
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTolerance against butanol stress by disrupting succinylglutamate desuccinylase in Escherichia coli†
Yuan Guo a,
Bo Lua,
Hongchi Tanga,
Dewu Bib,
Zhikai Zhangb,
Lihua Lin*a and
Hao Pang*a
a,
Bo Lua,
Hongchi Tanga,
Dewu Bib,
Zhikai Zhangb,
Lihua Lin*a and
Hao Pang*a
aGuangxi Academy of Sciences, Nanning 530007, China. E-mail: 1201guoyuan@163.com; lubo45@126.com; 346275185@qq.com; linlihua@gxas.cn; panghouse@126.com; Fax: +86-771-2503940; Tel: +86-771-2503973
bGuangxi University, Nanning 530004, China. E-mail: 514373062@qq.com; 1278463102@qq.com
First published on 15th April 2019
Abstract
Background: The four-carbon alcohol, butanol, is emerging as a promising biofuel and efforts have been undertaken to improve several microbial hosts for its production. However, most organisms have very low tolerance to n-butanol (up to 2% (v/v)), limiting the economic viability of butanol production. Although genomic tools (transcriptomics, proteomics, and metabolomics) have been widely used to investigate the cellular response to butanol stress, the existing knowledge of the molecular mechanisms involved in butanol tolerance is limited, and strain improvement is difficult due to the complexity of the regulatory network. Results: In this study, a butanol-tolerant Escherichia coli was constructed by disrupting gene astE (encoding succinylglutamate desuccinylase) to obtain higher butanol tolerance (increased by 34.6%). To clarify the tolerance mechanism, a metabolome analysis was also performed. As a result, a total of 73 metabolites (11 elevated and 62 decreased) were significantly changed. Most of the downregulated metabolites were mainly involved in the L-arginine degradation pathway, sulfate metabolic pathway, and 2-methylcitrate metabolic pathway. To further analyze the differential gene expression, a transcriptome was created. In total, 311 genes (113 upregulated and 198 downregulated) showed over a twofold difference and were associated with carbohydrate metabolism, energy metabolism, and ABC transporters. The integration of metabolomics and transcriptomics found that acid-activated glutaminase ybaS and the amino acid antiporter gadC were significantly up-regulated, but the levels of L-arginine and glutamate were not significantly increased and decreased. Therefore, the changes of amino acids between strains BW25113 and BW25113-ΔastE were measured by amino acid analysis. The ability of a mutant strain against acid stress was also measured by the growth experiment under various pH conditions in the absence of butanol. Conclusions: Based on the above experiments, it could be concluded that mutant BW25113-ΔastE mainly regulated intracellular pH-homeostasis to adapt to butanol stress, indicating the non-negligible impact of pH on microbial butanol tolerance, broadening our understanding of microbial butanol tolerance and providing a novel strategy for the rational engineering of a more robust butanol-producing host.
Background
Recently, global environmental problems and fuel crises have drawn public attention toward the development of microbial production for fuels and chemicals, such as ethanol, higher alcohols,1,2 biodiesel,3 and alkanes.4 However, most of them are toxic to microbial cells, leading to a low fermentation titer and high cost in downstream product recovery.5 For example, butanol is a promising biofuel but is highly toxic to microbes6 (such as Clostridia, Escherichia coli, and Saccharomyces cerevisiae),7 and only 13 g L−1 of butanol is sufficient to restrain cell growth, which is a major drawback in practical butanol production.8 Therefore, understanding the mechanisms of the response to butanol and improving microbial robustness will further ameliorate butanol toxicity, and maintain microbial stability and the dynamic homeostasis of butanol fermentation.9To date, several mechanisms underlying the physiological responses to solvent have been identified in eukaryotic and prokaryotic microorganisms. The hydrophobic nature and lipid solubility of butanol could: (i) alter cell membrane fluidity; (ii) denature membrane proteins; and (iii) impair membrane-related processes (e.g. nutrient transport, respiration, and photosynthetic electron transport).8 For example, butanol can effectively inhibit the activities of membrane-bound ATPases, resulting in a lower internal pH and the abolishment of a pH gradient across the membrane.10 Furthermore, n-butanol stress also promotes the release of autolysin, which can hydrolyze bacterial components by breaking down the b-1,4-bond between N-acetylmuramic acid and N-acetylglucosamine molecules.11 However, the butanol tolerance mechanism is complex and the existing knowledge of genes involved in butanol tolerance needs to be further expanded. The strategies for strain improvement should consider contributes synergistically to obtain a strain with the desired properties.12
At present, many strategies have been carried out to improve cellular robustness for butanol, including classical mutagenesis, metabolic engineering, and transcriptional engineering.13 For example, a genomic library enrichment strategy was performed for E. coli, in which approximately 270 candidate genes (enriched or deleted) were identified against n-butanol stress.14 Interestingly, the deletion of gene astE, encoding succinylglutamate desuccinylase, significantly enhanced n-butanol tolerance with increases of 48.7%,14 but the physiological mechanism has not been comprehensively elucidated (Fig. S1†). AstE gene encodes succinyl-glutamate desuccinylase, which catalyzes the fifth and final reaction in the ammonia-producing arginine catabolic pathway,15 can decompose succinyl-glutamate into succinic acid and glutamate, thus regulating the intracellular arginine concentration. Deletion of astE enhances tolerance to n-butanol.16 In this study, we aimed to comprehensively elucidate the tolerance mechanism for the disruption of the astE gene in E. coli. To this end, a butanol-tolerant strain was constructed by disrupting gene astE, to obtain a higher butanol tolerance (increase by 34.6%). In addition, to clarify the tolerance mechanism, metabolome and transcriptome analyses were performed to assess differential gene expression and related metabolic pathways. It was found that mutant BW25113-ΔastE had developed a special tolerance through regulating cellular pH-homeostasis. This expands the current knowledge on butanol tolerance and provides a novel strategy for the rational engineering of a more robust butanol-producing host.
Results
Effects of knocking-out astE on cellular robustness
To investigate the effect of knocking-out astE on cell growth under butanol stress, a spotting assay on agar plates containing different butanol concentrations was applied. Compared to the wild strain BW25113, the mutant BW25113-ΔastE could grow well on agar plates containing 8 g L−1 of butanol (Fig. S2†). Furthermore, cell tolerance was also evaluated in shake-flask cultures. As shown in Fig. 1a, under control conditions (0 g L−1 butanol), strain BW25113 and BW25113-ΔastE showed a similar growth ability, respectively, indicating that knocking-out gene astE had no effect on cell growth. However, when butanol stress was increased to 8 g L−1, the biomass of BW25113-ΔastE could reach 0.51 after cultivating for 24 h, which is an increase of 34.6% compared with that of BW25113 (Fig. 1b). Therefore, knocking-out gene astE could effectively improve cellular robustness, showing a higher butanol tolerance (increased by 34.6%), which agreed with a previous study.14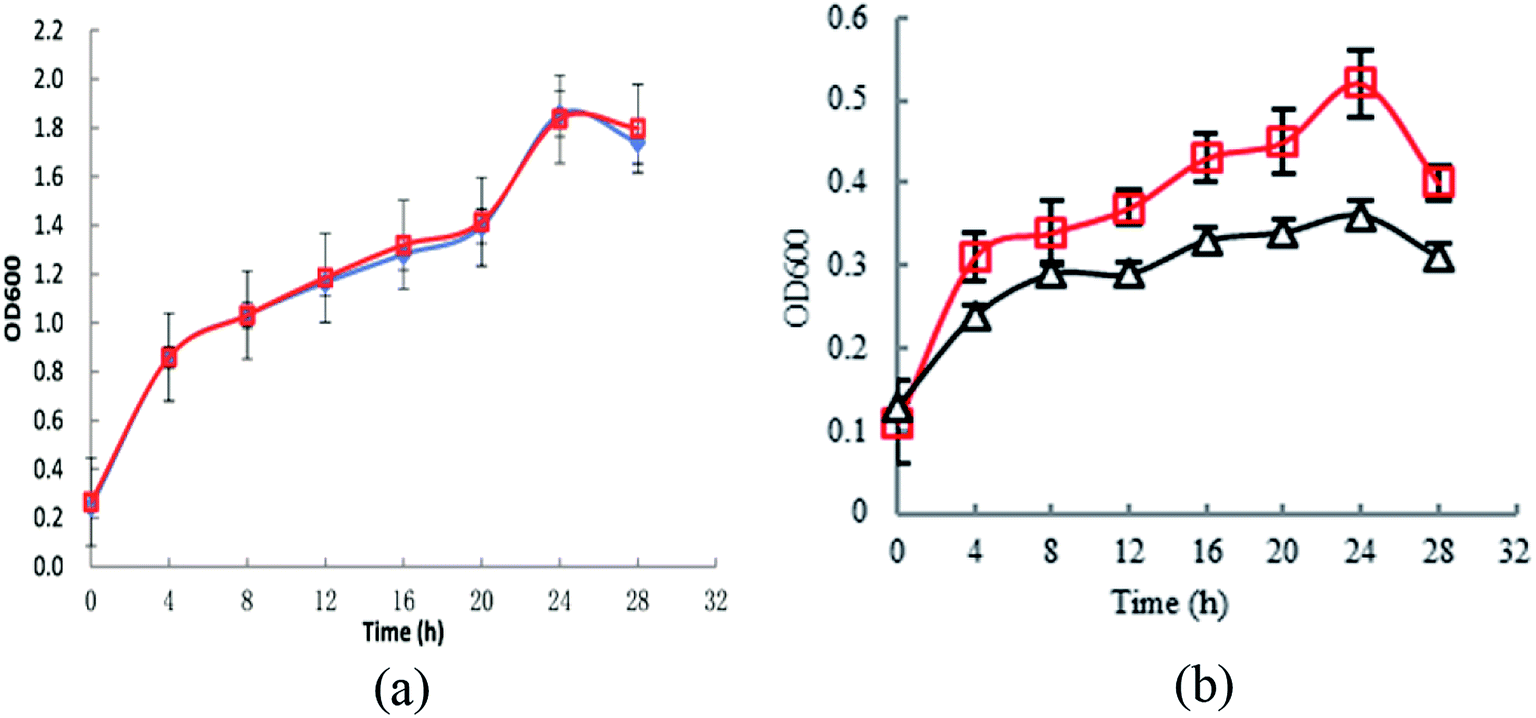 | ||
| Fig. 1 Effects of knocking-out gene astE on cell growth under different butanol concentrations: (a) 0 g L−1; (b) 8 g L−1 (△: BW25113; □: BW25113-ΔastE). | ||
GC-MS based metabolomics analysis
To further investigate the tolerance achieved by knocking-out gene astE, the difference in metabolic responses between BW25113 and BW25113-ΔastE after exposure to 0 g L−1 and 5 g L−1 butanol was determined with gas chromatography-mass spectrometry (GC-MS). Metabolite profiles were generated and data matrixes were analysis by the principal component analysis (PCA) (see Fig. 2). In which the differences of 76.7% in the metabolite profiles were explained by the first principal component. Compared to the wild strain, mutant strains under the stress of 0 g L−1 or 5 g L−1 butanol showed relatively concentrated at PCA analysis, indicating that mutant strain has better robustness under butanol stress.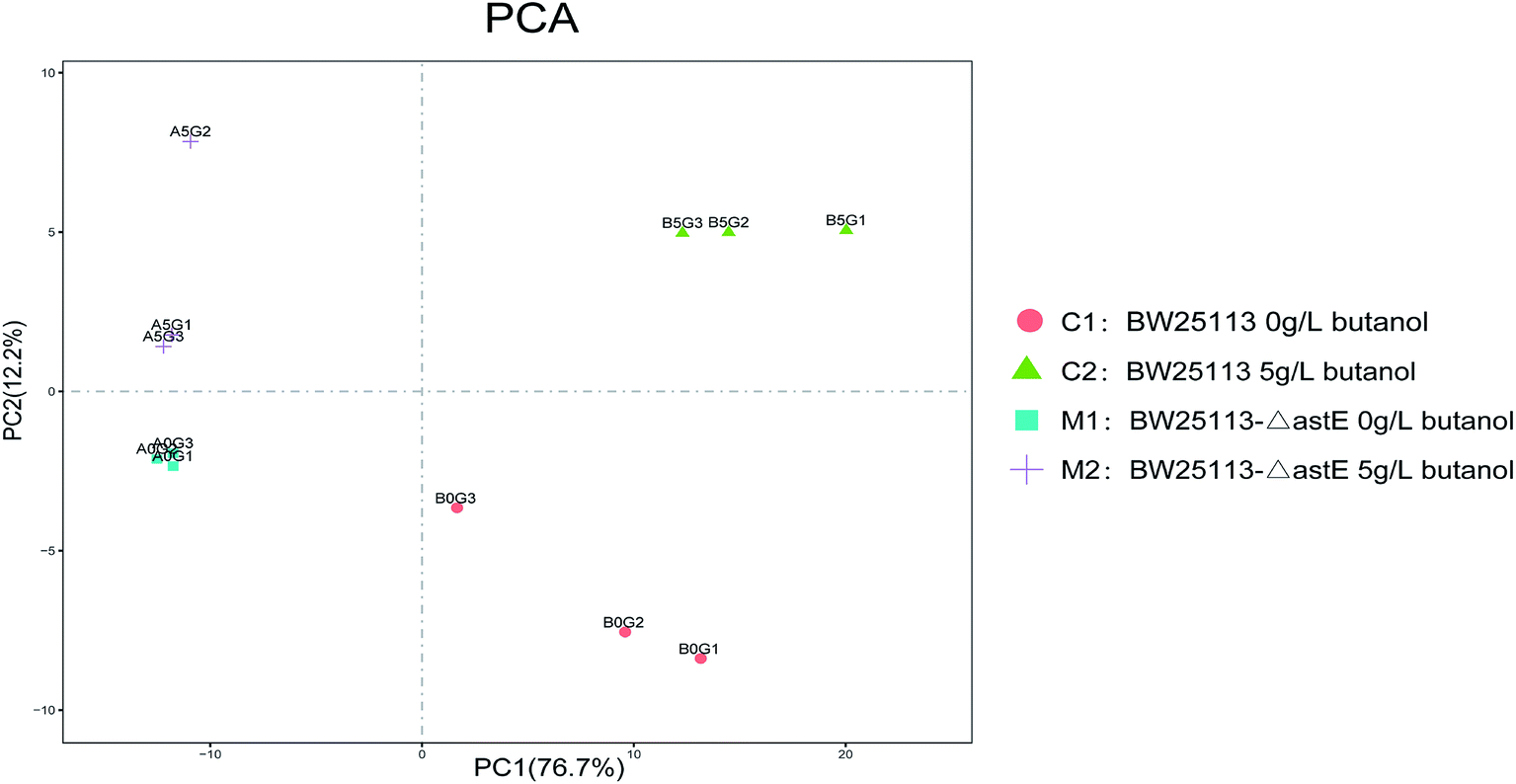 | ||
| Fig. 2 Principal component analysis of strains in the presence and absence of butanol. | ||
Meanwhile, under the 5 g L−1 butanol stress, there was a lots of differential metabolites between the wild strain and mutant strain based on the metabolomic cloud plot (Fig. S3†). The size of each bubble corresponds to the log fold change of the feature, the larger the bubble the bigger the log fold change. The colors of features with low p-values are brighter than which features with high p-values.
Therefore, we select the wild and mutant strain under the stress of 5 g L−1 butanol for detailed metabolite analysis and transcriptome analysis.
In order to get a better discrimination between BW25113 and BW25113-ΔastE after exposure to 5 g L−1 butanol, orthogonal partial least square-discriminant analysis (OPLS-DA) was applied in this study (Fig. 3). PCA and OPLS-DA analysis were performed using the OmicShare tools. The OPLS-DA showed a clear separation of samples into two distinct groups, indicating that BW25113 and BW25113-ΔastE had a significantly different metabolic profile. Score plots using the first two principal components were used to present a 2D representation of variations among the spectra. From the metabolic cloud plot in Fig. S3,† after exposure to 5 g L−1 butanol, a total of 73 metabolites were significantly changed between BW25113 and BW25113-ΔastE, of these 11 were significantly elevated, including 6-carboxy-5,6,7,8-tetrahydropterin, thymidine, 2-deoxycytidine, 7,8-dihydromonapterin, L-phenylalanine, 1-myristoyl-2-palmitoleoyl phosphatidate, L-leucine, γ-butyrobetaine, and L-valine, whereas 62 of them, such as 6-deoxy-6-sulfo-D-fructose 1-phosphate, and N-acetylmuramate, were significantly decreased (Table S1†).
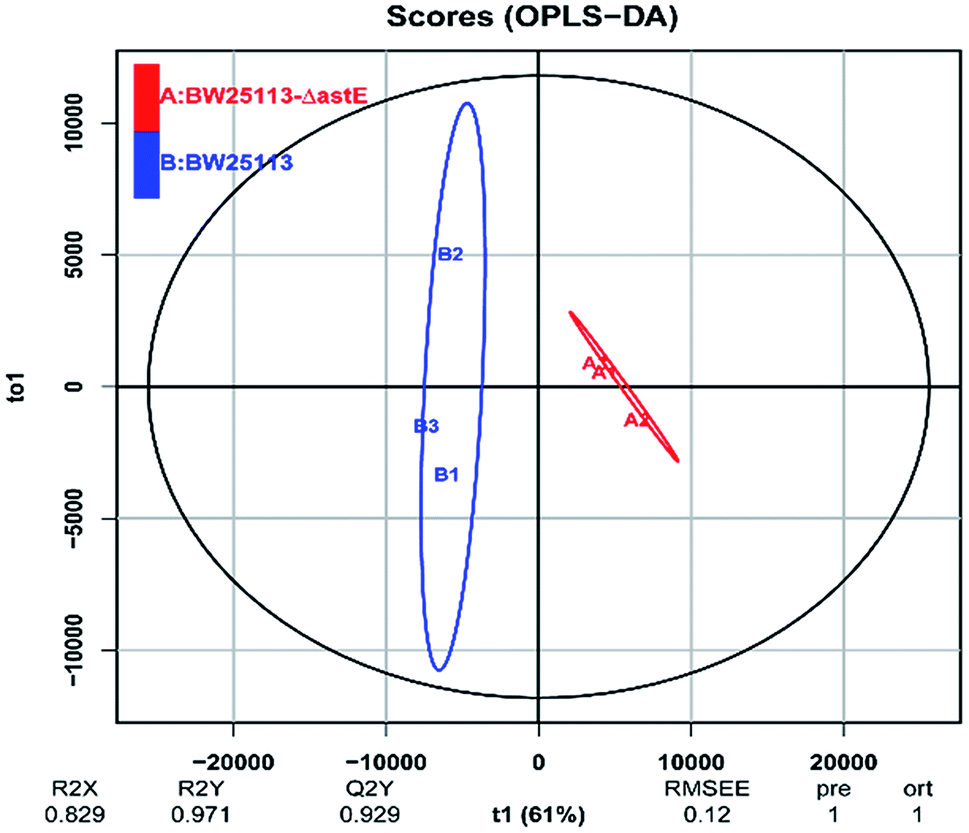 | ||
| Fig. 3 Orthogonal partial least square-discriminant analysis of strains BW25113 and BW25113-ΔastE after exposure to 5 g L−1 butanol. | ||
Investigation of the tolerance mechanism by transcriptomics
To further elucidate the tolerance mechanism of knocking out gene astE, transcriptomics of strains BW25113 and BW25113-ΔastE were analyzed after exposure to 5 g L−1 butanol stress. Reviewer/collaborator link to metadata: http://ftp://ftp-trace.ncbi.nlm.nih.gov/sra/review/SRP179828_20190117_093516_305cf1fb13b9539dcd317a0354c9ed61.As shown in Fig. S4,† based on the corrected P-value < 0.05, the results showed that 311 genes were considered to have a significantly different expression in response to butanol stress, of these, 113 genes were upregulated and 198 genes were downregulated.
GO enrichment analysis of the differentially expressed genes
GO enrichment analysis was performed to cluster genes with a significant difference in transcription level. The same types of genes were then gathered in a cluster with similar biological functions to intuitively understand the relationships and discrepancy of the samples (Fig. S5†). According to sequence homology, the differentially expressed genes were categorized into three groups: biological process, cellular component, and molecular function.In the biological process group, the enriched GO terms for downregulated genes included 17 subcategories. The maximum number of genes in one subcategory was found in the oxidation-reduction process, which included 25 genes that were significantly altered. Furthermore, the fatty acid metabolic process (involving eight genes) and mono-carboxylic-acid metabolic process (involving 10 genes) were also significantly downregulated. However, only 10 subcategories were found in the enriched GO terms for upregulated genes. Of these subcategories, the oxidation-reduction process, mono-carboxylic-acid metabolic process, and glutamine metabolic process were significantly upregulated, involving 12, 3, and 2 genes, respectively.
Additionally, the cellular component category, which included five subcategories, was also found in the enriched GO terms for different regulated genes. The altered genes related to cytoplasmic parts was significantly upregulated, involving 8 genes. Interestingly, in the molecular function group, upregulated genes were found in four subcategories: rRNA binding, squalene monooxygenase activity, oxidoreductase activity, and malate dehydrogenase activity. However, downregulated genes were only identified in three subcategories: oxidoreductase activity, malate dehydrogenase activity, and acyl-CoA dehydrogenase activity, and only a few genes were involved.
KEGG enrichment analysis of differentially expressed genes
To identify the function of differentially expressed genes, an enrichment analysis for specific metabolic pathways was carried out using the KEGG database. Differentially expressed genes were mainly involved in carbohydrate metabolism, energy metabolism, amino acid metabolism, ABC transporters, and microbial metabolism in diverse environments (Fig. S6†). As shown in Table 1, to adapt to butanol stress, most genes (such as cysH, cysC, cysN, cysJ, and cysD) involved in sulfur metabolism were downregulated by more than twofold (Fig. S7a†). In addition, genes related to propanoate metabolism were also significantly downregulated by over 8-fold, in which the most change was found in gene prpD (encoding 2-methylcitrate dehydratase) of over 16-fold (Fig. S7b†).| KEGG pathway | Gene name | Description | Corrected P-valuea | log 2-fold changeb |
|---|---|---|---|---|
| a A hypergeometric test was used for statistical analysis, and P-values have been corrected for multiple testing by the Benjamini and Hochberg adjustment method. A corrected P value of <0.05 is considered statistically significant.b log 2-fold change of differential expression; “+” means up-regulated genes, “−” means down-regulated genes. | ||||
| Transporters | fecC | Ferric citrate ABC transporter permease | 1.70 × 10−4 | +2.3 |
| sbp | Sulfate transporter subunit | 3.10 × 10−3 | −1.2 | |
| fecB | Ferric citrate ABC transporter periplasmic binding protein | 3.13 × 10−9 | +1.8 | |
| cysU | Sulfate/thiosulfate ABC transporter permease | 1.22 × 10−6 | −1.1 | |
| cysW | Sulfate/thiosulfate ABC transporter permease | 1.25 × 10−6 | −1.1 | |
| araG | L-arabinose ABC transporter ATPase | 3.28 × 10−2 | −1.0 | |
| znuC | Zinc ABC transporter ATPase | 5.11 × 10−3 | +1.0 | |
| ddA | D% 2CD-dipeptide ABC transporter periplasmic binding protein | 8.55 × 10−4 | −1.3 | |
| fadL | Long-chain fatty acid outer membrane transporter | 1.51 × 10−13 | −1.2 | |
| psuT | Putative nucleoside transporter | 3.76 × 10−5 | +1.3 | |
| ybaT | Putative amino acid transporter | 3.13 × 10−9 | +1.1 | |
| lplT | Lysophospholipid transporter | 1.90 × 10−5 | +1.0 | |
| ynfM | Putative arabinose efflux transporter | 1.17 × 10−12 | −1.2 | |
| uhpT | Hexose phosphate transporter | 1.19 × 10−3 | −1.2 | |
| yihO | Putative sulphoquinovose importer | 4.87 × 10−2 | −1.4 | |
| narU | Nitrate/nitrite transporter | 6.34 × 10−4 | −1.5 | |
| Membrane | yhiI | Putative membrane fusion protein (MFP) of efflux pump | 1.85 × 10−4 | +1.1 |
| frdC | Fumarate reductase (anaerobic)% 2C membrane anchor subunit | 1.79 × 10−3 | +1.3 | |
| mdtM | Multidrug efflux system protein | 6.48 × 10−5 | +1.5 | |
| fecA | TonB-dependent outer membrane ferric citrate transporter and signal transducer% 3B ferric citrate extracellular receptor% 3B FecR-interacting protein | 4.57 × 10−12 | +1.5 | |
| yjhB | Putative MFS transporter% 2C membrane protein | 3.24 × 10−2 | −1.5 | |
| slp | Outer membrane lipoprotein | 3.93 × 10−10 | +1.0 | |
| nlpA | Cytoplasmic membrane lipoprotein-28 | 1.79 × 10−4 | −1.5 | |
| ubiI | 2-Octaprenylphenol hydroxylase% 2C FAD-dependent | 6.13 × 10−3 | +1.0 | |
| chbC | N% 2CN′-diacetylchitobiose-specific enzyme IIC component of PTS | 1.34 × 10−10 | −1.9 | |
| cysN | Sulfate adenylyltransferase% 2C subunit 1 | 3.98 × 10−6 | −1.4 | |
| yjhB | Putative MFS transporter% 2C membrane protein | 3.24 × 10−2 | −1.5 | |
| Transcriptional | yebK | Putative DNA-binding transcriptional regulator | 1.74 × 10−6 | −1.2 |
| cbl | ssuEADCB/tauABCD operon transcriptional activator | 3.91 × 10−2 | −1.1 | |
| Pyruvate metabolism | aldB | Aldehyde dehydrogenase B | 1.38 × 10−10 | −1.0 |
| frdC | Fumarate reductase (anaerobic)% 2C membrane anchor subunit | 1.79 × 10−3 | +1.3 | |
| Propanoate metabolism | prpE | Propionate-CoA ligase | 1.33 × 10−13 | −3.0 |
| prpD | 2-Methylcitrate dehydratase | 5.84 × 10−7 | −4.0 | |
| prpB | 2-Methylisocitrate lyase | 3.82 × 10−32 | −3.6 | |
| prpC | 2-Methylcitrate synthase | 1.10 × 10−10 | −3.8 | |
| fadB | Fused 3-hydroxybutyryl-CoA epimerase/delta(3)-cis-delta(2)-trans-enoyl-CoA isomerase/enoyl-CoA hydratase/3-hydroxyacyl-CoA dehydrogenase | 2.47 × 10−5 | −1.1 | |
| Sulfur metabolism | cysN | Sulfate adenylyltransferase% 2C subunit 1 | 3.98 × 10−6 | −1.4 |
| cysC | Adenosine 5′-phosphosulfate kinase | 2.76 × 10−3 | −1.3 | |
| cysD | Sulfate adenylyltransferase% 2C subunit 2 | 3.33 × 10−7 | −1.8 | |
| cysU | Sulfate/thiosulfate ABC transporter permease | 1.22 × 10−6 | −1.1 | |
| cysW | Sulfate/thiosulfate ABC transporter permease | 1.25 × 10−6 | −1.0 | |
| cysH | Phosphoadenosine phosphosulfate reductase% 3B PAPS reductase% 2C thioredoxin dependent | 3.75 × 10−6 | −1.4 | |
| cysI | Sulfite reductase% 2C beta subunit% 2C NAD(P)-binding% 2C heme-binding | 1.07 × 10−15 | −1.5 | |
| cysJ | Sulfite reductase% 2C alpha subunit% 2C flavoprotein | 3.22 × 10−6 | −1.3 | |
| sbp | Sulfate transporter subunit | 3.10 × 10−3 | −1.2 | |
| Nucleotide metabolism | cysN | Sulfate adenylyltransferase% 2C subunit 1 | 3.98 × 10−6 | −1.4 |
| cysC | Adenosine 5′-phosphosulfate kinase | 2.76 × 10−3 | −1.3 | |
| cysD | Sulfate adenylyltransferase% 2C subunit 2 | 3.33 × 10−7 | −1.8 | |
| psuK | Pseudouridine kinase | 5.87 × 10−8 | +3.7 | |
| psuG | Pseudouridine 5′-phosphate glycosidase | 4.35 × 10−10 | +2.7 | |
| Acid resistance | gadC | glutamate:gamma-aminobutyric acid antiporter | 1.49 × 10−5 | +0.9 |
| patD | Gamma-aminobutyraldehyde dehydrogenase | 2.05 × 10−3 | −0.7 | |
| ariR | RcsB connector protein for regulation of biofilm and acid-resistance | 1.68 × 10−73 | −3.3 | |
| ybaS | Glutaminase 1 | 1.03 × 10−7 | +1.1 | |
In contrast, most genes (such as yhiI, frdC, mdtM, fecA, fecC, and fecB) involved in membrane metabolism and transporters were upregulated by more than twofold. More interestingly, genes, related to nucleotide metabolism psuK and psuG, and acid-activated glutaminase ybaS and the amino acid antiporter gadC, were significantly upregulated.
Validation of transcription results by RT-qPCR
Real-time quantitative PCR (RT-qPCR) was performed to confirm the transcription results using differentially expressed genes. A group of 7 upregulated and 13 downregulated genes involving different metabolism pathways was randomly selected for validation. As shown in Fig. 4, the RT-qPCR and transcription analysis results correlated well with each other, and with similar trends and expression levels between the transcription and RT-qPCR analysis, indicating that the RNA-Seq results were credible. | ||
| Fig. 4 Real-time qPCR analysis of the selected genes. | ||
Identification of significant metabolites and pathway analysis
To characterize the underlying metabolic impact of knocking-out gene astE under butanol stress, a hierarchical clustering was conducted to assist in accurately screening for marker metabolites (Fig. S8†), and then the related metabolic processes were investigated by integrating transcriptomics and metabolomics. As shown in Fig. 5, most of the differential metabolites were mainly involved in the L-arginine degradation pathway, sulfate metabolic pathway, and 2-methylcitrate metabolic pathway, and were downregulated to adapt to butanol stress. Unexpectedly, although the L-arginine degradation pathway was suppressed due to the knocking-out of gene astE, significantly decreasing most of the intermetabolites, the levels of L-arginine and glutamate did not appear to change, indicating that L-arginine and glutamate played an important role in improving cell robustness.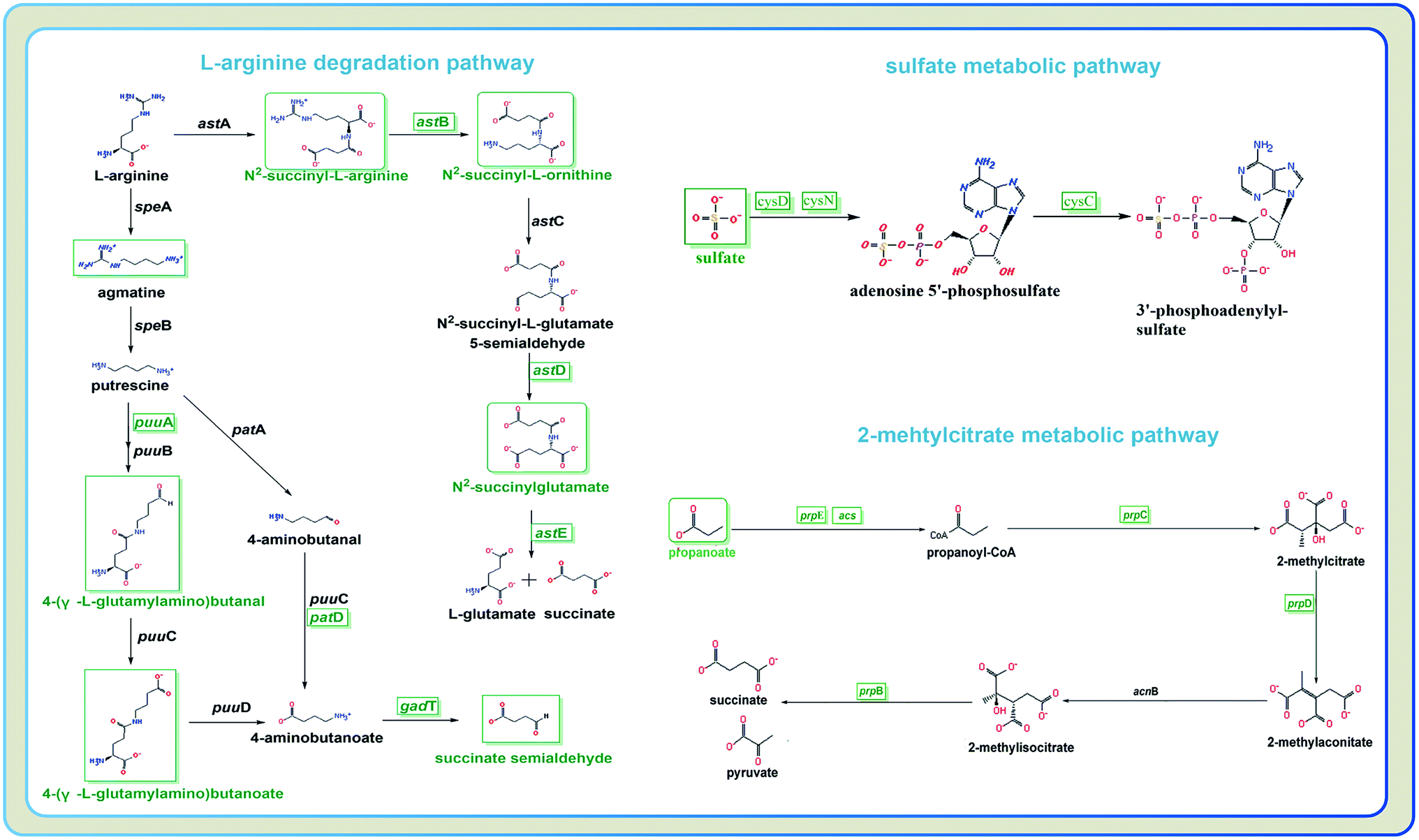 | ||
| Fig. 5 Different metabolites and genes in pathways: L-arginine degradation, sulfate metabolic pathway, and 2-methylcitrate metabolic pathway. | ||
However, interestingly, the acid-activated glutaminase ybaS and the amino acid antiporter gadC, mainly involved in the glutamine metabolic process, were significantly upregulated (Fig. 6). In the glutamine metabolic process, glutamine is converted into glutamate by ybaS, while NH3 is generated, which reacts with H+ to alkalize the environment. In addition, gadC, a glutamic acid γ-aminobutyrate antiporter, is part of the glutamate-dependent acid resistance system 2 (AR2) which confers resistance to extreme acid conditions.17 Insertional inactivation of the gadC gene results in sensitivity to extreme acid conditions (pH 2–3) and eliminates glutamic acid enhancement of acid resistance,18 which is similar to the acid resistance system for E. coli survival under an extremely acidic environment.19 To further demonstrate the hypothesis, the effect of knocking-out astE on amino acids were also investigated using the amino acid analyzer (Table S2†). Compared with strain BW25113, the mutant BW25113-ΔastE could significantly enhance the synthesis of some amino acids, such as Met, Val, and Tyr. It is interesting that the concentration of extracellular glutamate and free ammonia (NH3) in mutant BW25113-ΔastE was significantly increased. Furthermore, the growth experiment under various pH condition in absence of butanol was used to confirm the ability of mutant strain against acid stress. In the acid-stress experiment (Fig. 6), when extracellular pH decreased to 3.0, cell growth effectively improved in mutant BW25113-ΔastE and biomass reached 0.17 after 24 h of cultivation, which is an increase of 54.5% compared with that of BW25113.
 | ||
| Fig. 6 The upregulated genes mainly involved in glutamine metabolic process regulated the intracellular pH-homeostasis to adapt to butanol stress: L-glutamine degradation I; L-glutamate:4-aminobutyrate antiporter; impact of different pH stresses; acid resistance system. | ||
Discussion
During microbial fermentation, microorganisms are usually exposed to fluctuations in solvent concentration, osmotic pressure, pH, nutrient availability, and temperature.20 Particularly, in acetone–butanol–ethanol fermentation, butanol is highly toxic to microorganisms due to its hydrophobicity, and severely inhibits cell growth and the production of solvent.21 Therefore, understanding the mechanisms involved in the n-butanol response can help to facilitate the engineering of hosts with improved tolerance.22 In the present study, a butanol tolerance strain, with higher butanol tolerance (increased by 34.6%) than that of the control strain, was successfully engineered by the disruption of gene astE.To elucidate the mechanism of tolerance against butanol stress after deleting gene astE, metabolic responses were determined by using gas chromatography-mass spectrometry (GC-MS). As a result, a total of 73 metabolites were significantly changed between BW25113 and BW25113-ΔastE. Most of the differential metabolites were mainly involved in the L-arginine degradation pathway, sulfate metabolic pathway, and 2-methylcitrate metabolic pathway and were downregulated. Unexpectedly, although the L-arginine degradation pathway was suppressed due to the knocking-out of gene astE, decreasing most the intermetabolites significantly, the levels of L-arginine and glutamate did not appear to change.
To further elucidate the tolerance mechanism, a transcription analysis was also performed, and found that 311 genes (113 upregulated and 198 downregulated) showed different expression levels, which mainly involved carbohydrate metabolism, energy metabolism, nucleotide metabolism, amino acid metabolism, ABC transporters and microbial metabolism in diverse environments. For example, to adapt to butanol stress, most genes (such as cysH, cysC, cysN, cysJ, and cysD) involved in sulfur metabolism were downregulated by more than twofold. This was maybe due to the fact that butanol stress could effectively inhibit sulfur assimilation,23 and then further weakened by cysteine biosynthesis and the formation of cytomembrane.24 In contrast, most genes (such as yhiI, frdC, mdtM, fecA, fecC, and fecB) involved in membrane metabolism and transporters were upregulated by more than twofold, fecC encodes a hydrophobic inner membrane protein, increased external iron concentration and increased expression of iron transport genes (fecA, fecC, and fecB) improved E. coli butanol tolerance.25 Slp is believed to take part in acid resistance as its expression increased when cells were grown at pH 4.5 and 5.5 under conditions known to induce glutamate-dependent acid resistance.26 YhiI is a predicted bitopic inner membrane protein.27 Overexpression of yhiI leads to abnormal biofilm architecture.28 The MdtM proteins a multidrug efflux protein that belongs to the major facilitator superfamily (MFS) of transporters.29
In addition, genes related to transporters were also significantly upregulated by over twofold, suggesting that increased expression of the transporters was a physiological adaptation to stressful environments.30 In general, transporter systems consist of different transmembrane protein components and play roles in bacterial virulence, cell growth and development, and survival under various environments.31,32 Genes (such as psuK and psuG) related to nucleotide metabolism were also significantly upregulated by 6- to 15-fold. In microorganisms, nucleotides are obligatory metabolites and serve an important role in the regulation of numerous cellular processes, including cellular energy supply, signaling molecules, and are incorporated into cofactors (e.g., NAD and coenzyme A) and precursors (e.g., UDP-glucose and GDP-mannose).33 Particularly, nucleotide synthesis is phosphate consuming, involving the regulation of phosphate uptake and utilization,34 which then regulates the plasticity of the cell wall in bacteria.35
More importantly, the genes of ybaS and gadC involving in glutamine metabolism were significantly up-regulated. In E. coli, gadC, as the amino acid transporter protein, was responsible for the exchange of extracellular L-glutamine (Gln) with intracellular L-glutamic acid (Glu), and the glutamine enzyme ybaS was responsible for the conversion of Gln into Glu, releasing ammonia. As a result, the free ammonia could be used to neutralize intracellular protons, and then increased the intracellular pH to resist acidic environment. More interestingly, there was a typical acid resistance system in E. coli that relies on L-glutamine,19 in which glutamine is converted to L-glutamate by acid-activated glutaminase ybaS, with concomitant release of gaseous ammonia to neutralize protons, resulting in an elevated intracellular pH under an acidic environment. Therefore, the ybaS and the amino acid antiporter gadC, which exchanges extracellular glutamine with intracellular glutamate, together constitute an acid resistance system that is sufficient for E. coli survival under an extremely acidic environment, which was similar with our resistance system for E. coli survival under extreme butanol stress.
To further elucidate the physiological mechanism, the changes of amino acids between strains BW25113 and BW25113-ΔastE were measured by amino acid analyzer. It is interesting that the concentration of extracellular glutamate and free ammonia (NH3) in mutant BW25113-ΔastE was significantly increased. Furthermore, the mutant BW25113-ΔastE showed the better physiological performance against acid stress. When extracellular pH decreased to 3.0, cell growth of mutant increased 54.5% compared with BW25113. With the integration of amino acids analysis and an acid tolerance experiment, it could be concluded that, in the mutant BW25113-ΔastE, butanol can effectively inhibit the activities of membrane-bound ATPases, resulting in a lower internal pH.10 YbaS and GadC are activated by acidic pH. GadC exchanges intracellular L-glutamate and extracellular glutamine.19 Upon uptake into E. coli, glutamine (Gln) is converted to L-glutamate (Glu) by the acid-activated glutaminase YbaS, with concomitant release of gaseous ammonia.19 Then some of the free ammonia is transferred out of the cell, and some of the free ammonia neutralizes proton, resulting in elevated intracellular pH under acidic, thereby maintaining intracellular pH homeostasis to adapt to butanol stress.
At present, genomic tools (transcriptomics, proteomics, and metabolomics) have been widely used to investigate cellular response under butanol stress, and correspondingly, a series of strategies for improving cellular robustness was elucidated,36 including (i) metabolic detoxification; (ii) heat shock proteins; (iii) the proton motive force and associated energy production; (iv) molecular efflux pumps; (v) the changes of cell membrane composition and biophysics;37 and (vi) other transcriptional responses.38,39 However, the molecular mechanism underlying butanol tolerance is still not comprehensively understood for microorganisms, which has made strain improvement difficult due to the complexity of the regulatory network.40,41
Conclusion
In this study, a mutant strain with high butanol tolerance (increased by 34.6%) was constructed by disrupting gene astE. To clarify the tolerance mechanism, a transcriptome and metabolome were performed to analyze the differential gene expression and characterize the underlying metabolic impact. As a result, it was found that the mutant BW25113-ΔastE had developed a special tolerance mechanism by regulating intracellular pH-homeostasis to adapt to butanol stress. Our findings indicate the non-negligible impact of pH on microbial butanol tolerance, broadening the understanding on microbial butanol tolerance and also providing a novel strategy for the rational engineering of a more robust butanol-producing host.In the future, the correlation between cell growth and gene expression under the conditions of butanol and acid stresses would be investigated in detailed, providing some novel pathways for further improving cellular robustness and fermentation performance with metabolic engineering.
Materials and methods
Strains and plasmids
E. coli BW25113 was used as a receptor for gene deletion. More information about the plasmids and strains used in this study are presented in Table S3.†Construction of engineering strains
The primers were designed using E. coli k-12 MG1655 as the template and are given in Table S4.† Standard bacterial transformations were performed according to the procedures described by Sambrook.42 In all the cases, PCR was performed using TaKaRa PrimeSTAR® HS (TAKARA Bio Inc., Tokyo Japan). All the genes were sequenced to verify the insert prior to transformations. The engineered strains were transformed using electrical conversion method.43Culture medium and conditions
M9 medium: 6.78 g L−1 Na2HPO4, 3.0 g L−1 KH2PO4, 0.5 g L−1 NaCl, 1.0 g L−1 NH4Cl, 4 g L−1 glucose, 0.493 g L−1 MgSO4·7H2O, 0.011 g L−1 CaCl2;
Screened on synthetic complete (SC) medium: 20 g L−1 glucose, 7 g L−1 urea, 5 g L−1 KH2PO4, 0.8 g L−1 MgSO4·7H2O, 3 g L−1 sodium acetate, 15 g L−1 agar.
Butanol and acid tolerance curve of the BW25113 and the mutant BW25113-ΔastE: LB medium with different butanol concentrations or different PH (adjusted pH with 0.1 mol L−1 citric acid and 0.1 mol L−1 sodium citrate buffer) were prepared and inoculated with 10% (v/v) inoculum size of the BW25113 and the mutant BW25113-ΔastE, strains were inoculated and cultured at 37 °C for 24 h. OD600 was determined to detect the growth of the strains.
Fermentation was carried out in M9 medium with 0 g L−1 and 5 g L−1 butanol in a shake-flask culture (200 rpm, 37 °C) with 10% (v/v) of the inoculum size.
Analytical methods
Cell growth was determined by measuring the optical density at 600 nm (OD600) by using a UV-vis spectrophotometer (Beckman Coulter DU800). Cells were incubated to the exponential phase (OD600 = 0.6), different butanol or pH concentrations were added to the broth (200 rpm, 37 °C), and then the cell concentration was measured to represent cell growth.Spotting assay for evaluating butanol tolerance
To evaluate microbial tolerance, a spotting assay was applied in the presence of butanol.44 First, cells were incubated overnight in LB medium with shaking (200 rpm, 30 °C), and then collected by centrifugation (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g, 20 s), resuspended with sterilized water. Second, the suspension was serially diluted to an OD600 of 1 × 10−1, 1 × 10−2, 1 × 10−3, 1 × 10−4, 1 × 10−5, 1 × 10−6, and then spotted (5 μL each) onto agar plates containing different butanol concentrations. The plates were sealed with vinyl plastic tape to prevent evaporation of butanol and incubated at 37 °C.
000 × g, 20 s), resuspended with sterilized water. Second, the suspension was serially diluted to an OD600 of 1 × 10−1, 1 × 10−2, 1 × 10−3, 1 × 10−4, 1 × 10−5, 1 × 10−6, and then spotted (5 μL each) onto agar plates containing different butanol concentrations. The plates were sealed with vinyl plastic tape to prevent evaporation of butanol and incubated at 37 °C.
GC-MS-based metabolomics analysis
For metabolomics analysis, cells were collected after cultivating 12 h under different butanol concentration. For each sample, broth equivalent to 108 cells was collected by centrifugation at 8000 × g for 10 min at 4 °C. The pellets were immediately frozen by liquid nitrogen and then stored at −80 °C before using.The metabolomics analysis protocol was performed according to the following. (i) Sample preparation. Cells were thawed and resuspended in 5 mL of cold 2:2:1 (v/v/v) acetonitrile/methanol/H2O solution, and then vibrated with vortex oscillation blender for 1 min. Subsequently, cells were frozen in liquid nitrogen and thawed for five times. Supernatants were collected by centrifugation at 14000 × g for 3 min at 4 °C and dried by vacuum centrifugation. (ii) Sample derivatization. The samples were dissolved in 40 μL of 98% methoxyamine hydrochloride (40 mg mL−1 in pyridine), after shaking at 30 °C, 180 rpm for 90 min, 180 μL of N-methyl-N-(trimethylsilyl)trifluoroacetamide (MSTFA) was added and incubated at 37 °C, 180 rpm for 30 min to trimethylsilylate the polar functional groups. The derivates were then collected by centrifugation at 1, 4000 × g for 3 min, and the supernatant was used directly for GC/MS analysis. (iii) GC-MS analysis. The analysis was performed on an Agilent 5977E GC/MSD equipped with an HP-5MS capillary column (30 m × 250 mm × 0.25 μm), with 70 eV of electron impact ionization, in which 0.2 μL of sample was injected in splitless mode at 230 °C with a constant flow of 1 mL min−1 helium. The temperature program started isocratic at 45 °C for 2 min, followed by temperature ramping of 2 °C min to a final temperature of 250 °C, and then held constant for an additional 2 min (solvent delay: 6 min, scanning rate: 1250, and scanning method: full scanning). The range of mass scan was m/z 38–650. (iv) Data processing and statistical analysis. To identify the compounds, the mass fragmentation spectrum was analyzed using an automated mass spectral deconvolution and identification system (AMDIS)32 and the data was matched with the Fiehn Library 41 and the mass spectral library of the National Institute of Standards and Technology (NIST). Peak areas of all identified metabolites were normalized against the internal standard and the acquired relative abundances for each identified metabolite were used for data analysis. All metabolomics profile data were first normalized by the internal control and the cell numbers of the samples and then analyzed using xcms online and metaboanalyst.
Transcriptional analysis45
The cells of BW25113 and BW25113-ΔastE were collected after cultivating for 12 h under 0 g L−1 and 5 g L−1 butanol condition, washed with sterile water three times, then frozen in liquid nitrogen and stored at 80 °C for further RNA-seq and RT-qPCR analyses.RNA extraction and examination
The total RNA was extracted by grinding the cells in TRIzol reagent (TaKaRa, Japan) in liquid nitrogen, isolated with chloroform, and precipitated with isopropanol, and then the sediment was washed with 75% alcohol and dissolved in RNA-free distilled water. RNA degradation was detected on 1% agarose gels, and the purity was checked using a Nanodrop 2000C spectrophotometer. Furthermore, the concentration of RNA was measured using an RNA Assay Kit in Qubit (Life Technologies, CA, USA). The RNA integrity was assessed using an RNA Nano 6000 Assay Kit with the Bioanalyzer 2100 system (Agilent Technologies, CA, USA).Library preparation and transcriptome sequencing
A total amount of 3 μg RNA per sample was used as input material for the RNA sample preparations. Sequencing libraries were generated using the NEBNext Ultra™ RNA Library Prep Kit from Illumina (NEB, USA). First, the mRNA was separated from the total RNA using poly-T oligo-attached magnetic beads and fragmented using divalent cations under elevated temperature in NEBNext First-Strand Synthesis Reaction Buffer (5×). Second, the mRNA was reverse transcribed to synthesize double-stranded cDNA using random hexamer primers. Third, the base A and an adaptor with a hairpin loop structure were added to the 30 ends of the cDNA to prepare for hybridization. The library fragments were purified with the AMPure XP system (Beckman Coulter, Beverly, USA) to select fragments 150–200 bp in length. Then, PCR was performed with universal PCR primers and an Index (X) Primer. Finally, the PCR products were purified (AMPure XP system) and the library quality was assessed using the Agilent Bioanalyzer 2100 system. The library was loaded into a flow cell and the fragments hybridized to the flow cell surface. Each bound fragment was amplified into a clonal cluster through bridge amplification. Sequencing reagents including fluorescently labeled nucleotides were added and the first base was incorporated. The flow cell was imaged and the emission from each cluster was recorded. The emission wavelength and intensity were used to identify the base. This cycle was repeated 150 times to create a read length of 150 bases on an Illumina HiSeq platform at Novogene Bioinformatics Institute (Novogene, Beijing, China).Data analysis
Differential expression analysis of BW25113 and BW25113-ΔastE (three biological replicates under 5 g L−1 butanol stress) was performed using the DESeq R package (1.18.0). The resulting P-values were adjusted using the Benjamini and Hochberg's approach for controlling the false discovery rate. Genes with an adjusted P-value < 0.05 found by DESeq were assigned as differentially expressed.GO and KEGG enrichment analysis of differentially expressed genes
GO enrichment analysis of differentially expressed genes was implemented by the GOseq R package, in which gene length bias was corrected. GO terms with corrected P-value less than 0.05 were considered significantly enriched in differentially expressed genes.KEGG is a database resource for understanding high level functions and utilities of the biological system, such as the cell, the organism, and the ecosystem, from molecular-level information, especially large-scale molecular datasets generated by genome sequencing and other high-throughput experimental technologies (http://www.genome.jp/kegg/). We used KOBAS software to test the statistical enrichment of differential expression genes in KEGG pathways.
We then added the differentially expressed gene accession ID to the xcms online (https://xcmsonline.scripps.edu) multi-omics lookup database for integrating metabolic data to identify key differences.
Real-time quantitative PCR (RT-qPCR)
The expression levels of seven upregulated and 13 downregulated genes in two strains, which were selected randomly from different metabolism pathways, were detected by qPCR. Total RNA isolation was carried out by using an RNAprep pure Kit, and reverse transcription (cDNA synthesis) was performed according to the protocol of the PrimeScript® RT reagent kit Perfect Real Time (Takara Bio Inc, Shiga, Japan). Quantitative real-time PCR (RT-qPCR) was done using the rrsG gene as the internal control, and the primers used in qPCR are given in Table S5.† Each sample was tested in triplicate in a 96-well plate (Bio-Rad).Amino acid determination
When the cells had been incubated to the exponential phase (OD600 = 0.6), 5 g L−1 of butanol was added and cultivation was proceeded for 12 h (200 rpm, 37 °C). Cells were then collected and washed three times with sterilized water. Afterward, the concentration of amino acids from the collected cells and fermentation supernate were detected by a high speed amino acid analyzer (Hitachi L-8900, Japan), in which the test parameters were: sodium ion exchange column, visible light detector, separation column temperature for 57 °C, and a reaction column temperature of 135 °C.Consent for publication
All authors agreed to publish this article.Authors' contributions
YG and HP conceived of the study; YG, HP and LHL drafted the manuscript; YG, BL, DWB and CHT carried out the study; YG and ZKZ data analysis. All the authors read and approved the final manuscript.Authors' information
Yuan Guo, Bo Lu, Hongchi Tang, Lihua Lin, Hao Pang belong to National Engineering Research Center for Non-Food Biorefinery, State Key Laboratory of Non-Food Biomass and Enzyme Technology, Guangxi Key Laboratory of Bio-refinery, Guangxi Academy of Sciences, 98 Daling Road, Nanning, 530007, China.Dewu Bi, Zhikai Zhang, Lihua Lin, Hao Pang belong to College of Life Science and Technology, Guangxi University, Nanning 530004, China.
Funding
This research was financially supported by the National Natural Science Foundation of China-China (31560027), the Guangxi Scientific Research and Technological Development Program of China (AD16380016), the Guangxi Natural Science Foundation of China (2015GXNSFBA139048, 2015GXNSFBA139084, 2018GXNSFAA294047).Conflicts of interest
The authors declared that they have no competing interests.Abbreviations
| GO | Gene ontology |
| KEGG | Kyoto encyclopedia of genes and genomes |
| RT-PCR | Reverse-transcription PCR |
| GC-MS | Gas chromatography-mass spectrometry |
References
- Y. P. Zhang, Y. Li, C. Y. Du, M. Liu and Z. Cao, Inactivation of aldehyde dehydrogenase: a key factor for engineering 1,3-propanediol production by Klebsiella pneumoniae, Metab. Eng., 2006, 8(6), 578–586 CrossRef CAS PubMed.
- S. Atsumi, T. Hanai and J. C. Liao, Non-fermentative pathways for synthesis of branched-chain higher alcohols as biofuels, Nature, 2008, 451(7174), 86–89 CrossRef CAS PubMed.
- R. Kalscheuer, T. Stolting and A. Steinbuchel, Microdiesel: Escherichia coli engineered for fuel production, Microbiology, 2006, 152, 2529–2536 CrossRef CAS PubMed.
- A. Schirmer, M. A. Rude, X. Z. Li, E. Popova and S. B. del Cardayre, Microbial Biosynthesis of Alkanes, Science, 2010, 329(5991), 559–562 CrossRef CAS PubMed.
- S. A. Nicolaou, S. M. Gaida and E. T. Papoutsakis, A comparative view of metabolite and substrate stress and tolerance in microbial bioprocessing: from biofuels and chemicals, to biocatalysis and bioremediation, Metab. Eng., 2010, 12(4), 307–331 CrossRef CAS PubMed.
- R. Jana, S. Andreas and B. Lars Mathias, Selected Pseudomonas putida strains able to grow in the presence of high butanol concentrations, Appl. Environ. Microbiol., 2009, 75(13), 4653–4656 CrossRef PubMed.
- E. P. Knoshaug and M. Zhang, Butanol tolerance in a selection of microorganisms, Appl. Biochem. Biotechnol., 2009, 153(1–2), 13–20 CrossRef CAS PubMed.
- D. T. Jones and D. R. Woods, Acetone-butanol fermentation revisited, Microbiol. Rev., 1986, 50(4), 484 CAS.
- C. R. Fischer, D. Klein-Marcuschamer and G. Stephanopoulos, Selection and optimization of microbial hosts for biofuels production, Metab. Eng., 2008, 10(6), 295–304 CrossRef CAS PubMed.
- L. K. Bowles and W. L. Ellefson, Effects of butanol on Clostridium acetobutylicum, Appl. Environ. Microbiol., 1985, 50(5), 1165–1170 CAS.
- L. Yang, G. Bao, Y. Zhu, H. Dong, Y. Zhang and Y. Li, Discovery of a novel gene involved in autolysis of Clostridium cells, Protein Cell, 2013, 4(6), 467–474 CrossRef PubMed.
- J. R. Warner, R. Patnaik and R. T. Gill, Genomics enabled approaches in strain engineering, Curr. Opin. Microbiol., 2009, 12(3), 223–230 CrossRef CAS PubMed.
- H. Q. Chong, H. F. Geng, H. F. Zhang, H. Song, L. Huang and R. R. Jiang, Enhancing E. coli isobutanol tolerance through engineering its global transcription factor cAMP receptor protein (CRP), Biotechnol. Bioeng., 2014, 111(4), 700–708 CrossRef CAS PubMed.
- L. H. Reyes, M. P. Almario and K. C. Kao, Genomic library screens for genes involved in n-butanol tolerance in Escherichia coli, PLoS One, 2011, 6(3), 9 CrossRef PubMed.
- B. L. Schneider, A. K. Kiupakis and L. J. Reitzer, Arginine catabolism and the arginine succinyltransferase pathway in Escherichia coli, J. Bacteriol., 1998, 180(16), 4278–4286 CAS.
- L. H. Reyes, M. P. Almario and K. C. Kao, Genomic Library Screens for Genes Involved in n-Butanol Tolerance in Escherichia coli, PLoS One, 2011, 6(3), e17678 CrossRef CAS PubMed.
- H. Richard and J. W. Foster, Escherichia coli glutamate- and arginine-dependent acid resistance systems increase internal pH and reverse transmembrane potential, J. Bacteriol., 2004, 186(18), 6032–6041 CrossRef CAS PubMed.
- B. M. Hersh, F. T. Farooq, D. N. Barstad, D. L. Blankenhorn and J. L. Slonczewski, A glutamate-dependent acid resistance gene in Escherichia coli, J. Bacteriol., 1996, 178(13), 3978–3981 CrossRef CAS PubMed.
- P. Lu, M. Dan, Y. Chen, Y. Guo, G. Q. Chen and H. Deng, et al., L-glutamine provides acid resistance for Escherichia coli through enzymatic release of ammonia, Cell Res., 2013, 23(5), 635–644 CrossRef CAS PubMed.
- B. R. Gibson, S. J. Lawrence, J. P. R. Leclaire, C. D. Powell and K. A. Smart, Yeast responses to stresses associated with industrial brewery handling, FEMS Microbiol. Rev., 2007, 31(5), 535–569 CrossRef CAS PubMed.
- J. A. Cray, A. Stevenson, P. Ball, S. B. Bankar, E. C. Eleutherio and T. C. Ezeji, et al., Chaotropicity: a key factor in product tolerance of biofuel-producing microorganisms, Curr. Opin. Biotechnol., 2015, 33, 228–259 CrossRef CAS PubMed.
- F. Mingardon, C. Clement, K. Hirano, M. Nhan, E. G. Luning and A. Chanal, et al., Improving olefin tolerance and production in E. coli using native and evolved AcrB, Biotechnol. Bioeng., 2015, 112(5), 879–888 CrossRef CAS PubMed.
- M. M. Hryniewicz and N. M. Kredich, The cysP promoter of Salmonella typhimurium: characterization of two binding sites for CysB protein, studies of in vivo transcription initiation, and demonstration of the anti-inducer effects of thiosulfate, J. Bacteriol., 1991, 173(18), 5876–5886 CrossRef CAS PubMed.
- P. Singh, V. A. Ray, M. J. Mandel and K. L. Visick, CysK Plays a Role in Biofilm Formation and Colonization by Vibrio fischeri, Appl. Environ. Microbiol., 2015, 81(15), 5223–5234 CrossRef CAS PubMed.
- M. H. Rau, P. Calero, R. M. Lennen, K. S. Long and A. T. Nielsen, Genome-wide Escherichia coli stress response and improved tolerance towards industrially relevant chemicals, Microb. Cell Fact., 2016, 15(1), 176 CrossRef PubMed.
- D. L. Tucker, T. Nancy and C. Tyrrell, Gene expression profiling of the pH response in Escherichia coli, J. Bacteriol., 2002, 184(23), 6551 CrossRef CAS PubMed.
- A. L. Lomize, M. A. Lomize, S. R. Krolicki and I. D. Pogozheva, Membranome: a database for proteome-wide analysis of single-pass membrane proteins, Nucleic Acids Res., 2016, 45(D1), D250 CrossRef PubMed.
- E. Tenorio, T. Saeki, K. Fujita, M. Kitakawa, T. Baba and H. Mori, et al., Systematic characterization of Escherichia coli genes/ORFs affecting biofilm formation, FEMS Microbiol. Lett., 2003, 225(1), 107–114 CrossRef CAS PubMed.
- I. T. Paulsen, L. Nguyen, M. K. Sliwinski, R. Rabus and M. H. S. Jr, Microbial genome analyses: comparative transport capabilities in eighteen prokaryotes 1, J. Mol. Biol., 2000, 301(1), 75–100 CrossRef CAS PubMed.
- Z. Yan and T. C. Ezeji, Transcriptional analysis of Clostridium beijerinckii NCIMB 8052 to elucidate role of furfural stress during acetone butanol ethanol fermentation, Biotechnol. Biofuels, 2013, 6(1), 66 CrossRef PubMed.
- M. J. Fath and R. Kolter, ABC transporters: bacterial exporters, Microbiol. Rev., 1993, 57(4), 995–1017 CAS.
- K. Linton and C. Higgins, The Escherichia coli ATP-binding cassette (ABC) proteins, Mol. Microbiol., 1998, 28(1), 5–13 CrossRef CAS PubMed.
- A. Guillot, C. Gitton and P. Anglade, et al., Proteomic analysis of Lactococcus lactis, a lactic acid bacterium, Proteomics, 2003, 3(3), 337–354 CrossRef CAS PubMed.
- P. O. Ljungdahl and B. Daignanfornier, Regulation of amino acid, nucleotide, and phosphate metabolism in Saccharomyces cerevisiae, Genetics, 2012, 190(3), 885 CrossRef CAS PubMed.
- A. Solopova, C. Formosadague, P. Courtin, S. Furlan, P. Veiga and C. Péchoux, et al., Regulation of cell wall plasticity by nucleotide metabolism in Lactococcus lactis, J. Biol. Chem., 2016, 291(21), 11323–11336 CrossRef CAS PubMed.
- J. R. Borden and E. T. Papoutsakis, Dynamics of genomic-library enrichment and identification of solvent tolerance genes for Clostridium acetobutylicum, Appl. Environ. Microbiol., 2007, 73(9), 3061–3068 CrossRef CAS PubMed.
- K. Manabu, K. Taiki, T. Hideyuki, M. Yasuo, M. Xian-Ying and H. Tomoyuki, et al., Isolation of butanol- and isobutanol-tolerant bacteria and physiological characterization of their butanol tolerance, Appl. Environ. Microbiol., 2013, 79(22), 6998–7005 CrossRef PubMed.
- S. A. Nicolaou, S. M. Gaida and E. T. Papoutsakis, A comparative view of metabolite and substrate stress and tolerance in microbial bioprocessing: From biofuels and chemicals, to biocatalysis and bioremediation, Metab. Eng., 2010, 12(4), 307–331 CrossRef CAS PubMed.
- M. Ma and Z. L. Liu, Mechanisms of ethanol tolerance in Saccharomyces cerevisiae, Appl. Microbiol. Biotechnol., 2010, 87(3), 829–845 CrossRef CAS PubMed.
- C. Grimmler, H. Janssen, D. Krausse, R.-J. Fischer, H. Bahl and P. Duerre, et al., Genome-Wide Gene Expression Analysis of the Switch between Acidogenesis and Solventogenesis in Continuous Cultures of Clostridium acetobutylicum, J. Mol. Microbiol. Biotechnol., 2011, 20(1), 1–15 CrossRef CAS PubMed.
- S. Mao, Y. Luo, G. Bao, Y. Zhang, Y. Li and Y. Ma, Comparative analysis on the membrane proteome of Clostridium acetobutylicum wild type strain and its butanol-tolerant mutant, Mol. BioSyst., 2011, 7(5), 1660–1677 RSC.
- T. Maniatis, E. F. Fritsch and J. Sambrook, Molecular cloning: a laboratory manual, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY, 1982 Search PubMed.
- J. W. Zhou, Z. Y. Dong, L. M. Liu, G. C. Du and J. Chen, A reusable method for construction of non-marker large fragment deletion yeast auxotroph strains: a practice in Torulopsis glabrata, J. Microbiol. Methods, 2009, 76(1), 70–74 CrossRef CAS PubMed.
- N. Nishida, N. Ozato, K. Matsui, K. Kuroda and M. Ueda, ABC transporters and cell wall proteins involved in organic solvent tolerance in Saccharomyces cerevisiae, J. Biotechnol., 2013, 165(2), 145–152 CrossRef CAS PubMed.
- Y. Chen, Z. Lu, D. Chen, Y. Wei, X. Chen and J. Huang, et al., Transcriptomic analysis and driver mutant prioritization for differentially expressed genes from a Saccharomyces cerevisiae strain with high glucose tolerance generated by UV irradiation, RSC Adv., 2017, 7(62), 38784–38797 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra09711a |
| This journal is © The Royal Society of Chemistry 2019 |