
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTumour targeted contrast enhanced ultrasound imaging dual-modal microbubbles for diagnosis and treatment of triple negative breast cancer
Min Bai†
a,
Yang Dong†b,
Hui Huangb,
Hao Fub,
Yourong Duan b,
Qi Wang*c and
Lianfang Du*a
b,
Qi Wang*c and
Lianfang Du*a
aDepartment of Ultrasound, Shanghai General Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai 200080, China. E-mail: du_lf@163.com
bState Key Laboratory of Oncogenes and Related Genes, Shanghai Cancer Institute, Renji Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai 200032, China
cKey Laboratory for Advanced Materials and Institute of Fine Chemicals, Shanghai Key Laboratory of Functional Materials Chemistry, School of Chemistry and Molecular Engineering, East China University of Science and Technology, Shanghai 200237, China. E-mail: wangqi@ecust.edu.cn
First published on 15th February 2019
Abstract
At present, the treatment of triple negative breast cancer (TNBC) is a worldwide problem, urgently requiring early precise diagnosis and effective treatment methods. In our study, we designed a type of tumour targeted dual-modal microbubbles, paclitaxel (PTX)-loaded RGD-lipid microbubbles (PTX@RGD-MBs), combined with ultrasonic targeted microbubble destruction (UTMD) to precisely diagnose TNBC and to improve the curative effect. As the first-line drug, PTX, lacking specific tumour targeting and the ability to be effectively internalized by TNBC cells, is still not effective in killing TNBC cells. For this reason, we used the tumour active targeting peptide RGD to precisely guide MBs around TNBC cells through the high affinity to the integrin αvβ3 receptor, and use UTMD to generate shear stress on cells to open transient pores in the cellular membrane, so as to improve MB penetration into the cellular membrane. Sulfur hexafluoride (SF6) as the internal gas of MBs can greatly improve the resolution and sensitivity of conventional US images through nonlinear harmonics to enhance the comparison between lesions and surrounding normal tissues. The results in our study demonstrated that RGD-MBs with UTMD were internalized by TNBC cells more effectively, leading to significant increase in intercellular drug concentrations of TNBC cells, thus achieving the best inhibitory effect on TNBC cells in vitro. Ultrasonic experiment showed that PTX@RGD-MBs produced high quality contrast enhanced ultrasound (CEUS) images in vitro and in vivo, providing a better method for diagnosis and evaluation of the TNBC. Therefore, we conclude the advantages of excellent anti-tumour effect and CEUS imaging of PTX@RGD-MBs provided a better application for diagnosis and treatment of TNBC.
Introduction
Triple-negative breast cancer (TNBC) was first reported in the literature in 2005,1 which is characterized by the absence of estrogen receptor (ER), progesterone receptor (PR) and human epidermal growth factor receptor 2 (HER-2) expression, becoming one of the new hot spots in the research of breast cancer. TNBC is highly invasive and prone to recurrence and metastasis, accounting for about 10–20% of all breast cancers along with poor prognosis and a lack of standardized treatment guidelines.2–4 Therefore, early precise diagnosis and effective treatment methods are urgently required for TNBC nowadays.At present, cytotoxic chemotherapy remains the mainstay treatment for TNBC. In the National Comprehensive Cancer Network (NCCN) guidelines, paclitaxel (PTX)-based adjuvant chemotherapy is still recommended as a first-line regiment for treating patients with TNBC by interfering with the mitotic spindle, resulting in cell cycle arrest and ultimately apoptosis.5,6 However, paclitaxel faces the limitations of negative tumour targeting ability with strong side effect, and insufficient cellular internalization with weak lethality to TNBC cells. Herein, an effective drug carrier system for precisely delivering amount of PTX into TNBC cells is required to decrease the side effect and improve the therapeutic effect.7 With good biocompatibility, ultrasound (US) microbubbles (MBs) are reported to carry both drugs and inert gas into their lipid layer or hollow space, which provides a potential approach to combine the treatment and diagnosis.8–12 Moreover, tumour cells are inclined to internalize ultrasound microbubbles through ultrasound exposure, improving intercellular drugs concentration with stronger curative effect. However, the reported microbubbles are short of tumour targeting ability resulting to side effect.13,14 Furthermore, owing to the limited hollow space, it may generate the unbalance of drug loading effect with the gas encapsulation.15 Hence, there still a huge challenge of high-specificity dual-modal microbubbles for diagnosis and treatment of TNBC.16
In this work, we designed a drug carrier system, PTX-loaded RGD-lipid microbubbles (PTX@RGD-MBs) combining ultrasonic targeted microbubble destruction (UTMD), by the thin-film hydration-sonication method (Fig. 1), where RGD peptide (three peptides, arginine–glycine–aspartic acid) as tumour active targeting peptide to precisely guide microbubbles to TNBC cells,17–19 PTX in oil layer as chemotherapeutic agent to kill TNBC cells, sulfur hexafluoride (SF6) as internal gas of microbubbles to generate contrast enhanced ultrasound (CEUS) images through the nonlinear harmonics,20 along with UTMD to promote PTX's internalization.21 As shown in Fig. 1, PTX@RGD-MBs consisted of a gaseous core, a lipid layer with hydrophobic tails facing the gaseous core and the hydrophilic head groups facing the aqueous environment. This system had the following advantages: (i) the tumour active targeting peptide RGD could precisely guide MBs to TNBC tumour cells through the high affinity to integrin αvβ3 receptor which was highly expressed in TNBC cells and its angiogenesis, whereas poorly expressed in normal tissues. (ii) The cellular internalization was improved that because UTMD not only generated shear stress on cells to open transient pores on the membranes, so as to improve the microbubbles penetrating into the cellular membrane, but also enforced microbubbles bursting the internal gas to rapidly release PTX as needed.22–24 (iii) The SF6 as internal gas of MBs could greatly improve the resolution and sensitivity of traditional US images through the nonlinear harmonics so as to enhance the comparison between lesions and surrounding normal tissues.
 | ||
| Fig. 1 Schematic route of PTX@RGD-MBs. Synthetic steps: (i) uniform PTX-loaded RGD-lipid particles were prepared using ultrasonic-hydration method; (ii) freeze-drying to remove the inner water of microbubbles; (iii) blowing SF6 and re-dissolving the microbubbles, along with shaking the bottle vigorously until the freeze-dried powder was completely dispersed to obtain “PTX@RGD-MBs”. | ||
In this study, we reported tumour targeted ultrasound contrast enhanced US imaging dual-modal microbubbles PTX@RGD-MBs and demonstrated its higher internalization by TNBC cells with the help of tumour active targeted peptide RGD and UTMD, and further justified the improved therapeutic effect on TNBC cells in vitro. We then validated the excellent CEUS imaging of PTX@RGD-MBs in vitro and in vivo. In summary, the dual-modal microbubbles PTX@RGD-MBs provided a better method for diagnosing and treating the TNBC.
Results and discussion
Tumour targeted dual-modal microbubbles PTX@RGD-MBs
The carrier material of targeted dual-modal microbubbles was constructed with three biocompatible materials: RGD-PEG-DPPE (1,[2-dipalmitoyl-sn-glycerol-3-phosphoethanolamine]-N-[amino (polyethylene-glycol)]), DPPC (1,2-dipalmitoyl-sn-glycerol-3-phosphoethanolamine), and cholesterol. The DPPE, DPPC and cholesterol were the most important cells membrane composition, feasibly adopting by cells. PEG is a biodegradable biomaterial approved by FDA (Food and Drug Administration) for using in the human body.26 RGD peptide is composed of human body available amino acids.25 Therefore, RGD-lipid microbubbles were considered as a suitable biocompatible biomaterial to deliver drugs. First-line regiment PTX for TNBC and the inert gas SF6 were encapsulated into the microbubbles to realize the treatment and diagnosis of TNBC.The condensation reactions of carboxyl and amino groups could effectively combine the targeted peptides to organic materials with few by-products.26 The carboxyl groups of RGD peptide was reacted with the amine groups of DPPE-PEG-NH2 to form DPPE-PEG-RGD. As illustrated in 1H NMR spectra, both the proton peak of RGD (–CH3– at δ 1.4) and the PEG segment peaks (–CH2CH2O– at δ 3.7) from DPPE-PEG-NH2 was observed in the spectra of DPPE-PEG-RGD with a ratio of 188![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :24.39, demonstrating successful conjugation. The ideal ratio of proton peak from DPPE-PEG-NH2 (–CH2CH2O– at δ 3.7) and RGD (–CH3– at δ 1.4) was 188:26, so that 93.80% DPPE-PEG-NH2 was terminally modified with RGD peptides (Fig. 2A). The thin film hydration method was used to prepare PTX@RGD-MBs in three steps that (Fig. 1): first, three materials self-assemble to load PTX to formulate PTX-loaded RGD-MBs; second, freeze-drying to obtain hollow microbubbles; third, blowing SF6 and re-dissolving the microbubbles with shook to push the gas diffusing into MBs to obtain PTX@RGD-MBs.27 Due to the hydrophilic of PEG, the PTX@RGD-MBs was uniformly dissolved in the solution, along with spherical appearance and smooth surface without conjugation (Fig. 2B). The size and zeta potential of PTX@RGD-MBs were 1741.67 ± 67.72 nm (Fig. 2C) and −0.665 ± 0.033 mV (Fig. 2D) respectively. The polydispersity index (PDI) was 0.106. The PTX@RGD-MBs exhibited an excellent drug encapsulation efficiency (EE%) of 91.07% and a drug loading (DL%) of 4.01% for PTX (Table 1), suggesting the prevention from side effects created by unbound chemotherapeutic drugs.
:24.39, demonstrating successful conjugation. The ideal ratio of proton peak from DPPE-PEG-NH2 (–CH2CH2O– at δ 3.7) and RGD (–CH3– at δ 1.4) was 188:26, so that 93.80% DPPE-PEG-NH2 was terminally modified with RGD peptides (Fig. 2A). The thin film hydration method was used to prepare PTX@RGD-MBs in three steps that (Fig. 1): first, three materials self-assemble to load PTX to formulate PTX-loaded RGD-MBs; second, freeze-drying to obtain hollow microbubbles; third, blowing SF6 and re-dissolving the microbubbles with shook to push the gas diffusing into MBs to obtain PTX@RGD-MBs.27 Due to the hydrophilic of PEG, the PTX@RGD-MBs was uniformly dissolved in the solution, along with spherical appearance and smooth surface without conjugation (Fig. 2B). The size and zeta potential of PTX@RGD-MBs were 1741.67 ± 67.72 nm (Fig. 2C) and −0.665 ± 0.033 mV (Fig. 2D) respectively. The polydispersity index (PDI) was 0.106. The PTX@RGD-MBs exhibited an excellent drug encapsulation efficiency (EE%) of 91.07% and a drug loading (DL%) of 4.01% for PTX (Table 1), suggesting the prevention from side effects created by unbound chemotherapeutic drugs.
 | ||
| Fig. 2 Tumour targeted dual-modal microbubbles of PTX@RGD-MBs. (A) 1H NMR hydrogen spectrum analysis showed that DPPE-PEG-NH2 was successfully connected to RGD peptide. Compared with DPPE-PEG-NH2, DPPE-PEG-RGD appeared chemical displacement of RGD peptide' hydrogen (at δ 1.4) and PEG' hydrogen (at δ 3.7), indicating that the RGD peptide was successfully attached to DPPE-PEG. (B) PTX@RGD-MBs was uniformly spherical with good dispersion and no obvious agglomeration by TEM images. (C) The average particle size of PTX@RGD-MBs was 1741.67 ± 67.72 nm with the PDI of 0.106 ± 0.019. (D) Zeta potential of PTX@RGD-MBs was −0.665 ± 0.033 mV. | ||
| MBs | Particle size (nm) | PDI | Zeta potential (mV) | EE (%) | DL (%) |
|---|---|---|---|---|---|
| PTX-MBs | 1741.67 ± 67.72 | 0.106 | −0.665 ± 0.033 | 91.07 ± 2.51 | 4.01 ± 0.05 |
According to the structure of PTX@RGD-MBs, the exposed PEG hydrophilic chain can effectively enhance the stability of MBs and extend circulation time in blood. The peptide RGD, locating the surface of the MBs, guaranteed the MBs to recognize the integrin αvβ3 receptor of TNBC cells. Moreover, the cavity acoustic structure of lipid microbubbles not only provided a basis for the ultrasound contrast enhanced imaging properties, but also provided a precondition for UTMD to promote PTX@RGD-MBs to rapidly release PTX.
UTMD promoted PTX@RGD-MBs rupturing to release chemotherapeutic agent in vitro
Lipid bilayer of PTX@RGD-MBs was able to prevent the rapid release of PTX in circulation, so as to reduce the systemic toxic and side effects of PTX. Later, the active targeting peptide RGD would aid PTX@RGD-MBs to accumulate around tumour tissues and gradually release PTX. At this point, we applied UTMD with appropriate intensity at the tumour site, along with rapid release of PTX to increase the concentration in region as well as improve the cellular internalization.We conducted a drug releasing test in vitro to compare the PTX's release profile of PTX@RGD-MBs combining UTMD with those of PTX@RGD-MBs alone and free PTX (Fig. 3A). In our study, free PTX was released instantly that, more than 30% was released at 0.5 h, 80% at 12 h and almost completely released at 24 h. In contrast, the cumulative release amount of PTX from PTX@RGD-MBs was no more than 30% within 12 h and only 80% at 96 h, showing apparent sustained characteristic. In addition, PTX showed a biphasic release pattern in PTX@RGD-MBs + UTMD group, which was slowly released from the MBs initially (30% after 10 h) and instantly released after treating by UTMD at 10 h (from 30% to 70% within 2 h) similar to free PTX. This phenomenon demonstrated that the UTMD can induce the sonoporation effect to rupture and distort the shape of PTX@RGD-MBs into irregular damaged spherical structure (Fig. 3B), conducing the PTX's release. Hence, PTX@RGD-MBs can not only control the release rate of PTX, but also the release site combining UTMD.
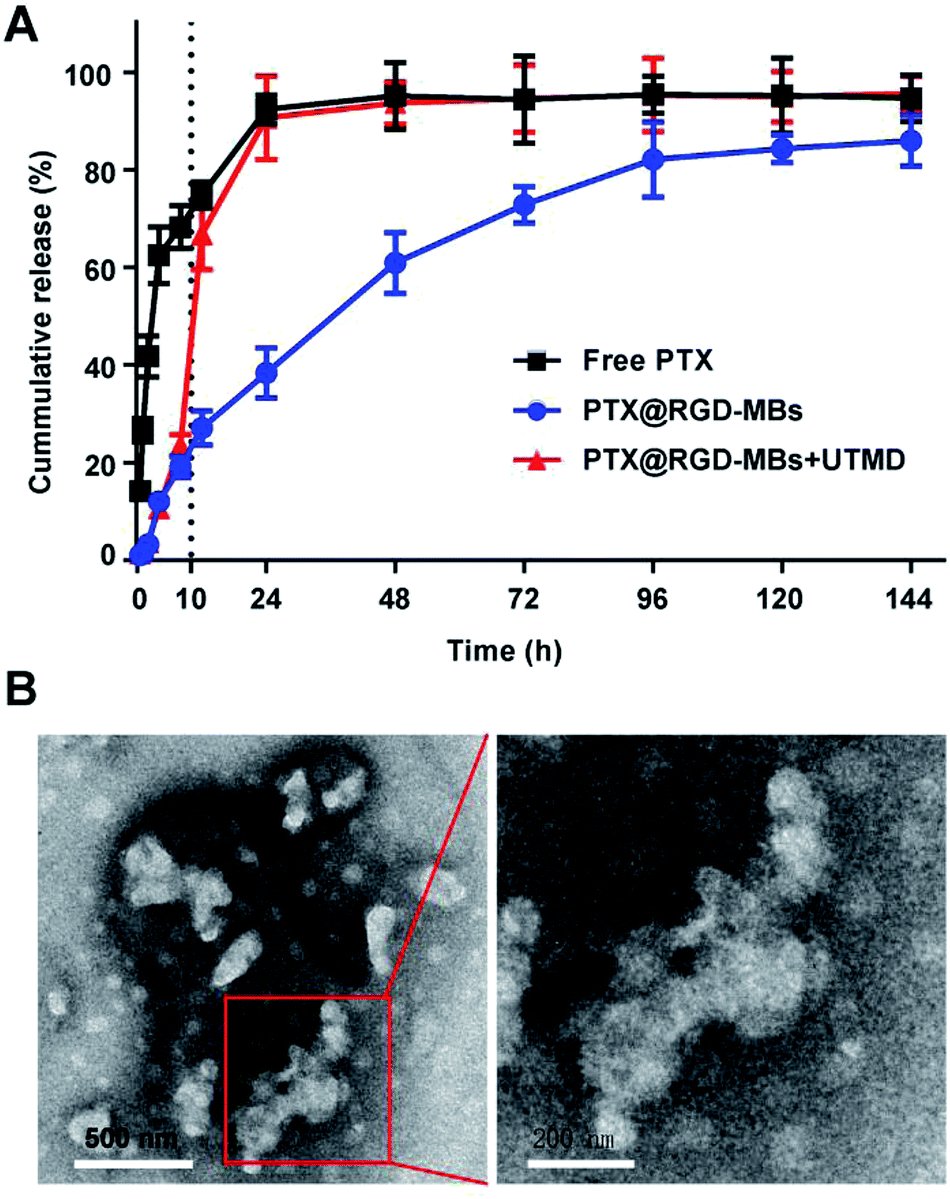 | ||
| Fig. 3 UTMD promoted PTX@RGD-MBs rupturing to release chemotherapeutic agent. (A) The release profiles of virial PTX formulations. PTX released much slower from PTX@RGD-MBs compared with unbounded PTX, indicating of sustained release property and protection during the circulation environment. UTMD pushed the microbubbles rupturing to release drug, illustrating as PTX@RGD-MBs + UTMD group at 10 h, suggesting drug accumulation at targeting site. (B) PTX@RGD-MBs became irregular compared with the original spherical structure by treating with UTMD. | ||
Compared with free PTX, the PTX@RGD-MBs was able to effectively slow down the release rate of PTX in circulation, thereby reducing toxicity to normal organs and alleviating systemic side effects. Moreover, UTMD could prompt the MBs to rupture and rapidly release PTX at the appropriate time and location, laying a theoretical foundation for the next step of anti-tumour efficiency experiment in vitro.
High stability and negative cytotoxicity of lipid microbubbles
The integrity and nontoxicity of MBs are the basic indexes which claim good stability and biocompatibility, affecting by the gas escape and the rupture of the inner membrane. Hence, in this work, several methods were conducted to stable the PTX@RGD-MBs: first, cholesterol was used to fill the space between the phospholipids, making them more tightly arranged to improve the stability of lipid microbubbles; second, the physical and chemical properties of inert gas SF6 were stable, reducing the escape diffusion of the gas and being beneficial to the stability of the MBs;28 third, DPPE-PEG-RGD with a long chain of hydrophilicity on the surface of the MBs was able to adsorb water molecules to form hydrated membranes, significantly reducing the surface tension of MBs, preventing collision and polymerization between MBs, and improving the stability of lipid microbubbles. Also, biodegradable DPPE-PEG materials ensured the excellent biocompatibility of MBs.29Then, the size and PDI were detected after dispersed the PTX@RGD-MBs into the solutions. As demonstrated, PTX@RGD-MBs can be stored at room temperature in PBS solution for at least 48 h without any obvious rupture, with the particle size of 1200–1600 nm and PDI < 0.3 (Fig. 4A). After continued storage for more than 72 h, the PTX@RGD-MBs gradually became cloudy and precipitated with decreased size and increased PDI. The results showed that the PTX@RGD-MBs solution was stable at room temperature for 48 h without appearance alteration, comparing to the SonoVue solution of 6 h at room temperature (SonoVue introduction), indicative of better stability of PTX@RGD-MBs solution for 48 h after dispersion.
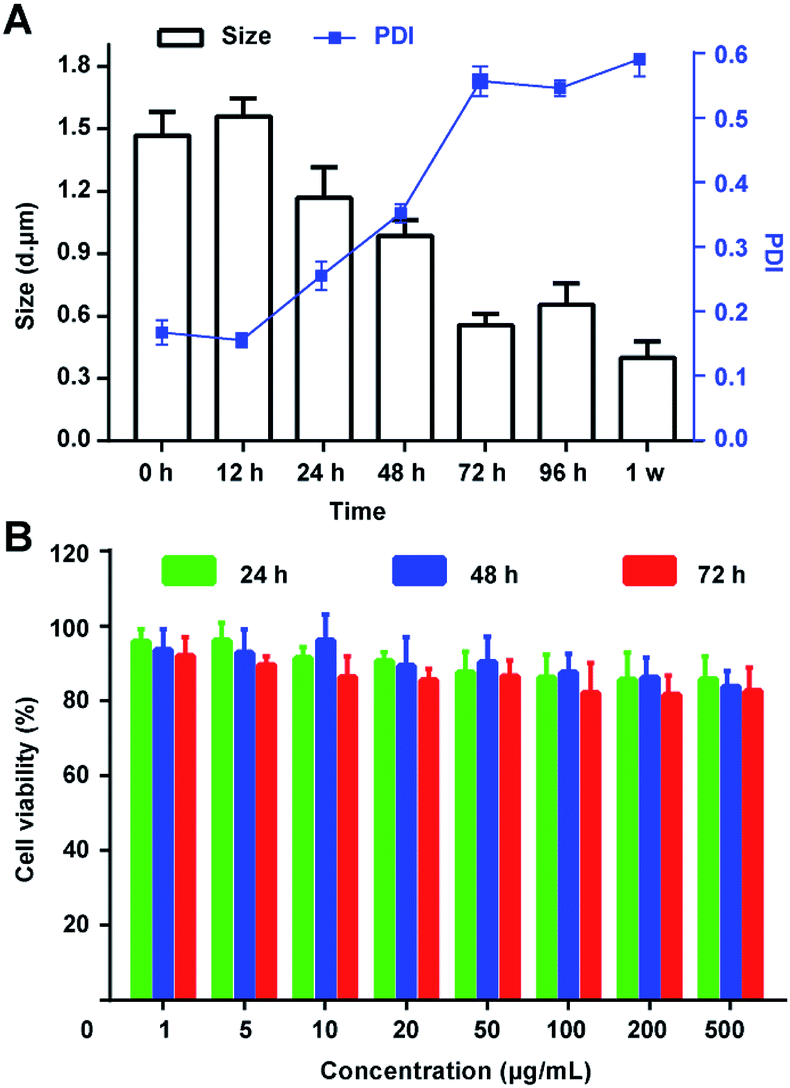 | ||
| Fig. 4 The stability of PTX@RGD-MBs and negative cytotoxicity of the blank lipid microbubbles. (A) PTX@RGD-MBs can be stored at room temperature in PBS for 24 h without obvious rupture, with particle size of 1200–1600 nm and PDI < 0.3. PTX@RGD-MBs would disintegrate after 72 h with decreased size. (B) 80% MDA-MB-231 breast cancer cells were survived after incubated with blank lipid microbubbles (1–500 μg mL−1) for 24 h, 48 h and 72 h, indicating negative toxicity of lipid microbubbles. | ||
The MTT method was used to test the relative proliferation of MDA-MB-231 cells (human TNBC cell line) after incubated with lipid MBs of various concentrations (0.001, 0.005, 0.01, 0.02, 0.05, 0.1, 0.2 and 0.5 mg mL−1) for 24, 48 and 72 h. The results showed that the survival rate of TNBC cells in all groups was above 80%, suggesting the lipid MBs has negligible effect on cells' viability. This result determined that lipid MBs had excellent biocompatibility and negligible toxicity toward TNBC cells (Fig. 4B).
Improved drug internalization and inhibited proliferation of TNBC cells by RGD-MBs + UTMD in vitro
Effective internalization by TNBC cells is a prerequisite for chemotherapy drugs to kill the tumour cells. In this work, the RGD-MBs + UTMD were designed to promote drugs internalization and further improve the treatment effect to TNBC cells by delivering more RGD-MBs around tumour cells through peptide RGD,17 and punching transient holes on the cells membrane surface by sonoporation effect inducing by UTMD.11The internalization and intracellular distribution of the RGD-MBs in TNBC cells (MDA-MB-231) were observed through fluorescence inverted microscope. The PTX was replaced by Rb (rhodamine B, red fluorescent probe) to prepare Rb@RGD-MBs. As shown in Fig. 5A and B, the intracellular fluorescence intensity in MDA-MB-231 cells for different groups was observed in the following order: Rb@RGD-MBs + UTMD > free Rb + UTMD > Rb@RGD-MBs > free Rb (Fig. 5B), with time dependence. Rb@RGD-MBs strongly enhanced the cellular internalization of drug (P < 0.01) compared to the free Rb. Also, UTMD aided the cells to intake more drugs by inducing the sonoporation effect to open transient pores on cells membrane, leading to stronger fluorescence intensity in free Rb + UTMD group than free Rb group (P < 0.01). In addition, the fluorescence intensity of Rb@RGD-MBs + UTMD group was obviously stronger than that of Rb@RGD-MBs (P < 0.05) and free Rb + UTMD group (P < 0.05) by combing the active targeting peptide RGD or UTMD alone. Therefore, the effective intracellular concentration of TNBC cells in Rb@RGD-MBs + UTMD group was significantly improved, which laid a foundation for future curative experiments.
 | ||
| Fig. 5 RGD-MBs + UTMD induced higher drug internalization, along with lower proliferation of MDA-MB-231 breast cancer cells. (A) Fluorescence images and (B) the statistics of the internalization ability of unbounded Rb and Rb@RGD-MBs by MDA-MB-231 cells. The internalized Rb ranged from more to less: Rb@RGD-MBs + UTMD > free Rb + UTMD > Rb@RGD-MBs > free Rb. Note: ***P < 0.001; **P < 0.01; *P < 0.05, compared with free Rb group. (C) The MDA-MB-231 cells treated with virial formulations (***P < 0.001; **P < 0.01; *P < 0.05, compared with free PTX group). The cell viability decreased in order of PTX@RGD-MBs + UTMD > PTX@MBs + UTMD > free PTX + UTMD > PTX@RGD-MBs > free PTX, which was consistent with the results of the TNBC cells internalization of drugs. | ||
Due to the excellent properties and characteristics, PTX@RGD-MBs + UTMD generated significant curative effect on TNBC cells. MTT test was carried out to reveal the curative effect by detecting the cell viability after incubated with PTX@RGD-MBs + UTMD, PTX@MBs + UTMD, PTX@RGD-MBs, free PTX + UTMD and free PTX for 24 h, 48 h and 72 h. The inhibition effect on the cells proliferation of different groups was in the following order: PTX@RGD-MBs + UTMD > PTX@MBs + UTMD > free PTX + UTMD > PTX@RGD-MBs > free PTX in a time-dependent manner (Fig. 5C), which was consistent with the results of the internalization of drugs in TNBC cells. PTX@RGD-MBs effectively inhibited cells proliferation compared to the free PTX (P < 0.01). Also, UTMD promoted the cellular internalization of PTX, leading to better treatment effect in free PTX + UTMD group than free PTX group (P < 0.01). In addition, the curative effects in PTX@RGD-MBs + UTMD group was manifestly stronger than all PTX@MBs + UTMD (P < 0.05), PTX@RGD-MBs (P < 0.01) and free PTX + UTMD groups (P < 0.01), suggesting combined application of RGD peptide and UTMD could achieve best therapeutic effect. Notably, either PTX@MBs + UTMD or PTX@RGD-MBs group alone could not achieve satisfactory therapeutic effect because peptide RGD alone could not effectively push internalization by TNBC cells, whereas UTMD alone could not precisely guide the PTX to TNBC cells.
In conclude, satisfactory therapeutic effect was achieved through combing active targeting peptide RGD to precisely delivery the drugs to tumour site, and UTMD to effectively push the drug internalization.
Bio-distribution evaluation and tumour targeting of PTX@RGD-MBs in nude mice TNBC xenografts
MBs was labelled by red fluorophore 1,1-dioctadecyl-3,3,3,3-tetramethylindotricarbocyaine iodide (Dir) to evaluate the bio-distribution in the subcutaneous TNBC tumour model (Fig. 6A). The Dir distributed around the whole body after 2 h and did not disappear after 48 h. The fluorescence intensities in tumours increased in the order of free Dir (D) < Dir-MBs + UTMD (DM + U) < Dir-MBs-RGD (DMR) < Dir-MBs-RGD + UTMD (DMR + U) in all time points (Fig. 6B). In the free Dir group, the fluorescence was mainly distributed in the abdominal cavity and barely observed in the tumour. The fluorescent was brighter in the tumour of DRM + U and DRM groups than that of free Dir group, indicating that the tumour targeting peptide RGD effectively induced the MBs to accumulate in the TNBC tumour. Besides, the accumulation of fluorescent signals in tumours of DMR + U group was stronger than that of DMR group, suggesting that UTMD was able to enhance the release of drugs from MBs to accumulate them in tumour tissues. After injection of various formulations for 48 h, the mice were sacrificed and the major organs were harvested to evaluate the bio-distributions. As shown in Fig. 6C and D, the Dir mainly distributed in liver organs in all groups, and the tumour's fluorescent intensity in the DMR + U group was the strongest among all groups, along with lowest intensities in other normal tissues and organs, which also demonstrated the improved tumour targeting ability. | ||
| Fig. 6 The distribution of tumor targeting MBs in nude mice TNBC xenografts. (A) The fluorescence images after injecting various formations. The Dir mainly distributed in the abdominal cavity in free Dir (D) and Dir-MBs + UTMD (DM + U) groups, while it was remained in tumor tissues (red circle) in Dir-MBs-RGD (DRM) and Dir-MBs-RGD + UTMD (DMR + U) groups, suggesting the tumor targeting ability of RGD-MBs-NPs. (B) Quantitative average fluorescent intensities in tumors. Fluorescent intensity in Dir-MBs-RGD + UTMD group was stronger than that of Dir-MBs-RGD group, suggesting UTMD was able to promote the release of drugs in tumor. *P < 0.05, **P < 0.01, ***P < 0.001, compared with free Dir group. (C) The ex vivo fluorescence images and (D) the quantitative intensities of the isolated tumors (T), hearts (H), livers (Li), spleens (S), lungs (Lu) and kidneys (K) after intravenous injection of various formations for 48 h. The fluorescent intensity of Dir-MBs-RGD + UTMD group in tumor was significantly stronger than other groups, indicative of higher tumor accumulation. | ||
Together, these results demonstrated that the tumor targeting peptide RGD and UTMD significantly increased the drugs' concentration in tumours. As a result, RGD-MBs with UTMD not only helped the drug to accumulate in tumor tissues for better therapeutic effect, but also effectively reduced the toxicity of the drug on normal tissues.
CEUS imaging of PTX@RGD-MBs for precise diagnosis and evaluation
Ultrasound microbubble contrast agent could significantly enhance the ultrasonic scattering signal through MBs' destruction inducing by sound wave, along with producing high quality CEUS images for better diagnosis and evaluation of TNBC tumour. The capability of PTX@RGD-MBs producing CEUS images was detected using peristaltic pump in vitro (Fig. 7A) and TNBC subcutaneous tumours in vivo (Fig. 7B). | ||
| Fig. 7 Enhanced contrast imaging of PTX@RGD-MBs in vitro and vivo. The ultrasonic imaging experiments (A) in vitro and (B) in vivo. The capability of PTX@RGD-MBs producing CEUS images was investigated using peristaltic pump in vitro and TNBC subcutaneous tumours in vivo. (C) Ultrasound contrast images of PTX@RGD-MBs in vitro. SonoVue and PTX@RGD-MBs showed strong point echo in both of the conventional B-mode and CEUS model, compared with echoless of the saline water. (D) Ultrasound contrast images of PTX@RGD-MBs in vivo. Obvious CEUS images were observed in both PTX@RGD-MBs and SonoVue groups with no significant differences, while no CEUS images were observed after the injection of saline, indicating that PTX@RGD-MBs with local injection have a satisfied development in vivo. | ||
Under the conventional B-mode model, the hose filled with PTX@RGD-MBs solution displayed small point with strong echo in vitro, whereas SonoVue displayed strong coarse point echo. In contrast, the hose filled with saline water was observed as black in vitro. Under the CEUS mode, the saline solution showed no echo, SonoVue displayed a significant amount of big-dotted echo, and the PTX@RGD-MBs displayed a large amount of small-dotted echo, which proved that PTX@RGD-MBs yielded effective CEUS images in vitro (Fig. 7C).
The subcutaneous tumour of TNBC nude mice model was utilized to explore the CEUS imaging by local injection of PTX@RGD-MBs in vivo. No contrast-enhanced images were captured in saline solution group, while excellent CEUS images were observed in either PTX@RGD-MBs or SonoVue solution with no significant differences in tumour site (Fig. 7D), indicating that CEUS images were satisfactory in breast cancer tumours with intratumoural administration of PTX@RGD-MBs. It was noted that,16,30 CEUS images in tumours were not captured after intravenous injection of PTX@RGD-MBs or SonoVue solution, because of the poorly distributed new vessels in the tumour parenchyma of the subcutaneous tumour in nude mice,31,32 which causing rarely MBs to immediately reach the interior of tumours as well as no CEUS images in tumour.
In conclude, PTX@RGD-MBs as a US contrast agent, produced high quality CEUS imaging in vitro and in vivo, and improved the resolution and sensitivity of conventional US images, which was particularly valuable in the diagnosis and evaluation of tumours.
