
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and characterization of MgF2–CoF2 binary fluorides. Influence of the treatment atmosphere and temperature on the structure and surface properties†
Mariusz Pietrowski *,
Michał Zieliński,
Emilia Alwin,
Agata Suchora and
Joanna Gawarecka
*,
Michał Zieliński,
Emilia Alwin,
Agata Suchora and
Joanna Gawarecka
Adam Mickiewicz University in Poznań, Faculty of Chemistry, Umultowska 89B, 61-614 Poznan, Poland. E-mail: mariop@amu.edu.pl
First published on 15th February 2019
Abstract
Research was carried out on the incorporation of divalent cobalt cations into the crystalline structure of MgF2 to form MgxCo1−xF2 binary fluorides, which had not been investigated before. The above fluorides were obtained by the precipitation from aqueous solution of magnesium and cobalt nitrates with ammonium fluoride. Binary fluorides containing 0.6, 7.5 and 37.7 mol% CoF2 were prepared. The effects of treatment temperature (300, 400 °C) and atmosphere (oxidizing or reducing) on the structure (XRD, TPR-H2, UV-Vis), texture (low-temperature N2 adsorption), surface composition (XPS) and surface acidity (NH3-TPD) of the binary fluorides were determined. It has been found that in MgxCo1−xF2 an isomorphic substitution occurs of Mg2+ cations by Co2+ cations which results in the formation of a rutile-type solid solution. The obtained binary fluorides are characterized by a mesoporous structure and relatively large surface area. It has been found that thermal treatment of the binary fluorides in oxidizing conditions results in the oxidation of CoF2 to Co3O4 even at 300 °C; therefore it is not possible to obtain pure MgxCo1−xF2 binary fluorides in the presence of air. The preparation of the latter requires reducing conditions, namely thermal treatment of dry precipitate at 300 °C in an atmosphere of hydrogen. If the treatment is conducted at a higher temperature (400 °C), CoF2 undergoes a partial reduction to metallic cobalt. An XPS study has shown the presence of hydroxyl groups in the investigated samples. However, these are solely surface groups because their presence was not detected by XRD measurements. The binary fluorides obtained by our method are characterized by a very narrow optical energy gap (5.31–3.50 eV), considerably narrower than that recorded for bulk fluorides. Measurements of temperature-programmed desorption of ammonia have shown that the incorporation of cobalt cations into the crystal structure of MgF2 results in a decrease in the surface acidity of the binary fluorides.
1 Introduction
The commercial importance of inorganic fluorides is mainly a result of their application in metallurgy (rare-earth metals, aluminum, hafnium, zirconium), isotope separation and the manufacture of optical materials such as antireflective coatings. However, recently an increase has been observed in the interest in the application of inorganic fluorides in such areas as catalysis,1–7 photonics,8–10 biosensing,11 reversible positive electrodes for rechargeable lithium batteries,12–18 high-temperature superconductor devices8 and supercapacitors.19 To tailor the properties of inorganic fluorides to the above applications, it is necessary to either use new ways of preparation15,16,20–27 or to modify already known materials, among other things by isomorphic substitution of cations in the structure of a host with cations of a guest.28–34 This strategy has been adopted for the modification of catalytic properties of magnesium fluoride which has been applied since many years as a catalyst support.1,2,35–39 It is characterized by quite an inert surface which contains only Lewis acid centers of weak and moderate acid strength as well as weak basic sites.40,41 Kemnitz et al.42 adapted to mixed metal fluorides the Tanabe's model43 explaining the Lewis acidity of the guest-host binary metal oxide systems. According to this model, isomorphic substitution of cations is possible, if sizes of host and guest cations are comparable. In the case when the positive charge of guest cation is greater than that of host cation, Lewis acidity is generated. By admixturing MgF2 with transition metal ions at the oxidation state of +3 (Fe3+, Cr3+, V3+, In3+, Ga3+), relatively strong Lewis acid sites are created.31,32,44–46 On the other hand, a change in the hydroxylation degree of high-surface area MgF2 (HS-MgF2) results in the formation of Brønsted acid centers, besides already mentioned Lewis acid sites, so the bi-acidic surface is obtained.47–49 The aforementioned modifications of MgF2 concerned only the introduction of trivalent cations. No research was carried out on the modification of MgF2 by divalent cations. Hence the idea to incorporate Co2+ cations into the crystal structure of MgF2. Cobalt(II) fluoride crystallizes in the rutile-type structure, as does magnesium fluoride, and ionic radii of both cations are comparable, therefore isomorphic substitution of Co2+ to replace Mg2+ and form a solid solution is perfectly possible.In the past, an interest in the application of CoF2 in practice was very small. Fu et al.50 were the first who applied cobalt(II) fluoride as an anode in lithium ion batteries (LIBs). Not long after that, researchers began studying CoF2 as a potential and promising anode candidate for LIBs.12,13,16–19,51–54
Literature data on binary fluorides with bivalent cobalt are extremely scant. The only available data concern nickel–cobalt binary fluorides.12,19 However, there are no data on magnesium-cobalt binary fluorides. Teng et al.12 obtained NixCo1−xF2 binary fluorides and applied them in LIBs. During the precipitation of binary fluorides from a solution of cobalt and nickel nitrates with ammonium fluoride, they have used oleic acid as a surfactant. XRD analysis showed that Ni–Co binary fluorides have tetragonal structure (as do NiF2 and CoF2 alone) and the unit cell parameters decrease regularly with the increase in nickel content in the binary fluorides. Similar materials were obtained by Ding et al.,19 who used the solvothermal method and their starting compounds were nickel and cobalt acetates and hydrofluoric acid.
The research presented in this paper concerns the preparation of MgxCo1−xF2 binary fluorides with different Co content by a simple precipitation method and their detailed structural (XRD, TPR-H2, UV-Vis), textural (BET, BJH) and surface (XPS, NH3-TPD) characterizations as well as the determination of the effect of thermal treatment in reducing and oxidizing atmospheres on the structure. The replacement of Mg2+ ions by d-electron Co2+ ions should result in a change in electronic properties, thereby in a change of surface properties as well.
2 Experimental
2.1. Preparation of MgxCo1−xF2 binary fluorides
The binary fluorides were prepared by adding an aqueous solution of Mg(NO3)2·6H2O and Co(NO3)2·6H2O to an aqueous solution of NH4F, followed by stirring with a magnetic stirrer for half an hour at room temperature. The formed precipitate was centrifuged and washed twice with distilled water. Then it was dried at 120 °C for 10 hours with temperature ramp of 0.1 °C min−1. The dried samples were subjected to heating for 5 h at 300 and 400 °C (5 °C min−1 ramp) in oxidizing atmosphere (air, static conditions, electric resistance furnace) and reducing atmosphere (99.999% purity hydrogen flow; 50 ml min−1, 5 °C min−1 ramp). The obtained samples with different cobalt content were denoted as MgCoX, where X stands for CoF2 content expressed in mol% (taking the total content of MgF2 + CoF2 as 100%). For example, the sample consisting of 37.7 mol% CoF2 and 62.3 mol% MgF2 is denoted as MgCo37.7. For pristine MgF2 the code is Mg100, and for pristine CoF2 it is Co100. The composition of the samples and their codes are presented in Table 1. The samples heated in air were denoted as Ox, e.g. MgCo37.7-Ox, whereas those reduced with hydrogen were labeled R, e.g. MgCo37.7-R. Temperatures of the thermal treatment were denoted as 3 – for 300 °C and 4 – for 400 °C, e.g. MgCo37.7-Ox3, MgCo37.7-R4.| Sample code | MgF2 [mol%] | CoF2 [mol%] | MgF2 [wt%] | CoF2 [wt%] | Standard deviation [wt%] |
|---|---|---|---|---|---|
| Mg100 | 100.0 | 0.0 | 100.0 | 0.0 | 0.5 |
| MgCo0.6 | 99.4 | 0.6 | 99.1 | 0.9 | 0.6 |
| MgCo7.5 | 82.5 | 7.5 | 88.9 | 11.1 | 0.9 |
| MgCo37.7 | 62.3 | 37.7 | 51.8 | 48.2 | 0.6 |
| Co100 | 0.0 | 100.0 | 0.0 | 100 | 0.5 |
2.2. X-ray diffraction measurements (XRD)
The XRD measurements of the samples were carried out in the 2θ range from 5 to 80° on a powder diffractometer manufactured by PANalytical Ltd., using CuKα1 radiation. The unit cell parameters were calculated using WinPlotr and UnitCellWin software. The crystallite sizes of the samples were calculated by Scherrer's equation on the basis of the strongest 3–6 reflections.2.3. X-ray photoelectron spectroscopy (XPS)
XPS spectra were obtained on a SPECS spectrometer equipped with non-monochromatic Al-Kα source emitting photons of energy 1486.61 eV and a hemispherical analyzer (PHOIBOS 150 MCD NAP) set to pass energies of 60 eV and 20 eV for survey and regions, respectively. The XPS system base pressure was 1 × 10−9 mbar. The internal calibration of the XPS spectra was performed according to C–C binding energy value of 284.6 eV.2.4. Scanning electron microscopy (SEM-EDS)
Scanning electron microscopy analysis was performed using a Quanta FEG 250 (FEI) microscope in high vacuum conditions at the acceleration voltage of 5 kV. The EDS analyses were conducted at beam acceleration voltage of 10 kV using an EDAX Octane SDD detector. To determine elemental compositions of samples, 9–15 EDS measurements were performed for 3–4 regions of each sample. The obtained results were averaged and measurement error was expressed as standard deviation.2.5. UV-Vis spectroscopy
A UV-Vis spectrophotometer (Varian Cary 100) with an integrating sphere attachment for diffuse reflectance measurements was used to determine the optical band gap. The optical absorption was measured in the range of 200–1200 nm. The optical band-gap (Eg) was estimated using Kubelka–Munk function from variation of (αhν)1/2 with photon energy plots.2.6. Temperature-programmed reduction (TPR-H2)
Temperature-programmed reduction with hydrogen (TPR-H2) was carried out by using an ASAP ChemiSorb 2705 instrument (Micromeritics). Catalyst samples of about 50 mg were reduced in a mixture of 10 vol% H2 in Ar (99.999%, Linde) at a flow rate of 30 cm3 min−1 and a heating rate of 10 K min−1. The hydrogen consumption was monitored with a thermal conductivity detector (TCD) and the signal intensity was normalized to 100 mg for each sample studied. The products of the reduction were retained by an isopropanol/liquid nitrogen cold trap at about −70 °C.2.7. BET surface area and porosity measurements
The Brunauer–Emmett–Teller surface areas were determined by nitrogen adsorption at −196 °C using a Micromeritics ASAP 2010 sorptometer. The total pore volumes and average pore diameters were determined by applying the Barrett–Joyner–Halenda (BJH) method to the isotherm desorption branch.2.8. Temperature-programmed desorption of NH3 (NH3-TPD)
The temperature-programmed desorption of NH3 (NH3-TPD) was carried out using an ASAP ChemiSorb 2705 instrument (Micromeritics). Catalyst samples of about 500 mg were activated in situ for 1 h at 300 °C in helium at a flow rate of 30 cm3 min−1. Then they were cooled down to 100 °C and contacted with NH3 for 10 min at a flow rate of 10 cm3 min−1 followed by flushing with helium for 30 min. Measurements of temperature-programmed desorption were conducted in helium flow (30 cm3 min−1) at a heating rate of 10 K min−1. The desorbing ammonia was monitored with a thermal conductivity detector (TCD) and the signal intensity was normalized to 1 g for each sample studied. The calibration was performed by dispensing 1 cm3 NH3 from a calibrated gas loop into the helium flow.3 Results and discussion
3.1. Visual analysis of the samples
The obtained gel-like precipitates of MgxCo1−xF2 binary fluorides were subjected to drying at 120 °C followed by grinding to powder in an agate mortar. Pictures of the samples are shown in Table S1 in ESI.† Pristine magnesium fluoride was white, whereas pristine cobalt(II) fluoride was pink-colored. On the other hand, binary fluorides assumed different hues of pink with the intensity increasing with the rise in CoF2 content.The dry samples were heated at 300 and 400 °C in either reducing (hydrogen) or oxidizing (air) atmosphere. Pristine MgF2 remained white irrespective of the kind of atmosphere and heating temperature. After the thermal treatment at 300 and 400 °C in air, the samples containing 7.5 and 37.7 mol% CoF2 changed their color to almost black which can indicate oxidation to cobalt oxides. On the other hand, after heating at 300 °C in hydrogen, no clear change in color was observed which suggests the preservation of the structure of binary fluorides with no admixture of other phases.
3.2. X-ray powder diffraction measurements – phase composition of binary fluorides
Fig. 1a shows XRD patterns of samples reduced at 300 °C. The location and intensity of reflections originating from Mg100 sample corresponds to the data contained in the PDF database for magnesium fluoride (sellaite, PDF 41-1443). No significant changes were observed in the position of reflections for MgCo0.6-R3 sample. At a greater Co content (the samples MgCo7.5-R3 and MgCo37.7-R3), the reflections became shifted towards smaller 2θ angles which indicates the replacement of Mg2+ ions by Co2+ ions. The shift of reflections towards smaller angles is indicative of an increase in experimental lattice parameters as the cobalt content is increased in the solid solution. The increase in the lattice parameters with increased cobalt content is due to the larger ionic radius of Co2+ compared to that of Mg2+ (0.735 and 0.72 Å, respectively55). Simultaneously, a decrease in intensities of the reflections occurs, which is particularly clear in the case of the MgCo37.7-R3 sample, i.e. it indicates a decline in the sample crystallinity. In Fig. 1b X-ray diffraction patterns are presented of samples reduced at 400 °C. Higher reduction temperature resulted in sharper reflections which became narrower and more intense and this points to a better crystallinity of the obtained samples. However, trace amounts of metallic cobalt (PDF 5-727) have been observed after reduction at 400 °C (it concerns Co100-R4 and MgCo37.7-R4 samples). | ||
| Fig. 1 Powder diffraction patterns of MgxCo1−xF2 binary fluorides reduced for 5 h with pure hydrogen at 300 °C (a) and 400 °C (b). | ||
Pristine MgF2 is oxygen-resistant at high temperatures and no presence of oxide phase (MgO) was detected even after calcination at 500 °C. CoF2, unlike MgF2, undergoes during the calcination a gradual transformation into oxofluorides and then into oxides as proved by XRD studies of samples calcined at 300 and 400 °C (ESI – Fig. S1†). That is why it was impossible to obtain pure-phase bifluorides during the treatment in oxidizing atmosphere.
The unit cell parameters of the samples reduced at 300 and 400 °C were determined using programs WinPlotr and UnitCellWin. According to different sources, the unit cell parameter a of MgF2 ranges from 4.621 to 4.628 Å and c parameter from 3.0159 to 3.0534 Å.56–58 Analogously, for CoF2 a 〈4.6950–4.6956〉 and c 〈3.1774–3.1817〉57,59,60 (Table S2†). Similarly, the c/a ratio changes in the range 0.6526–0.6606 for MgF2 and 0.6767–0.6777 for CoF2. Changes in the composition of investigated samples are accompanied by changes in the unit cell parameters (Fig. 2). It should be mentioned that these changes are not regular. Initially, a slight decrease in a and c parameters occurs with increasing Co content (MgCo0.6 and MgCo7.5 samples), however, only the incorporation of 37.7 mol% Co causes a significant increase of the above parameters. Crystallite sizes estimated on the basis of Scherrer's formula for Mg100-R3, MgCo0.6-R3 and MgCo7.5-R3 samples are of about 10 nm, whereas for MgCo37.7-R3 only 8 nm. No magnesium-containing sample, that is characterized by the narrowest reflection, contains crystallites of about 25 nm in size. The poor crystallinity of the MgCo37.7-R3 sample and large accumulation of defects can be an asset from the viewpoint of catalysis, because defects are often adsorption centers for reaction substrates and during the preparation of catalysts they play the role of adsorption sites for active phase. The aforementioned large amorphicity has also been reflected in high specific surface area of the above sample (see next subsection). Of course, an increase in the thermal treatment temperature leads to a rise in crystallite size.
 | ||
| Fig. 2 Changes in unit cell parameters (a and c) and crystallite sizes d for MgxCo1−xF2 binary fluorides reduced at 300 and 400 °C. | ||
Summing up, XRD measurements point to a possibility of obtaining MgxCo1−xF2 binary fluorides with rutile-type structure. The fact that no separate MgF2 and CoF2 phases have been found in the binary fluoride samples proves the formation of rutile-type structure in which Mg2+ and Co2+ ions are homogeneously dispersed and occupy the same lattice positions. However, it has to be emphasized that the preparation of high purity MgxCo1−xF2 binary fluoride systems requires using oxygen-free atmosphere (preferably reducing one) and relatively low temperature of about 300 °C (Table 2).
| Sample | Reduced | Oxidized | ||
|---|---|---|---|---|
| 300 °C | 400 °C | 300 °C | 400 °C | |
| Mg100 | MgF2 | MgF2 | MgF2 | MgF2 |
| MgCo0.6 | MgF2 | MgF2 | MgF2 | MgF2 |
| MgCo7.5 | MgxCo1−xF2 | MgxCo1−xF2 + Co0 | MgxCo1−xF2 + Co3O4 | MgxCo1−xF2 + Co3O4 |
| MgCo37.7 | MgxCo1−xF2 | MgxCo1−xF2 + Co0 | MgxCo1−xF2 + Co3O4 | MgxCo1−xF2 + Co3O4 |
| Co100 | CoF2 | CoF2 + Co0 | CoF2 + Co3O4 | Co3O4 |
The application of a higher temperature causes the separation of metallic cobalt phase, whereas the application of oxidizing atmosphere leads to the formation of Co3O4.
3.3. Temperature-programmed reduction (TPR-H2)
The measurements of temperature-programmed reduction with hydrogen (TPR-H2) provide information about reducibility of chemical compounds present in a sample and about their interactions, thus enabling to draw conclusions on the phase composition of a sample. The TPR-H2 measurements can confirm the presence of solid solution in the bifluoride samples. However, this should be preceded by the determination of reduction temperatures of cobalt ions present in the form of oxide and fluoride. To this end, TPR-H2 measurements of reduced and oxidized samples were carried out.According to literature data,61–63 the reduction of Co3O4 proceeds in two stages:
| Co3O4 → CoO | (1) |
| CoO → Co0 | (2) |
Fig. 3 shows TPR-H2 profiles of Co100 and MgCo37.7 samples heated in air at 300 and 400 °C. The two-stage reduction finds reflection in the shape of TPR-H2 profile of the Co100-Ox4 sample as two peaks at 355 and 398 °C.61 The shape of TPR-H2 curve of Co3O4 is very distinctive and was observed many times,61,63–66 including the Co3O4/MgF2 system.67 Therefore, the small signal at 457 °C should be ascribed to the reduction of CoF2. Similar signals are also visible in the TPR-H2 profile of the Co100-Ox3 sample subjected to oxidation at 300 °C. The presence of a small amount of cobalt oxide is indicated by the signal in the temperature range of 300–440 °C. Above this temperature, the reduction of CoF2 occurs. In Fig. 3b presented are TPR-H2 profiles of the MgCo37.7-Ox sample after its oxidation at 300 and 400 °C. As it was in the case of pristine CoF2 sample, the calcination at 400 °C results in the appearance of signals originating from the reduction of cobalt oxide (300–440 °C) and the reduction of CoF2 (550–570 °C). However, worth noting is the fact that the reduction of CoF2 in the binary fluorides proceeds at a considerably higher temperature than in the case of the Co100 sample. We believe that this is a result of a stabilization of CoF2 in the matrix formed by MgF2.
 | ||
| Fig. 3 TPR-H2 profiles of the samples Co100-Ox (a) and MgCo37.7-Ox (b), calcined at 300 and 400 °C (signal intensity was normalized to 100 mg). | ||
The TPR-H2 profiles of the MgCo7.5-Ox sample (not shown) have also indicated the presence of analogous signals, however, their intensity was several times smaller. No signals were observed in the case of Mg100-Ox and MgCo0.6-Ox samples, which is not surprising because magnesium fluoride is highly resistant to reduction.
The method of synthesis applied by us consists in adding magnesium and cobalt nitrates to a solution of ammonium fluoride which yields ammonium nitrate and a precipitate of binary fluorides. The obtained precipitate was thoroughly washed to remove NH4NO3 and TPR measurements show that the removal was complete. According to literature data,68–70 the thermal decomposition of ammonium nitrate proceeds in the range of 200–300 °C. In the TPR profiles of our binary fluorides no signals associated with the decomposition of NH4NO3 were detected, which proves high purity of the obtained materials.
To observe the reduction of CoF2 we have conducted TPR-H2 measurements of Co100, MgCo37.7 and MgCo7.5 samples heated in hydrogen at 300 °C (Fig. 4). Pristine cobalt fluoride (Co100-R3) undergoes reduction at 455 °C, whereas CoF2 dispersed in the MgF2 matrix (MgCo37.7 and MgCo7.5) shows considerably higher thermal stability – the peak maximum occurs at a significantly higher temperature (532 °C). Moreover, the shape of the profiles is different – in the case of Co100-R3 it is narrower and the reduction is completed around 500 °C, whereas in that of MgCo37.7-R3 the profile is very broad and the reduction lasts to almost 750 °C. We believe that such a large shift in the reduction temperature of CoF2 is a result of the stabilization of Co2+ ions in the binary fluoride structure due to the neighborhood with Mg2+ ions, which increases the resistance to reduction.
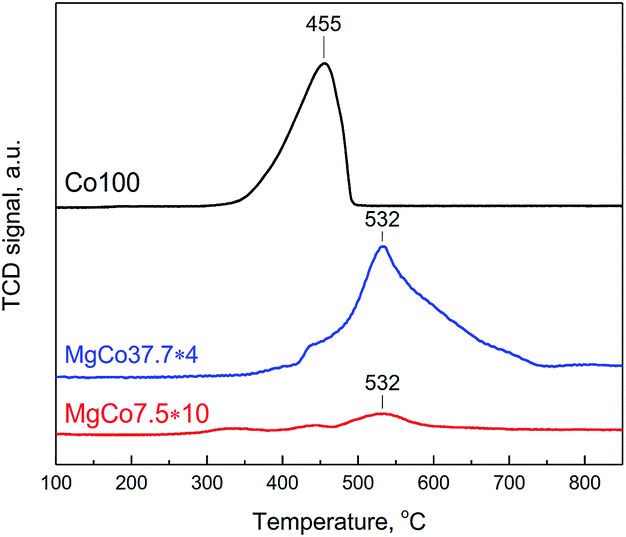 | ||
| Fig. 4 TPR-H2 profiles of Co100-R3, MgCo37.7-R3 and MgCo7.5-R3 samples (signal intensity was normalized to 100 mg and signal intensities for MgCo37.7-R3 and MgCo7.5-R3 were multiplied by 4, and 10, respectively). | ||
3.4. Textural analysis of the samples
Results presented so far clearly show that obtaining monophasic binary fluorides is possible only by thermal treatment in reducing conditions at a temperature not exceeding 300 °C. In oxidizing conditions, cobalt oxide phase is formed in addition to fluoride phase, whereas reduction at 400 °C results in the formation of metallic cobalt phase. Hence textural studies were performed for the samples reduced at 300 °C.Specific surface area and porous structure of the samples were determined from low-temperature nitrogen adsorption/desorption isotherms. Results of the measurements are presented in Table 3 and Fig. 5.
| Sample | Textural parameter | ||
|---|---|---|---|
| BET surface area, m2 g−1 | Average pore diameter, nm | Cumulative pore volume, cm3 g−1 | |
| Mg100 | 36.5(1) | 8.0 | 0.132 |
| MgCo0.6 | 40.9(1) | 5.9 | 0.116 |
| MgCo7.5 | 42.7(1) | 6.6 | 0.117 |
| MgCo37.7 | 49.0(2) | 3.9 | 0.096 |
| Co100 | 16.0(1) | 27.2 | 0.095 |
 | ||
| Fig. 5 Nitrogen physisorption isotherms (a) and pore volume distribution as a function of pore diameter (b) for the samples heated in hydrogen at 300 °C. The curves are shifted along Y axis for klarity. | ||
Specific surface area of reduced samples ranges from 16.0 to 49.0 m2 g−1. With increasing cobalt content in the samples of binary fluorides, a regular rise in surface areas was observed. The highest surface area (49.0 m2 g−1) was found for the MgCo37.7 sample. Simultaneously, a decrease in average pore diameter from 8.0 to 3.9 nm and in cumulative pore volume from 0.132 to 0.095 cm3 g−1 was observed (Table 3, Fig. 5). Only the sample Co100 has a considerably smaller surface area (16.0 m2 g−1) which is a result of the presence of much greater pores (27.2 nm).
All isotherms recorded for the studied samples belong to the type IV(a) according to the IUPAC classification,71 i.e. the type that is characteristic of mesoporous materials. The isotherms measured for the samples Mg100, MgCo0.6 and MgCo7.5, are similar one to another – their hysteresis loops start at relative pressures p/po of about 0.4 and belong to the type H2(b). Such a shape of hysteresis loop is characteristic of mesoporous adsorbents containing ink-bottle shaped pores with varied neck sizes. Somewhat different shape of hysteresis loop occurs in the case of the MgCo37.7 sample. It is characterized by a steep desorption branch, which is a characteristic feature of H2(a) loops, and can be attributed to either pore-blocking/percolation in a narrow range of pore necks or to cavitation-induced evaporation. The average pore size calculated by the BJH method on the basis of the isotherm desorption branch is 3.9 nm. However, this result (and also the pore size distribution curve – Fig. 5b) should be considered as an approximation only and not an accurate assessment, because in the case of the MgCo37.7 sample we most likely deal with cavitation-controlled evaporation, and in such a situation no quantitative information about the neck size and neck size distribution can be obtained.71 The Co100 sample, i.e. pristine cobalt fluoride heated in hydrogen at 300 °C, has considerably smaller surface area (16.0 m2 g−1) compared to the binary fluorides. The nitrogen physisorption isotherm remains of type IV(a), however, the hysteresis loop of type H1 indicates the presence of wide mesopores.
3.5. Band gap estimation
For the samples reduced at 300 °C, for which the presence was confirmed of monophasic binary fluorides MgxCo1−xF2, the optical band-gap energy (Table 4) was determined by the Kubelka–Munk method on the basis of UV-Vis reflectance spectra. The value obtained for pristine MgF2 was 5.31 eV. It was decreasing with increasing cobalt content, to reach 3.50 eV for pristine CoF2. The value of 5.31 eV obtained for pristine MgF2 seems to be significantly underestimated when confronted with literature data, according to which it is ∼10.8 eV (ref. 72) for bulk MgF2. However, for MgF2 in the form of thin films, the value of energy gap is 3.8 eV,73 whereas theoretical calculations give the value of 5.95 eV (ref. 72) or 6.83 eV.74 In the light of the above data the obtained by us value of 5.31 eV may be real. It is worth mentioning that MgF2 obtained in our study can hardly be called “bulk”, because XRD and porosity measurements showed that it is a mesoporous fine-crystalline material with crystallites as small as 10 nm.| Sample | Band gap, eV |
|---|---|
| Mg100 | 5.31 |
| MgCo0.6 | 5.40 |
| MgCo7.5 | 5.22 |
| MgCo37.7 | 4.85 |
| Co100 | 3.50 |
3.6. XPS spectroscopy
Information obtained from XRD and TPR-H2 measurements concerned bulk structure of binary fluorides. They showed that MgxCo1−xF2 binary fluorides have rutile-type structure and that high-temperature treatment (400 °C) in reducing conditions results in a partial reduction of cobalt cations to metallic cobalt, whereas in oxidizing conditions to the formation of some amount of Co3O4. These results should find confirmation by XPS measurements, albeit the latter provide information on surface elemental composition, because the depth of XPS analysis is of a few nanometers.The survey XPS spectra of MgF2, CoF2 and MgxCo1−xF2 binary fluorides are presented in Fig. S2.† In addition to the lines of main elements: fluorine, magnesium and cobalt, also carbon and oxygen were detected in the measurements.
Fig. 6a shows high-resolution XPS spectra of Mg 2p for MgxCo1−xF2 binary fluorides and pristine MgF2. For all the samples a single broad signal was detected in the range of 50.4–51.2 eV, which most likely can be ascribed to Mg2+ in MgF2. In the case of pristine MgF2, the signal occurs at 51.2 eV. The addition of cobalt results in a decrease of binding energy to 50.7–51.0 eV for MgCo37.7-R4 and both oxidized samples (MgCo37.7-Ox3 and MgCo37.7-Ox4), whereas in the case of the sample MgCo37.7-R4 to 50.4 eV. Various studies75–79 have shown that the position of the Mg 2p line provides no possibility to discriminate between magnesium in the form of oxide, hydroxide or fluoride. In our samples the fluoride is obviously the most likely form, however, it does not rule out a possibility of a coexistence of magnesium in the form of hydroxide or oxide. Fig. 6b shows XPS spectra of O 1s. A signal at binding energy (BE) of 531.0–532.5 eV has been observed for the samples heated at 300 °C (both binary fluorides and pristine MgF2). The signal can be ascribed neither to MgO for which O 1s binding energy is about 531 eV nor to Mg(OH)2 for which BE is 533.2 eV.75,76,78,80–82 A more likely interpretation can be made by analyzing the difference in O 1s – Mg 2p chemical shift. In the case of MgO the difference is by about 1.3 eV smaller than in that of Mg(OH)2. In Table 5, listed are chemical shifts and differences in the shifts (ΔE) for the studied binary fluorides, MgF2 and CoF2.
 | ||
| Fig. 6 XPS spectra of Mg 2p (a), O 1s (b), F 1s (c), Co 2p (d) photoemission line for MgxCo1−xF2 binary fluorides. | ||
| Sample | Binding energy (eV) | ΔE (eV) | |||||
|---|---|---|---|---|---|---|---|
| Mg 2p | F 1s | O 1s | O 1s – Mg 2p | Assignment | F 1s – Mg 2p | Assignment | |
| Mg100-R3 | 51.2 | 685.8 | 532.5 | 481.3 | Mg–OH | 634.6 | Mg(OH)2−x Fx |
| MgCo37.7-R3 | 50.4 | 684.9 | 531.4 | 481.0 | Mg–OH | 634.5 | Mg(OH)2−x Fx |
| MgCo37.7-R4 | 50.8 | 685.5 | 531.0 | 480.2 | Mg–O | 634.7 | Mg(OH)2−x Fx |
| MgCo37.7-Ox3 | 50.7 | 685.3 | 532.2 | 481.5 | Mg–OH | 634.6 | Mg(OH)2−x Fx |
| 529.1 | |||||||
| MgCo37.7-Ox4 | 51.0 | 685.6 | 531.5 | 480.5 | Mg–O | 634.6 | |
| 528.8 | |||||||
| Co100-R3 | — | 684.6 | 531.4 | — | — | — | Mg(OH)2−x Fx |
| 684.0 | |||||||
The analysis indicates the presence of hydroxyl groups on surfaces of binary fluorides and pristine MgF2 heated at 300 °C (irrespective of treatment atmosphere) which is proved by ΔE O 1s – Mg 2p values of 481.0–481.5 eV (ref. 79 and 83) and by many earlier reports on hydroxylated MgF2.1,35,36,40,47–49,84–87 Verdier et al.79,83 ascribed them to the Mg(OH)2−xFx species. The presence of surface complexes of such a type is indicated by the values of ΔE F 1s – Mg 2p that are equal to 634.5–634.7 eV, which in the case of crystalline MgF2 are about 633 eV and in that of Mg(OH)2−xFx they range between 634.5 and 635.0 eV.79 After reduction at 400 °C, the O 1s peak undergoes a shift towards lower energies (531.0 eV), which results in a decrease in the O 1s – Mg 2p difference to 480.2 eV and the latter value is characteristic of MgO.79,83 A similar situation was observed when oxidation was carried out at 400 °C. The appearance of the O 1s peak in the position corresponding to MgO results from the fact that the MgF2 surface undergoes dehydroxylation during heating at higher temperatures, according to the equation OH− + OH− → H2O + O2−. The above reaction leads to the gradual decay of hydroxyl groups and the appearance of O2− anions in place of them. The binary fluorides subjected to heating in the oxidizing atmosphere react in a similar way. After oxidation at 300 °C, the O 1s signal (Fig. 6b) appears at BE of 532.2 eV, which points to the presence of surface complexes of Mg(OH)2−xFx type, and after oxidation at 400 °C it undergoes a shift to 531.5 eV, i.e. the position characteristic of MgO.88 Moreover, a new signal around 529 eV appears in the spectra of calcined samples that can be ascribed to Co3O4.16,89–93 In the case of O 1s photoelectrons of pristine CoF2 subjected to reduction at 300 °C (Co100-R3 – Fig. 6b), the peak occurs at 531.4 eV. This signal cannot be attributed to cobalt oxide because in the case of the latter the discussed signal occurs around 529–530 eV.91,92 In this situation it would be justified to assume the interpretation analogous to that for MgF2, i.e. to ascribe the O 1s signal at 531.4 eV to the complex of Co(OH)2−xFx type.16 This means that the surface of CoF2, similarly to that of MgF2, when exposed to water vapor-containing environment, adsorbs water molecules that form surface OH groups. The XPS spectrum for F 1s is presented in Fig. 6c. For pristine MgF2 and all binary fluorides one broad peak with maximum in the range 685.8–684.9 eV has been observed, irrespective of the composition of binary fluorides, treatment temperature and atmosphere. The mentioned signal originates from fluorine bound in the complex of Mg(OH)2−xFx type. This is indicated both by the BE value itself and the difference ΔE F 1s – Mg 2p (Table 5). The binding energy difference from the fluorine core level and magnesium (ΔE F 1s – Mg 2p) is ∼634.6 eV. As already mentioned, according to Verdier et al.,79 this difference for MgF2 should be 632.9 eV.
The value observed in the case of our samples is larger and can be ascribed to Mg(OH)2−xFx – type magnesium hydroxyfluoride.94
Similarly, for pristine CoF2 a signal at 684.6 eV was observed, which by analogy to MgF2 can be attributed to complexes of Co(OH)2−xFx type. The Co 2p core level spectrum is characterized by two components appearing due to spin-orbital splitting Co 2p3/2 and Co 2p1/2, and shake-up satellites (Fig. 6d and 7). The high spin Co2+ compounds such as CoO, Co(OH)2 and CoF2 exhibit strong satellite lines which are located at about 5–6 eV above the photo line.54,89,95,96 The main Co 2p3/2 peak for Co2+ in compounds such as CoO and Co(OH)2 usually occurs around 780–781 eV.63,91,95 In the case of CoF2, it is shifted towards higher energies and occurs at about 783 eV as reported in ref. 17 and 54. All the recorded spectra have such a complex structure. The Co 2p3/2 feature around 783 eV and the intense shake-up satellite peak at 788 eV are visible in the obtained XPS spectra and originate from Co2+ in CoF2.17,54,96 Spin-orbital splitting between the Co 2p3/2 and Co 2p1/2 is at about 16 eV (Table 6) and is characteristic of Co2+.95,97 An additional signal at 778.5 eV appeared in the spectrum of the sample reduced at 400 °C. This feature can be unambiguously assigned to metallic cobalt.89 Such a conclusion is in agreement with results of XRD measurements which showed the presence of metallic cobalt in the MgCo37.7-R4 sample.
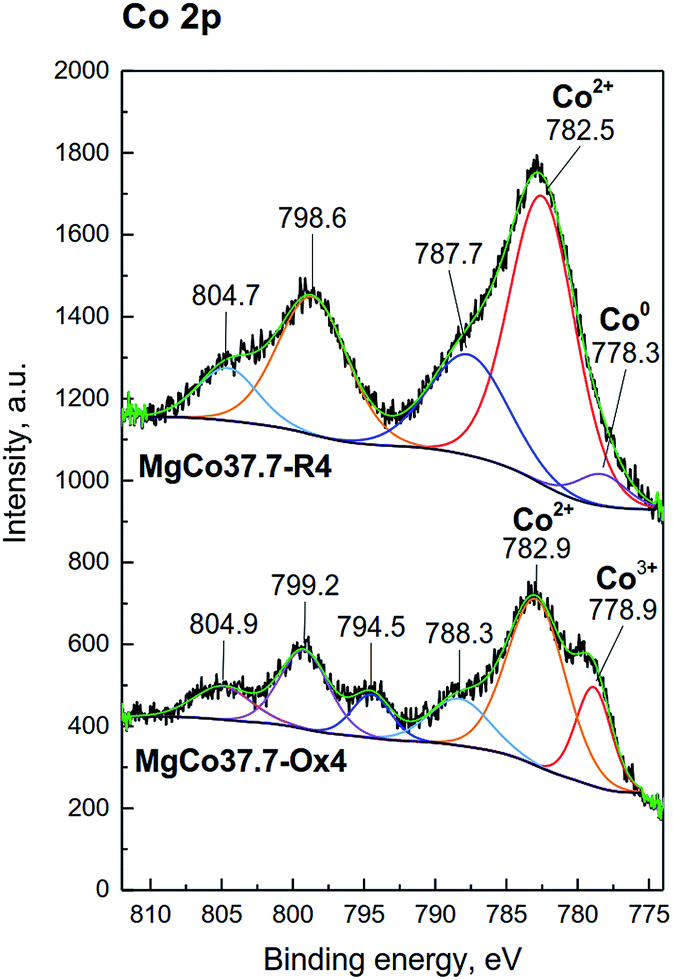 | ||
| Fig. 7 XPS spectra of the Co 2p photoemission line for MgCo37.7 samples reduced and oxidized at 400 °C. | ||
| Sample | Binding Energy (eV) | |||||||
|---|---|---|---|---|---|---|---|---|
| Co 2p3/2 | Satellite | Co 2p1/2 | Satellite | Orbital splitting 2p1/2–2p3/2 | ΔE Satellite-2p3/2 | |||
| Co100-R3 | — | 782.3 | 786.9 | — | 798.5 | 804.1 | 16.2 | 4.6 |
| MgCo37.7-R3 | — | 782.5 | 787.1 | — | 798.6 | 804.5 | 15.9 | 4.6 |
| MgCo37.7-R4 | 778.3 | 782.5 | 787.7 | — | 798.6 | 804.6 | 16.1 | 5.2 |
| MgCo37.7-Ox3 | 778.6 | 782.8 | 787.9 | 794.4 | 799.1 | 804.9 | 16.3 | 5.1 |
| MgCo37.7-Ox4 | 778.9 | 782.9 | 788.3 | 794.5 | 799.2 | 804.9 | 16.3 | 5.4 |
In the spectra of oxidized samples, the Co3+ 2p3/2 (at about 778.9 eV) and Co 2p1/2 (at about 794.4 eV) signals, attributed to Co3+ ions,63 are visible at lower BE values. In contradistinction to the high spin Co2+ compounds, the low spin Co3+ compounds, such as Co3O4, are characterized by a very weak satellite.91,95 In the case of our samples, due to the presence of intense signals from Co2+ in CoF2, satellite bands from Co3+ are practically invisible. The results of the XPS study are fully consistent with the results of XRD and TPR-H2 measurements which pointed to the presence of the Co3O4 phase in the calcined samples.
The XPS results prove that on surfaces of the binary fluorides (as well as Mg and Co fluorides alone) present are hydroxyl groups as indicated by the positions of F 1s and O 1s signals. The question arises why did not the presence of OH groups affect the crystalline structure of the binary fluorides? It can be a result of the fact that the effective ionic radii of F− and OH− do not differ much – the OH− ion is only slightly greater than F− ion (by about 0.3 nm (ref. 98)), hence any changes in the unit cell parameters caused by the replacement of F− by OH− would be slight. However, they should be noticeable because the introduction of OH− groups into the MgF2 structure results in a small increase in the lattice constant a and a decrease in the c constant, as proved by Booster et al.84 We have not observed such changes, hence our conclusion is that the proposed Mg(OH)2−xFx groups are the only surface complexes. A conclusion that similar complexes are present on the surface of CoF2 seems to be interesting. The XPS measurements have shown that in the case of CoF2 we deal with the phenomenon analogous to that of MgF2, i.e. the surface fluoride ions are replaced by hydroxyl groups. Such a phenomenon was not reported yet.
3.7. Surface acidity characterization by NH3-TPD measurements
The studies carried out by Kemnitz et al.31,32,42,44,46 have shown that the substitution of divalent cation with trivalent cation in the structure of MgF2 results in an increase in Lewis acidity of binary fluorides, as predicted by the Tanabe's model.43 The aforementioned model does not predict a rise in the acidity as a result of the substitution of cations with the same valence, hence in the case of the incorporation of Co2+ cation into the MgF2 lattice, no increase in the acidity should be expected. However, such an incorporation will modify electronic properties of the binary fluoride, because the presence of d orbitals in cobalt atom should affect charge distribution in the lattice of the binary fluoride, which play the role of Lewis acid centers. A certain diffusion of the charge, particularly as concerns the positive charge localized on cations, can be expected which will result in a reduction in Lewis acidity of binary systems.The above hypothesis was verified by determining the concentration of acid sites on the surface of binary fluorides using the NH3-TPD technique (Fig. 8). The measurements were conducted for the samples reduced at 300 °C. The shape of NH3-TPD profiles indicates that the acid centers differ in their strength which is clearly visible as a distinct inflection in the TPD curves. The concentration of acid centers on surfaces of the Mg100-R3 and MgCo0.6-R3 samples was almost the same. Hence, one can conclude that the incorporation of 0.6 mol% CoF2 into the structure of MgF2 has a negligible effect on the acid properties. A distinct change appears only for MgCo7.5-R3 and MgCo37.7-R3 samples, where the concentration of acid centers falls from the initial ∼0.3 mmol g−1 to 0.21 and 0.18 mmol g−1, respectively. The lowest acidity (0.07 mmol g−1) was found for pristine CoF2.
 | ||
| Fig. 8 NH3-TPD profiles of MgxCo1−xF2 binary fluorides after reduction at 300 °C. | ||
Summing up, the NH3-TPD measurements have shown that the substitution of Mg2+ ions with Co2+ ions in the structure of MgF2 leads to a decrease in the surface acidity which is probably caused by the presence of d orbitals in cobalt atoms and diffusion of the positive charge localized on surface cations being Lewis acid sites.
4 Conclusions
New binary fluoride systems consisting of magnesium fluoride doped with cobalt fluoride were synthesized and characterized in detail. The MgxCo1−xF2 binary fluorides with cobalt content from 0.6 to 37.7 mol% (1–50 wt%) were obtained for the first time by applying a simple method consisting in the precipitation from aqueous solutions of magnesium and cobalt nitrates with ammonium fluoride solution. All the obtained materials have tetragonal rutile-type structure (P42/mnm) in which the cationic positions of magnesium were isomorphically substituted by cobalt ions. The unit cell parameters (a and c) determined on the basis of XRD measurements increase with increasing cobalt content in the MgxCo1−xF2 binary fluorides and assume intermediate values between those characteristic of MgF2 and CoF2. As the CoF2 content in the binary fluorides rises, the degree of disorder of the structure increases which results in an increase in the porosity and surface area. The sample containing 37.7 mol% of CoF2, subjected to the reduction at 300 °C, was characterized by the largest surface area. It has been established that the preparation of pure phase of MgxCo1−xF2 binary fluorides requires reducing atmosphere during the thermal treatment, the temperature of which should not exceed 300 °C. At higher temperatures the separation of metallic cobalt phase occurs and in the case of oxidizing conditions, Co3O4 is formed. The presence of cobalt oxides in the calcined samples and the formation of metallic cobalt in the samples reduced at 400 °C has been confirmed by three independent techniques: XRD, TPR-H2 and XPS. An interesting fact is the detection of hydroxyl groups not only on the surfaces of pristine MgF2 and binary fluorides, but also on the surface of CoF2. High intensity of the O 1s peak in the XPS spectrum of pristine CoF2 indicates that the concentration of hydroxyl groups on its surface is even greater than on the surface of MgF2. Surface complexes of magnesium and cobalt hydroxyfluorides can be described by the following molecular formulae: Mg(OH)2−xFx and Co(OH)2−xFx. The UV-Vis measurements have shown that both binary fluorides and pristine fluorides (MgF2 and CoF2) are characterized by narrow optical energy gap (5.31–3.50 eV), considerably narrower than those recorded for bulk fluorides. The NH3-TPD measurements have shown that the substitution of Mg2+ ions with d-electron Co2+ ions results in a decrease in Lewis acidity of the binary fluorides.The binary fluorides, obtained and characterized in this work, can find application to many areas, among other things as catalysts and catalyst supports or as components of lithium-ion batteries.
Conflicts of interest
There are no conflicts of interest to declare.References
- M. Wojciechowska, M. Zielinski and M. Pietrowski, J. Fluorine Chem., 2003, 120, 1–11 CrossRef CAS
.
- E. Kemnitz, Catal. Sci. Technol., 2015, 5, 786–806 RSC
- M. Pietrowski, M. Zielinski and M. Wojciechowska, Chemcatchem, 2011, 3, 835–838 CrossRef CAS
- M. Pietrowski, Green Chem., 2011, 13, 1633–1635 RSC
- M. Zielinski, M. Pietrowski, A. Kiderys, M. Kot and E. Alwin, J. Fluorine Chem., 2017, 195, 18–25 CrossRef CAS
- M. Zielinski, A. Kiderys, M. Pietrowski, I. Tomska-Foralewska and M. Wojciechowska, Catal. Commun., 2016, 76, 54–57 CrossRef CAS
- M. Zielinski, M. Pietrowski and M. Wojciechowska, Chemcatchem, 2011, 3, 1653–1658 CrossRef CAS
- A. Tressaud and K. R. Poeppelmeier, Photonic and electronic properties of fluoride materials: progress in fluorine science series, Elsevier, 2016 Search PubMed
- S. Fujihara and K. Tokumo, J. Fluorine Chem., 2009, 130, 1106–1110 CrossRef CAS
- S. Sakka, Handbook of Sol-Gel Science and Technology Processing, Characterization and Applications, Kluwer Academic Publishers, Boston, Dordrecht, London, 2005 Search PubMed
- P. P. Fedorov, A. A. Luginina, S. V. Kuznetsov and V. V. Osiko, J. Fluorine Chem., 2011, 132, 1012–1039 CrossRef CAS
- Y. T. Teng, F. X. Wei and R. Yazami, J. Alloys Compd., 2015, 653, 434–443 CrossRef CAS
- Y. T. Teng, S. S. Pramana, J. F. Ding, T. Wu and R. Yazami, Electrochim. Acta, 2013, 107, 301–312 CrossRef CAS
- Y. Han, H. Li, J. Li, H. Si, W. Zhu and X. Qiu, ACS Appl. Mater. Interfaces, 2016, 8, 32869–32874 CrossRef CAS PubMed
- Y. Lu, Z. Y. Wen, J. Jin, K. Rui and X. W. Wu, Phys. Chem. Chem. Phys., 2014, 16, 8556–8562 RSC
- H. Groult, S. Neveu, S. Leclerc, A. G. Porras-Gutierrez, C. M. Julien, A. Tressaud, E. Durand, N. Penin and C. Labrugere, J. Fluorine Chem., 2017, 196, 117–127 CrossRef CAS
- J. L. Tan, L. Liu, S. P. Guo, H. Hu, Z. C. Yan, Q. Zhou, Z. F. Huang, H. B. Shu, X. K. Yang and X. Y. Wang, Electrochim. Acta, 2015, 168, 225–233 CrossRef CAS
- X. R. Wang, W. T. Gu, J. T. Lee, N. Nitta, J. Benson, A. Magasinski, M. W. Schauer and G. Yushin, Small, 2015, 11, 5164–5173 CrossRef CAS PubMed
- X. D. Li, R. Ding, W. Shi, Q. L. Xu and E. H. Liu, Chem.–Eur. J., 2017, 23, 6896–6904 CrossRef CAS PubMed
- D. S. Jacob, L. Bitton, J. Grinblat, I. Felner, Y. Koltypin and A. Gedanken, Chem. Mater., 2006, 18, 3162–3168 CrossRef CAS
- A. Saberi, Z. Negahdari, S. Bouazza and M. Willert-Porada, J. Fluorine Chem., 2010, 131, 1353–1355 CrossRef CAS
- G. Scholz, and E. Kemnitz, in Modern Synthesis Processes and Reactivity of Fluorinated Compounds: Progress in Fluorine Science, ed. H. Groult, F. Leroux, and A. Tressaud, Elsevier, 2017, pp. 609–649 Search PubMed
- E. Kemnitz and J. Noack, Dalton Trans., 2015, 44, 19411–19431 RSC
- T. Murata, H. Ishizawa, I. Motoyama and A. Tanaka, Appl. Opt., 2006, 45, 1465–1468 CrossRef CAS PubMed
- A. B. D. Nandiyanto, T. Ogi and K. Okuyama, ACS Appl. Mater. Interfaces, 2014, 6, 4418–4427 CrossRef CAS PubMed
- A. B. D. Nandiyanto, F. Iskandar, T. Ogi and K. Okuyama, Langmuir, 2010, 26, 12260–12266 CrossRef CAS PubMed
- Z. H. Li, Z. Jia, Y. X. Luan and T. C. Mu, Curr. Opin. Solid State Mater. Sci., 2008, 12, 1–8 CrossRef CAS
- T. Skapin, G. Tavcar, A. Bencan and Z. Mazej, J. Fluorine Chem., 2009, 130, 1086–1092 CrossRef CAS
- B. Ruprecht, M. Wilkening, S. Steuernagel and P. Heitjans, J. Mater. Chem., 2008, 18, 5412–5416 RSC
- S. Rüdiger, U. Gross and E. Kemnitz, J. Fluorine Chem., 2007, 128, 353–368 CrossRef
- J. K. Murthy, U. Gross, S. Rüdiger, E. Unveren and E. Kemnitz, J. Fluorine Chem., 2004, 125, 937–949 CrossRef CAS
- J. K. Murthy, U. Gross, S. Rüdiger and E. Kemnitz, Appl. Catal., A, 2004, 278, 133–138 CrossRef CAS
- S. N. Achary, S. J. Patwe and A. K. Tyagi, Mater. Res. Bull., 2000, 35, 1167–1169 CrossRef CAS
- M. Cao, Y. Wang, Y. Qi, C. Guo and C. Hu, J. Solid State Chem., 2004, 177, 2205–2209 CrossRef CAS
- J. Haber and M. Wojciechowska, J. Catal., 1988, 110, 23–36 CrossRef CAS
- M. Wojciechowska, B. Czajka, M. Pietrowski and M. Zielinski, Catal. Lett., 2000, 66, 147–153 CrossRef CAS
- E. Kemnitz, U. Gross, S. Rüdiger and C. S. Shekar, Angew. Chem., Int. Ed., 2003, 42, 4251–4254 CrossRef CAS PubMed
- E. Kemnitz, G. Scholz and S. Rüdiger, in Functionalized Inorganic Fluorides: Synthesis, Characterization & Properties of Nanostructured Solids, ed. A. Tressaud, John Wiley & Sons, 2010, pp. 1–38 Search PubMed
- S. Abbas, Y. Huang, J. Lin, A. Abbas, X. W. Xu, J. Li, S. Wang, X. Jin and C. C. Tang, RSC Adv., 2016, 6, 29818–29822 RSC
- S. Wutke, A. Vimont, J. C. Lavalley, M. Daturi and E. Kemnitz, J. Phys. Chem. C, 2010, 114, 5113–5120 CrossRef
- E. Kemnitz, S. Wuttke and S. M. Coman, Eur. J. Inorg. Chem., 2011, 31, 4773–4794 CrossRef
- E. Kemnitz, Y. Zhu and B. Adamczyk, J. Fluorine Chem., 2002, 114, 163–170 CrossRef CAS
- K. Tanabe, T. Sumiyoshi, K. Shibata, T. Kiyoura and J. Kitagawa, Bull. Chem. Soc. Jpn., 1974, 47, 1064–1066 CrossRef CAS
- B. Adamczyk, A. Hess and E. Kemnitz, J. Mater. Chem., 1996, 6, 1731–1735 RSC
- H. Lee, H. S. Kim, H. Kim, W. S. Jeong and I. Seo, J. Mol. Catal. A: Chem., 1998, 136, 85–89 CrossRef CAS
- J. Krishna Murthy, U. Gross, S. Rüdiger, E. Unveren, W. Ünger and E. Kemnitz, Appl. Catal., A, 2005, 282, 85–91 CrossRef CAS
- H. A. Prescott, Z. J. Li, E. Kemnitz, J. Deutsch and H. Lieske, J. Mater. Chem., 2005, 15, 4616–4628 RSC
- S. Wuttke, S. M. Coman, G. Scholz, H. Kirmse, A. Vimont, M. Daturi, S. L. M. Schroeder and E. Kemnitz, Chem.–Eur. J., 2008, 14, 11488–11499 CrossRef CAS PubMed
- S. Celerier and F. Richard, Catal. Commun., 2015, 67, 26–30 CrossRef CAS
- Z. W. Fu, C. L. Li, W. Y. Liu, J. Ma, Y. Wang and Q. Z. Qin, J. Electrochem. Soc., 2005, 152, E50–E55 CrossRef CAS
- M. J. Armstrong, A. Panneerselvam, C. O'Regan, M. A. Morris and J. D. Holmes, J. Mater. Chem., 2013, 1, 10667–10676 RSC
- S. B. Ni, J. J. Ma, J. C. Zhang, X. L. Yang and L. L. Zhang, Mater. Lett., 2015, 139, 138–140 CrossRef CAS
- Z. Y. Zhang, W. Y. Li, R. J. Zou, W. P. Kang, Y. S. Chui, M. F. Yuen, C. S. Lee and W. J. Zhang, J. Mater. Chem. A, 2015, 3, 6990–6997 RSC
- R. A. Quinlan, Y. C. Lu, D. Kwabi, Y. Shao-Horn and A. N. Mansour, J. Electrochem. Soc., 2016, 163, A300–A308 CrossRef CAS
- R. D. Shannon and C. T. Prewitt, Acta Crystallogr., Sect. B: Struct. Sci., 1969, B 25, 925–946 CrossRef
- K. V. K. Rao, S. V. N. Naidu and P. L. N. Setty, Acta Crystallogr., 1962, 15, 528–530 CrossRef CAS
- W. H. Baur and A. A. Khan, Acta Crystallogr., Sect. B: Struct. Sci., 1971, B 27, 2133–2139 CrossRef
- J. P. Vidal, G. Vidal-Valat, M. Galtier and K. Kurkisuonio, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1981, 37, 826–837 CrossRef
- J. W. Stout and S. A. Reed, J. Am. Chem. Soc., 1954, 76, 5279–5281 CrossRef CAS
- M. M. R. Costa, J. A. Paixao, M. J. M. Dealmeida and L. C. R. Andrade, Acta Crystallogr., Sect. B: Struct. Sci., 1993, 49, 591–599 CrossRef
- H. F. Gong, J. J. Zhu, K. L. Lv, P. Xiao and Y. X. Zhao, New J. Chem., 2015, 39, 9380–9388 RSC
- Y. H. Zhang, H. F. Xiong, K. Y. Liew and J. L. Li, J. Mol. Catal. A: Chem., 2005, 237, 172–181 CrossRef CAS
- L. F. Liotta, G. Di Carlo, G. Pantaleo, A. M. Venezia and G. Deganello, Appl. Catal., B, 2006, 66, 217–227 CrossRef CAS
- C. W. Tang, C. B. Wang and S. H. Chien, Thermochim. Acta, 2008, 473, 68–73 CrossRef CAS
- J. Li, G. Z. Lu, G. S. Wu, D. S. Mao, Y. L. Guo, Y. Q. Wanga and G. A. Yun, Catal. Sci. Technol., 2014, 4, 1268–1275 RSC
- A. Infantes-Molina, J. Mérida-Robles, E. Rodríguez-Castellón, J. L. G. Fierro and A. Jiménez-López, Appl. Catal., A, 2008, 341, 35–42 CrossRef CAS
- M. Wojciechowska, M. Zielinski, A. Malczewska, W. Przystajko and M. Pietrowski, Appl. Catal., A, 2006, 298, 225–231 CrossRef CAS
- J. C. Oxley, S. M. Kaushik and N. S. Gilson, Thermochim. Acta, 1989, 153, 269–286 CrossRef CAS
- J. C. Oxley, J. L. Smith, E. Rogers and M. Yu, Thermochim. Acta, 2002, 384, 23–45 CrossRef CAS
- K. R. Brower, J. C. Oxley and M. Tewari, J. Phys. Chem., 1989, 93, 4029–4033 CrossRef CAS
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CAS
- L. P. Wang, P. D. Han, Z. X. Zhang, C. L. Zhang and B. S. Xu, Comput. Mater. Sci., 2013, 77, 281–285 CrossRef CAS
- S. Ghahramani and H. Kangarlau, J. Nano- Electron. Phys., 2013, 5, 04051 Search PubMed
- K. R. Babu, C. B. Lingam, S. Auluck, S. P. Tewari and G. Vaitheeswaran, J. Solid State Chem., 2011, 184, 343–350 CrossRef CAS
- S. J. Splinter, N. S. McIntyre, W. N. Lennard, K. Griffiths and G. Palumbo, Surf. Sci., 1993, 292, 130–144 CrossRef CAS
- C. Chen, S. J. Splinter, T. Do and N. S. McIntyre, Surf. Sci., 1997, 382, L652–L657 CrossRef CAS
- N. S. McIntyre and C. Chen, Corros. Sci., 1998, 40, 1697–1709 CrossRef CAS
- H. B. Yao, Y. Li and A. T. S. Wee, Appl. Surf. Sci., 2000, 158, 112–119 CrossRef CAS
- S. Verdier, N. van der Laak, S. Delalande, J. Metson and F. Dalard, Appl. Surf. Sci., 2004, 235, 513–524 CrossRef CAS
- V. Fournier, P. Marcus and I. Olefjord, Surf. Interface Anal., 2002, 34, 494–497 CrossRef CAS
- P. A. Sermon and R. Badheka, J. Sol-Gel Sci. Technol., 2004, 32, 149–153 CrossRef CAS
- M. Santamaria, F. Di Quarto, S. Zanna and P. Marcus, Electrochim. Acta, 2007, 53, 1314–1324 CrossRef CAS
- S. Verdier, S. Delalande, N. van der Laak, J. Metson and F. Dalard, Surf. Interface Anal., 2005, 37, 509–516 CrossRef CAS
- J. L. Booster, J. H. L. Voncken, A. van Sandwijk and M. A. Reuter, Powder Diffr., 2002, 17, 112–118 CrossRef CAS
- S. Wuttke, S. M. Coman, J. Krohnert, F. C. Jentoft and E. Kemnitz, Catal. Today, 2010, 152, 2–10 CrossRef CAS
- W. A. Crichton, J. B. Parise, H. Mueller, J. Breger, W. G. Marshall and M. D. Welch, Mineral. Mag., 2012, 76, 25–36 CrossRef CAS
- A. Demourgues, N. Penin, D. Dambournet, R. Clarenc, A. Tressaud and E. Durand, J. Fluorine Chem., 2012, 134, 35–43 CrossRef CAS
- M. Liu, S. Zanna, H. Ardelean, I. Frateur, P. Schmutz, G. L. Song, A. Atrens and P. Marcus, Corros. Sci., 2009, 51, 1115–1127 CrossRef CAS
- N. S. McIntyre and M. G. Cook, Anal. Chem., 1975, 47, 2208–2213 CrossRef CAS
- F. Bensebaa, F. Zavaliche, P. L'Ecuyer, R. W. Cochrane and T. Veres, J. Colloid Interface Sci., 2004, 277, 104–110 CrossRef CAS PubMed
- S. C. Petitto, E. M. Marsh, G. A. Carson and M. A. Langell, J. Mol. Catal. A: Chem., 2008, 281, 49–58 CrossRef CAS
- J. Yang, H. W. Liu, W. N. Martens and R. L. Frost, J. Phys. Chem. C, 2010, 114, 111–119 CrossRef CAS
- J. J. Zhu, K. Kailasam, A. Fischer and A. Thomas, ACS Catal., 2011, 1, 342–347 CrossRef CAS
- L. X. Yang and B. Luan, J. Electrochem. Soc., 2005, 152, C474–C481 CrossRef CAS
- A. A. Khassin, T. M. Yurieva, V. V. Kaichev, V. I. Bukhtiyarov, A. A. Budneva, E. A. Paukshtis and V. N. Parmon, J. Mol. Catal. A: Chem., 2001, 175, 189–204 CrossRef CAS
- Y. C. Lu, A. N. Mansour, N. Yabuuchi and Y. Shao-Horn, Chem. Mater., 2009, 21, 4408–4424 CrossRef CAS
- J. S. Girardon, A. S. Lermontov, L. Gengembre, P. A. Chernavskii, A. Griboval-Constant and A. Y. Khodakov, J. Catal., 2005, 230, 339–352 CrossRef CAS
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra09365b |
| This journal is © The Royal Society of Chemistry 2019 |