
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntegrated optical and electrochemical detection of Cu2+ ions in water using a sandwich amino acid–gold nanoparticle-based nano-biosensor consisting of a transparent-conductive platform†
Mehdi Atapour ab,
Ghasem Amoabediny*ab and
Mojgan Ahmadzadeh-Rajibc
ab,
Ghasem Amoabediny*ab and
Mojgan Ahmadzadeh-Rajibc
aFaculty of Chemical Engineering, College of Engineering, University of Tehran, Enghelab St., P. O. Box 11155-4563, Tehran, Iran. E-mail: amoabediny@ut.ac.ir
bDepartment of Nanobiotechnology, Research Center for New Technologies in Life Science Engineering, University of Tehran, Tehran, Iran
cRastegar Research Lab, Faculty of Veterinary Medicine, University of Tehran, Tehran, Iran
First published on 18th March 2019
Abstract
In this paper, an optical-electrochemical nano-biosensor was introduced for measuring Cu2+ ion concentrations in water. A multi-step procedure was used to fabricate the transparent-conductive biosensor platform consisting of an L-cysteine–gold nanoparticle-based sandwich structure. First, colloidal gold nanoparticles (GNPs) were synthesized according to the Turkevich–Frens method with some modifications and then functionalized with L-cysteine molecules (GNP/L-cys). Then, cyclic voltammetry was preformed in buffered solutions containing HAuCl4·3H2O for gold nanoparticle electrodeposition on cleaned ITO glasses. The GNP-electrodeposited ITO glasses (ITO/GNPs) were thermally treated in air atmosphere for 1 hour at a temperature of 300 °C. Following the procedure, the gold nanoparticles on ITO/GNPs substrates were functionalized with L-cysteine to prepare ITO/GNPs/L-cys substrates. Finally, the sandwich-type substrates of ITO/GNPs/L-cys⋯Cu2+⋯L-cys/GNPs were fabricated by accumulation of Cu2+ ions using an open circuit technique performed in copper ion buffer solutions in the presence of previously produced colloidal GNP/L-cys nanoparticles. The effective parameters including GNP/L-cys solution volume, pre-concentration pH and pre-concentration time on the LSPR and SWV responses were investigated and optimized. The fabricated transparent-conductive platforms were successfully assessed as a nano-biosensor for detection of copper ions using two different methods of square wave voltammetry (SWV) and localized surface plasmon resonance (LSPR). As a result, the proposed biosensor showed a high sensitivity, selectivity and a wide detectable concentration range to copper ions. The total linear range and the limit of detection (LOD) of the nano-biosensor were 10–100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 nM (0.6–6354.6 ppb) and below 5 nM (0.3 ppb), respectively. The results demonstrated the potential of combining two different optical and electrochemical methods for quantitation of the single analyte on the same biosensor platform and obtaining richer data. Also, these results indicated that the developed LSPR-SWV biosensor was superior to many other copper biosensors presented in the literature in terms of linear range and LOD. The developed nano-biosensor was successfully applied in the determination of trace Cu2+ concentration in actual tap water samples.
000 nM (0.6–6354.6 ppb) and below 5 nM (0.3 ppb), respectively. The results demonstrated the potential of combining two different optical and electrochemical methods for quantitation of the single analyte on the same biosensor platform and obtaining richer data. Also, these results indicated that the developed LSPR-SWV biosensor was superior to many other copper biosensors presented in the literature in terms of linear range and LOD. The developed nano-biosensor was successfully applied in the determination of trace Cu2+ concentration in actual tap water samples.
1. Introduction
Heavy metals, similar to other potential pollutants such as pesticides, household chemicals and industrial compounds, are increasingly leading to serious human health and environmental problems. The determination of heavy metal ions in the environment is of great importance for the following reasons: (1) heavy metals are categorized as one of the main environmental pollutants, (2) heavy metals come from both natural sources and human-made sources, the concentration of the pollutants in the environment is drastically increasing, (3) the potential exposure of human beings, animals and plants to the metal pollutants is high because the metals can be widely dispersed into the water, air and soil, (4) the metal ions are highly toxic, non-biodegradable and can accumulate in the food chain, and may pose severe risks and hazards to humans and the ecosystem.1–3Many analytical methods can be used to determine trace amounts of heavy metals in water samples. The techniques such as Atomic Absorption Spectrometry (AAS), Inductively Coupled Plasma Mass Spectroscopy (ICP-MS), Inductively Coupled Plasma Optical Emission Spectrometry (ICP-OES), Atomic Emission/Fluorescence Spectrometry (AES/AFS), X-Ray Fluorescence (XRF) and Neutron Activation Analysis (NAA) are the most predominant ones. By employing the methods, quantitation of various heavy metal ions is performed with very high sensitivity and very low limit of detection. However, these techniques have several disadvantages. These methods are very costly and require highly-skilled operators and expensive instruments and materials. They are generally complex, suffer from several time-consuming steps of preparation and often, applying the supplementary techniques such as chromatography are necessary to obtain speciation results. In addition, the samples must be delivered to a qualified laboratory for analysis and the methods are unsuitable for on-site detection and monitoring of heavy metals.3,4 In contrast, sensors and biosensors, especially electrochemical and optical based ones due to their many advantages are increasingly appealing alternatives to the conventional methods. In other words, sensors and biosensors are operated based on simple and fast procedure. They are inexpensive and user-friendly approaches with much less consumption of chemicals and reagents for the detection of heavy metal ions. Also, they have great potential for miniaturization and hence using as portable devices for in situ detection.3,4
Many studies have been conducted on measurement of heavy metal ions using biosensors and several bio-receptors have been introduced in this field.5,6 In most of the studies, natural or genetically engineered whole cells, nucleic acids including DNA, and a variety of proteins such as enzymes, and antibodies have been utilized as biological receptors in heavy metal biosensors.1,2,5–7 Basically for a developing a heavy metal biosensor with higher selectivity, the high specificity between a biological element and a certain metal ion, is necessary. In this regard, amino acids and peptides were taken into consideration by researchers as well.8–11 Accordingly, some amino acids and peptides have been reported as potential bio-receptors. For example, it has been shown that L-cysteine (L-cys) based biosensors have a good selectivity to copper ions.12,13
On the other hand, researchers have focused on the application of various nano-structures such as nanoparticles, nanotubes, graphene, etc. in biosensors for detection of heavy metals ions. The nano-materials were often used as modifiers of the detection platforms. Indeed, nano-materials exhibit a number of special chemical, physical, electrical, optical and mechanical properties compared to bulk materials which make them in combination with the biological receptors extremely useful for biosensor construction. The unique properties of nano-materials lead to increased electrode surface area, fast electron transfer and the phenomenon called localized surface plasmon resonance, thus significantly improving biosensor performance parameters such as sensitivity and detection limits.6,14–16
Among the various transducers, electrochemical and optical transducers frequently used in the studies conducted on biosensor construction. Over the past decades, these methods have been developed separately. Electrochemical and optical biosensors present advantages of sensitive, real-time and sometimes label-free measurement of target analytes.17 Yet each of these techniques have their own unique advantages. Optical biosensors have high potential of miniaturization. The external factors such as ionic, electric and magnetic fields do not interfere with the signal of optical biosensors. Also, they are able to analyze simultaneously several target analytes. On the other hand, electrochemical biosensors present wide dynamic range and they are not affected by the light sources. Thus their combination, i.e. using two different electrochemical and optical methods to quantitation of the single analyte on the same biosensor platform, seems to be an exciting field of research. The combination could lead to combining benefits of the two methods and provide richer data including increased dynamics ranges.17 However, development of an optically-transparent and electrically-conductive substrate is the main drawback of performing the combination. It seems that indium tin oxide films deposited on glass substrates (ITO glasses) have such potential.
As far as we are concerned, electrochemical-optical nano-biosensors have not been reported in literatures published so far. Also, a few researches conducted in the field of sandwich-type nano-biosensors for detection of heavy metal ions in solutions. In this study, an L-cysteine–gold nanoparticle-based biosensor consisted of transparent-conductive platform for measuring of Cu2+ ion concentrations in aqueous solutions using both electrochemical and optical methods was developed. L-Cysteine was utilized due to good selectivity to Cu2+ ions. Furthermore, gold nanoparticles were used in the nano-biosensor design because of their unique properties including LSPR, conductivity and high surface area. In fact, the platform was composed of a new sandwich structure of GNP/L-cys⋯Cu2+⋯L-cys/GNP immobilized on ITO glass. Thus, using two different methods, i.e. SWV and LSPR, provided the possibility of copper ion measurement in water solution by the same platform.
2. Material and methods
2.1. Materials and equipment
Indium tin oxide coated glass slides (surface resistivity: 8–12 Ω per square; L × W × thickness: 25 mm × 25 mm × 1.1 mm; transmittance: 84%; refractive index: n20/D 1.517), gold(III) chloride trihydrate (HAuCl4·3H2O, ≥99.9%), potassium hexacyanoferrate(II) trihydrate (K4Fe(CN)6·3H2O, 99.0–102.0%) and potassium hexacyanoferrate(III) (K3Fe(CN)6, ≥99%) and perchloric acid (HClO4, 70%) were supplied from Sigma Aldrich, USA. Ultrapure water was obtained by passing tap water through Milli-Q Water Ultra-Purification System, Millipore Inc., Darmstadt, Germany. Double distilled water was supplied from Avisa Shimi Teb Co., a local company in Tehran, Iran. All other chemicals and solvents including copper(II) nitrate trihydrate (Cu(NO2)3·3H2O, ≥99.5%), L-cysteine (C3H7NO2S, ≥98%), acetone (C3H6O, ≥99.0%), 2-propanol (C3H8O, ≥99.8%), tri-sodium citrate anhydrous (C6H5Na3O7, 99.0–100.5%), citric acid anhydrous (C6H8O7, 99.5–100.5%), di-sodium hydrogen phosphate dihydrate (Na2HPO4·2H2O, ≥ 99.5%), ethylenedinitrilotetraacetic acid (EDTA, 99.4–100.6%) and phosphate-buffered saline (PBS) tablets were purchased from Merck, Germany. In this study, a solution of 5 mM [K3Fe(CN)6]/[K4Fe(CN)6] (in PBS, pH = 7.4) was used as a redox probe for electrochemical characterization.The ultraviolet-visible spectra were recorded using a T80 UV/VIS Spectrometer (PG Instruments Ltd., UK) equipped with UVWin 5 Spectrophotometer Software. Dynamic light scattering (DLS) particle size distributions and zeta potentials were determined using Malvern Zetasizer Nano ZS ZEN3600 (Malvern Instruments Ltd., Malvern, UK). The pH values of the solutions were measured by HI 8521 pH meter (HANNA Instruments Inc., USA). All electrochemical techniques including cyclic voltammetry, open circuit, chronoamperometry and square wave voltammetry were performed using a specially-designed set-up of three-electrode electrochemical cell controlled by IviumStat.XR electrochemical interface (Ivium Technologies USA Inc., Fernandina Beach, FL, USA). A modified ITO glass substrate was used as the working electrode. Also, for comparison, an IRI.2000-3 gold disk electrode (Azar Electrode Co., Tehran, Iran) with 2 mm in diameter, was also served as the working electrode. A platinum electrode as the counter electrode and an Ag/AgCl (saturated KCl) electrode as the reference electrode were applied. All experiments were performed at room temperature, unless otherwise stated. Also, a Universal 320R centrifuge (Hettich Zentrifugen, Germany), an S-4000 sonicator (Misonix Inc., Farmingdale, NY, USA) and KMH1-120W6501 ultrasonic cleaner (Guangdong GT Ultrasonic Co., Ltd., Guangdong, China) were used to carry out centrifugation and ultrasonic procedures, respectively. Filtration of GNPs-based solutions was done using 0.22 μm Syringe Driven Filters (Jet Biofil, Guangdong, China). Field emission scanning electron microscopy (FESEM) and energy-dispersive X-ray spectroscopy (EDX) were performed using a Nova NanoSEM 450 system (FEI Company, Oregon, USA). ImageJ as an open-source image processing software was used to analyze FESEM images to determine the size distributions of gold nanoparticles. Fourier-transform infrared spectroscopy (FT-IR) was performed using a Frontier FT-IR system (PerkinElmer Inc., USA). X-ray diffraction (XRD) patterns were determined using X'Pert Pro MPD X-ray diffractometer (Panalytical Ltd., Almelo, Netherlands). The atomic force microscopy (AFM) images was recorded using a NTEGRA (NT-MDT Spectrum Instruments, Moscow, Russia) equipped with Nova software.
2.2. Procedures
The general steps involved in the fabrication of the nano-biosensor platform and the electrochemical-optical detection of Cu2+ ions were: (A) colloidal GNPs synthesis, (B) colloidal GNPs functionalization with L-cysteine, (C) electrodeposition of GNPs on ITO glass, (D) ITO/GNP functionalization with L-cysteine, (E) preparation of Cu2+ buffer solution, (F) fabrication of transparent-conductive biosensor platform, (G) LSPR detection of copper ions, and (H) SWV detection of copper ions. The steps have been completely described in the following sections. Also, a schematic description of the whole procedure was given in Fig. S1.†500 rpm for 20 minutes. The supernatant was carefully removed and the resulting precipitates were redispersed in 25 mL of ultrapure water using an ultrasonic probe. The centrifugation-redispersion process repeated for 3 times to remove excess citrate ions. Then 10 mL of the final redispersed GNPs solution was transferred into a 50 mL glass beaker on top of a magnetic stirrer. A 0.3 mM solution of L-cysteine in ultrapure water was prepared and 100 μL of this solution was added slowly and dropwise to the GNPs solution under stirring for 2 hours at room temperature.20 The L-cysteine conjugated gold nanoparticles were filtered through 0.22 μm membrane filter and centrifuged at 11500 rpm for 20 minutes and the resulting precipitates were redispersed in ultrapure water. The centrifugation-redispersion steps repeated for three times and final precipitates were redispersed in citric acid–Na2HPO4 buffer at pH = 5.5. After characterization, the colloidal GNP/L-cys solution was stored in a light-tight bottle at 4 °C until next use.Following the procedure described above and for conducting electrochemical SWV technique, 50 mL of copper ion-free buffer (citric acid–Na2HPO4 solution, pH = 5.5) was transferred into the electrochemical cell and was deaerated with nitrogen gas for 10 minutes. Then, the previously fabricated ITO/GNPs/L-cys⋯Cu2+⋯L-cys/GNPs platform as working electrode was immersed in the buffer solution. Immediately SWV voltammetry was performed and the peak current attributed to the reduction of Cu(II) to Cu(I),21,22 was recorded. The optimal conditions of square wave voltammetry were as follows: a pulse amplitude of 50 mV, a frequency of 25 Hz, a potential step of 10 mV and a scan potential range from −0.2 to 0.7 V. The SWV analysis was repeated for platforms prepared using copper-ion standard solutions of different concentrations. Then the calibration plot of SWV peak current versus Cu2+ ion concentration was obtained. After each LSPR and SWV analysis, the biosensor was regenerated by immersing it into 0.1 M HClO4 solution in the electrochemical cell and conducting chronoamperometry technique at a fixed potential of +0.5 V for 30 seconds.21,22 After regeneration, the biosensor was washed with distilled water and dried using N2 gas stream.