Conclusions
The poor clinical therapy effect on TNBC was a worldwide problem, which required early precisely diagnosis and treatment method. Hence, we designed a tumour targeted dual-modal microbubbles PTX@RGD-MBs to improve the resolution and sensitivity of conventional US images for tumour diagnosis as well as to enhance the ability of killing tumour cells. This lipid microbubble consisted of the chemotherapeutic agent PTX for killing cancer cells, internal gas SF6 generating CEUS images through the nonlinear harmonics, lipid shell to encapsulate drug and gas, and peptide RGD for active tumour targeting. Under the UTMD, the ultrasound exposure could generate shear stress on TNBC cells to open transient pores on cell membranes, efficiently improve the penetration of MBs to the cells membrane with increased intracellular PTX for better treatment effect on TNBC cells. In addition, as ultrasound contrast agents, PTX@RGD-MBs produced high quality CEUS images in vitro and in vivo, improved the resolution and sensitivity of US imaging and enhanced the comparison between lesions and surrounding normal tissues. Thus, the combination of dual-modal microbubbles PTX@RGD-MBs and UTMD presented a wider range of applications in treatment and diagnosis of TNBC.Experimental section
Materials
1,2-Dipalmitoyl-sn-glycerol-3-phosphoethanolamine (DPPC) and 1,[2-dipalmitoyl-sn-glycerol-3-phosphoethanolamine]-N-[amino(polyethylene glycol)] (DPPE-PEG2000-NH2) were purchased from Ponsure Biotechnology (Shanghai, China, Lot). The commercial SonoVue®, diluting with 5 mL of saline to form MBs, was purchased from Bracco Diagnostics Inc (Geneva, Switzerland). PTX was purchased from Dalian Meilun Biology Technology Co., Ltd (Dalian, China). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and N-hydroxysuccinimide (NHS) were purchased from Sigma-Aldrich Co., Ltd (Shanghai, China). RGD peptide was purchased from CornerStone Therapeutics (Shanghai), Ltd (Shanghai, China). 1-(3-Dimethylamino-propyl)-3-ethylcarbodiimide hydrochloride (EDC) and cholesterol were purchased from Sinopharm Chemical Reagent Co., Ltd (China).MDA-MB-231 triple-negative breast cancer cell line was provided by the State Key Laboratory of Oncogenes and Related Genes, Shanghai Cancer Institute (Shanghai, China). BALB/c nu/nu female mice, weighing 18–20 g, were provided by the Animal Experiment Centre of Shanghai Cancer Institute. All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Shanghai Jiao Tong University and approved by the Animal Ethics Committee of Shanghai Cancer Institute (License no. SYXK (Hu) 2012-0001).
Mouse tumour model
The human MDA-MB-231 breast cancer cells suspension in the DMEM medium was subcutaneously injected into the right back of the nude mice (0.1 mL, 1 × 107 cells per mL per mouse). The mice with a tumour size of 0.6–0.8 cm were used for further experiments.33Synthesis and characterization of DPPE-PEG-RGD
First, RGD (6.62 mmol), EDC (7.28 mmol) and NHS (7.94 mmol) were dissolved in 10 mL DMSO solution. After the mixture was stirred for 1 h at 25 °C, the DPPE-PEG-NH2 (5.94 mmol) were added into the solution. The molar ratios of each component were that RGD:DPPE-PEG-NH2:EDC:NHS = 1:0.9:1.1:1.2. The mixed solution was stirred to react for 8 h at room temperature. Second, the solution was dialyzed against 1000 mL distilled water using a dialysis bag (Mw: 2000) to remove the left RGD peptides (Mw: 754). After drying under vacuum, DPPE-PEG-RGD was obtained as yellow powder and stored at −20 °C. The chemical structure of DPPE-PEG-RGD was confirmed by nuclear magnetic resonance (NMR) spectroscopy.
Preparation of PTX-loaded RGD-lipid microbubbles (PTX@RGD-MBs)
At first, the PTX@RGD-MBs was prepared using a thin film hydration method. Briefly, 2 mg DPPE-PEG-RGD, 250 μg cholesterol, 500 μg DPPC and 0.1 mg PTX were dissolved in 2 mL of trichloromethane solution (PTX 0.05 mg mL−1), in which the mixed film was formed by rotary evaporation. Then, 5 mL of PBS solution was added to hydrate the film for 30 min at 37 °C. Next, the solution was ultrasonic dispersion for 60 s (2–2 s 400 W) by an ultrasonic processor (JY92-2D Ultrasonic cell crusher, Ningbo SCIENTZ biotechnical Co., Ltd). The lipid particles solution was then freeze-dried and placed in a 20 mL vacuum ampere bottle. 10 mL SF6 and 5 mL saline were injected into the ampere bottle, followed by shaking the bottle vigorously until the freeze-dried powder was completely dispersed to obtain PTX@RGD-MBs solution (the concentration of PTX was 0.02 mg mL−1). To satisfy the demands of different experiments, during the preparation of PTX@RGD-MBs, the PTX was replaced by 50 μg Rb (rhodamine B, red fluorescent probe) to prepare Rb@RGD-MBs (the concentration of Rb was 10 μg mL−1).34Physical and chemical properties of PTX@RGD-MBs
The size and surface potential of MBs were evaluated using the Zetasizer IV analyzer (Malvern Zetasizer Nano ZS90, Malvern, UK). To observe the morphology of the MBs, the MBs solution was dropped onto a 300-mesh carbon-coated copper grid and the excess solution was removed using a filter paper. The grid was allowed to dry at room temperature and was observed using transmission electron microscopy (TEM) (H-800, Hitachi, Japan).The concentration of PTX was determined by HPLC (Agilent 1200, USA) with a C18 chromatographic column (Zorbax SB-C18, 150 × 4.6 mm, 5 μm). The mobile phase was acetonitrile: 10 mmol L−1 NH4Ac solution (pH = 5.0) 53:47 at a flow rate of 1.0 mL min−1, and the detection wavelength was 227 nm.
The PTX encapsulated in MBs was assessed using HLPC. Briefly, MBs solution was centrifuged (5000 rpm × 5 min) to remove precipitated PTX,35,36 and the concentration of PTX in the supernatant was determined by HLPC to obtain the unencapsulated amount. The MBs was broken using acetonitrile to release the inner PTX and detected by HPLC to obtain the total amount of PTX. The drug encapsulation efficiency (EE%) was expressed as the percentage of the amount of PTX encapsulated in the MBs (centrifuged MBs solution) in relation to the total amount of PTX initially added (non-centrifuged MBs solution). The drug loading (DL%) was expressed as the percentage of the amount of PTX encapsulated in the MBs with regards to the total amount of MBs.
| EE% = (the amount of the drug in MBs/the amount of total drug) × 100% |
| DL% = (the amount of the drug in MBs/the amount of material used) × 100% |
Ultrasound contrast imaging analysis of PTX@RGD-MBs in vitro
The 5 mL saline, SonoVue and PTX@RGD-MBs were injected into the transparent plastic hose without any visible bubbles. The speed of peristaltic pump (Baoding leifu fluid technology co. LTD, Hebei, China) was 60 rpm to simulate the internal circulation. The hose was immersed into water, and the images were collected by Ultrasonography (VEVO®2100 Small animal ultrasonic imaging platform, Visual Sonics co. LTD, Hong Kong, China) with the conventional B-mode model and the CEUS mode model. Note: the model of ultrasonic probe was MX250S; ultrasonic frequency was 12.5 MHz; the pulse duration was 1 min.Ultrasound contrast imaging analysis of PTX@RGD-MBs in vivo
The anesthetized tumour-bearing nude mice were randomly divided into three groups. By the real-time guidance of ultrasound, the subcutaneous tumours were punctured with a syringe, along with intratumoural injection of 0.5 mL saline, SonoVue and PTX@RGD-MBs solution. Ultrasonography was used to capture the pre- and post-injection images of tumours under the conventional B-mode model and the CEUS model. The post-injection images of tumours were acquired immediately after the injection of either PTX@RGD-MBs or SonoVue solution.Release profiles of PTX@RGD-MBs in vitro
Release profiles of PTX from PTX@RGD-MBs in vitro were investigated using dialysis bag (Mw cutoff: 3500; Millipore, USA). PBS buffer (pH 7.4, with 1 M sodium salicylate) was used as the medium at 37 °C. PTX@RGD-MBs were suspended to 2 mL and dialyzed against 18 mL PBS using a dialysis bag in a constant-temperature shaker with 80 rpm. The PTX@RGD-MBs groups were randomly divided into two groups. For one of PTX@RGD-MBs groups, the PTX@RGD-MBs solution and medium were treated by UTMD (therapeutic ultrasound apparatus topteam161, Chattanooga Company, USA) with a frequency of 1.5 MHz for 1 min at 10 h. In addition, free PTX was tested as the control. For these three groups, 200 μL of dialyzed solution were collected and the same volume of fresh buffer was added at 0.5, 1, 2, 4, 8, 12, 24, 48, 72, 96, 120, 144 h. The concentrations of PTX were determined using HPLC.Cell viability analysis by MTT assay
The cytotoxicity of lipid microbubbles on MBA-MD-231 breast cancer cells was evaluated by the MTT assay. The MBA-MD-231 breast cancer cells were seeded into 96-well plates and cultured overnight. And then, the medium was replaced with a series of concentrations of lipid microbubbles (1–500 μg mL−1). After further cultured for 24, 48 and 72 h, fresh DMEM with MTT (0.5 mg mL−1) was added into each well and incubated for 4 h. After incubation, the resulting formazan was dissolved in 150 μL of DMSO and detected at 490 nm using a Microplate Reader (Bio-Rad Laboratories Inc. Hercules, CA, USA). We also tested the cytotoxicity of PTX@RGD-MBs against MBA-MD-231 cells (PTX 10 μg mL−1).Cellular uptake assay
MBA-MD-231 breast cancer cells were seeded in six-wells culture plate and then exposed to free Rb and Rb@RGD-MBs (Rb: 5 μg mL−1). They were incubated for 2, 6 and 12 h, respectively. The free Rb and Rb@RGD-MBs groups were randomly divided into two groups. One of free Rb and Rb@RGD-MBs groups were treated using UTMD with a frequency of 1.5 MHz for 30 s at 0, 4 and 10 h. The cells were washed 3 times with PBS and stained with Hoechst33342 for 5 min. The intracellular distribution of Rb was visualized with fluorescence inverted microscope (Huarui Chemical Instrument Inc. Guangdong, China).Live imaging using an in vivo fluorescence imaging system
The mice were divided into four groups randomly (n = 3). Free Dir, Dir-MBs + UTMD, Dir-MBs-RGD and Dir-MBs-RGD + UTMD were administered by intravenous injection when the volume of the tumors reached about 100 mm3. Then, at 2, 4, 8, 24 and 48 h post-injection, the nude mice were anesthetized, and fluorescence was observed using In Vivo Imaging Apparatus (LB983, Berthold Technologies Gmbh & Co. KG, Bad Wildbad, Germany). After the experiment, the nude mice were separately sacrificed. Their hearts, livers, spleens, lungs, kidneys, brains and tumors were harvested and also observed with the In Vivo Imaging Apparatus (Ex was 649 nm, Em was 666 nm). UTMD was used with a frequency of 1.5 MHz for 30 s at 1, 3, 7, 23 and 47 h in Dir-MBs + UTMD and Dir-MBs-RGD + UTMD groups.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors acknowledge financial support from NSFC/China (81671687, 81771839 and 81602718), Fundamental Research Funds for the Central Universities (222201814013).Notes and references
- J. D. Brenton, L. A. Carey, A. A. Ahmed and C. Caldas, J. Clin. Oncol., 2005, 23, 7350–7360 CrossRef CAS PubMed.
- S. P. Shah, A. Roth, R. Goya, A. Oloumi, G. Ha, Y. Zhao, G. Turashvili, J. Ding, K. Tse, G. Haffari, A. Bashashati, L. M. Prentice, J. Khattra, A. Burleigh, D. Yap, V. Bernard, A. McPherson, K. Shumansky, A. Crisan, R. Giuliany, A. Heravi-Moussavi, J. Rosner, D. Lai, I. Birol, R. Varhol, A. Tam, N. Dhalla, T. Zeng, K. Ma, S. K. Chan, M. Griffith, A. Moradian, S. W. Cheng, G. B. Morin, P. Watson, K. Gelmon, S. Chia, S. F. Chin, C. Curtis, O. M. Rueda, P. D. Pharoah, S. Damaraju, J. Mackey, K. Hoon, T. Harkins, V. Tadigotla, M. Sigaroudinia, P. Gascard, T. Tlsty, J. F. Costello, I. M. Meyer, C. J. Eaves, W. W. Wasserman, S. Jones, D. Huntsman, M. Hirst, C. Caldas, M. A. Marra and S. Aparicio, Nature, 2012, 486, 395–399 CrossRef CAS PubMed.
- B. D. Lehmann and J. A. Pietenpol, J. Pathol., 2014, 232, 142–150 CrossRef PubMed.
- E. A. Rakha and I. O. Ellis, Pathology, 2009, 41, 40–47 CrossRef PubMed.
- W. J. Gradishar, B. O. Anderson, R. Balassanian, S. L. Blair, H. J. Burstein, A. Cyr, A. D. Elias, W. B. Farrar, A. Forero, S. H. Giordano, M. Goetz, L. J. Goldstein, C. A. Hudis, S. J. Isakoff, P. K. Marcom, I. A. Mayer, B. McCormick, M. Moran, S. A. Patel, L. J. Pierce, E. C. Reed, K. E. Salerno, L. S. Schwartzberg, K. L. Smith, M. L. Smith, H. Soliman, G. Somlo, M. Telli, J. H. Ward, D. A. Shead and R. Kumar, J. Natl. Compr. Cancer Network, 2015, 13, 1475–1485 CrossRef CAS.
- E. Hamilton, G. Kimmick, J. Hopkins, P. K. Marcom, G. Rocha, R. Welch, G. Broadwater and K. Blackwell, Clin. Breast Cancer, 2013, 13, 416–420 CrossRef CAS PubMed.
- H. Fu, X. Zhang, Q. Wang, Y. Sun, L. Liu, L. Huang, L. Ding, M. Shen, L. Zhang and Y. Duan, Biomater. Sci., 2018, 6, 2667–2680 RSC.
- D. M. Skyba, R. J. Price, A. Z. Linka, T. C. Skalak and S. Kaul, Circulation, 1998, 98, 290–293 CrossRef CAS PubMed.
- J. Ren, C. Xu, Z. Zhou, Y. Zhang, X. Li, Y. Zheng, H. Ran and Z. Wang, Acad. Radiol., 2009, 16, 1457–1465 CrossRef PubMed.
- A. Kheirolomoom, P. A. Dayton, A. F. Lum, E. Little, E. E. Paoli, H. Zheng and K. W. Ferrara, J. Control. Release, 2007, 118, 275–284 CrossRef CAS PubMed.
- S. M. Fix, M. A. Borden and P. A. Dayton, J. Control. Release, 2015, 209, 139–149 CrossRef CAS PubMed.
- S. L. Huang, Adv. Drug Delivery Rev., 2008, 60, 1167–1176 CrossRef CAS PubMed.
- S. Qin, C. F. Caskey and K. W. Ferrara, Phys. Med. Biol., 2009, 54, R27–R57 CrossRef PubMed.
- Y. I. Yoon, Y. S. Kwon, H. S. Cho, S. H. Heo, K. S. Park, S. G. Park, S. H. Lee, S. I. Hwang, Y. I. Kim, H. J. Jae, G. J. Ahn, Y. S. Cho, H. Lee, H. J. Lee and T. J. Yoon, Theranostics, 2014, 4, 1133–1144 CrossRef CAS PubMed.
- C. de Gracia Lux, A. M. Vezeridis, J. Lux, A. M. Armstrong, S. R. Sirsi, K. Hoyt and R. F. Mattrey, RSC Adv., 2017, 7, 48561–48568 RSC.
- Y. Zhao, W. Song, D. Wang, H. Ran, R. Wang, Y. Yao, Z. Wang, Y. Zheng and P. Li, ACS Appl. Mater. Interfaces, 2015, 7, 14231–14242 CrossRef CAS PubMed.
- P. Zhong, X. Gu, R. Cheng, C. Deng, F. Meng and Z. Zhong, Int. J. Nanomed., 2017, 12, 7913–7921 CrossRef CAS PubMed.
- B. A. Othman, C. Greenwood, A. F. Abuelela, A. A. Bharath, S. Chen, I. Theodorou, T. Douglas, M. Uchida, M. Ryan, J. S. Merzaban and A. E. Porter, Adv. Healthcare Mater., 2016, 5, 1310–1325 CrossRef CAS PubMed.
- J. L. Wagstaff, M. L. Rowe, S. J. Hsieh, D. DiCara, J. F. Marshall, R. A. Williamson and M. J. Howard, RSC Adv., 2012, 2, 11019–11028 RSC.
- C. Nicolau, V. Catala, R. Vilana, R. Gilabert, L. Bianchi, M. Sole, M. Pages and C. Bru, Eur. J. Radiol., 2004, 14, 1092–1099 CrossRef PubMed.
- L. Lin, Y. Fan, F. Gao, L. Jin, D. Li, W. Sun, F. Li, P. Qin, Q. Shi, X. Shi and L. Du, Theranostics, 2018, 8, 1923–1939 CrossRef CAS PubMed.
- K. Tachibana, T. Uchida, K. Ogawa, N. Yamashita and K. Tamura, Lancet, 1999, 353, 1409 CrossRef CAS.
- A. van Wamel, K. Kooiman, M. Harteveld, M. Emmer, F. J. ten Cate, M. Versluis and N. de Jong, J. Control. Release, 2006, 112, 149–155 CrossRef CAS PubMed.
- S. Mehier-Humbert, T. Bettinger, F. Yan and R. H. Guy, J. Control. Release, 2005, 104, 213–222 CrossRef CAS PubMed.
- Q. Wang, P. Liu, Y. Sun, T. Gong, M. Zhu, X. Sun, Z. Zhang and Y. Duan, Nanotoxicology, 2015, 9, 190–200 CrossRef CAS PubMed.
- C. Lim, J. Moon, T. Sim, N. H. Hoang, W. R. Won, E. S. Lee, Y. S. Youn, H. G. Choi, K. Oh and K. T. Oh, Int. J. Nanomed., 2018, 13, 4627–4639 CrossRef PubMed.
- T. Yue, H. L. Xu, P. P. Chen, L. Zheng, Q. Huang, W. S. Sheng, Y. D. Zhuang, L. Z. Jiao, T. T. Chi, D. L. Zhu Ge, J. J. Liu, Y. Z. Zhao and L. Lan, Int. J. Pharm., 2017, 528, 664–674 CrossRef CAS PubMed.
- Q. Shi, P. Liu, Y. Sun, H. Zhang, J. Du, F. Li, L. Du and Y. Duan, J. Biomed. Nanotechnol., 2014, 10, 436–444 CrossRef CAS PubMed.
- Y. Zhang, H. F. Chan and K. W. Leong, Adv. Drug Delivery Rev., 2013, 65, 104–120 CrossRef CAS PubMed.
- R. Wang, Y. Zhou, P. Zhang, Y. Chen, W. Gao, J. Xu, H. Chen, X. Cai, K. Zhang, P. Li, Z. Wang, B. Hu, T. Ying and Y. Zheng, Theranostics, 2017, 7, 846–854 CrossRef CAS PubMed.
- E. Provenzano, V. Bossuyt, G. Viale, D. Cameron, S. Badve, C. Denkert, G. MacGrogan, F. Penault-Llorca, J. Boughey, G. Curigliano, J. M. Dixon, L. Esserman, G. Fastner, T. Kuehn, F. Peintinger, G. von Minckwitz and J. White, Mod. Pathol., 2015, 28, 1185–1201 CrossRef PubMed.
- S. Elzamly, N. Badri, O. Padilla, A. K. Dwivedi, L. A. Alvarado, M. Hamilton, N. Diab, C. Rock, A. Elfar, M. Teleb, L. Sanchez and Z. Nahleh, Breast Cancer: Basic Clin. Res., 2018, 12, 1178223418788074 Search PubMed.
- S. Liu, J. Liu, Q. Ma, L. Cao, R. J. Fattah, Z. Yu, T. H. Bugge, T. Finkel and S. H. Leppla, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E4079–E4087 CrossRef CAS PubMed.
- L. Xing, Q. Shi, K. Zheng, M. Shen, J. Ma, F. Li, Y. Liu, L. Lin, W. Tu, Y. Duan and L. Du, Theranostics, 2016, 6, 1573–1587 CrossRef CAS PubMed.
- A. Upadhyay, B. Yagnik, P. Desai and S. V. Dalvi, ACS Omega, 2018, 3, 12824–12831 CrossRef CAS PubMed.
- H. K. Chen, S. M. Zhang, J. L. Chang, H. C. Chen, Y. C. Lin, C. P. Shih, H. K. Sytwu, M. C. Fang, Y. Y. Lin, C. Y. Kuo, A. H. Liao, Y. H. Chu and C. H. Wang, Cancers, 2018, 10(9), 311 CrossRef PubMed.
Footnote |
| † The two authors have contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |