
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn situ preparation of C–SiCxOy coatings with controllable composition on continuous oxygen-enriched SiC fibres
Haonan Xu ab,
Qi Wangab,
Hongrui Xiaoab,
Xiangdong Liab,
Xiaohui Suab,
Ming Tangab,
Lifu Chenab and
Siwei Li*ab
ab,
Qi Wangab,
Hongrui Xiaoab,
Xiangdong Liab,
Xiaohui Suab,
Ming Tangab,
Lifu Chenab and
Siwei Li*ab
aCollege of Materials, Key Laboratory of High Performance Ceramic Fibers, Xiamen University, Xiamen 361005, China. E-mail: swli@xmu.edu.cn
bFujian Key Laboratory of Advanced Materials, Key Laboratory of High Performance Ceramic Fibers of Ministry of Education, College of Materials, Xiamen University, Xiamen 361005, China
First published on 10th January 2019
Abstract
Oxygen-enriched SiC fibres derived from polycarbosilane are exposed to chlorine at 500 to 750 °C for up to 60 min. After chlorination, uniform coatings with controllable thickness are formed in situ on the SiC fibres. Evolution of the coating composition and its relation to the electrical properties are investigated. Results indicate that the porous nature of the coating realizes a reaction-controlling mechanism of the coating growth. By enhancing the chlorination temperature within the selected range, SiC, SiCO3 and SiC2O2 phases in the fibre can be successively etched as free carbon to give C–SiCxOy hybrid coatings with increased content of C and decreased content of O. Accordingly, the specific resistivity of the coated SiC fibre declines continuously from 5.01 × 106 to 0.39 Ω cm.
1 Introduction
With the advantages of thermal stability, low density, high strength and excellent oxidation resistance,1–3 polymer derived SiC-based fibres are widely used for fabricating thermal structural composites with specific aims. Nowadays, the most important application of SiC-based fibres is to toughen ceramics through mechanisms of interfacial debonding and microcrack deflecting.4–6 In this case, fibre layers are required to generate controllable weak bonding between the SiC fibre and the ceramic matrix.7,8 For the SiC-based fibres, the most popular species of surface layers are known as pyrolytic carbon (PyC) and boron nitride (BN),9–11 which are commonly prepared by a costly approach of chemical vapor infiltration (CVI). Y. G. Gogotsi. et al. developed a less expensive method to prepare carbide-derived carbon (CDC) coatings on SiC fibres by using hydrothermal treatment or halogen etching.12 Uniform carbon coatings are obtained with a good adherence to the SiC fibres. Mechanical properties of the SiC fibres are even improved after formation of the carbon coating with optimized thickness13 Moreover, thanks to the porous nature, the CDC layers can also be used as templates for yielding BN coatings via inexpensive chemical routes.14–17A. Delcamp et al. reported the kinetics of the growth of the CDC coating on two types of the SiC-based fibres, Tyranno ZMI and Nicalon.18 In the temperature range of 550–675 °C, the coating thickness is found to increase linearly with the etching time (in Cl2 environment), suggesting that the coating growth is controlled by interfacial reaction. It should be noted that oxygen of ∼9 at% and ∼14 at% is present in the Tyranno ZMI and Nicalon fibre, respectively. But compositions of the resultant CDC coatings on both fibres are all considered as pure carbon. Assisted by thermodynamic calculation, the authors suggested that oxygen impurities would be extracted as gases during chlorination by reacting with residual carbon then with Cl2. However, the equilibrium results may not represent the real case as they do not account for the kinetics of the process. Also, the calculations could not be responsible for the evolution of the metaphases, which may influence the composition and function of the SiC fibre to a great extent. We recently found that if an oxygen-enriched SiC fibre (oxygen content is comparable to Nicalon fibre) was exposed to Cl2 at above 500 °C, the oxygen content in the resultant CDC coating decreased gradually with elevated temperatures. This suggests that the tetrahedral metaphases of SiCxOy (SiC3O, SiC2O2, SiCO3) in the SiC fibre can be selectively etched into free carbon. Therefore, the coating formed at the fibre surface is not pure carbon but actually C–SiCxOy hybrid phases with variable C/SiCxOy ratios. Change of the coating composition offers great possibilities for modifying the behaviour of interfacial debonding and fibre pullout, and may also endow the composites new functions, such as controllable resistivity and dielectric performance. Moreover, the doping of SiCxOy in the fibre coating may enhance the oxidation resistance of the fibre as compared with the pure carbon coatings. The current work focuses on the influence of chlorination on microstructure of the C–SiCxOy hybrid coatings. Controllability of the electrical resistivity of the coating by the chlorination is also discussed.
2 Materials preparation
2.1 Preparation of the oxygen-enriched SiC fibre
The oxygen-enriched SiC fibre is prepared following Yajima's route.1 Polycarbosilane (PCS) with an average molecular weight (M) of 1230 and softening point of 198 °C is used as the precursor. Continuous SiC fibres with a Si/C/O mass ratio of 54/30/16 are obtainable after the processes of melt-spinning, oxidation curing and pyrolysis.2.2 Chlorination of the SiC fibre
The as-pyrolyzed SiC fibres were placed in a graphite boat and then put into a horizontal quartz tube furnace with inner tube diameter of 30 mm. The fibres were first treated in high purity N2 (99.999%, 100 ml min−1) from room temperature to 500 °C, the N2 was cut down and the Cl2 gas (99.999%, 60 ml min−1) was blown into the furnace tube in the temperature range of 500–750 °C, holding for 5–60 minutes. After the chlorination, the Cl2 was immediately diluted by the N2, and the furnace was cooled down in the N2. An NaOH solution with a concentration of 30 mol% was used to capture the reaction products of SiCl4 and residual Cl2 gas.2.3 Characterization
Scanning electron microscopy (SEM, SU70, Hitachi, Japan) was used to observe the fracture morphology and the coating thickness. Transmission electron microscopy (TEM, JEM2100, JEOL, Japan) was used to observe the microstructure of the fibre. X-ray diffraction (XRD; Bruker-AXS, USA) was used for phase identification. Auger electron spectroscopy (AES; PHI660, USA) with an electron beam of 5 keV (100 nA) was used for analysing the coating composition. The sputtering rate was estimated to be 11 nm min−1 (for SiO2). X-ray photoelectron spectroscopy (XPS; PHI Quantum 2000, USA) analysis was carried out to give chemical states by employing AlKα1,2 radiation of 1486.60 eV. Resistance (R) of SiC fibre was measured using the two-points testing method.19 Specific resistivity (ρ) was calculated by the formula ρ = RS/L.The pull-out tests were conducted on SiCf/SiOC composites. The as-received SiC fibres coated with C–SiCxOy coatings were used as the reinforcement for this study. Commercial available poly-methylsilsesquioxane (MK resin, Wacker-Chemie GmbH, München, Germany), with relative molecular mass 9100 g mol−1 and softening point 35–55 °C, was used as the precursor to SiOC glasses. The composites were fabricated by precursor infiltration and pyrolysis (PIP) process.