3. Results and discussion
3.1. Characterization of GNP and L-cysteine caped GNP
The photograph of the synthesized GNP and the L-cysteine conjugated GNP solutions is shown in Fig. S3.† As illustrated in the figure and mentioned in literatures,20,23,24 the ruby-red color of GNP solution did not change noticeably during functionalization with L-cysteine. The GNP and the L-cysteine functionalized GNP were characterized by using UV-Vis spectroscopy, dynamic light scattering (DLS), zeta potential analysis, field-emission scanning electron microscopy, Fourier-transform infrared spectroscopy. In Table 1, some characterization data of the synthesized GNP and L-cysteine caped GNP are presented. The pH values of the GNP and GNP/L-cys solutions were measured to be 5.6 and 5.4, respectively. The UV-Vis absorption spectra of GNP and GNP/L-cys solutions are shown in Fig. 1. The surface plasmon resonance absorption peak of GNP and GNP/L-cys solutions appeared at about 521 nm and 522 nm wavelengths, respectively which is characteristic of colloidal GNP-based solutions. So that the SPR peak position is dependent on local refractive index and size and shape of gold nanoparticles.24,25 As shown in the figure, at around the peak wavelength, the intensity of GNP/L-cys absorption is decreased in comparison with GNP absorption intensity. The results are compatible with those of other researchers.20,24,25 The concentrations of the synthesized colloidal GNP and GNP/L-cys solutions were calculated from UV-Vis spectra data and tabular material obtained by Haiss et al.26 As calculated, the gold nanoparticle contents in the final GNP and GNP/L-cys solutions were 6.03 and 2.92 nM, respectively. Dynamic light scattering size distribution curve, size distribution histogram and zeta potential distribution of GNP and GNP/L-cys solutions are illustrated in Fig. S4a–f.† The average hydrodynamic (Z-average) diameter of GNP and GNP/L-cys nanoparticles is observed to be 32.1 nm and 34.6 nm, respectively. The increasing of GNP/L-cys hydrodynamic size compared to the GNP average size is due to surface functionalization of GNPs with L-cysteine amino acid.24,25 The zeta potentials of GNP and GNP/L-cys were −29.1 and −21.0 mV, respectively which caused by enough negative charge on the surface of nanoparticle and revealed the highly stability of the nanoparticles.27| Colloidal nano-particle | pH | SPR peak wavelength (nm) | Zeta potential (mV) | Z-Average (nm) | Average diametera (nm) | Average diameterb (nm) | Concentrationb (nM) |
|---|---|---|---|---|---|---|---|
| a Determined by analysis of FE-SEM images using ImageJ software.b Calculated from UV-Vis spectra data and tabular material (see ESI Tables S-2 and S-3, respectively) presented by Haiss et al.26c Calculated from UV-Vis spectra data and see ESI Table S-3 (ref. 26) for 15 nm GNP/L-cys. | |||||||
| GNP | 5.6 | 521 | −29.1 | 32.1 | 13.9 ± 3.9 | 12 | 6.03 |
| GNP/L-cys | 5.4 | 522 | −21.0 | 34.6 | 15.2 ± 3.4 | — | 2.92c |
 | ||
| Fig. 1 UV-Vis absorption spectra of GNP and GNP/L-cys solutions. The insets show the corresponding schematic images of GNP (1) and GNP/L-cys (2). | ||
The Fig. S5† shows the FE-SEM images of synthesized and functionalized gold nanoparticles and their size distribution histograms and analysis carried out using ImageJ software. As seen in the figure, the nanoparticles had nearly spherical shape with a relatively uniform size distribution. The average diameter of GNP and GNP/L-cys nanoparticles were 13.89 ± 3.94 and 15.19 ± 3.45 nm, respectively.
In order to investigate and verify whether L-cysteine amino acids were bound to the surface of gold nanoparticles, FT-IR spectra of L-cysteine and L-cysteine functionalized GNP were carried out. As shown in Fig. 2, the FT-IR spectrum of L-cysteine is a typical amino acid spectrum. The pure L-cysteine shows a peak at 2549 cm−1, while the peak disappeared in the GNP/L-cys spectrum and this is the most significant difference between L-cys and GNP/L-cys spectra. The peak attribute to S–H bond stretch. The disappearance of the peak in the FT-IR spectrum of GNP/L-cys indicated L-cysteine S–H bonds were broken and L-cysteine molecules were successfully attached onto gold nanoparticle surface through S–Au bond. Meanwhile, the spectrum of L-GNP/L-cys presents some peaks which are similar to those of L-cysteine spectrum which indicated other reason for functionalization of gold nanoparticle surface with L-cysteine amino acid. These results are consistent with those of other studies.20,24,28
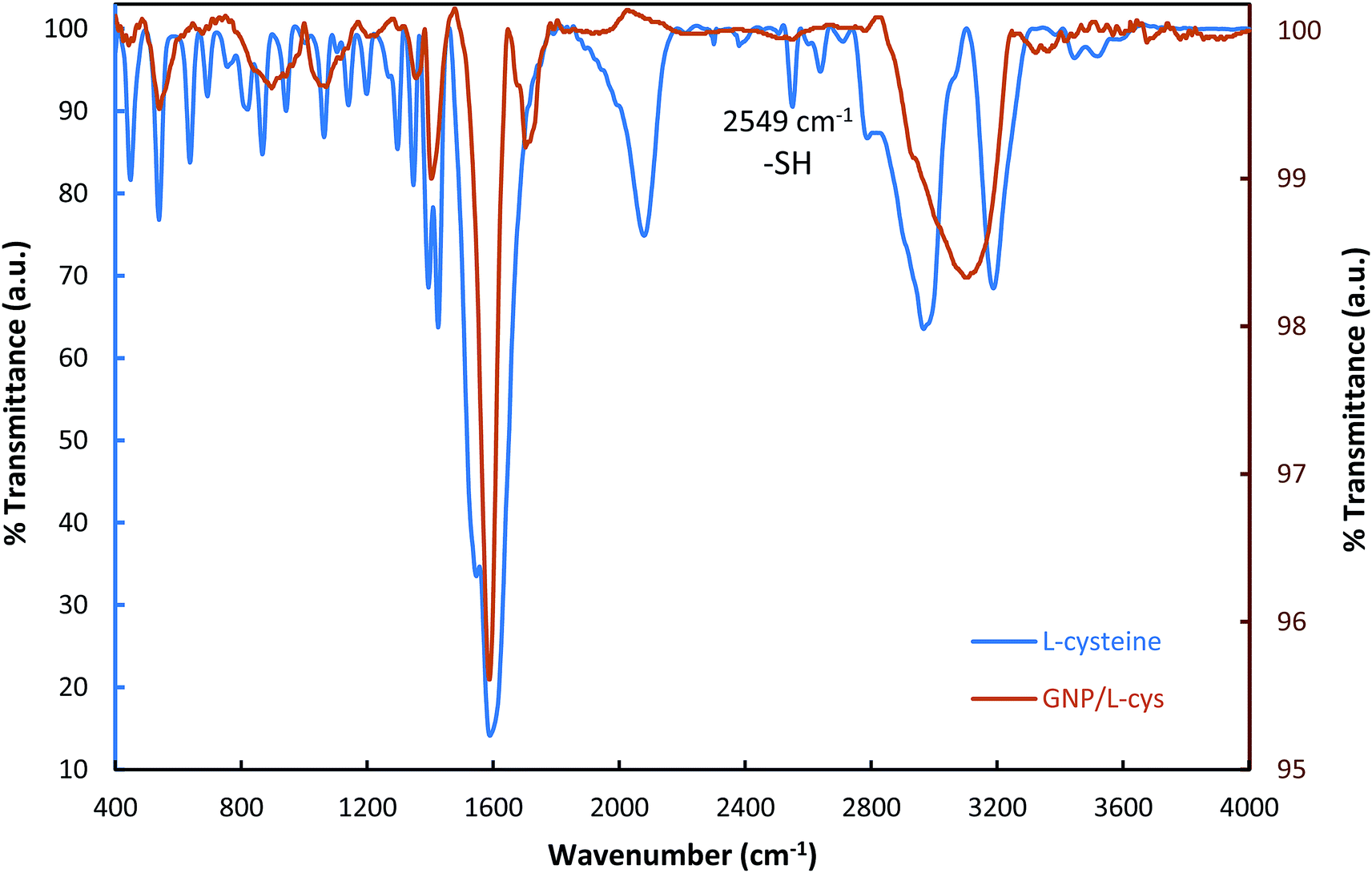 | ||
| Fig. 2 FT-IR spectra of L-cysteine and L-cysteine modified GNP. | ||
3.2. Characterization of transparent-conductive ITO and modified ITO
The photographic image of bare ITO, ITO/GNPs, ITO/GNPs/L-cys and ITO/GNPs/L-cys⋯Cu2+⋯L-cys/GNPs substrates is shown in Fig. S6,† from left to right, respectively. It is worth noting that the sandwich ITO/GNPs/L-cys⋯Cu2+⋯L-cys/GNPs structure in the Fig. S6d† was produced using copper ion concentration of 1000 nM. Fig. 3 shows the UV-Vis absorption spectra of bare ITO, ITO/GNPs, ITO/GNPs/L-cys and ITO/GNPs/L-cys⋯Cu2+⋯L-cys/GNPs substrates. As illustrated in the figure, a weak absorption peak at 526 nm in the UV-Vis spectrum of ITO/GNPs substrate is clearly observable which is ascribed to localized surface plasmon resonance of gold nanoparticles electrodeposited on the substrate surface. However, no peak appeared in spectrum of bare ITO substrate. After functionalization of gold nanoparticles electrodeposited on the substrate surface with L-cysteine through the S–Au covalent bond, the LSPR peak of the ITO/GNPs/L-cys substrate was red-shifted from 526 nm to 530 nm. This result, which was attributed to the interaction between gold nanoparticles and L-cysteine molecules and changes in the nanoparticle's local dielectric environment, is compatible with those of other researchers.29,30 When complexation of L-cysteine with copper ions occurred and sandwich ITO/GNPs/L-cys⋯Cu2+⋯L-cys/GNPs produced, the LSPR peak was further red-shifted of 14 nm. The LSPR peak shift in the presence of copper ions was the basis for the Cu(II) concentration measurements using optical technique, since an increase in copper ion concentration leads to a limited linear increasing in the LSPR peak shift. It should be noted that the sandwich ITO/GNPs/L-cys⋯Cu2+⋯L-cys/GNPs structure in the Fig. 3 was produced using copper ion concentration of 2200 nM.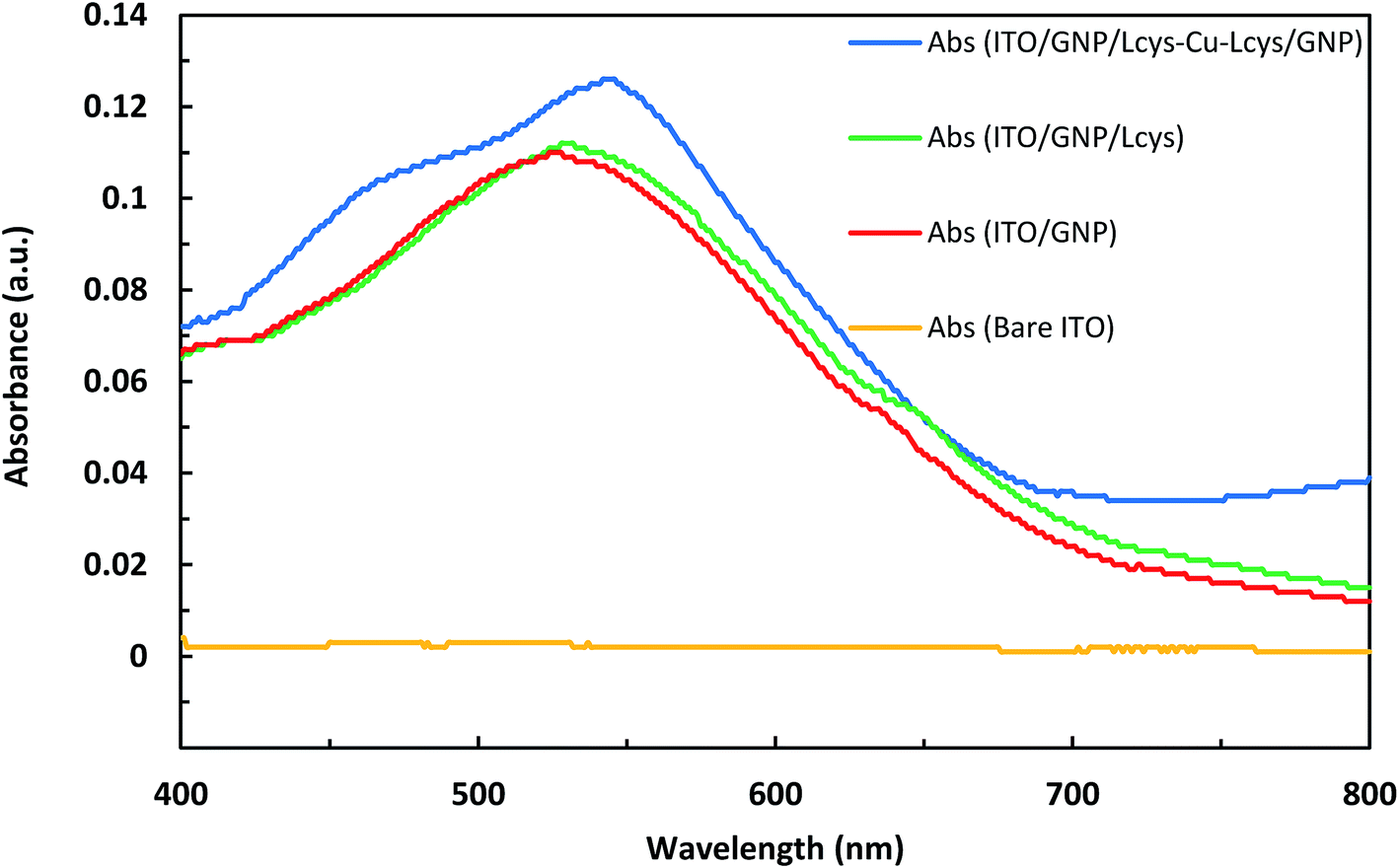 | ||
| Fig. 3 UV-Vis absorption spectra of bare ITO, ITO/GNPs, ITO/GNPs/L-cys and ITO/GNPs/L-cys⋯Cu2+⋯L-cys/GNPs substrates. | ||
To assess the electrochemical behavior of bare ITO, ITO/GNPs and ITO/GNPs/L-cys substrates, cyclic voltammetry technique with the redox couple of [K3Fe(CN)6]/[K4Fe(CN)6] as the electrochemical probe was used (Fig. 4). The optimized parameters of potential scan rate, potential step, potential range and number of cycles for the CV technique were 50 mV s−1, 5 mV, −0.3 to 0.8 V and 5 cycles. As shown in the figure, the cyclic voltammograms of bare and modified ITO substrates showed a typical, quasi-reversible redox reaction. After the functionalization of the ITO with gold nanoparticles, the redox peak current significantly increased. However, the redox peak current notably decreased after the functionalization of the gold nanoparticles deposited on the ITO surface with L-cysteine amino acid. The event was attributed to the electrostatic repulsion between the negatively charge of [Fe(CN)6]3−/4− redox probe and the COO– group of L-cysteine on the surface of gold nanoparticles.31 It was clearly revealed that L-cysteine was successfully immobilized on ITO/GNPs surface. As mentioned earlier, the gold disk electrode with the active surface area of about 0.03 cm2 was also characterized by CV for comparison. The redox peak potential separation and the general shape of the cyclic voltammogram of ITO/GNPs were more similar to those of gold electrode in comparison with bare ITO. In other words, the peak potential separation drastically decreased when GNPs electrodeposited on the ITO surface. The results was obviously attributed to the electron conductivity improvement of the ITO substrate due to GNPs electrodeposition on the ITO surface.32 It is worth noting that the active surface area of the ITO electrodes in the electrolyte was 0.6 cm2.
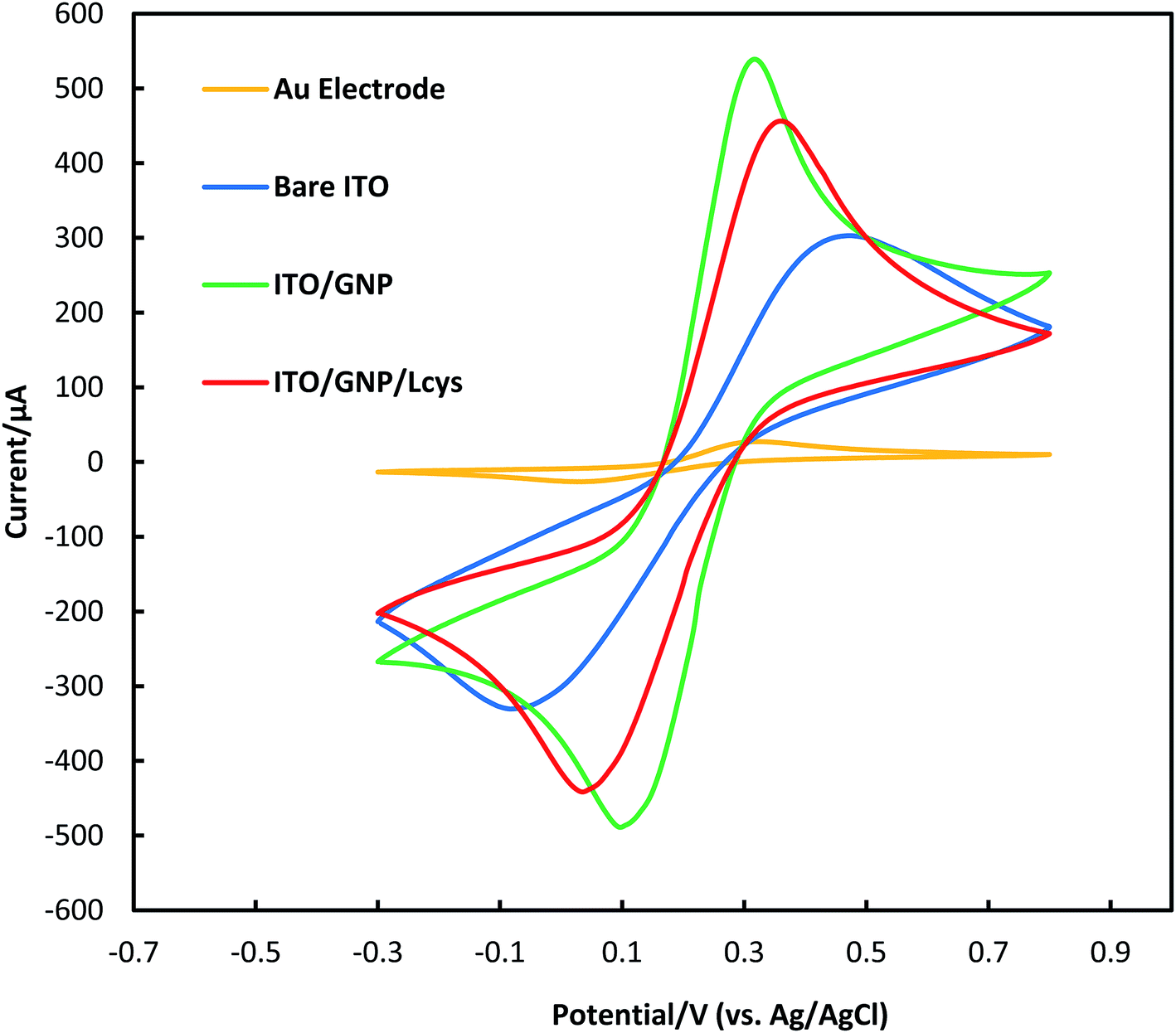 | ||
| Fig. 4 Electrochemical characterization of a gold disk electrode, bare ITO, ITO/GNPs and ITO/GNPs/L-cys substrates using CV technique. | ||
The Fig. S7† shows the XRD patterns of GNPs electrodeposited on ITO surface after thermal treatment at 300 °C for 1 hour. As shown in the figure, the peaks located at 2θ = 38.24°, 44.42°, 64.67° and 77.59° correspond to the (1 1 1), (2 0 0), (2 2 0) and (3 1 1) planes of the crystal structure of gold nanoparticles, respectively. The peak at 2θ = 38.24° which corresponds to the (1 1 1) facet of the FCC crystals is relatively intensive one. However, the intensities of the other peaks of gold nanoparticles are relatively weak. Also, the observed peaks at 2θ = 21.39°, 30.33°, 35.3°, 37.49°, 45.41°, 50.68° and 60.26° correspond to the (2 1 1), (2 2 2), (4 0 0), (4 1 1), (4 3 1), (4 4 0) and (6 2 2) planes of the ITO film, respectively. The results are in accordance with those of other researchers.33,34
Fig. S8† shows the FE-SEM images of bare ITO, ITO/GNPs, ITO/GNPs/L-cys and ITO/GNPs/L-cys⋯Cu2+⋯L-cys/GNPs (copper ion concentration of 1000 nM) substrates. The scanning electron microscopy results verified the formation of the ITO/GNPs/L-cys⋯Cu2+⋯L-cys/GNPs structure. In other words, prior to exposure the ITO/GNPs/L-cys substrate to copper ion solution and the formation of cysteine⋯Cu2+⋯cysteine coordination complexes and thus the formation of the sandwich structure of GNPs/L-cys⋯Cu2+⋯L-cys/GNPs, the gold nanoparticles were relatively uniformly distributed and almost isolated from each other across the surface of the ITO glasses. However, as shown in Fig. S8g,† the aggregation of the nanoparticles on the surface of GNPs/L-cys⋯Cu2+⋯L-cys/GNPs substrate is clearly observed. Also, the size distribution histograms of ITO/GNPs, ITO/GNPs/L-cys and ITO/GNPs/L-cys⋯Cu2+⋯L-cys/GNPs substrates has been illustrated in this figure. As shown in the figure, the size distribution of the gold-based nanoparticles stabilized on ITO/GNPs, ITO/GNPs/L-cys and ITO/GNPs/L-cys⋯Cu2+⋯L-cys/GNPs substrates gives an average diameter of = 20.0 ± 6.4 nm, 21.8 ± 6.1 nm and 31.4 ± 15.2 nm, respectively. The nanoparticle size distributions on ITO/GNPs and ITO/GNPs/L-cys substrates are much more uniform than those of ITO/GNPs/L-cys⋯Cu2+⋯L-cys/GNPs substrate. The fact and also the significantly increase of the nanoparticle average diameter from about 21 to about 31 nm are other reasons of the formation of the sandwich structure of GNPs/L-cys⋯Cu2+⋯L-cys/GNPs. For further investigation, ITO/GNPs, ITO/GNPs/L-cys and ITO/GNPs/L-cys⋯Cu2+⋯L-cys/GNPs substrates were analyzed using energy-dispersive X-ray spectroscopy and atomic force microscopy and the results are depicted in Fig. S9 and S10,† respectively. The EDX spectrum of all substrates demonstrated the strong signals of gold witch confirmed that gold nanoparticles were clearly immobilized on the substrates. According the EDX spectra, gold content of ITO/GNPs, ITO/GNPs/L-cys and ITO/GNPs/L-cys⋯Cu2+⋯L-cys/GNPs substrates were 8.26%, 7.92% and 11.97%, respectively. Also, the strong signals of indium, silicon and oxygen elements from ITO glass substrate are clearly observable in the EDX spectra. The presence of carbon, nitrogen and oxygen in EDX spectra of ITO/GNPs/L-cys and ITO/GNPs/L-cys⋯Cu2+⋯L-cys/GNPs substrate revealed the functionalization of gold nanoparticles with L-cysteine. Fig. S10† shows 2D and 3D AFM topographic images along with height distribution histograms of bare ITO, ITO/GNPs, ITO/GNPs/L-cys and ITO/GNPs/L-cys⋯Cu2+⋯L-cys/GNPs substrates. Also, in Table 2, some quantitative data obtained from AFM analyses of the substrates are presented. As illustrated in the table, the arithmetic average roughness of bare ITO, ITO/GNPs, ITO/GNPs/L-cys and ITO/GNPs/L-cys⋯Cu2+⋯L-cys/GNPs substrates were 0.80 nm, 3.44 nm, 3.64 nm and 5.14 nm, respectively. Also, the root mean square roughness (RSM) of bare ITO, ITO/GNPs, ITO/GNPs/L-cys and ITO/GNPs/L-cys⋯Cu2+⋯L-cys/GNPs substrates were 1.03 nm, 4.41 nm, 4.51 nm and 6.66 nm, respectively. The values of the surface roughness parameters gradually increased with successive modification of bare ITO surface. The average height values (17.87, 18.76 and 25.97) of nanoparticles obtained from AFM analysis were smaller than their corresponding average diameter values (20.0, 21.8, 31.4 nm) acquired from FE-SEM analysis in all three substrates. This is likely due to two reasons. First, the gold nanoparticles electrodeposited on ITO, were not exactly spherical. Second, the difference between average height and average diameter of nanoparticles were originated from AFM artifacts.35
| Substrate | Maximum peak height (nm) | Average peak height (nm) | Average roughness, Sa (nm) | Root mean square roughness, Sq (nm) |
|---|---|---|---|---|
| Bare ITO | 12.56 | 4.24 | 0.80 | 1.03 |
| ITO/GNPs | 44.22 | 17.87 | 3.44 | 4.41 |
| ITO/GNPs/L-cys | 57.96 | 18.76 | 3.64 | 4.51 |
| ITO/GNPs/L-cys⋯Cu2+⋯L-cys/GNPs | 79.57 | 25.97 | 5.14 | 6.66 |
3.3. Optimization of effective parameters on the biosensor response
The influence of volume of GNP/L-cys solution, pre-concentration pH and pre-concentration time on the LSPR and SWV responses of the nano-biosensor were investigated and optimized. The effect of GNP/L-cys solution volume on the LSPR peak wavelength red-shift and SWV cathodic peak current is shown in Fig. S11.† Experiments were carried out using GNP/L-cys solution volume in the range of 0–20 mL, while other variables including pre-concentration pH and pre-concentration time were set at 5.5 and 12 min, respectively. It was observed that with increasing GNP/L-cys solution volume from 0 to about 5 mL (or 10 mL in SWV detection), the biosensor response increased. Further increase in the solution volume had a little effect on the product yield. However, with further increase in the solution, the response decreased. The effect of pre-concentration pH on the LSPR and SWV responses of the biosensor is depicted in Fig. S12.† Experiments were performed using pre-concentration pH in the range of 2.5–7.5, while the GNP/L-cys solution volume and pre-concentration time were set at 5 mL and 12 min, respectively. As shown in the figure, with increasing pH value from 2.5 to 5.5, the biosensor responses constantly increased. The maximum values of both LSPR and SWV responses observed at pH 5.5, which is near to the isoelectric point of L-cysteine i.e., 5.02 at 25 °C. The lower response values at pH level less than 5.5 might be related to the chelating ability reduction of L-cysteine toward Cu2+ ions due to carboxylate protonation.13 Further increase in pH value, resulted in decreased biosensor responses. At higher pH values, strong interaction between the amino nitrogen of L-cysteine and the gold nanoparticle surface led to reduce the ligation strength of the amino acid toward Cu2+ ions.13 The effect of the pre-concentration time on the LSPR and SWV responses of the biosensor is represented in Fig. S13.† Experiments were accomplished utilizing a pre-concentration time in the range of 0–40 min, while the GNP/L-cys solution volume and pre-concentration pH were set at 5 mL and 5.5, respectively. As demonstrated in Fig. S13,† the SWV peak current of the biosensor rapidly increased with increasing pre-concentration time and reached a constant pattern at about 12 min. Also, the LSPR peak wavelength of the biosensor gradually red-shifted with increasing pre-concentration time until about 24 min and then showed a relatively fluctuating pattern.3.4. LSPR and SWV detection of copper ions
Under the optimized experimental conditions, the LSPR and SWV detection of copper ion concentrations was performed. Fig. 5a shows UV-Vis absorption spectra of the ITO/GNPs/L-cys⋯Cu2+⋯L-cys/GNPs substrates prepared by accumulation of different concentrations of Cu2+ on the ITO/GNPs/L-cys substrates in the presence of GNP/L-cys nanoparticles, as described above. The absorption peak wavelength shift in the presence of copper ions was the basis of the measurement of the copper concentration using LSPR optical method. Indeed, the strong complexation of Cu2+ with two L-cysteine molecules and formation of the sandwich structure resulted in the red-shift of the LSPR peak wavelength due to the change in the refractive index of the environment.29,30 It can be seen that increase in concentration of Cu2+ ions led to more red-shift of LSPR peak wavelength, so that with increasing the concentrations of Cu2+ from 1000 to 100000 nM, the LSPR peak wavelength increased from 532.5 to 579.5 nm. Also, Fig. 5b presents the linear calibration curve describing the relation between the LSPR peak wavelength red-shifts and the different concentrations of Cu2+ ions. As illustrated in the figure, the LSPR peak wavelength red-shift (Δλ) has a good linear relationship with the logarithm of the concentration of Cu2+ ions (Δλ (nm) = 23.592 × log[Cu2+] (mol L−1) + 147.07) in the range of 1000 to 100000 nM (63.5 to 6354.6 ppb) and with a correlation coefficient (R2) of 0.9814. The sensitivity of the LSPR biosensor witch is defined as the slope of the LSPR calibration curve, was 23.592 nm per decade. The LOD of the LSPR biosensor was calculated to be blow 621 nM. The square wave voltammograms of ITO/GNPs/L-cys⋯Cu2+⋯L-cys/GNPs electrodes produced in the presence of different concentrations of Cu2+ have been presented in Fig. 6a. As illustrated in the figure, a clear cathodic peak at the potential of around 0.2 V due to the reduction of Cu2+ to Cu1+ ions observed. However, as the concentration of copper ions increased, the peak potential shifted slightly from negative to positive potential side with respect to Ag/AgCl reference electrode. The cathodic peak current increased linearly with increasing the concentration of Cu2+ ions bound to the L-cysteine molecules witch was the basis of the determination of the copper concentration using SWV electrochemical technique. In fact, more reduction of the Cu2+ ions led to higher peak current. So that with increasing the concentrations of Cu2+ from 10 to 3300 nM, the SWV peak current increased from 2.21 to 118.05 μA. The Fig. 6b represents the linear calibration curve of the SWV cathodic peak current versus the concentrations of Cu2+. The figure indicated a well-defined linear relationship between the SWV peak current and the concentration of Cu2+ ions (current (μA) = 0.037 × [Cu2+] (nM) + 0.031) over the range of 10 to 3300 nM (0.64 to 209.7 ppb) with a correlation coefficient (R2) of 0.9938. The sensitivity of the SWV biosensor witch is defined as the slope of the calibration curve, was 0.037 μA nM−1. Also, the limit of detection of the SWV biosensor witch is defined as 3σ/S (where σ is the standard deviation of the ten blank measurements and S is the slope of the SWV calibration curve)36 was calculated to be blow 5 nM (0.31 ppb). It should be noted that the LOD was much lower than the maximum copper limit of WHO (2 mg L−1 or 31.5 μM),37 EU (2 mg L−1)38 and EPA (1.3 mg L−1)39 standards in drinking water. Analytical figures of the developed LSPR-SWV biosensor for detection of copper ions are summarized in Table 3.
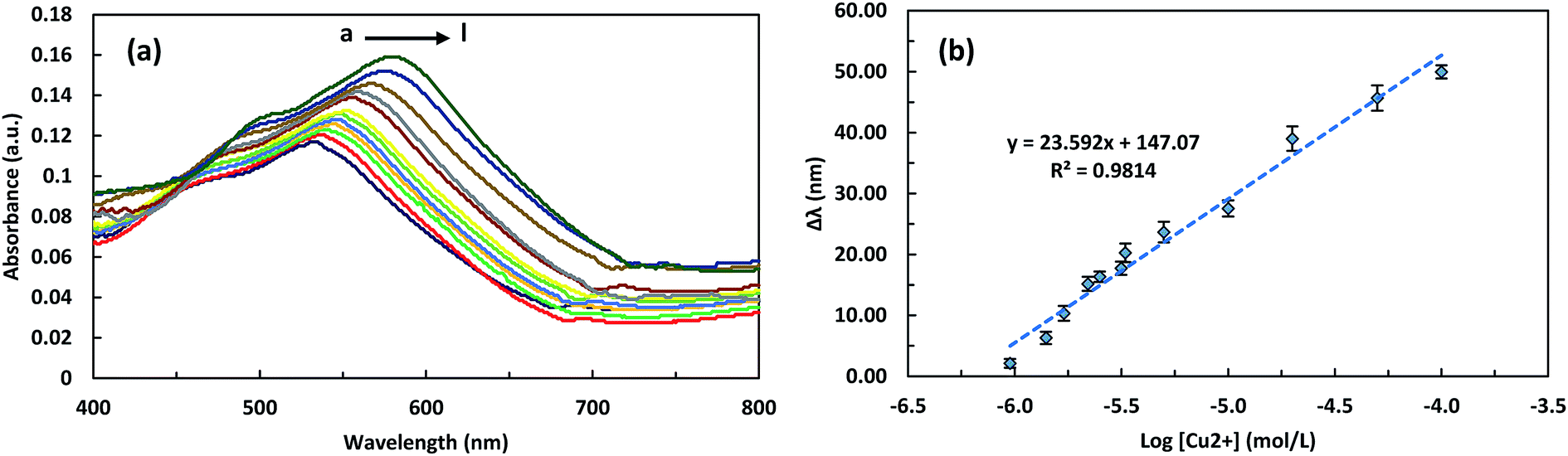 | ||
| Fig. 5 UV-Vis absorption spectra of ITO/GNPs/L-cys⋯Cu2+⋯L-cys/GNPs produced in the presence of different concentrations of Cu2+ (from a to l): 1000, 1400, 1700, 2200, 2500, 3000, 3300, 5000, 10000, 20000, 50000 and 100000 nM, respectively (a), and the linear calibration curve describing the relation between the LSPR peak wavelength red-shifts and the concentrations of Cu2+ (b). The data points and the error bars represent the average values and the standard deviation of three independent measurements, respectively. | ||
 | ||
| Fig. 6 Square wave voltammograms of ITO/GNPs/L-cys⋯Cu2+⋯L-cys/GNPs electrodes produced in the presence of different concentrations of Cu2+ (from a to l): 10, 50, 100, 300, 600, 1000, 1400, 1700, 2200, 2500, 3000, 3300 nM, respectively (a), and the linear calibration curve describing the relation between the SWV cathodic peak current and the concentrations of Cu2+ (b). The data points and the error bars show the mean values and the standard deviation of three independent experiments, respectively. | ||
| Calibration equation (LSPR) | (Δλ (nm) = 23.592 × log[Cu2+] (mol L−1) + 147.07) |
| Calibration equation (SWV) | Current (μA) = 0.037 × [Cu2+] (nM) + 0.031 |
| Correlation coefficient (R2) | 0.9814 (LSPR); 0.9938 (SWV) |
| Total linear range | 10–100000 nM (0.64–6354.6 ppb) |
| Limit of detection | Blow 5 nM (0.31 ppb) |
To assess the selectivity of the developed nano-biosensor toward copper ions, the effects of other metal cations including Ca2+, Mg2+, Mn2+, Zn2+, Cd2+, Co2+, Ni2+, Pb2+, Ag+ and Hg2+ on the LSPR and SWV responses were investigated under the optimized experimental conditions. The concentration of interfering metal cations (10000 nM) was chosen about 5 times compared to the concentration of copper ions (2200 nM). The results are illustrated in Fig. S14.† As shown in this figure, the proposed nano-biosensor has higher selectivity to Cu2+ ions than the other metal ions.
The linear range and the limit of detection of the optical-electrochemical biosensor for detection of copper ions were compared with that of reported values by other researchers.40–59 This comparison is summarized in Table 4. According to the table, the developed LSPR-SWV biosensor was superior to many other methods in terms of linear range and LOD.
| Biosensor | Method | Liner range (μM) | LOD (ppb) | Ref. |
|---|---|---|---|---|
| a Au: gold disc electrode.b MPA: 3-mercaptopropionic acid, LAsp: poly-L-aspartic acid.c NA: not available.d Its chemical composition and structure have been presented in the paper.42 | ||||
| Au/L-cysa | Electrochemical | 0.1–1000 | 6.3 | 40, 1999 |
| Au/MPA/PLAspb | Electrochemical | 0.005–0.02 | 0.2 | 41, 2001 |
| GNPs/DNAzyme | Colorimetric | 0.625–15 | 18.4 | 52, 2010 |
| Gold nanoclusters/BSA | Fluorescent | 100–5000 | 3177 | 47, 2011 |
| p-Cresol/hydrogen peroxide | Fluorescent | 0.3–50 | 0.64 | 49, 2011 |
| Gold microelectrode/peptide nanofibrils | Electrochemical | 15–50 | 1906 | 55, 2011 |
| SPE/GNPs/L-cys | Electrochemical | 0.08–157.4 | 8 | 58, 2011 |
| Gold nanorod/L-cys | Colorimetric | 1–100 | 21.6 | 53, 2011 |
| Coumarin derivative | Fluorescent | 0.1–1.0 | 0.95 | 57, 2012 |
| GNPs/BSA | Colorimetric | 0.1–5 | 6.4 | 54, 2012 |
| In situ formation of silver nanoparticles | Colorimetric | 0.25–2 | 15.9 | 51, 2012 |
| Gold microelectrode/GNPs/L-cys/L-asp | Electrochemical | 0.08–31.5 | 1 | 59, 2012 |
| Luminescent porous silicon nanoparticles | Fluorescent | 1–10 | NAc | 50, 2013 |
| Amplex® UltraRed/hydrogen peroxide | Fluorescent | 1.6–12 | 10.8 | 48, 2013 |
| GNPs/Peptide | Colorimetric | 10–150 | 1.3 | 46, 2014 |
| Receptor 1d | Colorimetric | 7.5–21.0 | 23.5 | 42, 2015 |
| C. metallidurans cell | Fluorescent | 0–100 | 1544.2 | 43, 2017 |
| Pyoverdine | Fluorescent | 0.2–10 | 3.2 | 56, 2016 |
| Salicylaldehyde Schiff-base derivative | Fluorescent | 0–20 | 14.6 | 44, 2017 |
| GC-NKB-CNTs/PDDA-ABTS | Electrochemical | 0–10 | 2.5 | 45, 2018 |
| ITO/GNPs/L-cys⋯Cu2+⋯L-cys/GNPs | LSPR-SWV | 0.01–100 | 0.3 | Current study |
3.5. Practical application
The developed nano-biosensor was applied to determine the concentrations of Cu2+ ions in two actual samples of tap water collected locally. The results are summarized in Table 5. As presented in the table, there is a good agreement between the results obtained by the AAS method and detection results of both LSPR and SWV biosensors. The data points and the error bars represent the average values and the standard deviation of three independent measurements, respectively. The LSPR and SWV results are presented in Table 5 as the average value ± the standard deviation of three independent measurements.4. Conclusion
In summary, the potential of combining two different optical and electrochemical methods to quantitation of the single analyte on the same biosensor platform, was demonstrated. In other words, the transparent-conductive platforms of ITO/GNPs/L-cys⋯Cu2+⋯L-cys/GNPs were successfully developed and assessed as a biosensor for detection of copper ions using two different methods of square wave voltammetry and localized surface plasmon resonance. In this regard, the sandwich-type structures of ITO/GNPs/L-cys⋯Cu2+⋯L-cys/GNPs were fabricated from two previously produced structures of ITO/GNPs/L-cys and GNPs/L-cys using open circuit technique. The total linear range and the LOD of the biosensor were 10–100000 nM (0.64–6354.6 ppb) and blow 5 nM (0.31 ppb), respectively. The application of the fabricated biosensor for measuring of Cu2+ ion concentrations in water samples demonstrated high sensitivity and high selectivity to copper ions. Also, there is a good agreement between the results obtained by the developed nano-biosensor and those obtained by the conventional AAS method. Therefore, it may have a good potential application for the measurement of Cu2+ concentration in water samples.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors would like to thank the Research Center for New Technologies in Life Science Engineering, University of Tehran for its financial support.References
- G. March, T. D. Nguyen and B. Piro, Biosensors, 2015, 5, 241–275 CrossRef CAS PubMed.
- O. Domínguez-Renedo, M. A. Alonso-Lomillo and M. J. Arcos-Martínez, Crit. Rev. Environ. Sci. Technol., 2013, 43, 1042–1073 CrossRef.
- M. Li, H. Gou, I. Al-Ogaidi and N. Wu, J. Am. Chem. Soc., 2013, 1, 713–723 CAS.
- L. Pujol, D. Evrard, K. Groenen-Serrano, M. Freyssinier, A. Ruffien-Cizsak and P. Gros, Front. Chem., 2014, 2, 1–24 CAS.
- N. Verma and M. Singh, BioMetals, 2005, 18, 121–129 CrossRef CAS.
- L. Cui, J. Wu and H. Ju, Biosens. Bioelectron., 2015, 63, 276–286 CrossRef CAS PubMed.
- F. Li, Y. Feng, C. Zhao and B. Tang, Chem. Commun., 2011, 47, 11909–11911 RSC.
- M. R. A. Manap, N. Yusof, S. M. M. Nor and F. B. J. Ahmad, Orient. J. Chem., 2010, 26, 23–29 Search PubMed.
- E. Chow and J. J. Gooding, Electroanalysis, 2006, 18, 1437–1448 CrossRef CAS.
- W. Yang, D. Jaramillo, J. J. Gooding, D. B. Hibbert, R. Zhang, G. D. Willett and K. J. Fisher, Chem. Commun., 2001, 0, 1982–1983 RSC.
- E. Chow, D. B. Hibbert and J. J. Gooding, Analyst, 2005, 130, 831–837 RSC.
- A. Foroushani, Y. Zhang, D. Li, M. Mathesh, H. Wang, F. Yan, C. J. Barrow, J. He and W. Yang, Chem. Commun., 2015, 51, 2921–2924 RSC.
- A. C. Liu, D. C. Chen, C. C. Lin, H. H. Chou and C. H. Chen, Anal. Chem., 1999, 71, 1549–1552 CrossRef CAS.
- G. Aragay and A. Merkoçi, Electrochim. Acta, 2012, 84, 49–61 CrossRef CAS.
- L. Wang, W. Ma, L. Xu, W. Chen, Y. Zhu, C. Xu and N. A. Kotov, Mater. Sci. Eng., R, 2010, 70, 265–274 CrossRef.
- L. Zhang and M. Fang, Nano Today, 2010, 5, 128–142 CrossRef CAS.
- E. Eltzov, S. Cosnier and R. S. Marks, Expert Rev. Mol. Diagn., 2011, 11, 533–546 CrossRef CAS.
- J. Turkevich, P. C. Stevenson and J. Hillier, Discuss. Faraday Soc., 1951, 11, 55–75 RSC.
- G. Frens, Nature: Physical science., 1973, 241, 20–22 CAS.
- L. Zhang, C. Xu, G. Song and B. Li, RSC Adv, 2015, 5, 27003–27008 RSC.
- W. Yang, J. J. Gooding and D. B. Hibbert, J. Electroanal. Chem., 2001, 516, 10–16 CrossRef CAS.
- W. Yang, E. Chow, G. D. Willett, D. B. Hibbert and J. J. Gooding, Analyst, 2003, 128, 712–718 RSC.
- F. Keshvari, M. Bahram and A. A. Farshid, Anal. Methods, 2015, 7, 4560–4567 RSC.
- S. Devi, B. Singh, A. Paul and S. Tyagi, Anal. Methods, 2016, 8, 4398–4405 RSC.
- M. Ashjari, S. Dehfuly, D. Fatehi, R. Shabani and M. Koruji, RSC Adv, 2015, 5, 104621–104628 RSC.
- W. Haiss, N. T. Thanh, J. Aveyard and D. G. Fernig, Anal. Chem., 2007, 79, 4215–4221 CrossRef CAS PubMed.
- A. Rossi, S. Donati, L. Fontana, F. Porcaro, C. Battocchio, E. Proietti, I. Venditti, L. Bracci and I. Fratoddi, RSC Adv, 2016, 6, 99016–99022 RSC.
- S. Wang and D. Du, Sensors, 2002, 2, 41–49 CrossRef CAS.
- B. Feng, R. Zhu, S. Xu, Y. Chen and J. Di, RSC Adv, 2018, 8, 4049–4056 RSC.
- P. Dong, Y. Lin, J. Deng and J. Di, ACS Appl. Mater. Inter., 2013, 5, 2392–2399 CrossRef CAS.
- J. I. A. Rashid, N. A. Yusof, J. Abdullah, U. Hashim and R. Hajian, Mater. Sci. Eng. C, 2014, 45, 270–276 CrossRef CAS PubMed.
- J. Wang, L. Wang, J. Di and Y. Tu, Talanta, 2009, 77, 1454–1459 CrossRef CAS PubMed.
- J. Du and J. Di, Adv. Mat. Res., 2011, 287–290, 2271–2274 CAS.
- V. G. Praig, G. Piret, M. Manesse, X. Castel, R. Boukherroub and S. Szunerits, Electrochim. Acta, 2008, 53, 7838–7844 CrossRef CAS.
- X. Sun and H. Li, Nanotechnology, 2013, 24, 355706–355715 CrossRef PubMed.
- J. Mocak, A. Bond, S. Mitchell and G. Scollary, Pure Appl. Chem., 1997, 69, 297–328 CAS.
- World Health Organization, Guidelines for Drinking-Water Quality, Geneva, 4th edn, 2011 Search PubMed.
- World Health Organization, European Standards for Drinking Water, Geneva, 2nd edn, 1970 Search PubMed.
- USEPA, National Primary Drinking Water Regulations EPA 816-F-09-004, United States Environmental Protection Agency, 2009 Search PubMed.
- D. W. M. Arrigan and L. L. Bihan, Analyst, 1999, 124, 1645–1649 RSC.
- W. Yang, J. J. Gooding and D. B. Hibbert, Analyst, 2001, 126, 1573–1577 RSC.
- Y. S. Kim, G. J. Park, S. A. Lee and C. Kim, RSC Adv, 2015, 5, 31179–31188 RSC.
- P.-H. Chen, C. Lin, K.-H. Guo and Y.-C. Yeh, RSC Adv, 2017, 7, 29302–29305 RSC.
- X. Yang, W. Zhang, Z. Yi, H. Xu, J. Wei and L. Hao, New J. Chem., 2017, 41, 11079–11088 RSC.
- Y. Yu, P. Wang, X. Zhu, Q. Peng, Y. Zhou, T. Yin, Y. Liang and X. Yin, Analyst, 2018, 143, 323–331 RSC.
- H. Chen, J. Zhang, X. Liu, Y. Gao, Z. Ye and G. Li, Anal. Methods, 2014, 6, 2580–2585 RSC.
- C. Durgadas, C. Sharma and K. Sreenivasan, Analyst, 2011, 136, 933–940 RSC.
- C.-Y. Tsai and Y.-W. Lin, Analyst, 2013, 138, 1232–1238 RSC.
- H. Cao, W. Shi, J. Xie and Y. Huang, Anal. Methods, 2011, 3, 2102–2107 RSC.
- B. Xia, W. Zhang, J. Shi and S. Xiao, Analyst, 2013, 138, 3629–3632 RSC.
- X. Yuan and Y. Chen, Analyst, 2012, 137, 4516–4523 RSC.
- Y. Wang, F. Yang and X. Yang, Nanotechnology, 2010, 21, 205502–205508 CrossRef.
- J.-M. Liu, H.-F. Wang and X.-P. Yan, Analyst, 2011, 136, 3904–3910 RSC.
- Y.-F. Lee, T.-W. Deng, W.-J. Chiu, T.-Y. Wei, P. Roy and C.-C. Huang, Analyst, 2012, 137, 1800–1806 RSC.
- B. Viguier, K. Zór, E. Kasotakis, A. Mitraki, C. H. Clausen, W. E. Svendsen and J. Castillo-León, ACS Appl. Mater. Inter., 2011, 3, 1594–1600 CrossRef CAS PubMed.
- K. Yin, Y. Wu, S. Wang and L. Chen, Sens. Actuators, B, 2016, 232, 257–263 CrossRef CAS.
- K. Li, N. Li, X. Chen and A. Tong, Anal. Chim. Acta, 2012, 712, 115–119 CrossRef CAS.
- W. Pooi See, S. Nathan and L. Y. Heng, J. Sensors, 2011, 2011, 1–5 CrossRef.
- J. Wang, C. Bian, J. Tong, J. Sun and S. Xia, Thin Solid Films, 2012, 520, 6658–6663 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra09659g |
| This journal is © The Royal Society of Chemistry 2019 |