Polym. Bull., 2016, 5, 1–11 Search PubMed.
- H. Sun, COMPASS: an ab initio force-field optimized for condensed-phase applications overview with details on alkane and benzene compounds, J. Phys. Chem. B, 1998, 102(38), 7338–7364 CrossRef CAS.
- M. J. Kamlet and S. J. Jacobs, Chemistry of detonations. I. A simple method for calculating detonation properties of C–H–N–O explosives, J. Chem. Phys., 1968, 48(1), 23–35 CrossRef CAS.
- M. J. Kamlet and J. E. Ablard, Chemistry of detonations. II Buffered equilibria, J. Chem. Phys., 1968, 48(1), 36–42 CrossRef CAS.
- Y. Lu, Y. Shu, N. Liu, Y. Shu, K. Wang, Z. Wu, X. Wang and X. Ding, Theoretical simulations on the glass transition temperatures and mechanical properties of modified glycidyl azide polymer, Comput. Mater. Sci., 2017, 139, 132–139 CrossRef CAS.
- J. K. Nair, T. S. Reddy, R. S. Satpute, T. Mukundan and S. N. Asthana, Synthesis and characterization of energetic thermoplastic elastomers (ETPEs) based on 3,3-bis (azidomethyl) oxetane (BAMO)-3-azidomethyl-3-methyloxetane (AMMO) copolymers, J. Polym. Mater., 2004, 21(2), 205–212 CAS.
- J. Jutier, A. De Gunzbourg and R. E. Prud'Homme, Synthesis and characterization of poly (3,3 bis (azidomethyl) oxetane-co-ε-caprolactone)s, J. Polym. Sci., Part A: Polym. Chem., 1999, 37(7), 1027–1039 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |