
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRapid selection of aptamers based on protein microarray†
Fang Yua,
Hui Li*a,
Wei Sunb,
Danke Xu *a and
Fuchu He*ab
*a and
Fuchu He*ab
aState Key Laboratory of Analytical Chemistry for Life Science, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing, Jiangsu 210046, China. E-mail: lihui@nju.edu.cn; xudanke@nju.edu.cn
bState Key Laboratory of Proteomics, National Center for Protein Sciences Beijing, Beijing Institute of Radiation Medicine, Beijing 102206, China, hefc@nic.bmi.ac.cn
First published on 27th March 2019
Abstract
We report a novel method for the efficient screening of aptamers from a complex ssDNA library based on a microarray chip, which was named Microarray-SELEX. In this method, the target protein (lactoferrin) and negative proteins (α-lactalbumin, β-lactoglobulin, bovine serum albumin, and casein) were each dotted and immobilized on a slide to form a protein microarray. Moreover, the library was added to this chip to interact with negative proteins, then with the target protein. The process of SELEX could be monitored on-line using a fluorescent microarray scanner and the whole process was performed in only six rounds. Finally, five aptamers (YFL-1, YFL-4, YFL-5, YFL-6 and YFL-7) were obtained, which showed good specificity towards lactoferrin in the presence of negative proteins. The equilibrium dissociation constants (Kd) of the aptamers were in the nanomolar range. Briefly, Microarray-SELEX is a rapid, easy, sensitive and efficient method for screening aptamers.
Introduction
Aptamers are oligonucleotide molecules (either RNA or DNA) isolated through SELEX (Systematic Evolution of Ligands by Exponential enrichment) from a very large random oligonucleotide library.1–3 Their applications are extensive, such as the detection of disease biomarkers, cell imaging, drug delivery and cancer therapy.4–7 Many methods have been developed for the rapid and efficient screening of aptamers,8,9 including traditional bead-based SELEX,10,11 capillary electrophoresis SELEX (CE-SELEX),12,13 flow cytometry SELEX,14,15 surface plasmon resonance SELEX (SPR-SELEX),16 and microfluidic SELEX.17,18 These methods have their advantages and disadvantages. Among them, microfluidic SELEX has the advantages of high separation efficiency and high-throughput. However, SPR-SELEX is known for its function to monitor the interaction between the target and library. The flow cytometry-based approach specializes in aptamer screening for cell membrane proteins on the cell surface. Most methods require the targets to be immobilized on a specific vector, such as beads-based SELEX, SPR-SELEX, flow cytometry SELEX and microfluidic SELEX. However, we established a new screening method named Microarray-SELEX, which is easy and has simple experimental operations.Microarray chips are generally manufactured by printing a small volume of biological macromolecules, for example, nucleic acids, polypeptide, tissue sections, cells and other biological samples, on a carrier (such as slides and nylon membrane) with a robotic microarray. They form a dense two-dimensional array of molecules. Next, the biological macromolecules are commonly reacted with the labeled target molecules, and both of them are measured by a specific instrument, namely a laser confocal scanner or a charge-coupled camera. The number of target molecules in a sample can be detected and analysed quickly and efficiently based on the signal. Furthermore, a large number of interactions can be detected simultaneously. Besides, with the development of computer technology and the miniaturization of instruments, microarray analysis can be performed in small academic laboratories. Microarray technology is divided into several categories, including RNA microarray,19 protein microarray,20 glycon microarray,21 peptide microarray22 and small molecule microarray (SMM).23 The development of high-throughput microarrays offers a potential solution to solve the logjam of biomaterial discovery. Microarrays have revolutionized the discovery of drugs and genomics, enabling the rapid, parallel and economical screening of thousands of microscopic spots printed on a carrier.24 Moreover, they are widely used for many researches including research on gene expression,25 cell–ligand,26 protein–protein,27 protein–carbohydrate28 and protein–small molecule interactions.29 For SELEX, many researchers immobilized the library on a carrier. Cho et al.30 performed microfluidic-SELEX with high-throughput sequencing to obtain the enriched aptamers. Then, they synthesized an array based on these sequences to identify the highest-affinity aptamer for human angiopoietin-2. This aptamer was used to block the primary binding epitopes on the target. Next, these complexes were applied for screening with the same array to identify aptamers that bind secondary sites. They immobilized the aptamers on the chip to form a microarray for the screening of aptamers. Besides, Martin et al.31 performed Microarray-SELEX to identify the highest-affinity aptamer targeting thrombin in a single round. They immobilized the aptamers on an array chip with a 3′-T10 spacer. After incubation and washing, the arrays were scanned and analysed using the Agilent Scan Control software and Agilent Feature Extraction software version 10.7.3.1, individually. Statistics were used for the foundation of potential binders to compare with control sequences, which were identified by microarray.31 Both of the above methods performed aptamer screening through immobilizing the library on a chip. However, they required many pre-experiments to obtain the preliminary sequences or needed some commercial products to build a screening method. We built a simple and efficient screening method by immobilizing the proteins on a chip. In this method, lactoferrin was the target protein, which could bind iron and have powerful biological functions during infancy.32 Firstly, these proteins were dotted on a chip to form a microarray by physical adsorption with good reproducibility. Secondly, in situ monitoring and real-time evaluation were used to control the selection process, avoid blind selection, and shorten the selection time. Lastly, the microarray consisted of a set of mature instruments, including a Smart Arrayer and Microarray Scanner, which made the entire SELEX process simple. Also, it was high-throughput, repeatable and time-saving, where 1 round could be finished in 1 day and the whole process finished in six rounds. Microarray-SELEX was built based on a protein microarray chip and the whole SELEX was done twice independently to ensure the effectiveness of the screening.
Prior to this, our research group established two screening platforms, which were named microfluidic-SELEX33 and SPR-Imaging-SELEX.34 First, microfluidic-SELEX screened aptamers for lactoferrin by combining microfluidics with a protein chip. The entire screening process took seven rounds. Second, SPR-Imaging-SELEX combined SPR and nanosilver signal amplification to screen aptamers for lactoferrin within six rounds. The advantages of microfluidic-SELEX include high efficiency and high-throughput, while SPR-Imaging-SELEX can monitor the interaction between the target and library on-line. However, the disadvantages of the above two methods are that the chips used in the screening are complicated to manufacture, expensive, have complicated instrument operation, and high in operator requirements. Finally, we provided a new screening method, which is fast, simple, inexpensive and efficient. In our approach, the chip fabrication and experimental operation were simple. Moreover, using the same number of screening rounds, the same screening results as the previous two methods were obtained. In addition, we could choose the appropriate screening method for different situations.
Experimental
Materials and reagents
Lactoferrin (Lac), β-lactoglobulin (β-Lac), α-lactalbumin (α-Lac), casein (Cas) from bovine milk and bovine serum albumin (BSA) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Streptavidin Magnesphere® paramagnetic particles (SA-MPs) were purchased from Promega Corporation (Madison, USA). 2× SYBR® Premix Ex Taq™ II (Takara Bio Inc., Japan) and 2× Taq Master Mix (Tiangen Biotech Co., Ltd.) were used for PCR amplification. The following phosphate buffers were used in the experiments: 1× PBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4·12H2O, 2 mM KH2PO4), 1× PBSM (1× PBS with 1 mM MgCl2, pH 7.4), and 1× PBST (1× PBS with 0.05% Tween-20, pH 7.4). 1× TBE was used for 30% polyacrylamide gel electrophoresis, which consisted of 89 mmol L−1 Tris, 89 mmol L−1 boric acid and 2 mmol L−1 EDTA.Apparatus
Fluorescence intensity and ultraviolet-visible (UV-Vis) spectra were measured on a BioTek instrument (Synergy H1, USA). A Smart Arrayer was used for the formation of proteins microarrays. Microarrays were scanned using a Luxscan-10K/A Microarray Scanner (532 nm laser source for TAMRA, Beijing CapitalBio Co., China). An Applied Biosystems SimpliAmp™ Thermal Cycler (Thermo Fisher Scientific Inc., USA) was used for PCR amplification. A GEN4T Thermal Cycler (Hangzhou Bio-gener Technology Co., Ltd., China) was applied to heat the microarray chip to 95 °C to elute the target-bound ssDNA.DNA and library preparation
All oligonucleotides used in this study were synthesized according to Yang's group35 by Sangon Biotech Co. Ltd. (Shanghai, China). Each single-stranded DNA (ssDNA) library member consisted of 40 nucleotide (nt) randomized sequences and 20 nt primer sites (5′-TAMRA-GACAGGCAGGACACCGTAAC-N40-CTGCTACCTCCCTCCTCTTC-3′). The library was modified with TAMRA and named Lib. The primers for PCR amplification were TAMRA-modified forward primer (TAMRA-FP) and biotinylated-modified reverse primer (Biotin-RP). TAMRA-FP: 5′-TAMRA-GACAGGCAGGACACCGTAAC-3′ and Biotin-RP: 5′-biotin-GAAGAGGAGGGAGGTAGCAG-3′. Identically, the unlabeled primers were used for the PCR sample for cloning and sequencing, which included forward primer (named FP, GACAGGCAGGACACCGTAAC) and reverse primer (named RP, GAAGAGGAGGGAGGTAGCAG).Selecting process by microarrays
To increase physical adsorption and hydrophilicity, glass slides were treated with NaOH and washed with ultrapure water. Then, 2.5 mg mL−1 target protein (Lac) and negative proteins (2.5 mg mL−1 α-Lac, 2.5 mg mL−1 β-Lac, 2.5 mg mL−1 Cas and 2.5 mg mL−1 BSA) were dotted on a glass slide with a Smart Arrayer. Subsequently, these proteins were immobilized at 37 °C for 2 h and blocked with 10 mg mL−1 BSA and 0.01 mM random short ssDNA (20 nt) at 37 °C for 1 h. After that, it was washed with 1× PBST (200 μL) three times. The initial ssDNA library (1 nmol) was dissolved in 125 μL 1× PBSM, reacted with negative proteins at 37 °C without shaking for 1 h, and then reacted with Lac under the same conditions. After washing with 1× PBST (200 μL) three times, the slide was scanned with a Luxscan-10K/A Microarray Scanner. Finally, the aptamer-bound protein microarray was heated to 95 °C for 5 min and the bound aptamers were eluted with 450 μL DEPC water. The slide was scanned for a second time to ensure that the bound aptamers were completely eluted. The volume of solution was reduced to 92 μL by nitrogen blow drying. Also, it was used for PCR amplification. SA-MPs and NaOH were used to turn PCR products to ssDNA according to our previous reports.33 Only the sixth-round PCR product of the first cycle selection was cloned by Sangon Bio-Technology.For the second round of selection, negative selection was introduced from the third round, and the obtained ssDNA from the second round was preferentially incubated with negative proteins at 37 °C for 1 h to remove nonspecific binding sequences. Thereafter, the unbound aptamers were added to interact with the target protein for positive selection. Finally, one part of the collected supernatant from the sixth, seventh and eighth round was PCR amplified with unlabeled primers, and these PCR products were cloned by Sangon Bio-Technology Co., Ltd (Shanghai, China). A hundred colonies of every round were randomly picked and sequenced.
PCR amplification
The collected supernatants were divided into four parts and each part (23 μL collected supernatants) was mixed with 25 μL 2× Taq Master Mix, 1 μL TAMRA-FP (20 μM), and 1 μL Biotin-RP (20 μM). PCR was carried out at 94 °C for 5 min, 94 °C for 30 s, 60.5 °C for 30 s, and 72 °C for 30 s, followed by 72 °C for 5 min in the final extension step. Subsequently, 5 μL amplified PCR product was used as a template. 25 μL 2× SYBR® Premix Ex Taq™ II, 1 μL TAMRA-FP (20 μM), 1 μL Biotin-RP (20 μM) and 18 μL DEPC water were added to this template to be reamplified according to the above process. Moreover, 10 μL of the reamplified PCR products from each cycle was used to detect the fluorescence intensity with a BioTek Synergy H1 to determine the optimal PCR cycle number. Finally, the remaining PCR products were reamplified at the optimal cycle number with 2× Taq Master Mix and primers.ssDNA generation
To separate dsDNA from the PCR mixtures, the PCR products were incubated with SA-MPs at room temperature for 1 h with violent shaking. Then, SA-MPs were washed with 1× PBS two times, and incubated with 50 mM NaOH (25 μL) at room temperature for 5 min to generate ssDNA. ssDNA was collected and neutralized with 100 mM HCl (12.5 μL). Lastly, to achieve the same salt concentration as 1× PBSMC, 25 μL DEPC water and 62.5 μL 2× PBSMC (254 mM NaCl, 5.4 mM KCl, 20 mM Na2HPO4·12H2O, 4 mM KH2PO4, 2 mM MgCl2 and 4 mM CaCl2) were added to this solution. The mixture was quantified and used as an enriched library for the following SELEX.Cloning and sequencing
Selection was completed when a significant fluorescence intensity difference was observed. Then, one part of the PCR product was cloned by Sangon Bio-Technology Co., Ltd (Shanghai, China), which was amplified with unlabeled primers. 100 colonies of each round were randomly picked and sequenced. Besides, the secondary structures of the selected aptamers were analyzed with IDT (http://sg.idtdna.com/calc/analyzer).Specificity of selected aptamers toward different proteins
5 mg mL−1 each protein (Lac, α-Lac, β-Lac, Cas and BSA) were dotted on a FisherFinest Premium glass slide to form a 6 × 6 microarray and immobilized at 37 °C for 2 h. Then, the slide was blocked with 10 mg mL−1 BSA and 0.01 mM random short ssDNA (20 nt) at 37 °C for 1 h and washed with 1× PBST for three times. Subsequently, 1 μM TAMRA-labeled aptamers (YFL-1, YFL-4, YFL-5, YFL-6, and YFL-7) and 10 μM Lib were added and reacted at 37 °C for 1 h. Followed by washing with 1× PBST, the microarray was scanned and data was collected using a Luxscan-10K/A Microarray Scanner.Results and discussion
Microarray-SELEX for aptamer selection against lactoferrin
The process of Microarray-SELEX is demonstrated in Scheme 1. First, the target protein (Lac) and negative proteins (α-Lac, β-Lac, Cas and BSA) were dotted on a glass slide to form a protein-microarray. A 12 × 12 microarray was fabricated on a slide using a Smart Arrayer. For the target microarray, the 12 × 12 microarray consisted of Lac. Four proteins made up the negative microarray, and 12 × 3 for each negative protein. Then, the initial ssDNA library was incubated with negative proteins for 1 h at 37 °C, and the unbound ssDNA was added to the target protein and incubated under the same conditions. After washing with 1× PBST, the slide was scanned using a Luxscan-10K/A Microarray Scanner. Afterwards, the reacted aptamers were eluted with DEPC water at 95 °C for 5 min and used for PCR amplification. The dsDNA was separated from the PCR products by SA-MPs. Finally, ssDNA was produced using NaOH for the next round of screening.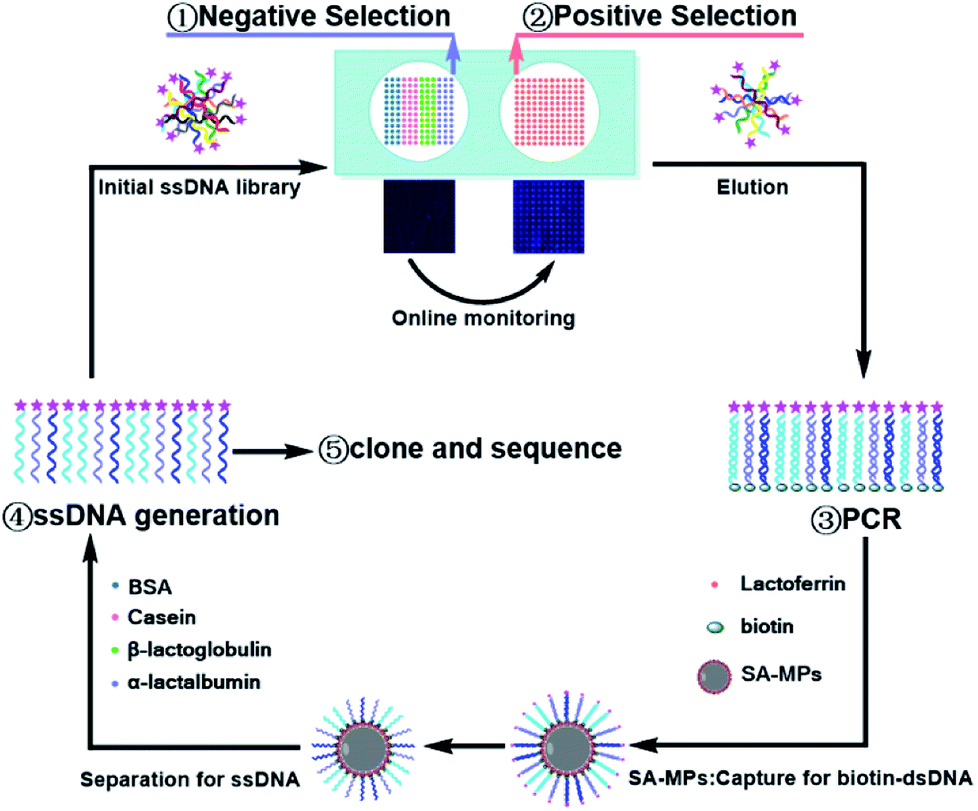 | ||
| Scheme 1 Principle of Microarray-SELEX for aptamer screening. | ||
The reaction between the target and ssDNA was performed under static conditions. Channels and flow paths were not needed in this method. It was only necessary to transfer the library to the chip via a pipette. However, the results for this static reaction mode demonstrated it could achieve the same effect as channel screening (dynamic screening). Besides, we used this method to obtain as many aptamer candidates as other dynamic methods. Moreover, an array in this method was necessary. Firstly, the enrichment of the library was monitored in situ for every round of screening to control the entire screening process and avoid the blindness of screening. Without these arrays, the enrichment of the library would not have been visible. Besides, the operation of protein immobilization was very simple. Proteins were only spotted on a chip using a spotter and immobilized by physical adsorption. Secondly, no microbeads and other carriers were needed to simplify the operation. Placing microspheres with fixed targets or libraries in a regular array was a very difficult task. Thirdly, different target proteins could be spotted on one array for high-throughput screening. Several proteins were screened simultaneously on one chip by monitoring the fluorescence signals of different proteins from an array. Thus, this method was a general screening method.
The results of Microarray-SELEX are shown in Fig. 1. The fluorescence intensities of every round increase with an increase in the number of selection rounds. The Lib in each round of selection were generated by NaOH and their fluorescence intensities are shown in Fig. 1b. The screening procedure for Lac was monitored on-line using a Luxscan-10K/A Microarray Scanner (Fig. 1c). It was seen that the fluorescence intensity of the Lac-aptamers increased with the number of screening rounds. To verify complete elution of the enriched aptamer, the fluorescent images after DEPC elution are shown in Fig. 1d. However, the fluorescence images of the enriched aptamers on the negative microarrays in each round are also shown in Fig. S1.† The fluorescence intensity increased gradually from round 1 to round 3, then surged from round 4 to round 6, and reached a plateau in round 7 and round 8 (Fig. 1a). This indicated the enrichment of specific aptamers. Thus, the sixth round PCR product was cloned and sequenced. Finally, 16 sequences, which were repeated more than two times, were obtained. Information about them is shown in Table S1.† Especially, the sequences from YFL-1 to YFL-14 were the same as the sequences in PMM-SELEX,33 which showed the reliability of Microarray-SELEX.
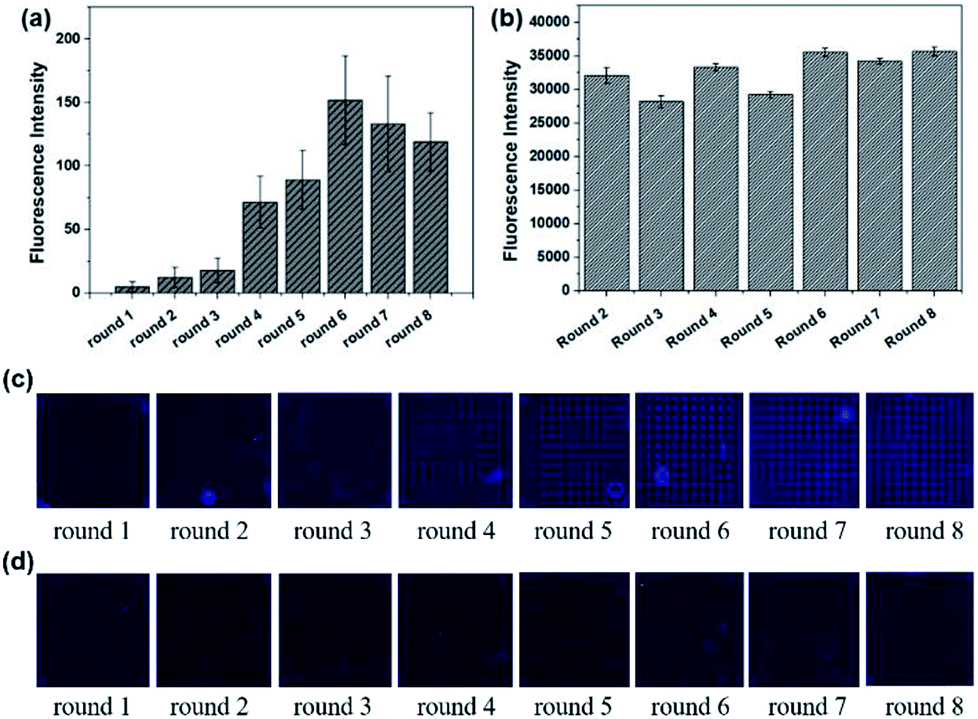 | ||
| Fig. 1 Signals in the first circle selection from round 1 to round 8. (a) Fluorescence intensities of ssDNA bound to Lac on microarrays. (b) Fluorescence intensities for the evolved Lib in each round. (c) Fluorescence images of the enriched aptamers on Lac microarrays before elution. (d) Fluorescence images for the enriched aptamers on Lac microarrays after elution by DEPC water. | ||
The whole process was repeated one more time to test the stability and repeatability of Microarray-SELEX. We changed some parameters to improve the fluorescence signals on the chip and prove our assumption. For the second cycle of Microarray-SELEX, the concentration of Lac was 5 mg mL−1, and only positive selection was performed in the first and second round. As shown in Fig. 2a, negative selection was introduced from the third round and the fluorescence intensity was slightly lower than that of the second round. Besides, the fluorescence intensity increased gradually from round 1 to round 2, then reduced slightly in round 3, increased from round 4 to round 6, and reached a plateau in the last four rounds. The fluorescence images of the negative microarrays in each round are shown in Fig. S2.† Thus, the PCR products of the sixth, seventh and eighth round were cloned and sequenced. Information about the sequences is shown in Table S2,† where 18 sequences, which were repeated more than three times, were selected. Eight aptamers were identical to the sequences in the first selection. Eleven sequences were the same with that in PMM-SELEX.33 This fully proved the reliability and repeatability of our screening method. Additionally, the selected number of aptamers was related to the fluorescence intensity on the microarrays, where YFL-10, YFL-11 and YFL-14 appeared more frequently than the first selection. Besides, the fluorescence intensity of round 5 from the second selection was higher than that of round 6 from the first selection, which was 260 and 151, respectively. Thus, we believed that the second screening is sufficient in five rounds. The fluorescence intensities of Lib generated in each round are shown in Fig. 2b, which were higher than that in the first selection. The fluorescence images from round 1 to round 10 before and after elution are shown in Fig. 2c and d, respectively. As the concentration of Lib in each round increased, the fluorescence intensities in the second screening were higher than that of the first time screening.
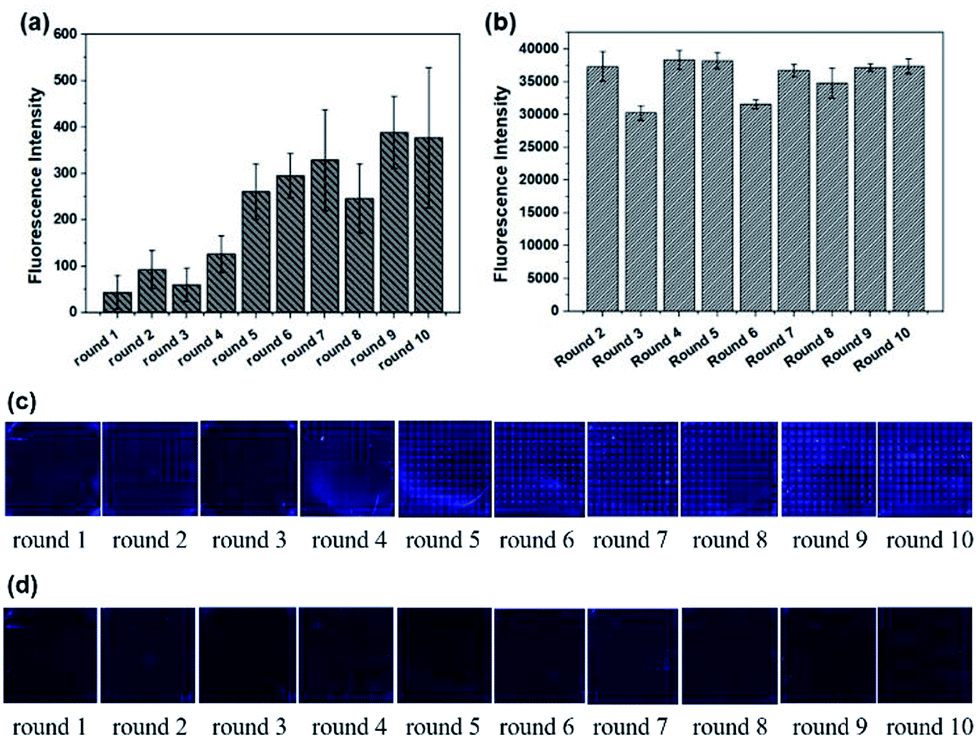 | ||
| Fig. 2 Signals in the second circle selection from round 1 to round 10. (a) Fluorescence intensities for ssDNA bound to Lac on microarrays. (b) Fluorescence intensities of the evolved Lib in each round. (c) Fluorescence images for the enriched aptamers on Lac microarrays before elution. (d) Fluorescence images of these enriched aptamers on Lac microarrays after elution by DEPC water. | ||
Five sequences (YFL-1, YFL-4, YFL-5, YFL-6 and YFL-7) appeared in both cycles of Microarray-SELEX, which were repeated 5-fold, 10-fold, 4-fold, 8-fold and 8-fold, respectively. They were the same as the sequences in PMM-SELEX,33 which were named Lac-6a, Lac-A1, Lac-A3, Lac-A4 and Lac-A6, respectively. Thus, these sequences were used for further study, including structure and specificity towards the target.
Quantitative analyses of every parameter is very important because many rounds of selection may introduce undesirable biases. PCR is an important parameter in the SELEX procedure. During the screening, we monitored the PCR process using the fluorescence intensity of SYBR. Also, the feasibility of SYBR-PCR was confirmed by comparison with polyacrylamide gel electrophoresis (PAGE). In the present work, standard PCR was used to verify the feasibility of SYBR-PCR. Lib (10 fM and 0 fM) was firstly pre-amplified with 10 cycles, then the PCR products were used as templates and re-amplified with 2× SYBR® Premix Ex Taq™. The fluorescence intensities of 10 fM re-amplified PCR products increased from 2 to 6, and then reduced from 8 to 16 cycles slowly (Fig. 3a). Byproducts were formed after 8 cycles of re-amplified PCR, which were also detected in the PAGE (Fig. 3b). Thus, the optimal PCR circle produced by SYBR corresponded with gel electrophoresis. SYBR-PCR was reliable and time-saving, and used for further selection. Also, the fluorescence intensities of 0 fM re-amplified PCR products increased after 8 cycles (Fig. 3c). This further confirmed that by-products were generated as the number of PCR increased. Meanwhile, the enriched Lib obtained in the first round was amplified in the same way. The fluorescence spectra of the re-amplified PCR products from 5 to 9 cycles are shown in Fig. 3d. The optimal PCR circle was 7 cycles for re-amplification. Then, SA-MPs was used to separate dsDNA from the other products and NaOH was added to transform dsDNA into ssDNA.
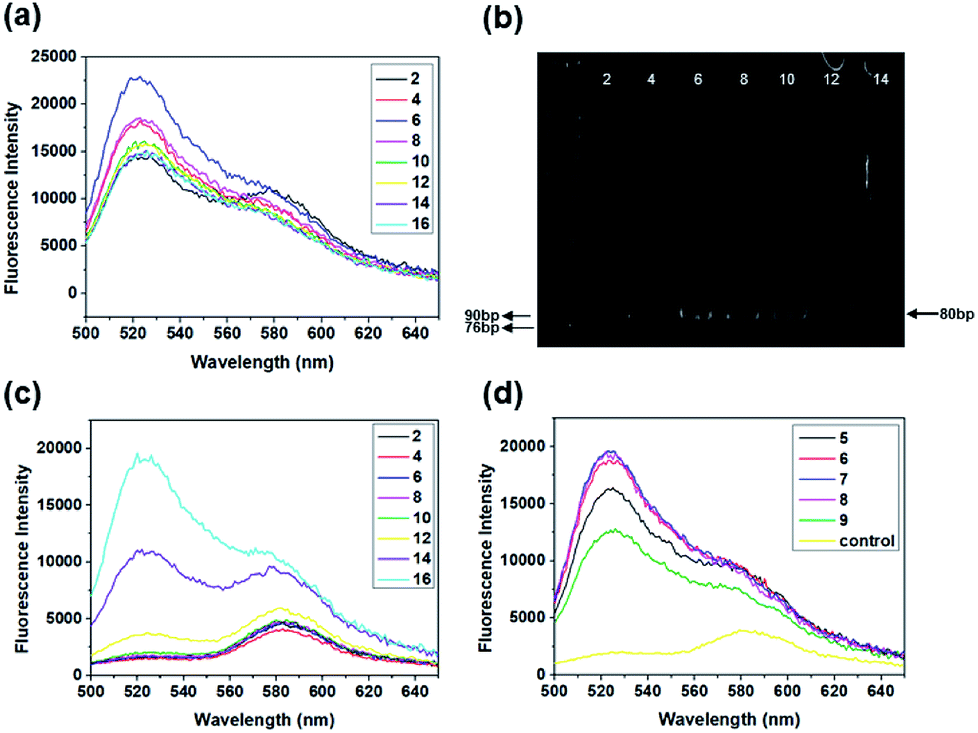 | ||
| Fig. 3 Fluorescence spectra of the standard re-amplified PCR products from different PCR cycles (from 2 to 16 cycles). (a) 10 fM library and (c) 0 fM library. (b) Gel electrophoretogram of the re-amplified PCR products. (d) Fluorescence spectra of selected re-amplified PCR products collected from 5 cycles to 9 cycles, and the control. | ||
Information of selected aptamer sequences
Five aptamers (YFL-1, YFL-4, YFL-5, YFL-6 and YFL-7) were chosen because they appeared in two circle screenings and had the lowest ΔG. We performed a preliminary simulation of their structures and calculated their ΔG on the IDT website.33,34 The bases were truncated to make aptamers with a lower ΔG, which did not affect the secondary structures. In addition, the truncated sequences were used for further specificity verification. As shown in Table S2† and Fig. 4, five aptamers had a lower ΔG and had a relatively stable structure. Besides, their secondary structures were very similar. The characteristic stems and loops were found in these sequences. Moreover, stems and loops were necessary to bind with Lac, which was tested before.34 Furthermore, the secondary structures of the other six sequences with the lowest ΔG are listed in Fig. S3,† which were used for the comparison with these five sequences. It was seen that the structures were roughly divided into two categories. The secondary structures of YFL-13, YFL-22 and YFL-24 were similar to that of YFL-1 and YFL-6. All of them had two small rings; whereas, YFL-16, YFL-21 and YFL-26 had three small rings, which were similar to the structures of YFL-4, YFL-5, and YFL-7. This result also demonstrated that the repeated five aptamers were representative. All aptamers in both circles are shown in Fig. 4f. Furthermore, the repeated eight aptamers in the middle showed the repeatability and reliability of this screening method.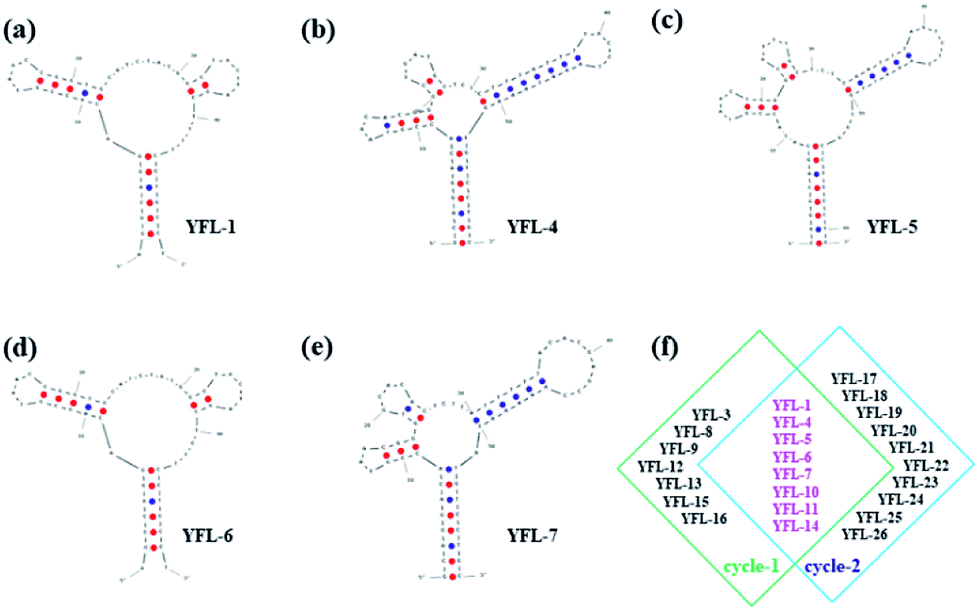 | ||
| Fig. 4 Secondary structures of five selected aptamers were simulated with IDT at the minimum ΔG. (a) YFL-1, (b) YFL-4, (c) YFL-5, (d) YFL-6, and (e) YFL-7. (f) Selected aptamers from both the first and second cycle of selection. | ||
Specificity of aptamers
To study the specificity and selectivity of the selected sequences towards Lac, a microarray chip was used to differentiate the target protein and negative proteins using their fluorescent signals. The use of a microarray chip is a simple, rapid and reliable method. 5 mg mL−1 proteins was dotted on a slide to form a microarray, and a schematic is shown in Fig. 5a. After immobilizing, blocking and washing, 1 μM TAMRA-labeled aptamers (YFL-1, YFL-4, YFL-5, YFL-6, and YFL-7) and 10 μM Lib were added to the chip and reacted with proteins at 37 °C for 1 h. As shown in Fig. 5b, the fluorescence intensities of aptamers-Lac were obviously higher than that of the aptamer-negative proteins. In addition, the negative proteins showed almost no binding with the selected aptamers, and their fluorescence intensities ranged from 1 to 22. However, the fluorescence intensities of sequences-Lac were 1775, 2154, 2236, 2842, 2489 and 320 for YFL-1, YFL-4, YFL-5, YFL-6, YFL-7 and Lib, respectively. Furthermore, the fluorescence signals of aptamers-Lac were at least five times more than that of Lib-Lac. These results indicated the high specificity and good selectivity of the aptamers towards Lac.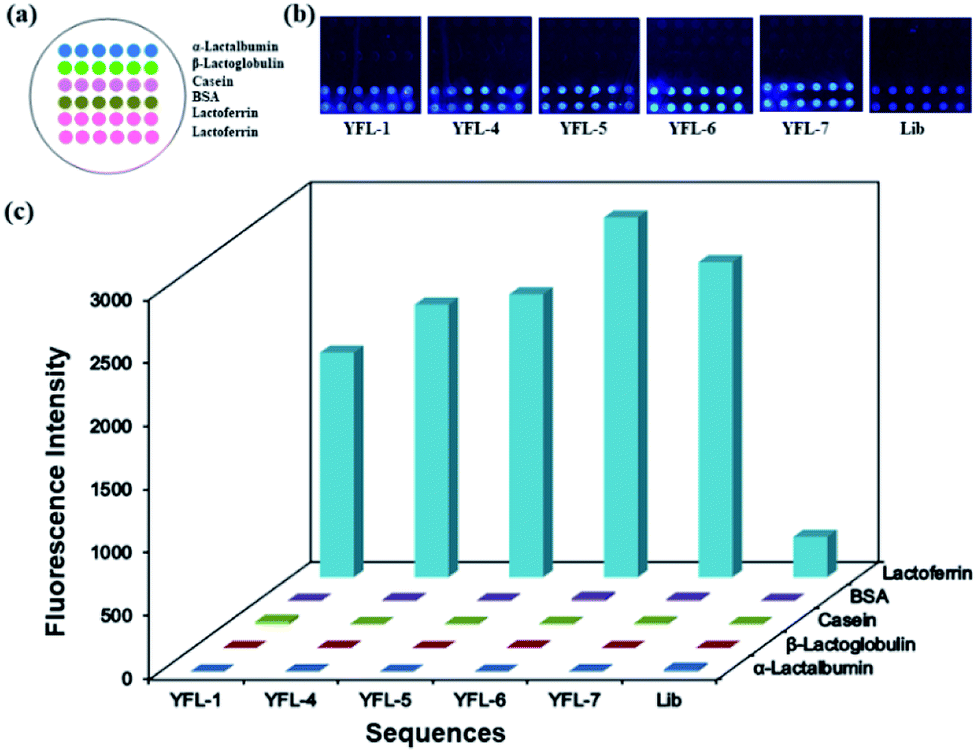 | ||
| Fig. 5 (a) Schematic of microarray chip for protein immobilization, (b) fluorescence images for the selected aptamers on microarrays and (c) fluorescence intensities of the selected aptamers to Lac and negative proteins. | ||
Analysis of aptamer sequences
Some aptamers obtained by our method were identical to the aptamer sequences previously screened by other methods, namely PMM-SELEX and SPRI-SELEX. These sequences (YFL-1, YFL-4, YFL-5, YFL-6 and YFL-7) were the same as Lac-6a, Lac-A1, Lac-A3, Lac-A4 and Lac-A6 in PMM-SELEX,33 respectively. YFL-4 and YFL-5 were consistent with Lac1-4 and Lac8-2, respectively, which were obtained in SPRI-SELEX.34 The same sequences were sufficient to demonstrate the reliability and reproducibility of Microarray-SELEX. Moreover, compared to previous screening methods, our method is simple to operate and easy to promote. The affinity between the screened aptamers and Lac was verified using our previous methods. The equilibrium dissociation constants (Kd) of YFL-1, YFL-4, YFL-5 and YFL-6 were 1.04 ± 0.50 nM, 0.953 ± 0.11 nM, 8.91 ± 1.29 nM and 4.97 ± 0.46 nM, respectively.33,36 All these values were detected by SPR and based on the formula Kd = Koff/Kon. The Kd values of these four aptamers were in the low nanomolar range, from 0.953 nM to 8.91 nM, demonstrating the strong binding of these aptamers to Lac.Conclusions
In summary, we established a new Microarray-SELEX method and it was applied to screen aptamers for Lac. Microarray-SELEX possesses some remarkable features. First, the chip affords a relatively static form of interaction for screening, where the target and ssDNA interact without channel liquid flow. The entire process can be monitored in situ with fluorescence. Moreover, we obtained the same aptamers with channel dynamic methods and the amount of aptamers and their affinity were sufficient. Second, no complicated instruments and devices were needed, such as SPR imaging and microfluidic chips. Third, this method saved time and could complete 1 to 2 rounds in 1 day. Finally, the aptamers exhibited high specificity towards Lac, which was proven using a microarray chip. Ultimately, our work provides a standard method to screen aptamers for any protein, which is versatile, efficient and stable. It can be further developed into a high-throughput SELEX method by simultaneously spotting several proteins on a positive selection area. These targets can be simultaneously screened on a single chip. After several rounds of screening, we can obtain aptamers against different targets. Thus, our method is general and easy to promote.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We acknowledge the financial support of National Natural Foundation of China (Grant No. 21405077, 21227009, 21475060, 21775068).References
- A. D. Ellington and J. W. Szostak, Nature, 1990, 346, 818–822 CrossRef CAS.
- C. Tuerk and L. Gold, Science, 1990, 249, 505–510 CrossRef CAS.
- W. Y. Zhang, W. H. Zhang, Z. Y. Liu, C. Li, Z. Zhu and C. Y. Yang, Anal. Chem., 2012, 84, 350–355 CrossRef CAS.
- L. Xu, W. Yan, W. Ma, H. Kuang, X. Wu, L. Liu, Y. Zhao, L. Wang and C. Xu, Adv. Mater., 2015, 27, 1706–1711 CrossRef CAS.
- J. S. Paige, T. Nguyen-Duc, W. J. Song and S. R. Jaffrey, Science, 2012, 335, 1194 CrossRef CAS.
- R. Mo, T. Y. Jiang, R. DiSanto, W. Y. Tai and Z. Gu, Nat. Commun., 2014, 5, 3364 CrossRef.
- G. Zhu, J. Zheng, E. Song, M. Donovan, K. Zhang, C. Liu and W. Tan, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7998–8003 CrossRef CAS.
- M. Darmostuk, S. Rimpelova, H. Gbelcova and T. RumL, Biotechnol. Adv., 2015, 33, 1141–1161 CrossRef CAS.
- M. Yüce, N. Ullah and H. Budak, Analyst, 2015, 140, 5379–5399 RSC.
- Y. L. Song, Z. Zhu, Y. An, W. T. Zhang, H. M. Zhang, D. Liu, C. D. Yu, W. Duan and C. J. Yang, Anal. Chem., 2013, 85, 4141–4149 CrossRef CAS.
- N. Duan, W. H. Gong, S. J. Wu and Z. P. Wang, Anal. Chim. Acta, 2017, 961, 100–105 CrossRef CAS.
- M. Berezovski, A. Drabovich, S. M. Krylova, M. Musheev, V. Okhonin, A. Petrov and S. N. Krylov, J. Am. Chem. Soc., 2005, 127, 3165–3171 CrossRef CAS.
- S. D. Mendonsa and M. T. Bowser, J. Am. Chem. Soc., 2004, 126, 20–21 CrossRef CAS.
- J. P. Wang, J. W. Yu, Q. Yang, J. McDermott, A. Scott, M. Vukovich, R. Lagrois, Q. Gong, W. Greenleaf, M. Eisenstein, B. S. Ferguson and H. T. Soh, Angew. Chem., Int. Ed., 2017, 56, 744–747 CrossRef CAS.
- G. Mayer, M. S. Ahmed, A. Dolf, E. Endl, P. A. Knolle and M. Famulok, Nat. Protoc., 2010, 5, 1993–2004 CrossRef CAS.
- T. S. Misono and P. K. Kumar, Anal. Biochem., 2005, 342, 312–317 CrossRef CAS.
- X. Lou, J. Qian, Y. Xiao, L. Viel, A. E. Gerdon, E. T. Lagally, P. Atzberger, T. M. Tarasow, A. J. Heeger and H. T. Soh, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 2989–2994 CrossRef CAS.
- M. Cho, S. Soo, O. J. Nie, R. Stewart, M. Eisenstein, J. Chambers, J. D. Marth, F. Walker, J. A. Thomson and H. T. Soh, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 18460–18465 CrossRef CAS.
- Y. Li, H. J. Lee and R. M. Corn, Anal. Chem., 2007, 79, 1082–1088 CrossRef CAS.
- G. MacBeath, Nat. Genet., 2002, 32, 526–532 CrossRef CAS.
- A. Nand, V. Singh, P. Wang, J. Na and J. Zhu, Anal. Biochem., 2014, 465, 114–120 CrossRef CAS.
- Y. Li, H. Wen, Y. Xi, K. Tanaka, H. Wang, D. Peng, Y. Ren, Q. Jin, S. Y. R. Dent, W. Li, H. Li and X. Shi, Cell, 2014, 159, 558–571 CrossRef CAS.
- F. G. Kuruvilla, A. F. Shamji, S. M. Sternson, P. J. Hergenrother and S. L. Schreiber, Nature, 2002, 416, 653–657 CrossRef CAS.
- M. J. Heller, Annu. Rev. Biomed. Eng., 2002, 4, 129–153 CrossRef CAS.
- M. Schena, D. Shalon, R. W. Davis and P. O. Brown, Science, 1995, 270, 467–470 CrossRef CAS.
- O. J. Barrett, J. L. Childs and M. D. Disney, ChemBioChem, 2006, 7, 1882–1885 CrossRef CAS.
- G. MacBeath and S. L. Schreiber, Science, 2000, 289, 1760–1763 CAS.
- B. T. Houseman and M. Mrksich, Chem. Biol., 2002, 9, 400–401 CrossRef.
- J. E. Bradner, O. M. McPherson and A. N. Koehler, Nat. Protoc., 2006, 1, 2344–2352 CrossRef CAS.
- M. Cho, S. S. Oh, J. Nie, R. Stewart, M. J. Radeke, M. Eisenstein, P. J. Coffey, J. A. Thomson and H. T. Soh, Anal. Chem., 2015, 87, 821–828 CrossRef CAS.
- J. A. Martin, P. A. Mirau, Y. Chushak, J. L. Chávez, R. R. Naik, J. A. Hagen and N. K. Loughnane, J. Anal. Methods Chem., 2015, 2015, 137489 Search PubMed.
- P. Pomastowski, M. Sprynskyy, P. Zuvela, K. Rafinska, M. Milanowski, J. J. Liu, M. Yi and B. Buszewski, J. Am. Chem. Soc., 2016, 138, 7899–7909 CrossRef CAS.
- X. H. Liu, H. Li, W. C. Jia, Z. Chen and D. K. Xu, Lab Chip, 2017, 17, 178–185 RSC.
- W. C. Jia, H. Li, T. Wilkop, X. H. Liu, X. D. Yu, Q. Cheng, D. K. Xu and H. Y. Chen, Biosens. Bioelectron., 2018, 109, 206–213 CrossRef CAS.
- Q. Wang, W. Liu, Y. Q. Xing, X. H. Yang, K. M. Wang, R. Jiang, P. Wang and Q. Zhao, Anal. Chem., 2014, 86, 6572–6579 CrossRef CAS.
- Z. Chen, H. Li, W. C. Jia, X. H. Liu, Z. M. Li, F. Wen, N. Zheng, J. D. Jiang and D. K. Xu, Bivalent Aptasensor Based on Silver-Enhanced Fluorescence Polarization for Rapid Detection of Lactoferrin in Milk, Anal. Chem., 2017, 89, 5900–5908 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra09232j |
| This journal is © The Royal Society of Chemistry 2019 |