3 Results and discussion
3.1 Effect of chlorination on the microstructure of the C–SiCxOy hybrid coating
A series of samples are obtained by controlling the chlorination temperature and soaking time. Weight loss and coating thickness of the treated SiC fibres are shown in Table 1. It can be seen that the deviations of the coating thickness measured for all treatments are quite small, indicating a homogeneous infiltration of Cl2 with within fibre tows during reaction. The coating thickness evidently increases with the increase in temperature and prolonged soaking time. After chlorination at 500 °C for 5 minutes, the weight loss is identified as 0.8 wt%, revealing that the Cl2 starts to react with the fibre. At this time, the resultant coating is too thin to be observed in SEM (see Fig. 1). After chlorination at 600 °C for 5 min, the coating grows intensively to an average thickness of 591 nm. In case of treating at 750 °C for 5 minutes, the chlorination goes through the whole fibre, thus the interface of the fibre/coating disappears.| Sample | Temperature (°C) | Soaking time (min) | Weight loss (wt%) | Coating thickness (μm) |
|---|---|---|---|---|
| 500-5 | 500 | 5 | 0.80 | Invisible in SEM |
| 500-30 | 500 | 30 | 3.30 | 0.141 ± 0.015 |
| 500-60 | 500 | 60 | 5.54 | 0.326 ± 0.013 |
| 550-5 | 550 | 5 | 7.82 | 0.426 ± 0.017 |
| 550-30 | 550 | 30 | 19.46 | 1.016 ± 0.027 |
| 550-60 | 550 | 60 | 38.82 | 1.603 ± 0.015 |
| 600-5 | 600 | 5 | 18.01 | 0.591 ± 0.017 |
| 600-30 | 600 | 30 | 70.79 | 1.954 ± 0.093 |
| 600-60 | 600 | 60 | 80.49 | Go through the fibre |
| 650-5 | 650 | 5 | 40.55 | 1.285 ± 0.039 |
| 700-5 | 700 | 5 | 72.99 | 4.382 ± 0.045 |
| 750-5 | 750 | 5 | 78.77 | Go through the fibre |
 | ||
| Fig. 1 Cross-section morphology of the fibres before and after chlorination. (a) The as-pyrolyzed SiC fibre, (b) 600-5, (c) 650-5, (d) 750-5. | ||
Fig. 2a shows the change of fibre weight and coating thickness as function of chlorination temperature. The loss of the fibre weight has a trend similar to the growth of the fibre coating. For both curves, a rapid enhancement is observed at above 600 °C, indicating that the chlorination is accelerated at this point. It is worth noting that weight loss of the 750-5 sample and the 600-60 sample reach 79 wt% and 80 wt% respectively. Both values are much higher than the Si content (54 wt%) of the as-prepared SiC fibre, indicating that more elements in addition to Si are extracted from fibre. It is thus concluded that the presence of the SiCxOy phases lead to removal of C and O in the Cl2 environment. The possible reactions are as follows.20,21
| SiC(s) + 2Cl2(g) = C(s) + SiCl4(g) | (1) |
| SiCxOy(s) + 2Cl2(g) = (x − y)C(s) + SiCl4(g) + yCO(g) | (2) |
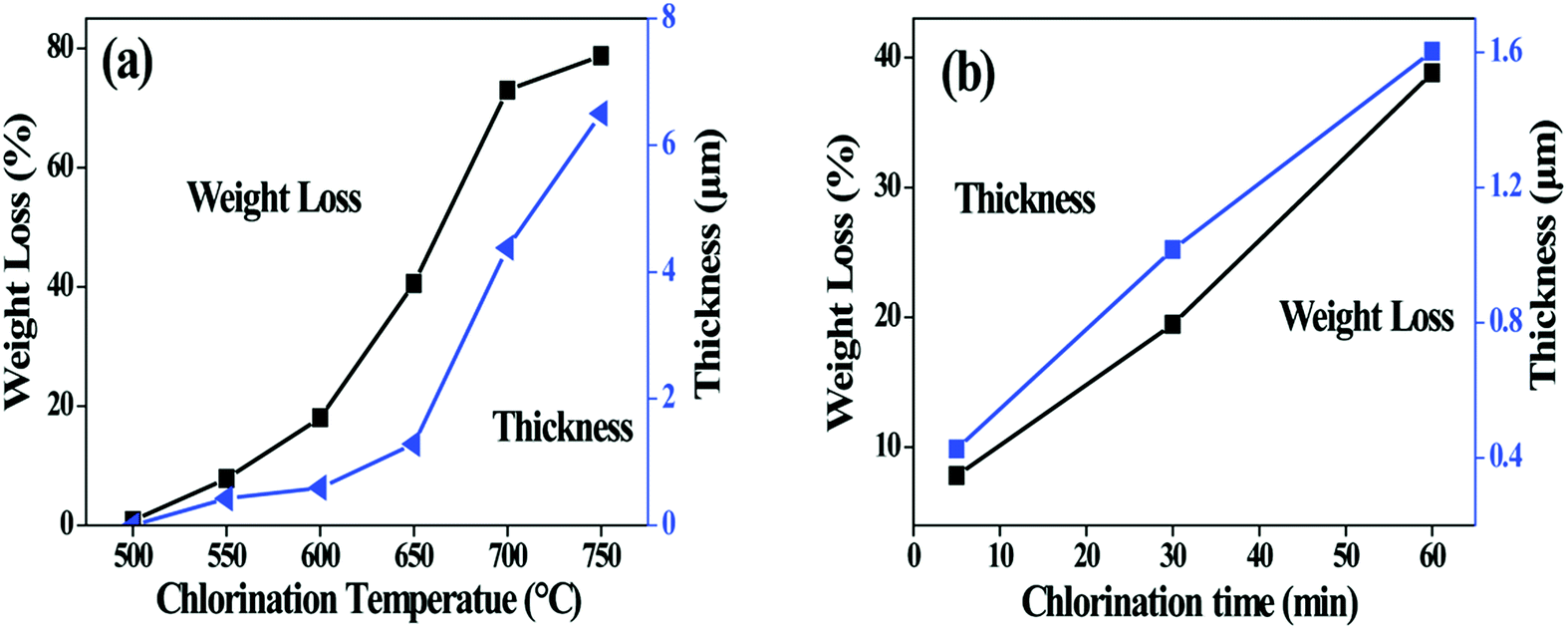 | ||
| Fig. 2 Change in fibre weight and coating thickness with (a) chlorination temperature and (b) soaking time. The holding time for (a) is 5 min, and the chlorination temperature for (b) is 550 °C. | ||
Fig. 2b shows the influence of isothermal process on the fibre weight and the coating thickness. The thickness increases almost linearly with the soaking time, suggesting that the kinetics of coating growth is controlled by interfacial reaction rather than diffusion of the Cl2.
The porous structure of CDC coating is characterized using low-temperature nitrogen sorption method. The isotherm curve of nitrogen adsorption displayed in Fig. 3a presents an increasing uptake at relative pressure between 0.05 and 0.3 (a type I form according to IUPAC classification),22 indicating the existence of micropores in the CDC coating. Specific surface area (SSA) is calculated using the BET equation (Fig. 3b). The SSA of the coating increases with elevating temperature, which is in accordance with the results in ref. 18. However, since the thickness of the CDC coatings obtained at various temperatures is different, the true weight percentage of the porous region (CDC coating in the fibre) is changeable and unmeasurable for different samples. This will introduce errors to the BET calculation. Taking this into account, different fibres (600-60 and 750-5) with whole body of CDC are used to determine the BET characters to eliminate the influence of coating thickness. The SSA of the 750-5 is identified to be much higher than that of the 600-60, while the average pore width of the former (1.56 nm) is close to that of the latter (1.62 nm). It is obvious that more phases in the SiC fibre are consumed as gases at higher temperatures. As a result, more micropores are generated to enhance the SSA. According to the BET results, there should be evident differences in the reactivity of Cl2 with different phases in the SiC fibre.
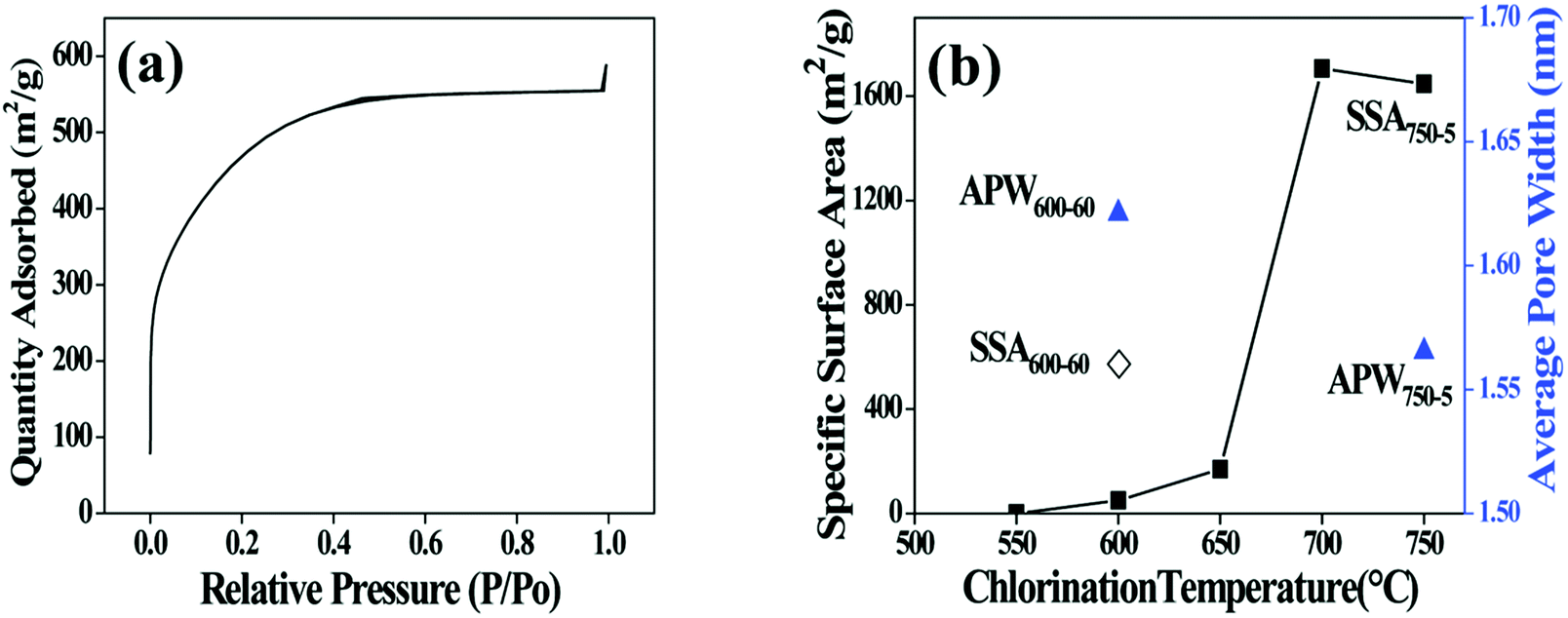 | ||
| Fig. 3 (a) N2 adsorption and desorption isotherm linear plot of 700-5 sample. (b) Calculated specific surface area (SSA) and average pore width (APW) of the chlorinated fibres. (◇) represents SSA of 600-60. (■) represent SSA of the fibre chlorinated at different temperatures for 5 min. (▲) represent PW of 600-60 and 750-5. | ||
Surface composition of the fibre coating is examined by AES. After chlorination, the Si peaks at fibre surface are significantly weakened whereas the C peaks are intensified (Fig. 4a), proving that Si atoms are diminished from the fibre. The chlorination is found to proceed evenly as the growth of the coating since the elements distribute uniformly along the thickness direction, as shown in the depth-profile of 550-5 sample (Fig. 4b). It is also seen form Fig. 4b that the O content of the coating is very close to that of the fibre core, revealing that the oxygen containing phases in the fibre are relatively more stable than the SiC domains. The Si and C gradients of 200 nm observed between coating and fibre are most likely induced by dilution of Cl2 and reduction of reactivity as it moves inward the fibre during the cooling process. Fig. 5 shows variations of the coating composition with chlorination temperature and soaking time. At elevated temperatures, the content of Si and O gradually decreased while the content of C correspondingly increased. Nevertheless, the isothermal process has little impact on the elemental concentration. Combining Fig. 2b, it is evident that reaction temperature is the key factor to control the coating composition, while changing the soaking time is mainly related to variation of the coating thickness. To meet different purposes, the composition and thickness of the coating can be independently regulated.
 | ||
| Fig. 4 (a) AES spectra on the surface of as-pyrolyzed SiC fibre and 700-5 sample. (b) AES depth-profile of the 550-5 sample. | ||
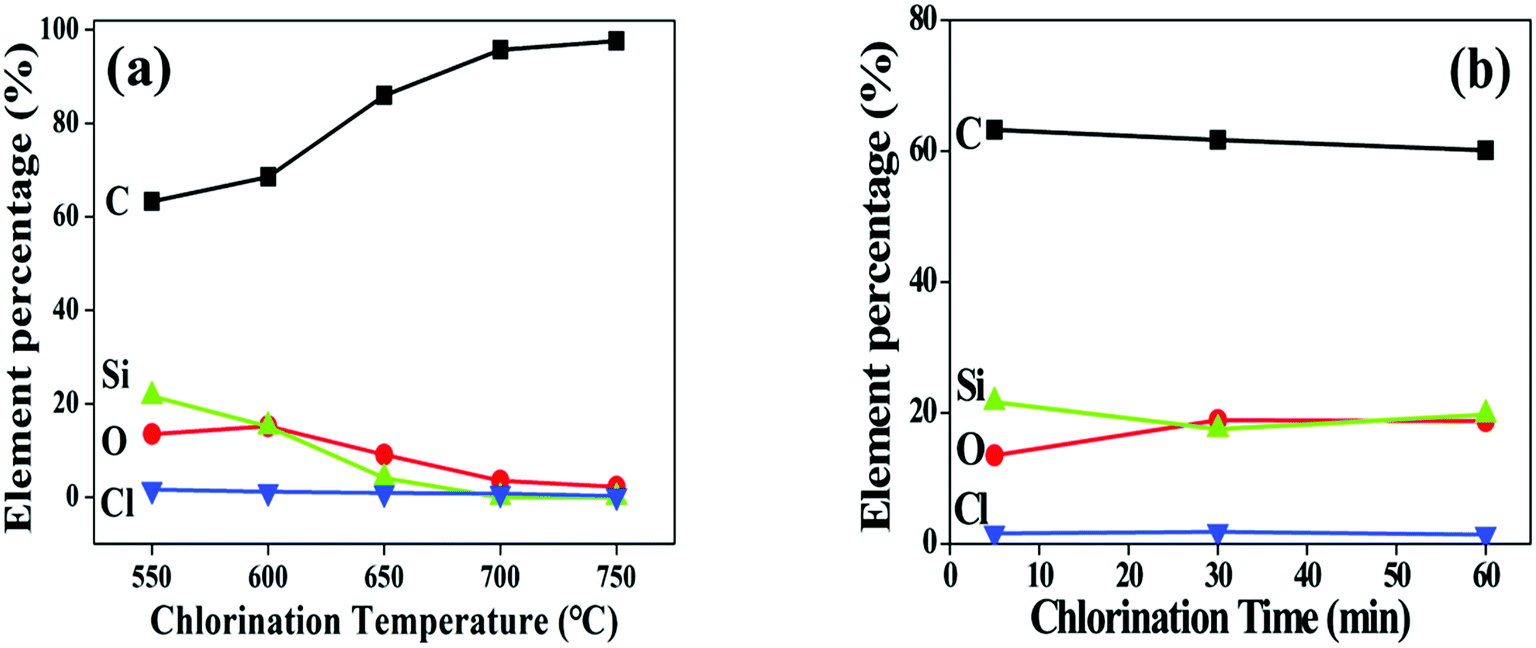 | ||
| Fig. 5 Variations of coating composition as function of chlorination temperature and soaking time. (a) Chlorination for 5 min with temperature range from 550 to 750 °C. (b) Chlorination at 550 °C for 5 to 60 minutes. | ||
The chemical state of the fibre coatings is characterized by XPS. Fig. 6 illustrates the spectra of Si 2p, C 1s, O 1s and the fitted results. Based on the structural characters of the PCS-SiC fibres, the Si peak is fitted into the following five sub-peaks: SiC4 (refers to SiC phase), SiC3O, SiC2O2, SiCO3 and SiO4 (refers to SiO2 phase) with binding energies of 100.7, 101.6, 102.6, 103.4 and 104.3 eV respectively.23–25 Accordingly, the C spectra are fitted into four peaks of C–Si, C–C, C–O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (refers to residual gas product of CO) with binding energies of 283.0, 284.5, 285.8 and 287.0 eV respectively.26,27 The O spectra is fitted as three peaks of O–C, OC and O–Si with binding energies of 531.2, 532.1 and 533.4 eV, respectively.28–30 The relative content of the tetrahedral phases is calculated based on the fitted results, as listed in Table 2. With increasing temperature, the relative contents of SiC and SiC3O decreased evidently. Also, SiC2O2 is lowered in concentration but in a slower speed. It is obvious that the Cl2 reacts with SiC and SiC3O much more intense than with SiC2O2. Considering that the bonding energy of Si–Cl is smaller than Si–O but larger than SiC, the erosiveness of Cl2 to SiCxOy should decline significantly as the increase of the O/Si (y/x) ratio. This is verified by the fact that relative content of the SiO2 and SiCO3 in the coating increased with the elevated chlorination temperature. The XRD graphs also detect the residue of SiO2, wherein a broad peak corresponding to amorphous SiO2 appears in 750-5 sample.
O (refers to residual gas product of CO) with binding energies of 283.0, 284.5, 285.8 and 287.0 eV respectively.26,27 The O spectra is fitted as three peaks of O–C, OC and O–Si with binding energies of 531.2, 532.1 and 533.4 eV, respectively.28–30 The relative content of the tetrahedral phases is calculated based on the fitted results, as listed in Table 2. With increasing temperature, the relative contents of SiC and SiC3O decreased evidently. Also, SiC2O2 is lowered in concentration but in a slower speed. It is obvious that the Cl2 reacts with SiC and SiC3O much more intense than with SiC2O2. Considering that the bonding energy of Si–Cl is smaller than Si–O but larger than SiC, the erosiveness of Cl2 to SiCxOy should decline significantly as the increase of the O/Si (y/x) ratio. This is verified by the fact that relative content of the SiO2 and SiCO3 in the coating increased with the elevated chlorination temperature. The XRD graphs also detect the residue of SiO2, wherein a broad peak corresponding to amorphous SiO2 appears in 750-5 sample.
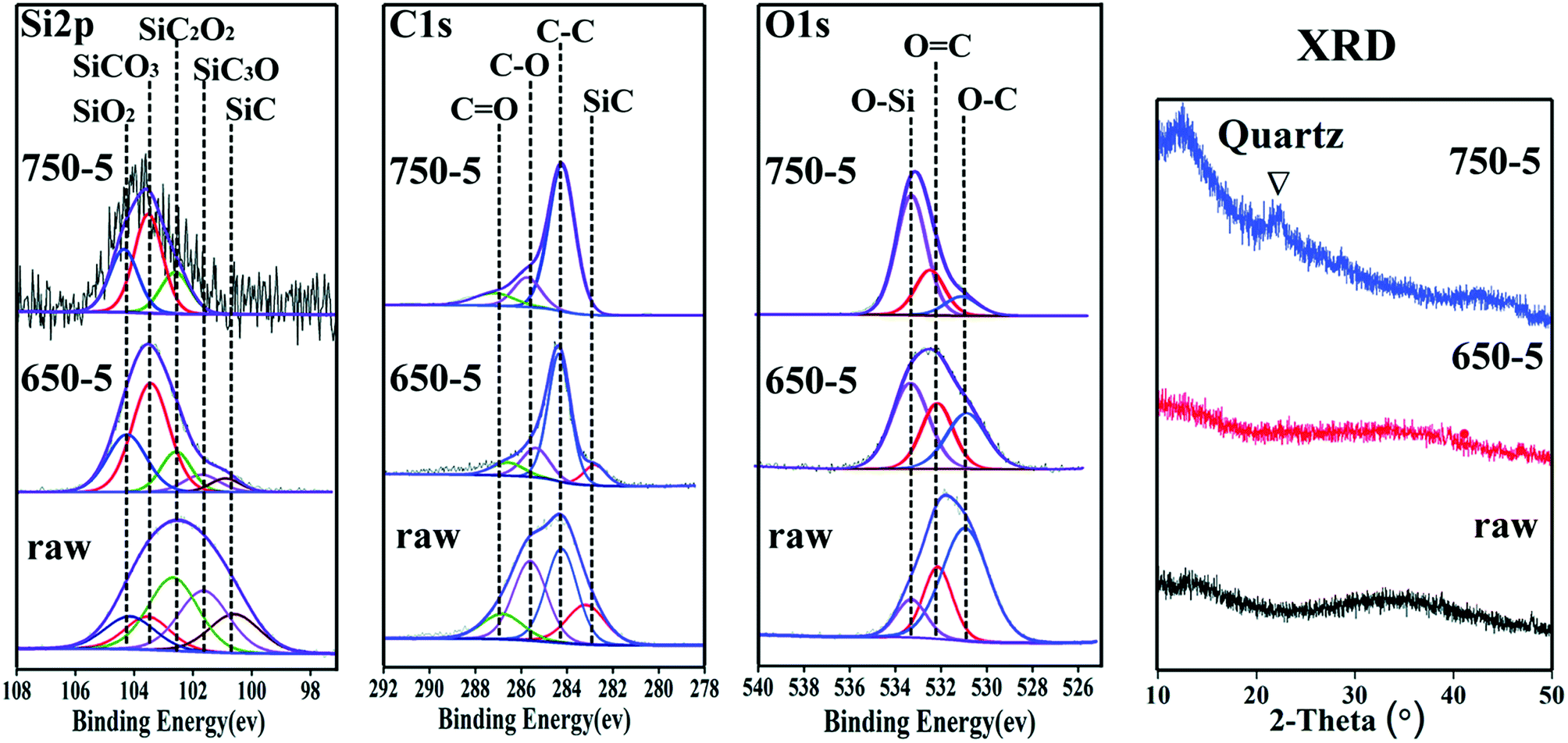 | ||
| Fig. 6 The fitted XPS spectra of Si 2p, C 1s, O 1s and the related XRD pattern. | ||
| Si (mol%) | O (mol%) | C (mol%) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SiC | SiC3O | SiC2O2 | SiCO3 | SiO2 | O–Si | OC |
O–C | C–Si | C–C | C–O | CO |
|
| Raw | 16.7 | 25.9 | 30.9 | 12.6 | 13.9 | 18.3 | 18.6 | 63.1 | 18.4 | 36.1 | 32.9 | 12.6 |
| 500-5 | 10.7 | 16.4 | 30.5 | 24.1 | 18.3 | 20.1 | 21.3 | 58.6 | 17.4 | 38.8 | 31.3 | 12.5 |
| 550-5 | 10.4 | 15.4 | 24.4 | 28 | 21.8 | 28.3 | 20.4 | 51.3 | 17.3 | 40.3 | 27.7 | 14.7 |
| 600-5 | 7.4 | 9.3 | 24.1 | 31 | 28.2 | 40.6 | 21.8 | 37.6 | 12.9 | 58.8 | 21.2 | 7.1 |
| 650-5 | 4.8 | 7.1 | 14.3 | 47.6 | 26.2 | 44.6 | 25.3 | 30.1 | 10.2 | 63.7 | 16.2 | 9.9 |
| 750-5 | 0 | 0 | 21.2 | 48.1 | 30.7 | 51.5 | 24.7 | 23.8 | 0 | 74.6 | 13.6 | 11.8 |
To further evidence the microstructural evolution, TEM observation is conducted and shown in Fig. 7. The as-pyrolyzed SiC fibre shows dense (Fig. 7a) and amorphous structure doped by nano-sized SiC grains (Fig. 7b). Although the chlorinated fibre of 750-5 is still amorphous, the porous structure facilitates a totally different contrast in the TEM bright field image with more loosely packed nanophases (Fig. 7c). By enlarging this region, large amount of turbostratic carbon clusters can be observed (Fig. 7d). It should be noted that the free carbon in the C–SiCxOy hybrid phases is actually present in two forms of CDC and PyC. The former is filled with micropores that generated during halogen etching, while the latter is dense since the pores induced by decomposition of the PCS fibre have already closed during the significant volume shrinkage in pyrolysis.2 The content of PyC is unlikely to be changed by the current treatments since the chlorination temperatures are much lower than the decomposition point of SiCxOy. Therefore, the carbon clusters in Fig. 7d should be identified as CDC rather than PyC since they are invisible in the fibre before chlorination.
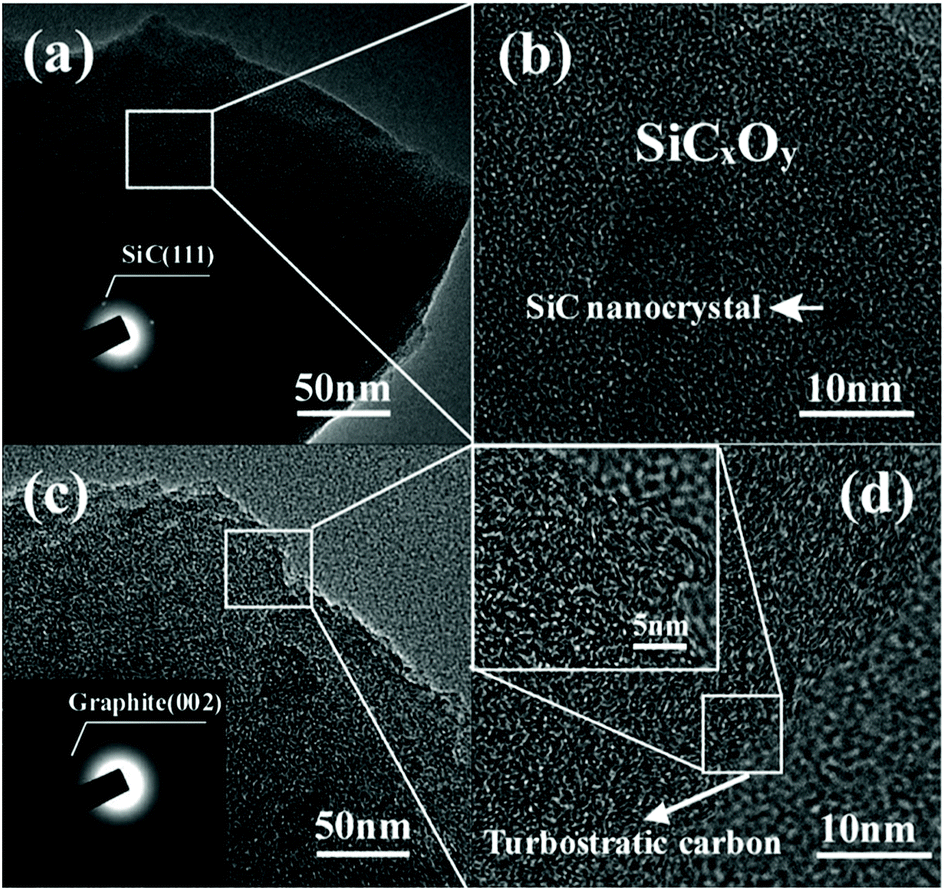 | ||
| Fig. 7 TEM observations on the fibre before and after chlorination. (a) Low and (b) high magnification image of as-pyrolyzed fibre. (c) Low and (d) high magnification image of 750-5 sample. | ||
It is interesting to note that the reactivity of Cl2 with sintered SiC particles of micro size31 is identified to be much lower as compared with the current experiment. This is probably due to the fact the surface energy of the SiC nano grains in the fibre is much higher than that of the large SiC particles. Moreover, the polymer–ceramic conversion endows the SiC fibres with active sites (such as dangling bonds induced by cleavage of the Si–H) of much higher concentration than sintered SiC, which may also contribute to enhancing the reactivity of the fibre to the Cl2.
Considering that the SiCxOy phases behave as insulators while the free carbon is much more conductive,32,33 the change of the C/SiCxOy ratio in the coating should also be reflected by the evolution of the specific resistivity of the coated SiC fibres (ρf). The influence of the treating parameters on the ρf is listed in Table 3 and also shown in Fig. 8. For soaking time of 5 min, ρf decreases steadily over 7 orders of magnitude (from 5.01 × 106 to 0.39 Ω cm) as the temperature increasing from 500 to 750 °C. According to conductor skin effects, the current may only go through the C–SiCxOy coatings because of its much lower resistance compared with the unreacted fibre core,3 especially as the carbon yield of the coating is enhanced. In consideration of this, the specific resistivity of the bare C–SiCxOy coatings (ρc) is also calculated based on its hollow cylinder shape. With temperature increasing, ρc declines in a trend similar to ρf, and approaches ρf because of the increase in the coating thickness. The huge change in ρc should be induced by the change of C/SiCxOy ratios through consumption of oxygen and formation of CDC. With the depletion of SiC3O and SiC2O2 at above 750 °C, the resistivity of the C–SiCxOy coating is close to that of the amorphous carbon,34 indicating that the isolated carbon islands tend to connect as a conductive network. Fig. 8b shows that the isothermal process has ignorable influence on the coating resistivity, which is consisting with the AES/XPS results that the composition of C–SiCxOy coating is almost independent of treating time.
| 500 °C | 550 °C | 600 °C | 650 °C | 700 °C | 750 °C | |
|---|---|---|---|---|---|---|
| a The data out of the brackets show resistivity of the fibres (ρf) while that in the brackets represent resistivity of bare surface coatings (ρc). | ||||||
| 5 min | 4.81 × 106 (NA) | 5.0 × 104 (5.9 × 103) | 83.91 (13.57) | 7.8 (2.60) | 1.34 (1.15) | 0.39 (0.39) |
| 30 min | 3.01 × 106 (1.2 × 105) | 2.03 × 104 (5.5 × 103) | 31.79 (15.27) | NA | NA | NA |
| 60 min | 1.30 × 106 (1.2 × 105) | 1.28 × 104 (5.2 × 103) | 5.26 (5.26) | NA | NA | NA |
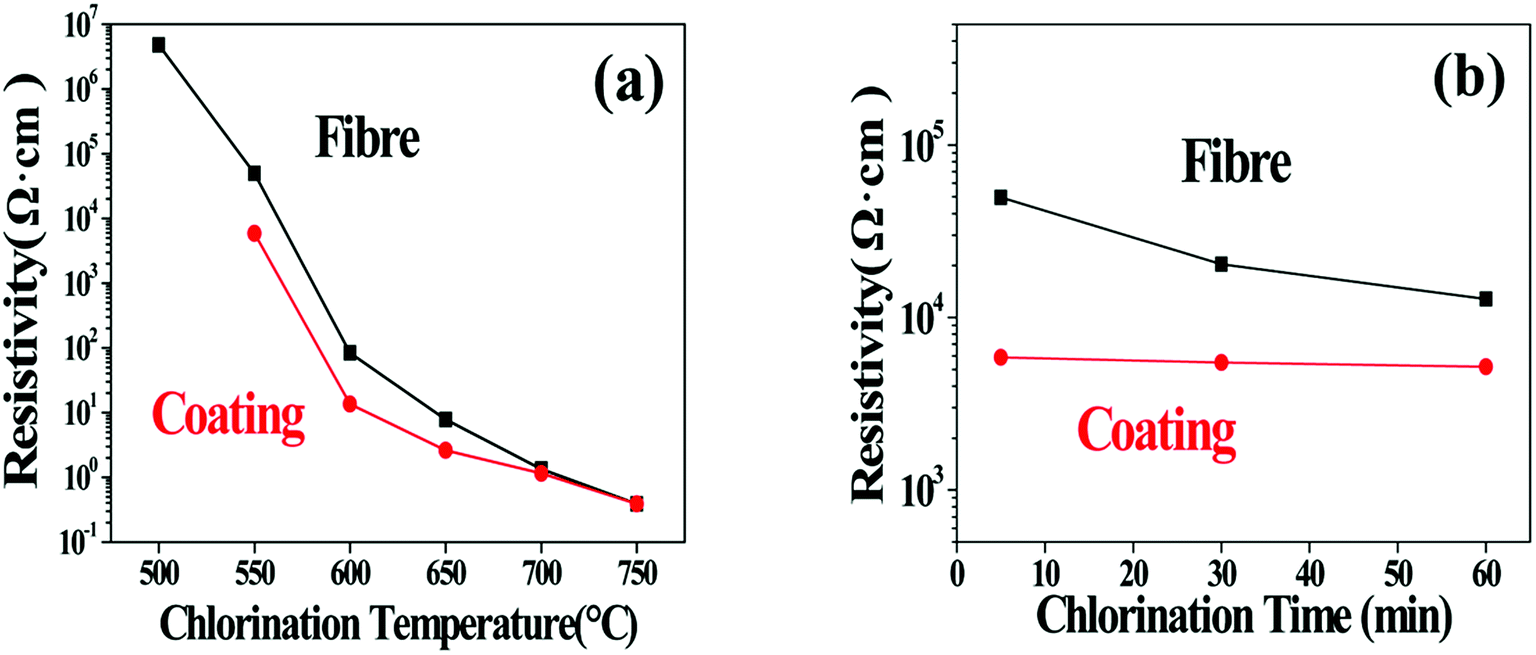 | ||
| Fig. 8 (a) Resistivity of fibres chlorinated for 5 min at temperature range of 500–750 °C. (b) Resistivity of fibres chlorinated for 5, 30, 60 min at 550 °C. | ||
In order to know the feasibility of the C–SiCxOy coatings as interphase layers, two kinds of coated fibres (550-30 and 650-5) with approximate thickness are selected to fabricate SiCf/SiOC ceramic composites. Fig. 9 shows the fracture surface of the SiCf/SiOC composites reinforced with 550-30 fibre and 650-5 fibre respectively. Both composites show obvious interface debonding and fibre pullout. Length of the fibre pullout in the 650-5 composite is larger than that in the 550-30 composite, suggesting that the bonding at the fibre/matrix interface is weakened as the formation temperature of the C–SiCxOy coating is elevated and the CDC content of the coating is increased.
 | ||
| Fig. 9 Fracture surface of the SiCf/SiOC ceramic composites. (a) SiCf/SiOC reinforced with 500-30 fibres and (b) the enlarged image of a fractured fibre. (c) SiCf/SiOC reinforced with 650-5 fibres and (d) the enlarged image of a fractured fibre. | ||
In general, the chlorination offers flexibility in adjusting the microstructure of the C–SiCxOy coatings on oxygen-enriched SiC fibres. By adjusting the coating composition, interfacial debonding state of the ceramic composites can be changed and mechanical performance of the composite can be easily modified. Considering the great variability of resistivity, the coated SiC fibres may generate controllable dielectric losses in electromagnetic wave (EMW), which thus has potential for preparing advanced composites for stealth devices.
4 Conclusion
The current work focuses on preparation and characterization of C–SiCxOy coatings formed on oxygen-enriched SiC fibres. The main conclusions are summarized as follows:(1) Formation of the C–SiCxOy coatings is identified to be a result of the select etching of the SiCxOy metaphases by Cl2. The chlorination temperature is a key factor to change the coating composition, while the soaking time is mainly related to variation of the coating thickness. Thus, the composition and the thickness of the coatings can be independently regulated to meet different purposes.
(2) SiOC ceramic composites reinforced with the coated fibres show obvious interface debonding and fibre pullout. The fibre pullout length in the 650-5 composite is larger than that in the 550-30 composite, suggesting that the bonding at the fibre/matrix interface is weakened as the increase of the CDC content in the C–SiCxOy coating.
(3) Through changing the composition of the C–SiCxOy coating, the resistivity of the coated SiC fibres changed over 7 orders of magnitude. Combing the superiorities of high strength retain ratio, the C–SiCxOy coated SiC fibres are promising for the development of newly structural composites with electric and dielectric functions.
Conflicts of interest
We declare that we do not have any commercial or associative interest that represents a conflict of interest in connection with the work submitted.Acknowledgements
We thank the National Science Foundation of China (NSFC) (grant number 51302234), Creative Research Foundation of Science and Technology on Thermostructural Composite Materials Laboratory (China) (grant number 6142911040113) for financial support.References
- S. Yajima, J. Hayashi and M. Omori, Nature, 1976, 261, 683–685 CrossRef CAS.
- A. R. Bunsell and A. Piant, J. Mater. Sci., 2006, 41, 823–839 CrossRef CAS.
- T. Shikawa, Compos. Sci. Technol., 1994, 51, 135–144 CrossRef.
- N. P. Bansal, Mater. Today, 2005, 8, 57 Search PubMed.
- E. Buet, C. Sauder and S. Poissonnet, J. Eur. Ceram. Soc., 2012, 32, 547–557 CrossRef CAS.
- R. Naslain, Compos. Sci. Technol., 2004, 64, 155–170 CrossRef CAS.
- N. Carrère, E. Martin and J. Lamon, Composites, Part A, 2000, 31, 1179–1190 CrossRef.
- J. P. Singh, D. Singh and M. Sutaria, Composites, Part A, 1999, 30, 445–450 CrossRef.
- K. Shimoda, J. S. Park and T. Hinoki, Compos. Sci. Technol., 2008, 68, 98–105 CrossRef CAS.
- H. Araki, W. Yang, H. Suzuki and Q. Hu, J. Nucl. Mater., 2004, 329, 567–571 CrossRef.
- S. Jacques, A. Lopez-Marure and C. Vincent, J. Eur. Ceram. Soc., 2000, 20, 1929–1938 CrossRef CAS.
- Y. G. Gogotsi and M. Yoshimura, Nature, 1994, 367, 628–630 CrossRef CAS.
- L. Chen, G. Behlau and Y. Gogotsi, Carbide derived carbon (CDC) coatings for Tyranno ZMI SiC Fibers, John Wiley & Sons, 2003, vol. 4, pp. 57–62 Search PubMed.
- M. J. Yuan, T. Zhou and J. He, Appl. Surf. Sci., 2016, 382, 27–33 CrossRef CAS.
- L. Chen, H. Ye, Y. Gogotsi and M. J. McNallan, J. Am. Ceram. Soc., 2003, 86, 1830–1837 CrossRef CAS.
- L. Chen, H. Ye and Y. Gogotsi, J. Am. Ceram. Soc., 2004, 87, 147–151 CrossRef CAS.
- L. Shen, B. J. Tan, W. S. Willis, F. S. Galasso and S. L. Suib, J. Am. Ceram. Soc., 1994, 77, 1011–1016 CrossRef CAS.
- A. Delcamp, et al., Surf. Coat. Technol., 2010, 205, 703–709 CrossRef CAS.
- G. Chollon, R. Pailler and R. Canet, J. Eur. Ceram. Soc., 1998, 18, 725–733 CrossRef CAS.
- A. Jänes, T. Thomberg and E. Lust, Carbon, 2007, 45, 2717–2722 CrossRef.
- S. H. Yeon, P. Reddington and Y. Gogotsi, Carbon, 2010, 48, 201–210 CrossRef CAS.
- K. S. W. Sing, Pure Appl. Chem., 1985, 57, 603–619 CAS.
- S. Yu, R. Tu and T. Goto, J. Eur. Ceram. Soc., 2016, 36, 403–409 CrossRef CAS.
- M. A. Rooke and P. M. A. Sherwood, Carbon, 1995, 33, 375–380 CrossRef CAS.
- A. Avila, I. Montero and L. Galan, J. Appl. Phys., 2001, 89, 212–216 CrossRef CAS.
- B. Su, H. Liang and G. Liu, J. Eur. Ceram. Soc., 2018, 38, 2289–2296 CrossRef CAS.
- S. Stankovich, D. A. Dikin and R. D. Piner, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- A. P. Dušan, J. Marschall and M. R. George, J. Eur. Ceram. Soc., 2010, 30, 2289–2300 CrossRef.
- C. Laffon, A. M. Flank and P. Lagarde, J. Mater. Sci., 1989, 24, 1503–1512 CrossRef CAS.
- G. Honstein, C. Chatillon and F. Baillet, J. Eur. Ceram. Soc., 2012, 32, 1117–1135 CrossRef CAS.
- Y. G. Gogotsi, J. Mater. Chem., 1997, 7, 1841–1848 RSC.
- M. S. Cao, W. L. Song and Z. L. Hou, Carbon, 2010, 48, 788–796 CrossRef CAS.
- R. Bodet, N. Jia and R. E. Tressler, J. Eur. Ceram. Soc., 1996, 16, 653–664 CrossRef CAS.
- F. Carmona, P. Delhaes and G. Keryer, Solid State Commun., 1974, 14, 1183–1187 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |