
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTotal synthesis of diazaquinomycins H and J using double Knorr cyclization in the presence of triisopropylsilane†
Allan M. Prior and
Dianqing Sun *
*
Department of Pharmaceutical Sciences, The Daniel K. Inouye College of Pharmacy, University of Hawaii at Hilo, 34 Rainbow Drive, Hilo, Hawaii 96720, USA. E-mail: dianqing@hawaii.edu; Fax: +1 808 933 2974; Tel: +1 808 933 2960
First published on 14th January 2019
Abstract
The first total synthesis of diazaquinomycins H (1) and J (2), which are promising anti-tuberculosis natural product leads, has been achieved via selective amidation of diamine 6 with Meldrum's acid derivatives, subsequent EDC coupling with 3-oxobutanoic acid, followed by double Knorr cyclization in the presence of triisopropylsilane (TIPS). We found that the addition of TIPS was crucial to obtain pure diazaquinomycins H and J, while preventing isomerization of the terminal iso-branched tail in sulfuric acid. Our developed synthesis provided diazaquinomycins H (1) and J (2) in 8 steps from commercially available starting materials in 25% and 21% overall yields, respectively. The spectroscopic data of synthetic diazaquinomycins H (1) and J (2) agreed very favorably with those of reported natural products.
Introduction
Diazaquinomycins (DAQs) are secondary metabolites produced by terrestrial and aquatic Actinobacteria, predominantly from the genus Streptomyces sp.1–5 Spanning the last three and a half decades, nine DAQs have been reported, however, diazaquinomycins D, F, and G were not obtained in pure form (Fig. 1).1–5 Specifically, diazaquinomycin D was a trace component in a sample of diazaquinomycin C that was produced by Streptomyces sp. GW48/1497 when cultivated in soybean flour and mannitol.3 Its structure was assigned using (−)-ESI mass spectrometry.3 The isomeric diazaquinomycins F and G were isolated as an inseparable mixture;2 their structures were established using a combination of NMR and UV spectroscopy, HRMS, and X-ray crystallography.2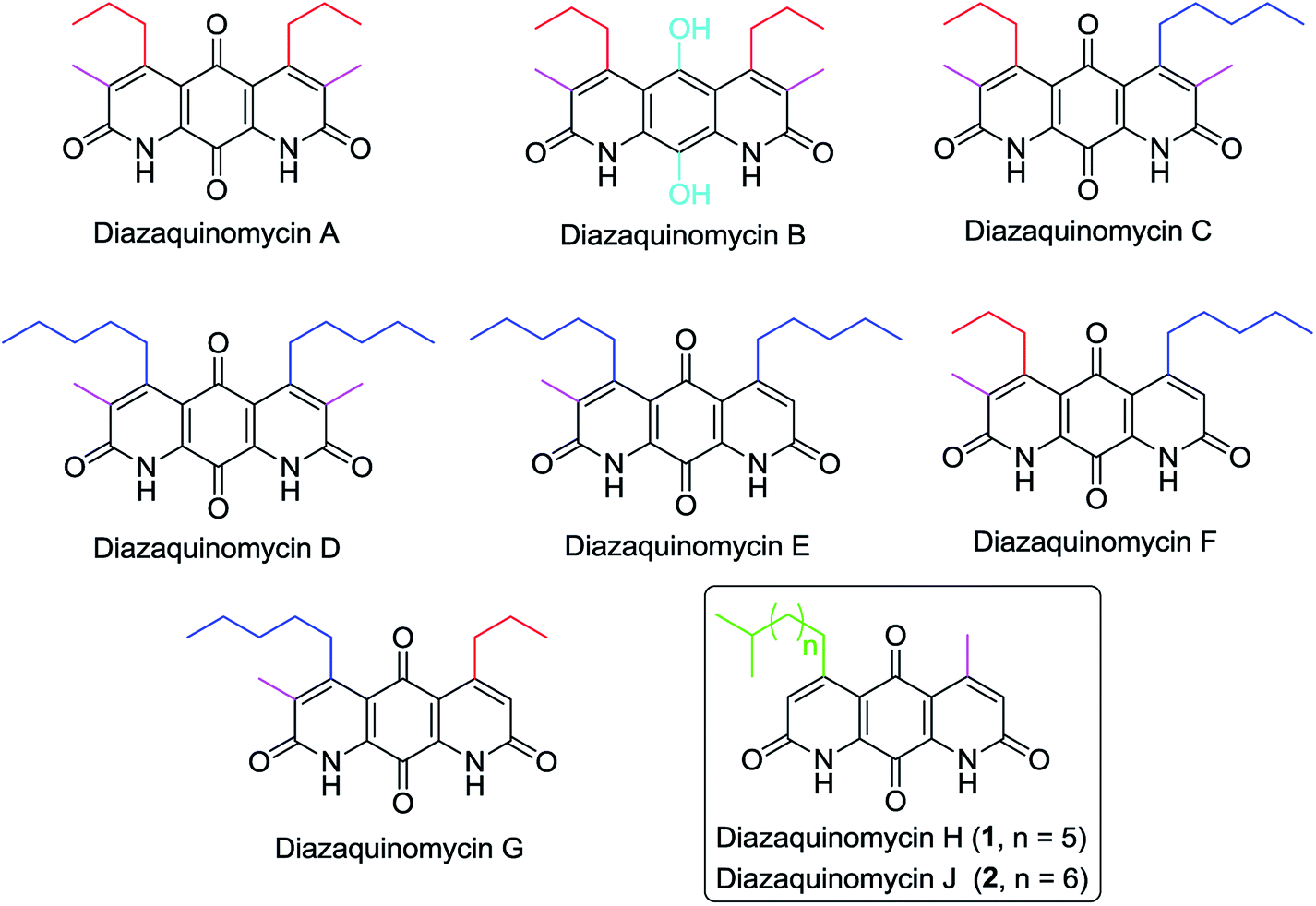 | ||
| Fig. 1 Chemical structures of naturally occurring diazaquinomycins A–H and J. | ||
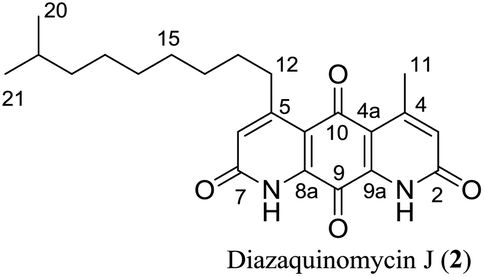
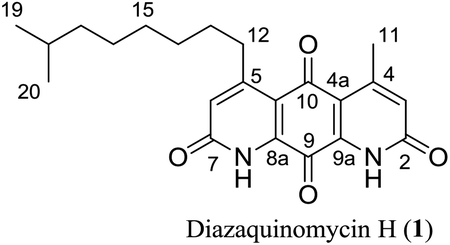
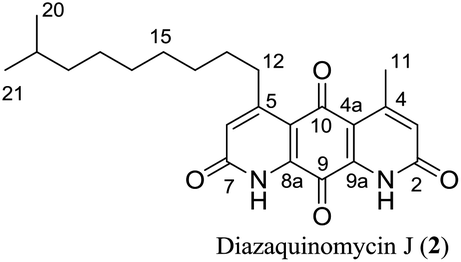
Diazaquinomycins H (1) and J (2), bearing a long lipophilic side chain with a unique terminal isopropyl functionality, are the latest additions to this family and were isolated by Mullowney et al. from fresh water-derived Actinobacteria in Lake Michigan.1 Notably, diazaquinomycins H and J along with diazaquinomycin A showed excellent in vitro antibacterial activity against Mycobacterium tuberculosis (M. tuberculosis) with minimum inhibitory concentration (MIC) values of 0.04, 0.07, and 0.10 μg mL−1, respectively.1 In addition, these analogues did not display mammalian cytotoxicity against Vero cells when tested at 28 μM, however, the limited quantity (0.3 mg) of isolated diazaquinomycins H and J prevented further antimicrobial assessment.1 More extensive evaluation and profiling using diazaquinomycin A revealed that it also retained potent anti-tuberculosis activity (MIC = 0.06–0.27 μg mL−1) against drug resistant M. tuberculosis strains with known resistance to rifampicin, isoniazid, streptomycin, kanamycin, or cycloserine.1 The specific mechanism of action of diazaquinomycin A against M. tuberculosis remains unclear, though it was previously reported to interfere with the folate pathway,6 acting as a competitive inhibitor of thymidylate synthase.7,8 However, M. tuberculosis does not encode human thymidylate synthase and M. tuberculosis thymidylate synthase enzymes (ThyA and ThyX) were not targeted by diazaquinomycin A.1
Diazaquinomycins A and B were first synthesized in 1988 by Kelly et al. via a double Knorr cyclization of a symmetrical di-β-ketoanilide hydroquinone precursor.9 In 1990, Lee et al. reported the synthesis of a diazaanthraquinone scaffold using hetero Diels–Alder methodology by coupling a dienophile with a 1-azadiene, followed by dibromohydrin formation and oxidation.10 In 1998 and 2000, two alternative syntheses of diazaquinomycin A were reported by Perez et al. and similarly made use of hetero Diels–Alder chemistry to construct the tricyclic core.11,12 Subsequent aromatization, N-oxide formation, and tosyl chloride mediated N-oxide rearrangement afforded diazaquinomycin A.11,12 The Rinehart group reported the synthesis of deoxynyboquinone (DNQ) possessing a 1,8-diazaanthraquinone scaffold via the oxidation of natural product deoxynybomycin in hot nitric acid.13 Hergenrother and coworkers reported the synthesis of DNQ analogues starting from a monocyclic dipinacolborane.14,15 Their synthetic route used Suzuki–Miyaura cross coupling, followed by Buchwald–Hartwig amidation as key ring forming methodology. Final oxidation of the penultimate tricyclic methoxy intermediate to quinone was done using nitric acid15 or a sequence of hydrobromic acid, followed by salcomine in the presence of oxygen.14
We became interested in diazaquinomycins due to their unique 1,8-diazaanthraquinone scaffold and promising antitubercular and antibacterial activities.1,4 Recently, we expanded the scope of an underexplored double Knorr cyclization and demonstrated that this methodology can serve as a general and expedient route to symmetrical or unsymmetrical 1,8-diazaanthraquinone scaffolds (Scheme 1).16 We established that the 5-hydroxy substituent on the di-β-ketoanilide hydroquinone substrate is essential for successful double Knorr cyclization. On the other hand, sterically hindered di-β-ketoanilides with benzyl or isopropyl substitution on the side chains adjacent to β-carbonyl were poor substrates in this reaction.16 In this work, we extend this methodology to further develop an efficient and first chemical synthesis of diazaquinomycins H (1) and J (2) with a long iso-branched aliphatic side chain.
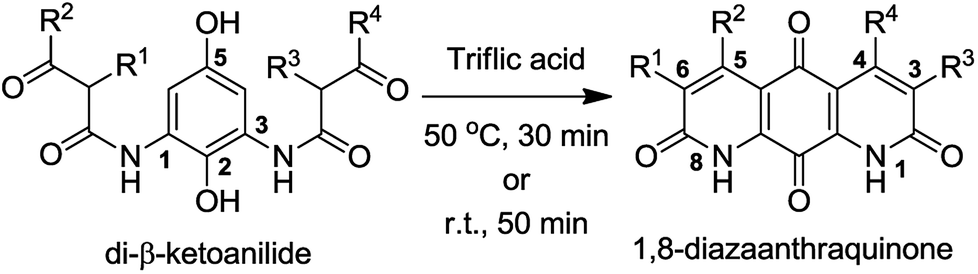 | ||
| Scheme 1 Double Knorr cyclization of di-β-ketoanilides to 1,8-diazaanthraquinones. | ||
Results and discussion
Retrosynthetic analysis
The retrosynthetic approach to diazaquinomycins H (1) and J (2) is outlined in Scheme 2. A key synthetic step would involve the double Knorr cyclization of di-β-ketoanilide hydroquinones 3a and 3b having their long iso-branched side chain preinstalled. Hydroquinones 3a and 3b will be obtained from 4a and 4b after removal of MOM protecting groups. The di-β-ketoanilides 4a and 4b will be obtained in two sequential steps, firstly amidation of diamine 6 with Meldrum's acid derivatives (7a or 7b), then 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) coupling with 3-oxobutanoic acid. The Meldrum's acid derivatives can be obtained through coupling commercially available iso-branched carboxylic acids 8a and 8b with Meldrum's acid (9). We envisioned that challenges may reside in the double Knorr cyclization of di-β-ketoanilide precursor 3a or 3b having a long, iso-branched aliphatic side chain, as well as the mono-amidation of diamine intermediate 6.16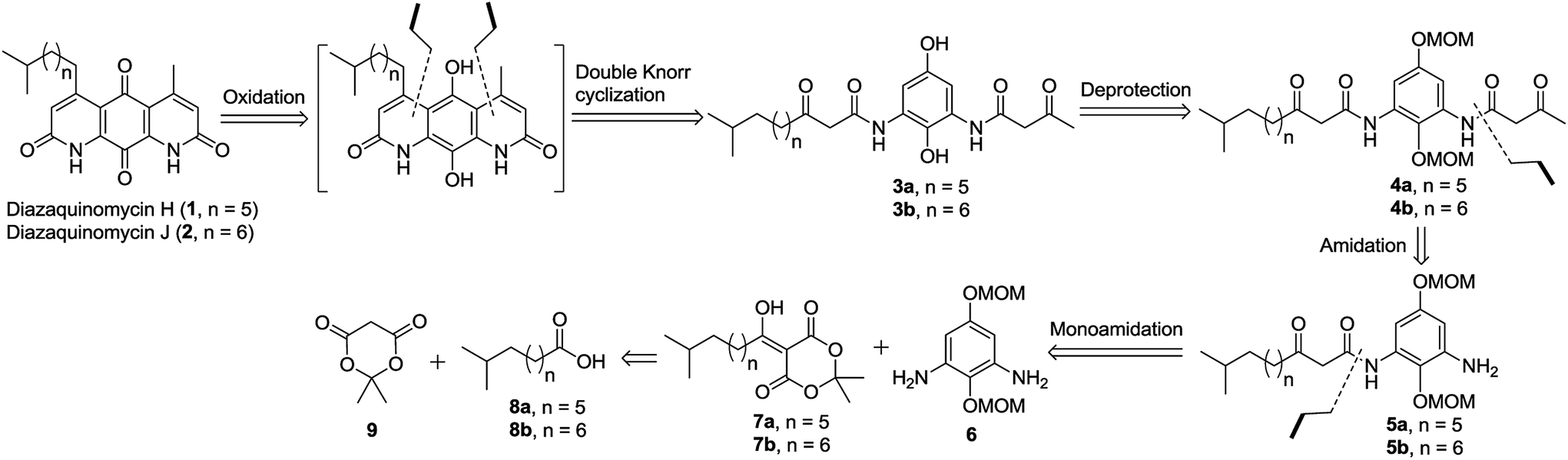 | ||
| Scheme 2 Retrosynthetic analysis to diazaquinomycins H and J. | ||
Synthesis of key di-β-ketoanilide intermediates 3a–b via selective amidation of 6 with Meldrum's acid derivatives
The synthesis of di-β-ketoanilide hydroquinone substrates 3a and 3b started from commercially available 1,4-diacetoxybenzene (10) and iso-branched 8-methylnonanoic acid (8a) or 9-methyldecanoic acid (8b) (Scheme 3). Diamine 6 was obtained in 4 synthetic steps from 10 following our reported synthesis.16 Meldrum's acid derivatives 7a and 7b were synthesized by coupling 8a and 8b with Meldrum's acid (9) using N,N′-dicyclohexylcarbodiimide (DCC) and DMAP in DCM in 78% and 84% yields, respectively, following a modified literature protocol.17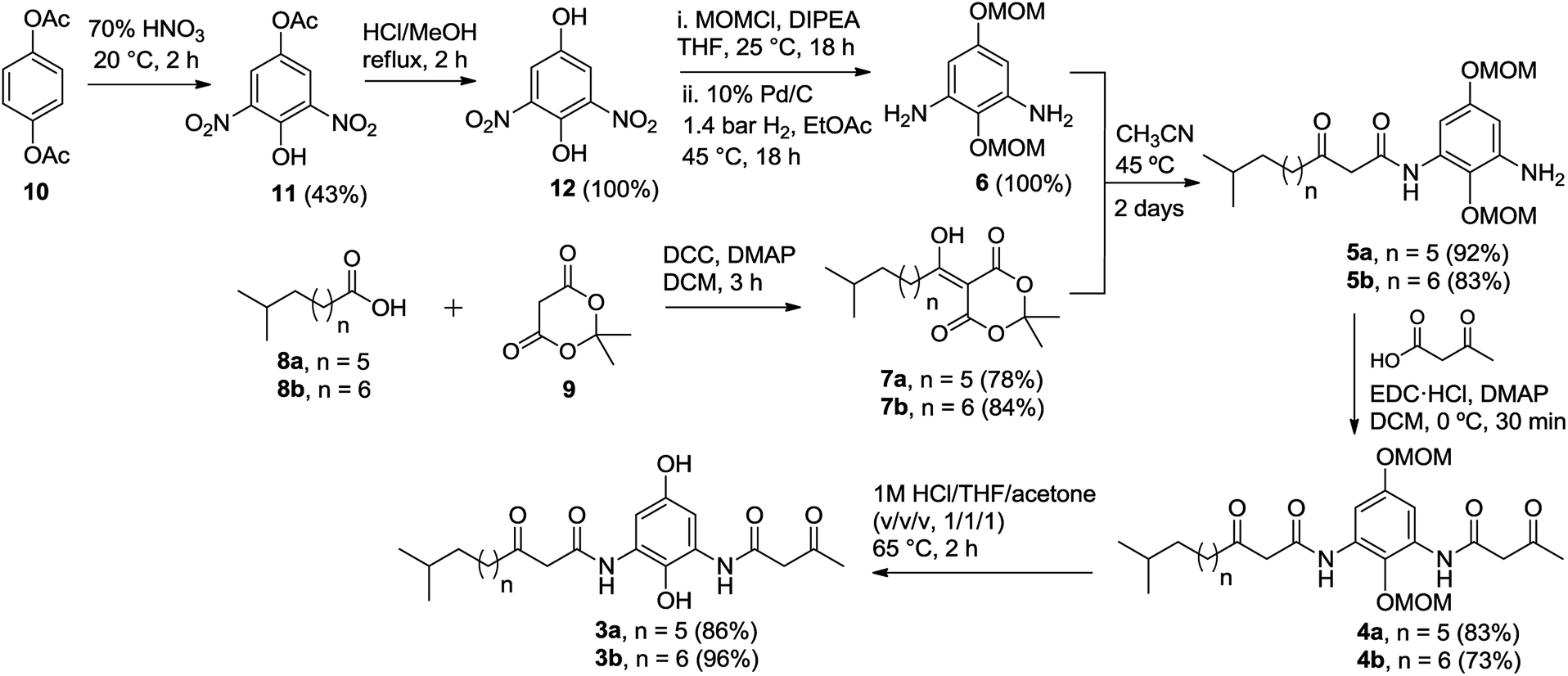 | ||
| Scheme 3 Synthesis of unsymmetrical di-β-ketoanilide hydroquinones 3a–b. | ||
With diamine 6 and Meldrum's acid derivatives 7a and 7b in hand, we evaluated the synthesis towards mono-β-ketoanilides 5a and 5b. Our previous synthesis of a mono-β-ketoanilide analogue via EDC coupling of 6 with a β-ketoacid was successful, but resulted in poor isolated yield (25%).16 Therefore, an improved synthesis was developed by coupling diamine 6 with Meldrum's acid derivative 7a or 7b to form mono-β-ketoanilide 5a or 5b, respectively. Initially, 6 was treated with 0.7 molar equivalents of Meldrum's acid derivative at 45 °C in dry acetonitrile. After 2 days the Meldrum's acid derivative was consumed and HPLC analysis at 254 nm showed a mono- to di-β-ketoanilide ratio of 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, along with unreacted 6. When 0.6 equivalents of Meldrum's acid derivative was used relative to 6, the mono- to di-β-ketoanilide ratio increased to 9:1. This improvement greatly facilitated purification of mono-β-ketoanilides using flash column chromatography, affording 5a and 5b in 92% and 83% isolated yields, respectively. It is noteworthy to mention that the yields of 5a and 5b were calculated based on Meldrum's acid derivative 7a or 7b, which was used as the limiting reagent in order to favor the formation of desired mono-β-ketoanilide, as well as due to the valuable nature of carboxylic acid precursors 8a and 8b (approx. $900 per gram). Unreacted diamine 6 could also be recovered from column purification. To the best of our knowledge, this represents the first example reporting the synthesis of mono-β-ketoanilides directly from a diamine precursor using Meldrum's acid derivatives. In the literature, it was reported that β-ketoanilides or β-ketoamides can form when 5-substituted 2,2-dimethyl-1,3-dioxane-4,6-dione (acyl Meldrum's acid) derivatives were treated with anilines18,19 or amines,20,21 in solvent such as benzene18,19 or acetonitrile.21,22
:1, along with unreacted 6. When 0.6 equivalents of Meldrum's acid derivative was used relative to 6, the mono- to di-β-ketoanilide ratio increased to 9:1. This improvement greatly facilitated purification of mono-β-ketoanilides using flash column chromatography, affording 5a and 5b in 92% and 83% isolated yields, respectively. It is noteworthy to mention that the yields of 5a and 5b were calculated based on Meldrum's acid derivative 7a or 7b, which was used as the limiting reagent in order to favor the formation of desired mono-β-ketoanilide, as well as due to the valuable nature of carboxylic acid precursors 8a and 8b (approx. $900 per gram). Unreacted diamine 6 could also be recovered from column purification. To the best of our knowledge, this represents the first example reporting the synthesis of mono-β-ketoanilides directly from a diamine precursor using Meldrum's acid derivatives. In the literature, it was reported that β-ketoanilides or β-ketoamides can form when 5-substituted 2,2-dimethyl-1,3-dioxane-4,6-dione (acyl Meldrum's acid) derivatives were treated with anilines18,19 or amines,20,21 in solvent such as benzene18,19 or acetonitrile.21,22
Once 5a–b were synthesized, the second β-ketoanilide side chain was installed via EDC coupling of 5a or 5b with 3-oxobutanoic acid in dichloromethane at 0 °C. We found that a cold reaction temperature was necessary to slow the decomposition of 3-oxobutanoic acid (via decarboxylation), allowing for reaction completion. This afforded the unsymmetrical di-β-ketoanilides 4a and 4b in 83% and 73% yields, respectively, after flash column chromatography. Alternatively, crude product 5a or 5b containing unreacted 6 could be used directly in EDC coupling with excess 3-oxobutanoic acid since the resulting di-β-ketoanilide reaction mixture can be easily separated by flash column chromatography. Next, the MOM protecting groups were removed by treating 4a or 4b with 1 M HCl:THF:acetone (1:1:1) at 65 °C for 2 h to afford 3a or 3b in 86% and 96% yields, respectively, after trituration with diethyl ether. The trituration step was introduced because in our hands the purification of 3a or 3b by flash column chromatography on silica gel led to poor isolated yields. This observation was in contrast to linear chain analogues, which could be isolated in good yields using flash column chromatography, suggesting the iso-branched functionality in 3a or 3b may play a major role in decomposition on silica gel.
Double Knorr cyclization and optimization studies
Using our previously optimized conditions,16 3a or 3b was subjected to double Knorr cyclization in an effort to synthesize diazaquinomycins H (1) and J (2). Very surprisingly, double Knorr cyclization of 3a or 3b in triflic acid at 50 °C resulted in a complex reaction mixture, and diazaquinomycins H (1) or J (2) could not be detected and isolated (Table 1, entries 1–2). This revealed that the longer (n = 5, 6) and/or iso-branched side chain significantly impacted this reaction outcome. Presumably, when cyclization rate is lowered, decomposition could occur through anilide hydrolysis,9,16 4-pyrone formation or heterolytic cleavage of anilide.23 Additionally, hydrocarbons have been shown to undergo isomerization24–26 under certain acidic conditions. Moreover, the length of aliphatic side chain can have an effect in these processes, with longer chains showing faster conversion rates.27,28 To probe the effect of branched functionality on reaction outcome as well as given the precious nature of 3a–b, we next synthesized model substrate 3c with a linear side chain (n = 5) for comparison and further optimization studies (Table 1, entries 3–10). Interestingly, when the double Knorr cyclization was performed using 3c in triflic acid, the reaction yielded a mixture of tricyclic hydroquinone (HQ) and 13 in 33% and 33% HPLC yields, respectively, along with unidentified decomposition byproducts (Table 1, entry 3). Nevertheless, the fact that linear chain analogue 3c could be cyclized using triflic acid demonstrated that decomposition of 3a and 3b in triflic acid is probably accelerated by having an iso-branched side chain, as this is the only structural difference between these substrates. Furthermore, the observation of HQ intermediates in this study was striking because they were not previously detected in the double Knorr cyclization of shorter chain analogues.16 The slower rate of oxidation from HQ intermediates to desired quinone products was likely due to steric effect of the adjacent longer aliphatic chain at the 5 position.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Substrate | Reagent/solvent | Temp (°C) | Time (min) | HPLC yield (%) | |
| HQ intermediatea | Productb | |||||
| a Hydroquinone (HQ) intermediates were observed using HPLC and MS and not isolated.b Isolated yield in parentheses.c The reaction was performed in sulfuric acid for 30 min, then diluted in methanol, and continued to stir for 2–3 h.d LC-MS and 13C NMR revealed that 1 or 2 contained its corresponding branched side chain isomer(s). | ||||||
| 1 | 3a | TfOH | 50 | 30 | 0 | 0 |
| 2 | 3b | TfOH | 50 | 30 | 0 | 0 |
| 3 | 3c | TfOH | 50 | 30 | 33 | 33 |
| 4 | 3c | H2SO4 | 50 | 30 | 60 | 40 |
| 5 | 3c | H2SO4 | 50 | 3 h | 50 | 50 |
| 6 | 3c | H2SO4 | 50 | 18 h | 33 | 67 |
| 7 | 3c | H2SO4 | 110 | 30 | 10 | 80 |
| 8 | 3c | H2SO4 | 110 | 60 | 1 | 90 (91) |
| 9 | 3c | H2SO4 | 25 | 15 | 89 | 11 |
| 10c | 3c | H2SO4 | 25 | 30 | 0 | 100 (99) |
| 11c | 3a | H2SO4 | 25 | 30 | 0 | 100 (99)d |
| 12c | 3b | H2SO4 | 25 | 30 | 0 | 100 (99)d |
Considering that sulfuric acid is much milder acid than triflic acid, we next investigated if sulfuric acid can perform better in the double Knorr cyclization of long-chain (e.g., n = 5 or 6) di-β-ketoanilide substrates, while potentially minimizing the byproduct formation. In contrast to triflic acid, cyclization of 3c using sulfuric acid at 50 °C for 30 min gave a clean reaction profile and afforded its corresponding HQ intermediate and 13 in 60% and 40% HPLC yields, respectively (Table 1, entry 4). Continued stirring at 50 °C for 3 h (Table 1, entry 5) and 18 h (Table 1, entry 6) could not complete the oxidation of HQ into 13. At 110 °C in sulfuric acid, the oxidation of HQ into 13 was greatly accelerated, affording 13 in 91% isolated yield after 1 h (Table 1, entries 7–8). The rate limiting step for this transformation was the oxidation of HQ intermediate to 13 since starting material 3c was already consumed after 2 min based on HPLC monitoring. When the double Knorr cyclization of 3c was performed at room temperature in sulfuric acid, an 8:1 mixture of HQ and 13 was formed after 15 min (Table 1, entry 9) and additional stirring at room temperature could not complete the oxidation into 13. In the literature, structurally similar tricyclic hydroquinone scaffolds have been shown to spontaneously be oxidized to anthraquinone in the presence of oxygen.29–33 We re-analyzed some previous HPLC samples and found that the HQ intermediate can be oxidized into 13 within 2 h when diluted in methanol. Based on our finding, a milder and optimized protocol was developed, which involved stirring 3c in sulfuric acid at room temperature for 30 min, followed by dilution in methanol and continued stirring open to air until the oxidation of HQ into 13 was complete (Table 1, entry 10). This provided 13 in 99% isolated yield as a red solid after removing methanol in vacuo, precipitation with water, and collecting solids by centrifugation.
Double Knorr cyclization of 3a or 3b in sulfuric acid leading to C-5 tail isomerization
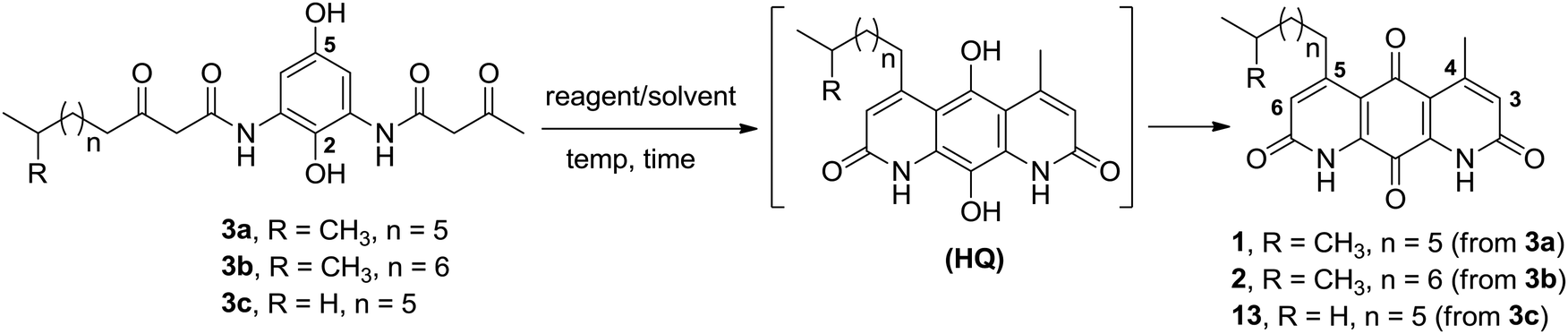
Having established a milder double Knorr cyclization protocol, we proceeded to synthesize diazaquinomycins H (1) and J (2) from 3a or 3b, respectively. Di-β-ketoanilide 3a or 3b was treated in sulfuric acid at room temperature for 30 min, then diluted with methanol and stirred at room temperature to complete the oxidation of HQ intermediates to diazaquinomycins H (1) and J (2) (Table 1, entries 11 and 12). In both cases, reactions went to completion and red solids were obtained in corresponding to 99% isolated yields. Based on their analytical HPLC profiles and 1H NMR spectra, the purity of 1 and 2 appeared to be excellent. However, additional carbon signals were observed in their 13C NMR spectra, especially in the C-5 aliphatic side chain region. Further LC-MS analysis revealed that both synthesized diazaquinomycins H (1) and J (2) samples also contained corresponding C-5 aliphatic side chain isomers. The representative HPLC chromatogram of the isomeric mixture of 1 showed a single peak (Fig. 2a). However, using a longer HPLC column under optimized mobile phase conditions, we were able to detect and partially separate 1 and its side chain isomers, which were confirmed by (+)-ESI mass spectrometry (Fig. 2b). To further investigate the isomerization rate during the double Knorr cyclization from 3a to 1, we closely monitored the reaction using LC-MS and the results are shown in Fig. 2c. For clarity and improving the peak resolution, the mass range on the MS detector was set to exclusively monitor m/z = 383 ± 2. Our LC-MS reaction monitoring data showed the presence of the C-5 chain isomer even after 2 min, which continued to increase over the course of 60 min.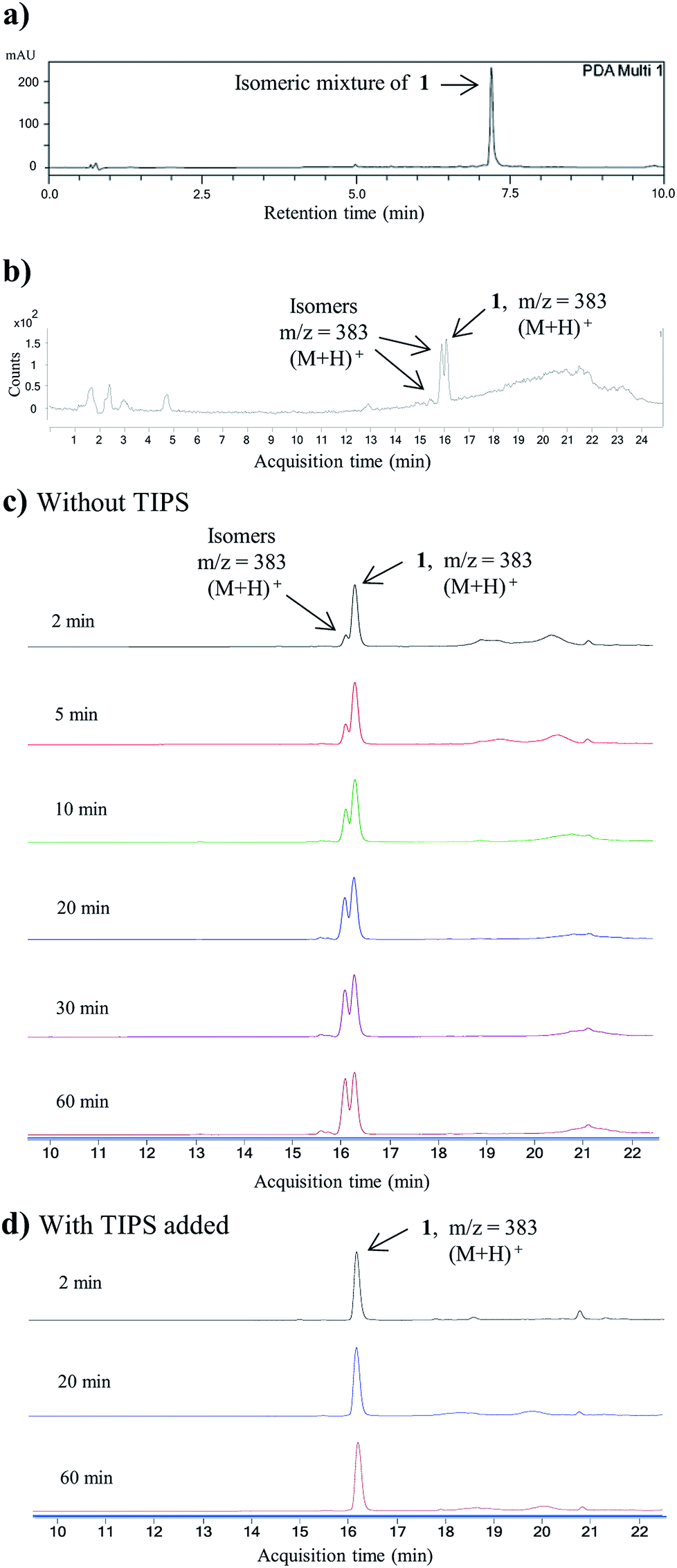 | ||
| Fig. 2 (a) HPLC chromatogram for the isomeric mixture of 1. (b) LC-MS chromatogram of the isomeric mixture of 1. (c) LC-MS reaction monitoring of double Knorr cyclization of 3a to 1, which was performed in sulfuric acid only at room temperature. (d) LC-MS reaction monitoring of double Knorr cyclization of 3a to 1 in sulfuric acid at room temperature with TIPS added. For clarity, LC-MS analyses shown in Fig. (c) and (d) were recorded with MS detector set to monitor m/z = 383 ± 2. | ||
It was concluded that the C-5 distal iso-branched side chain of diazaquinomycins H (1) or J (2) must play a critical role in isomerization since our linear chain model compound 13 was obtained in pure form. In addition, we found that the length of the iso-branched alkyl chain also played an important role in this isomerization process, with a longer iso-branched alkyl chain being more prone to isomerization. For example, for comparison to 2, we also synthesized an iso-branched four-carbon shorter alkyl chain analogue 14 from 3d (Scheme S1, ESI†), which did not undergo isomerization during double Knorr cyclization in H2SO4. Moreover, in a side by side comparison, 9-methyldecanoic acid (8b) showed notable isomerization after being treated in conc. H2SO4 for 30 min at room temperature (Fig. S1, ESI†), whereas 4-methylpentanoic acid with a five-carbon shorter chain was stable under the same conditions even after 18 h (Fig. S2, ESI†).
The isomerization of iso-branched analogues may result from carbenium ion formation near the terminal end of the C-5 aliphatic chain in sulfuric acid, followed by hydride or methide migration.24,25 A proposed isomerization mechanism for representative diazaquinomycin J (2) is shown in Scheme 4. Unlike expected, the isomerization pathway probably proceeds via the initial formation of a secondary carbocation center, in which subsequent rearrangement results in anteiso and propyl-methyl isomers. In the case of initial formation of a tertiary carbocation center at the iso-branched tail, rearrangement may not take place due to its potential to generate a less favorable primary carbocation intermediate. Therefore, the secondary carbenium ion formation in the C-5 side chain is induced instead, upon exposure of 2 to concentrated sulfuric acid. Skeletal rearrangement of the carbenium ion then takes place via a non-classical protonated cyclopropane intermediate25,27,34,35 to form a rearranged carbenium ion. The newly formed carbenium ion further undergoes intermolecular hydride transfer with a reactant alkane to form an isomerized alkane (anteiso-isomer), while generating another carbenium ion and making the process catalytic. In a similar manner, the formed anteiso-isomer can also be rearranged to form additional isomers (e.g., propyl-methyl isomer).
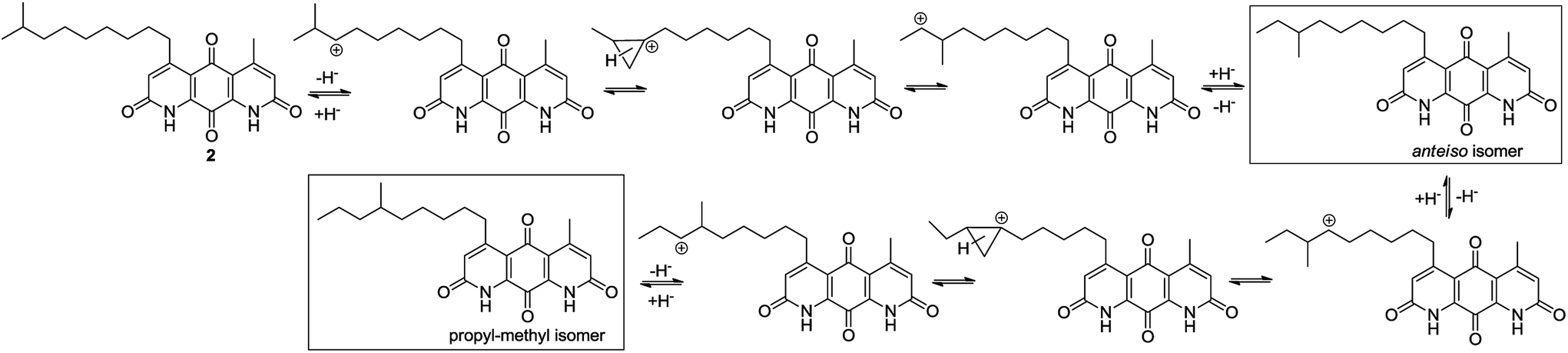 | ||
| Scheme 4 Proposed isomerization mechanism for diazaquinomycin J (2) in sulfuric acid. | ||
Adding TIPS effectively preventing isomerization in double Knorr cyclization of 3a or 3b in sulfuric acid
As shown in Fig. 2b, c and 3b, the double Knorr cyclization of 3a and 3b in sulfuric acid resulted in significant isomerization of the terminal C-5 iso-branched aliphatic chain. Attempts to prevent isomerization by lowering the reaction temperature or shortening the exposure time to sulfuric acid were unsuccessful. To overcome this problem, we envisioned that adding a carbocation scavenger such as triisopropylsilane (TIPS) in the reaction may eliminate isomerization, since the isomerization process in acid presumably took place via carbenium ion formation.25 This hypothesis led to an extremely successful outcome and adding TIPS in the reaction completely suppressed the isomerization of 1 or 2 in sulfuric acid, and no isomerization products were detected up to 60 min (Fig. 2d and 3c). In short, 3a or 3b was treated with H2SO4 and TIPS (5:1) at room temperature for 15 min, followed by stirring in methanol for 2 h to complete the oxidation to diazaquinomycin scaffolds (Scheme 5). This provided pure samples of diazaquinomycins H (1) and J (2) in 89% and 84% isolated yields, respectively.
 | ||
| Fig. 3 (a) Structures of diazaquinomycin J (2) and proposed anteiso and propyl-methyl isomers. (b) LC-MS chromatogram of isomeric mixture of 2; (c) LC-MS chromatogram of pure synthetic 2; (d) zoomed 1H NMR spectrum of isomeric mixture of 2; (e) zoomed 1H NMR spectrum of pure synthetic 2; (f) zoomed 13C NMR spectrum of isomeric mixture of 2; (g) zoomed 13C NMR spectrum of pure synthetic 2. For clarity, LC-MS analyses shown in (b) and (c) were recorded with MS detector set to monitor m/z = 397 ± 2. | ||
 | ||
| Scheme 5 Optimized synthesis of diazaquinomycins H (1) and J (2). | ||
A direct comparison of 1H and 13C NMR spectra for isomeric mixture and pure diazaquinomycin J (2) was made and is shown in Fig. 3d–g. When only sulfuric acid was used in double Knorr cyclization, 2 and its isomers were produced. The doublet for the C-5 iso-branch (H-20/H-21) at 0.87 ppm was seen overlapping with a multiplet (Fig. 3d). In contrast, when TIPS was included in the double Knorr cyclization of 3b in sulfuric acid, pure diazaquinomycin J (2) was obtained, and the multiplet was absent from the 1H NMR spectrum with a clean doublet at 0.87 ppm observed (Fig. 3e). The 13C NMR spectrum for the isomeric mixture of 2 showed multiple sets of carbon signals, one of which corresponded to those of reported natural diazaquinomycin J (2)1 (Fig. 3f). When the double Knorr cyclization was carried out in the presence of TIPS, the extra carbon peaks in the 13C NMR spectrum of isolated pure diazaquinomycin J (2) were absent (Fig. 3g). It should be noted, the additional carbon signals in the isomeric mixture (Fig. 3f) could not be completely assigned; however, characteristic chemical shift patterns in the up field region (<21 ppm) matched extremely well with those of anteiso and propyl-methyl isomers (Fig. 3a). The presence of carbon signals at 11 and 19 ppm strongly suggested the presence of an anteiso-type36–39 substitution pattern and the presence of 14, 19 and 20 ppm signals strongly suggested the presence of a propyl-methyl isomer form.36,39 In each case, the terminal methyl group migrated to the C-18 or C-17 position of the alkyl chain, respectively. The direct comparison of NMR spectra for the isomeric mixture of 1 and pure synthetic 1 is provided in ESI (Fig. S3†).
Comparison of 1H and 13C NMR data of natural and synthetic diazaquinomycins H (1) and J (2)
Finally, the 1H and 13C NMR spectroscopic data of synthesized diazaquinomycins H (1) and J (2) compared very favorably with reported data for the isolated natural products1 (Tables 2 and 3). It should also be noted, NH resonance was not observed in our 1H NMR spectra, presumably due to overlapping with CF3CO2D co-solvent or hydrogen/deuterium exchange (Table 2). In addition, for synthetic diazaquinomycin H (1), protons H-3 and H-6 resonate at 7.00 ppm, agreeing with reported value for 11. However, for the natural sample of diazaquinomycin J (2), protons H-3 and H-6 were reported at 6.93 ppm.1 Given the volatile nature of added CF3CO2D co-solvent, we believe, this slight up field shift is likely due to a difference in percentage of CF3CO2D in CDCl3. In our hands, we observed notable variations to the chemical shifts of protons that reside close to the 1,8-anthraquinone core (H-3, H-6, H-11, and H-12) when the percentage of CF3CO2D varied from 0.4 to 2% (ESI, Fig. S4†).| Position |
|
|
||||
|---|---|---|---|---|---|---|
| Naturala 11 δ (ppm) | Syntheticb 1 δ (ppm) | Δδ (ppm) | Naturala 21 δ (ppm) | Syntheticb 2 δ (ppm) | Δδ (ppm) | |
| a 900 MHz (CDCl3/1% CF3CO2D).b 400 MHz (CDCl3/1% CF3CO2D).c Hydrogen/deuterium exchange. | ||||||
| 1, 8 | 8.05 (s) | H/D exc.c | — | 8.03 (s) | H/D exc.c | — |
| 3, 6 | 7.00 (s) | 7.00 (s) | 0.00 | 6.93 (s) | 7.00 (s) | 0.07 |
| 12 | 3.13 (t) | 3.14 (t) | 0.01 | 3.10 (t) | 3.13 (t) | 0.03 |
| 11 | 2.77 (s) | 2.77 (s) | 0.00 | 2.74 (s) | 2.77 (s) | 0.03 |
| 13 | 1.60 (p) | 1.66–1.57 (m) | −0.01 | 1.59 (m) | 1.66–1.57 (m) | 0.02 |
| 18 | 1.53 (m) | 1.57–1.50 (m) | 0.00 | 1.17 (m) | 1.21–1.13 (m) | 0.00 |
| 14 | 1.47 (p) | 1.50–1.43 (m) | 0.00 | 1.45 (m) | 1.50–1.42 (m) | 0.01 |
| 15 | 1.35 (m) | 1.40–1.27 (m) | −0.01 | 1.35 (m) | 1.40–1.34 (m) | 0.02 |
| 16 | 1.31 (m) | 1.40–1.27 (m) | 0.03 | 1.31 (m) | 1.33–1.27 (m) | −0.01 |
| 17 | 1.19 (q) | 1.22–1.15 (m) | 0.00 | 1.28 (m) | 1.33–1.27 (m) | 0.02 |
| 19 | 0.87 (d) | 0.88 (d) | 0.01 | 1.52 (m) | 1.57–1.50 (m) | 0.01 |
| 20 | 0.87 (d) | 0.88 (d) | 0.01 | 0.86 (d) | 0.87 (d) | 0.01 |
| 21 | 0.86 (d) | 0.87 (d) | 0.01 | |||
As shown in Table 3, the 13C NMR data for synthetic diazaquinomycin H and J samples also compared favorably with reported data for the isolated natural products.1 However, an inconsistency was noticed for the reported chemical shift of amide carbons (C-2/C-7) for 2 that was up field shifted (Δ ≈ 2.5 and 2.6 ppm), when compared to both synthetic samples of 1 and 2 as well as natural sample of 1. It is worthwhile mentioning, these reported chemical shift values overlap with the CF3CO2D co-solvent peaks, which could make their precise assignments challenging. In our hands, the signals for amide carbons (C-2/C-7) in 2 clearly resonate at 163.6 or 163.4 ppm and are consistent with those of other diazaquinomycin samples.16 It appears that the signal intensity for amide carbons (C-2/C-7) in diazaquinomycin J (2) is significantly lower in our DEPTQ spectrum relative to 13C NMR spectrum (ESI†). Notably, these signals were almost completely absent in our DEPTQ spectrum for diazaquinomycin J (2). In this light, it is possible that the amide carbons (C-2/C-7) were not observed in the reported DEPTQ spectrum of 2 and therefore were assumed to overlap with the CF3CO2D co-solvent. More extensive two dimensional HMBC, HSQC and COSY NMR studies (ESI†) of synthetic diazaquinomycins H (1) and J (2) further confirmed their structures and also matched well with reported spectra of isolated natural products.1
| Position |
|
|
||||
|---|---|---|---|---|---|---|
| Naturala 11 δ (ppm) | Syntheticb 1 δ (ppm) | Δδ (ppm) | Naturala 21 δ (ppm) | Syntheticb 2 δ (ppm) | Δδ (ppm) | |
| a 226.2 MHz (CDCl3/1% CF3CO2D).b 100 MHz (CDCl3/2% CF3CO2D).c Overlapping with TFA co-solvent peak. | ||||||
| 10 | 180.1 | 180.1 | 0.0 | 180.0 | 180.0 | 0.0 |
| 9 | 172.9 | 172.9 | 0.0 | 173.0 | 172.8 | −0.2 |
| 2 | 163.2 | 163.7 | 0.5 | 161.0c | 163.6 | 2.6 |
| 7 | 163.0 | 163.5 | 0.5 | 160.9c | 163.4 | 2.5 |
| 5 | 160.1c | 160.8c | 0.7 | 160.2c | 161.2c | 1.0 |
| 4 | 155.6 | 156.6 | 1.0 | 154.6 | 156.6 | 2.0 |
| 9a | 136.8 | 137.0 | 0.2 | 136.8 | 136.9 | 0.1 |
| 8a | 136.3 | 136.5 | 0.2 | 136.2 | 136.4 | 0.2 |
| 3 | 128.6 | 128.3 | −0.3 | 128.9 | 128.3 | −0.6 |
| 6 | 127.6 | 127.3 | −0.3 | 128.0 | 127.3 | −0.7 |
| 4a | 118.0 | 118.6 | 0.6 | 118.3 | 118.6 | 0.3 |
| 10a | 117.5 | 118.1 | 0.6 | 117.8 | 118.1 | 0.3 |
| 17 | 39.1 | 39.2 | 0.1 | 27.5 | 27.6 | 0.1 |
| 12 | 35.0 | 35.1 | 0.1 | 35.0 | 35.1 | 0.1 |
| 13 | 29.7 | 29.9 | 0.2 | 29.9 | 29.9 | 0.0 |
| 14 | 29.7 | 29.8 | 0.1 | 29.8 | 29.9 | 0.1 |
| 15 | 29.7 | 29.8 | 0.1 | 29.7 | 29.7 | 0.0 |
| 18 | 28.1 | 28.2 | 0.1 | 39.1 | 39.2 | 0.1 |
| 16 | 27.4 | 27.4 | 0.0 | 29.6 | 29.5 | −0.1 |
| 11 | 23.1 | 23.1 | 0.0 | 23.0 | 23.2 | 0.2 |
| 19 | 22.8 | 22.8 | 0.0 | 28.1 | 28.2 | 0.1 |
| 20 | 22.8 | 22.8 | 0.0 | 22.8 | 22.8 | 0.0 |
| 21 | 22.8 | 22.8 | 0.0 | |||
Experimental section
General methods
Solvents and reagents were purchased from Sigma-Aldrich, Acros, or Fisher Scientific, and used without further purification. 8-Methylnonanoic acid (8a) and 9-methyldecanoic acid (8b) were purchased from BOC Sciences and Synnovator Inc, respectively. Reactions were monitored either by thin-layer chromatography (TLC) or by analytical high performance liquid chromatography (HPLC) employing a Shimadzu LC-20A series HPLC system. 1H and 13C NMR spectra were recorded on a Bruker Avance III HD-400 (ultrashield) spectrometer (400 MHz and 100 MHz, respectively). All chemical shifts are given as δ value (ppm) and coupling constants, J, are reported in hertz (Hz). NMR solvent peaks were referenced as follows: (1H NMR) CDCl3: 7.27 ppm, DMSO-d6: 2.50 ppm. (13C NMR) CDCl3: 77.23 ppm, DMSO-d6: 39.51 ppm. s = singlet; t = triplet; q = quartet; p = pentet; m = multiplet. High-resolution mass spectroscopy (HRMS) data was obtained using an Agilent 6530 Q-TOF LC/MS. Melting points were determined in open capillary tube using a Buchi B-540 melting point apparatus. Compounds were purified by flash column chromatography on silica gel using a Biotage Isolera One system. The purity of compounds was determined by analytical HPLC (Shimadzu LC-20A series) using a Gemini, 3 μm, C18, 110 Å column (50 mm × 4.6 mm, Phenomenex) and flow rate of 1 mL min−1. Gradient conditions: solvent A (0.1% trifluoroacetic acid in water) and solvent B (acetonitrile): 0–2.0 min 100% A, 2.0–7.0 min 0–100% B (linear gradient), 7.0–8.0 min 100% B, UV detection at 254 nm and 220 nm. LC-MS data was obtained using an Agilent 1260 Infinity HPLC system coupled with an Agilent G6120A quadrupole mass spectrometer. HPLC separation was performed using a SunFire C18 column (Waters), 100 Å, 3.5 μm, 2.1 mm × 150 mm, and flow rate of 0.2 mL min−1. Gradient conditions: solvent A (0.1% formic acid in water) and solvent B (0.1% formic acid in acetonitrile): 0–5.0 min 40% B, 5.0–15.0 min 40–100% B (linear gradient), 15.0–18.0 min 100% B. The mass spectrometer was run in (+)-ESI mode with a scan range (m/z) of 100–1700.Double Knorr cyclization using H2SO4/TIPS; syntheses of diazaquinomycins H (1) and J (2)
:1); mp > 200 °C (dec); 1H NMR (400 MHz, CDCl3/1% CF3CO2D): δ = 7.00 (s, 2H), 3.14 (t, J = 7.3 Hz, 2H), 2.77 (s, 3H), 1.66–1.57 (m, 2H), 1.57–1.50 (m, 1H), 1.50–1.43 (m, 2H), 1.40–1.27 (m, 4H), 1.22–1.15 (m, 2H), 0.88 (d, J = 6.7 Hz, 6H); 1H NMR (400 MHz, CDCl3/2% CF3CO2D): δ = 7.04 (s, 2H), 3.16 (t, J = 7.3 Hz, 2H), 2.79 (s, 3H), 1.66–1.57 (m, 2H), 1.57–1.50 (m, 1H), 1.50–1.44 (m, 2H), 1.40–1.27 (m, 4H), 1.22–1.15 (m, 2H), 0.88 (d, J = 6.7 Hz, 6H); 13C NMR (100 MHz, CDCl3/2% CF3CO2D): δ = 180.1, 172.9, 163.7, 163.5, 160.8, 156.6, 137.0, 136.5, 128.3, 127.3, 118.6, 118.1, 39.2, 35.1, 29.9, 29.8, 29.8, 28.2, 27.4, 23.1, 22.8; HRMS (ESI): m/z [M + H]+ calcd for C22H27N2O4+: 383.1965, found: 383.1969; HPLC purity: 100% (254 nm), 100% (220 nm), tR: 7.18 min.:1); mp > 200 °C (dec); 1H NMR (400 MHz, CDCl3/1% CF3CO2D): δ = 7.00 (s, 2H), 3.13 (t, J = 7.3 Hz, 2H), 2.77 (s, 3H), 1.66–1.57 (m, 2H), 1.57–1.50 (m, 1H), 1.50–1.42 (m, 2H), 1.40–1.34 (m, 2H), 1.33–1.27 (m, 4H), 1.21–1.13 (m, 2H), 0.87 (d, J = 6.7 Hz, 6H); 1H NMR (400 MHz, CDCl3/2% CF3CO2D): δ = 7.02 (s, 2H), 3.15 (t, J = 7.3 Hz, 2H), 2.79 (s, 3H), 1.66–1.57 (m, 2H), 1.57–1.50 (m, 1H), 1.50–1.43 (m, 2H), 1.41–1.35 (m, 2H), 1.33–1.25 (m, 4H), 1.21–1.13 (m, 2H), 0.87 (d, J = 6.7 Hz, 6H); 13C NMR (100 MHz, CDCl3/2% CF3CO2D): δ = 180.0, 172.8, 163.6, 163.4, 161.2, 156.7, 136.9, 136.4, 128.3, 127.3, 118.6, 118.1, 39.2, 35.1, 30.0, 29.9, 29.7, 29.5, 28.2, 27.6, 23.2, 22.8; HRMS (ESI): m/z [M + H]+ calcd for C23H29N2O4+: 397.2122, found: 397.2126; HPLC purity: 100% (254 nm), 100% (220 nm), tR: 7.40 min.Typical procedure for removal of MOM protecting groups
:1:1, 70 mL) and stirred at 65 °C for 2 h under nitrogen protection. The reaction was cooled, diluted with EtOAc (200 mL) and washed with water (100 mL × 3). The organic layer was dried (anhydrous Na2SO4), filtered, and concentrated to give 3a (160 mg, 0.381 mmol, 86%) as an off white solid after trituration with diethyl ether (5 mL). Rf = 0.11 (EtOAc/hexanes, 1:1); mp > 150 °C (dec); 1H NMR (400 MHz, DMSO-d6): δ = 9.78 (s, 2H), 9.02 (s, 1H), 8.74 (s, 1H), 6.99 (d, J = 2.9 Hz, 1H), 6.97 (d, J = 2.9 Hz, 1H), 3.67 (s, 2H), 3.65 (s, 2H), 2.53 (t, J = 7.2 Hz, 2H), 2.19 (s, 3H), 1.53–1.43 (m, 3H), 1.26–1.20 (m, 6H), 1.17–1.09 (m, 2H), 0.84 (d, J = 6.6 Hz, 6H); 13C NMR (100 MHz, DMSO-d6): δ = 205.1, 203.1, 166.0, 165.9, 149.9, 131.1, 127.9, 104.8, 104.7, 51.5, 50.8, 42.2, 38.4, 30.2, 29.1, 28.5, 27.4, 26.6, 22.9, 22.5; HRMS (ESI): m/z [M + H]+ calcd for C22H33N2O6+: 421.2333, found: 421.2330; [M + Na]+ calcd for C22H32N2NaO6+: 443.2153, found: 443.2151; HPLC purity: 100% (254 nm), 100% (220 nm), tR: 7.00 min.:1); mp > 150 °C (dec); 1H NMR (400 MHz, DMSO-d6): δ = 9.77 (s, 2H), 9.00 (s, 1H), 8.75 (s, 1H), 6.99 (d, J = 2.8 Hz, 1H), 6.98 (d, J = 2.8 Hz, 1H), 3.67 (s, 2H), 3.65 (s, 2H), 2.53 (t, J = 7.2 Hz, 2H), 2.20 (s, 3H), 1.53–1.43 (m, 3H), 1.27–1.20 (m, 8H), 1.16–1.09 (m, 2H), 0.84 (d, J = 6.6 Hz, 6H); 13C NMR (100 MHz, DMSO-d6): δ = 205.1, 203.1, 166.00, 165.95, 149.9, 131.1, 127.9, 104.8, 104.7, 51.6, 50.8, 42.2, 38.5, 30.2, 29.2, 28.9, 28.5, 27.4, 26.8, 22.9, 22.5; HRMS (ESI): m/z [M + H]+ calcd for C23H35N2O6+: 435.2490, found: 435.2495; [M + Na]+ calcd for C23H34N2NaO6+: 457.2309, found: 457.2320; HPLC purity: 100% (254 nm), 100% (220 nm), tR: 7.19 min.:1 to 1:2, v/v). Rf = 0.11 (EtOAc/hexanes, 1:1); mp 159–160 °C; 1H NMR (400 MHz, DMSO-d6): δ = 9.77 (s, 2H), 9.01 (s, 1H), 8.74 (s, 1H), 6.99 (d, J = 2.9 Hz, 1H), 6.97 (d, J = 2.9 Hz, 1H), 3.67 (s, 2H), 3.65 (s, 2H), 2.53 (t, J = 7.2 Hz, 2H), 2.19 (s, 3H), 1.52–1.43 (m, 2H), 1.29–1.19 (m, 10H), 0.85 (t, J = 7.0 Hz, 3H); 13C NMR (100 MHz, DMSO-d6): δ = 205.1, 203.1, 166.0, 165.9, 149.9, 131.1, 127.87, 127.85, 104.77, 104.74, 51.5, 50.8, 42.2, 31.2, 30.2, 28.8, 28.6, 28.5, 22.9, 22.1, 13.9; HRMS (ESI): m/z [M + H]+ calcd for C21H31N2O6+: 407.2177, found: 407.2179; [M + Na]+ calcd for C21H30N2NaO6+: 429.1996, found: 429.1998; HPLC purity: 98.3% (254 nm), 98.5% (220 nm), tR: 6.83 min.Typical EDC coupling procedure for the synthesis of di-β-ketoanilides
:1 to 3:2, v/v). Rf = 0.23 (EtOAc/hexanes, 1:1); 1H NMR (400 MHz, CDCl3): δ = 9.42 (s, 1H), 9.37 (s, 1H), 7.79 (s, 2H), 5.15 (s, 2H), 5.07 (s, 2H), 3.65 (s, 3H), 3.58 (s, 2H), 3.55 (s, 2H), 3.47 (s, 3H), 2.58 (t, J = 7.5 Hz, 2H), 2.33 (s, 3H), 1.66–1.56 (m, 2H), 1.55–1.45 (m, 1H), 1.32–1.23 (m, 6H), 1.18–1.10 (m, 2H), 0.85 (d, J = 6.5 Hz, 6H); 13C NMR (100 MHz, CDCl3): δ = 206.8, 204.1, 163.8, 163.7, 154.2, 132.4, 131.83, 131.78, 105.0, 104.9, 101.3, 94.9, 58.4, 56.4, 51.3, 50.4, 44.2, 39.1, 31.2, 29.8, 29.2, 28.1, 27.4, 23.6, 22.8; HRMS (ESI): m/z [M + H]+ calcd for C26H41N2O8+: 509.2857, found: 509.2846; [M + Na]+ calcd for C26H40N2NaO8+: 531.2677, found: 531.2673; HPLC purity: 99.6% (254 nm), 99.8% (220 nm), tR: 7.40 min.:1 to 3:2, v/v). Rf = 0.26 (EtOAc/hexanes, 1:1); 1H NMR (400 MHz, CDCl3): δ = 9.44 (s, 1H), 9.39 (s, 1H), 7.80 (s, 2H), 5.15 (s, 2H), 5.07 (s, 2H), 3.66 (s, 3H), 3.59 (s, 2H), 3.56 (s, 2H), 3.47 (s, 3H), 2.59 (t, J = 7.5 Hz, 2H), 2.34 (s, 3H), 1.66–1.57 (m, 2H), 1.56–1.45 (m, 1H), 1.33–1.22 (m, 8H), 1.18–1.10 (m, 2H), 0.86 (d, J = 6.5 Hz, 6H); 13C NMR (100 MHz, CDCl3): δ = 206.8, 204.2, 163.8, 163.7, 154.1, 132.4, 131.81, 131.77, 105.0, 104.9, 101.3, 94.9, 58.4, 56.4, 51.3, 50.4, 44.2, 39.2, 31.2, 29.9, 29.6, 29.2, 28.1, 27.5, 23.6, 22.8; HRMS (ESI): m/z [M + H]+ calcd for C27H43N2O8+: 523.3014, found: 523.3012; [M + Na]+ calcd for C27H42N2NaO8+: 545.2833, found: 545.2844; HPLC purity: 100% (254 nm), 100% (220 nm), tR: 7.59 min.Typical procedure for the synthesis of mono-β-ketoanilides
:1 to 4:1, v/v), affording 5a (205 mg, 0.483 mmol, 92%) as a clear oil. Rf = 0.45 (EtOAc/Hexanes, 1:1); 1H NMR (400 MHz, CDCl3): δ = 9.34 (s, 1H), 7.43 (d, J = 2.9 Hz, 1H), 6.23 (d, J = 2.9 Hz, 1H), 5.09 (s, 2H), 5.00 (s, 2H), 3.95 (br s, 2H), 3.62 (s, 3H), 3.53 (s, 2H), 3.45 (s, 3H), 2.57 (t, J = 7.5 Hz, 2H), 1.65–1.55 (m, 2H), 1.54–1.44 (m, 1H), 1.32–1.23 (m, 6H), 1.17–1.10 (m, 2H), 0.85 (d, J = 6.7 Hz, 6H); 13C NMR (100 MHz, CDCl3): δ = 206.8, 163.6, 154.6, 140.4, 132.3, 130.2, 100.3, 99.9, 99.2, 94.8, 58.0, 56.2, 50.5, 44.2, 39.1, 29.7, 29.2, 28.1, 27.3, 23.6, 22.8; HRMS (ESI): m/z [M + H]+ calcd for C22H37N2O6+: 425.2646, found: 425.2645; [M + Na]+ calcd for C22H36N2NaO6+: 447.2466, found: 447.2460; HPLC purity: 100% (254 nm), 100% (220 nm), tR: 7.30 min.:1); mp 64–65 °C; 1H NMR (400 MHz, CDCl3): δ = 9.35 (s, 1H), 7.44 (d, J = 2.9 Hz, 1H), 6.24 (d, J = 2.9 Hz, 1H), 5.10 (s, 2H), 5.01 (s, 2H), 3.98 (br s, 2H), 3.63 (s, 3H), 3.54 (s, 2H), 3.46 (s, 3H), 2.58 (t, J = 7.5 Hz, 2H), 1.65–1.56 (m, 2H), 1.56–1.45 (m, 1H), 1.33–1.21 (m, 8H), 1.18–1.10 (m, 2H), 0.86 (d, J = 6.7 Hz, 6H); 13C NMR (100 MHz, CDCl3): δ = 206.9, 163.6, 154.6, 140.4, 132.4, 130.3, 100.4, 100.0, 99.3, 94.9, 58.0, 56.2, 50.5, 44.2, 39.2, 29.9, 29.6, 29.2, 28.1, 27.5, 23.6, 22.8; HRMS (ESI): m/z [M + H]+ calcd for C23H39N2O6+: 439.2803, found: 439.2782; [M + Na]+ calcd for C23H38N2NaO6+: 461.2622, found: 461.2599; HPLC purity: 100% (254 nm), 100% (220 nm), tR: 7.52 min.:1 to 6:1, v/v). Rf = 0.27 (EtOAc/hexanes, 1:4). 1H NMR (400 MHz, CDCl3): δ = 15.29 (s, 1H), 3.09–3.03 (m, 2H), 1.73 (s, 6H), 1.72–1.65 (m, 2H), 1.57–1.46 (m, 1H), 1.45–1.36 (m, 2H), 1.33–1.24 (m, 4H), 1.18–1.11 (m, 2H), 0.86 (d, J = 6.6 Hz, 6H); 13C NMR (100 MHz, CDCl3): δ = 198.5, 170.8, 160.4, 104.9, 91.4, 39.1, 35.9, 29.7, 29.6, 28.1, 27.3, 27.0, 26.4, 22.8; HRMS (ESI): m/z [M − H]− calcd for C16H25O5−: 297.1707, found: 297.1709; HPLC purity: 100% (254 nm), 100% (220 nm), tR: 7.99 min.:1 to 6:1, v/v). Rf = 0.28 (EtOAc/hexanes, 1:4). 1H NMR (400 MHz, CDCl3): δ = 15.29 (s, 1H), 3.09–3.03 (m, 2H), 1.73 (s, 6H), 1.72–1.64 (m, 2H), 1.55–1.45 (m, 1H), 1.44–1.35 (m, 2H), 1.35–1.29 (m, 2H), 1.29–1.23 (m, 4H), 1.18–1.10 (m, 2H), 0.86 (d, J = 6.6 Hz, 6H); 13C NMR (100 MHz, CDCl3): δ = 198.5, 170.8, 160.4, 104.9, 91.4, 39.2, 35.9, 29.9, 29.6, 29.5, 28.1, 27.5, 27.0, 26.4, 22.8; HRMS (ESI): m/z [M − H]− calcd for C17H27O5−: 311.1864, found: 311.1860; HPLC purity: 100% (254 nm), 100% (220 nm), tR: 8.16 min.:1); mp > 200 °C (dec); 1H NMR (400 MHz, CDCl3/1% CF3CO2D): δ = 7.01 (s, 2H), 3.14 (t, J = 7.3 Hz, 2H), 2.78 (s, 3H), 1.65–1.57 (m, 2H), 1.51–1.43 (m, 2H), 1.41–1.28 (m, 8H), 0.90 (t, J = 7.0 Hz, 3H); 1H NMR (400 MHz, CDCl3/2% CF3CO2D): δ = 7.09 (s, 2H), 3.15 (t, J = 7.3 Hz, 2H), 2.79 (s, 3H), 1.66–1.57 (m, 2H), 1.52–1.43 (m, 2H), 1.41–1.28 (m, 8H), 0.90 (t, J = 7.0 Hz, 3H); 13C NMR (100 MHz, CDCl3/2% CF3CO2D): δ = 180.0, 172.8, 164.0, 163.9, 162.1, 157.5, 137.0, 136.5, 128.2, 127.2, 119.0, 118.5, 35.3, 32.1, 30.0, 29.8, 29.5, 29.4, 23.1, 22.9, 14.1; HRMS (ESI): m/z [M + H]+ calcd for C21H25N2O4+: 369.1809, found: 369.1816; HPLC purity: 99.2% (254 nm), 99.2% (220 nm), tR: 7.03 min.Conclusions
In summary, the first total synthesis of anti-tuberculosis diazaquinomycins H (1) and J (2) has been accomplished via selective amidation of diamine 6 with Meldrum's acid derivatives, subsequent EDC coupling with 3-oxobutanoic acid, and double Knorr cyclization in the presence of TIPS. Adding TIPS during the double Knorr cyclization was crucial in preventing isomerization of the C-5 iso-branched tail in sulfuric acid. Diazaquinomycins H (1) and J (2) were obtained in 8 steps from commercially available starting materials in 25% and 21% overall yields, respectively. In contrast to triflic acid, sulfuric acid gave much cleaner double Knorr cyclization product for long-chain (n = 5 or 6) di-β-ketoanilide substrates. In addition to a terminal isopropyl functionality, we found that the length of the iso-branched alkyl chain also played an important role in the isomerization process in sulfuric acid, with a longer iso-branched alkyl chain being more prone to isomerization. We observed tricyclic hydroquinone intermediates by HPLC and mass spectrometry, indicating that oxidation to diazaquinomycin scaffold was notably slower for long-chain analogues compared to short-chain analogues. Moreover, oxidation of the hydroquinone intermediates to diazaquinomycins was greatly accelerated by simply stirring in methanol open to air. The spectroscopic data for synthesized diazaquinomycins H (1) and J (2) compared very well with reported data for the natural products, with minor discrepancies noted.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Hawaii Community Foundation LE'AHI FUND for Pulmonary Research (16ADVC-78728) and UHH DKICP RTRF fund. We thank Justin Reinicke for his expert support for developing LC-MS method and acquiring low resolution LC-MS and HRMS data. We also thank Skyla Lee for her assistance with the synthesis of intermediate compound 6.Notes and references
- M. W. Mullowney, C. H. Hwang, A. G. Newsome, X. Wei, U. Tanouye, B. Wan, S. Carlson, N. J. Barranis, E. Ó hAinmhire, W.-L. Chen, K. Krishnamoorthy, J. White, R. Blair, H. Lee, J. E. Burdette, P. K. Rathod, T. Parish, S. Cho, S. G. Franzblau and B. T. Murphy, ACS Infect. Dis., 2015, 1, 168–174 CrossRef CAS PubMed.
- M. W. Mullowney, E. Ó hAinmhire, A. Shaikh, X. Wei, U. Tanouye, B. D. Santarsiero, J. E. Burdette and B. T. Murphy, Mar. Drugs, 2014, 12, 3574–3586 CrossRef PubMed.
- R. P. Maskey, I. Grün-Wollny and H. Laatsch, Nat. Prod. Res., 2005, 19, 137–142 CrossRef CAS PubMed.
- S. Ōmura, Y. Iwai, K. Hinotozawa, H. Tanaka, Y. Takahashi and A. Nakagawa, J. Antibiot., 1982, 35, 1425–1429 CrossRef.
- S. Ōmura, A. Nakagawa, H. Aoyama, K. Hinotozawa and H. Sano, Tetrahedron Lett., 1983, 24, 3643–3646 CrossRef.
- S. Ōmura, M. Murata, K. Kimura, S. Matsukura, T. Nishihara and H. Tanaka, J. Antibiot., 1985, 38, 1016–1024 CrossRef.
- M. Murata, T. Miyasaka, H. Tanaka and S. Ōmura, J. Antibiot., 1985, 38, 1025–1033 CrossRef CAS PubMed.
- K. Tsuzuki, T. Yokozuka, M. Murata, H. Tanaka and S. Ōmura, J. Antibiot., 1989, 42, 727–737 CrossRef CAS PubMed.
- T. R. Kelly, J. A. Field and Q. Li, Tetrahedron Lett., 1988, 29, 3545–3546 CrossRef CAS.
- H. Lee and W. K. Anderson, Tetrahedron Lett., 1990, 31, 4405–4408 CrossRef CAS.
- J. M. Pérez, P. López-Alvarado, C. Avendaño and J. C. Menéndez, Tetrahedron Lett., 1998, 39, 673–676 CrossRef.
- J. M. Pérez, P. López-Alvarado, E. Pascual-Alfonso, C. Avendaño and J. C. Menéndez, Tetrahedron, 2000, 56, 4575–4583 CrossRef.
- K. L. Rinehart Jr and H. B. Renfroe, J. Am. Chem. Soc., 1961, 83, 3729–3731 CrossRef.
- E. I. Parkinson, J. S. Bair, M. Cismesia and P. J. Hergenrother, ACS Chem. Biol., 2013, 8, 2173–2183 CrossRef CAS PubMed.
- J. S. Bair, R. Palchaudhuri and P. J. Hergenrother, J. Am. Chem. Soc., 2010, 132, 5469–5478 CrossRef CAS PubMed.
- A. M. Prior and D. Sun, Synthesis, 2018, 50, 859–871 CrossRef CAS.
- T. Knoth, K. Warburg, C. Katzka, A. Rai, A. Wolf, A. Brockmeyer, P. Janning, T. F. Reubold, S. Eschenburg, D. J. Manstein, K. Hübel, M. Kaiser and H. Waldmann, Angew. Chem., Int. Ed., 2009, 48, 7240–7245 CrossRef CAS PubMed.
- M. T. Huggins, P. S. Barber, D. Florian and W. Howton, Synth. Commun., 2008, 38, 4226–4239 CrossRef CAS.
- C. S. Pak, H. C. Yang and E. B. Choi, Synthesis, 1992, 1992, 1213–1214 CrossRef.
- H. Kikuchi, K. Sasaki, J. Sekiya, Y. Maeda, A. Amagai, Y. Kubohara and Y. Oshima, Bioorg. Med. Chem., 2004, 12, 3203–3214 CrossRef CAS PubMed.
- M. Syrpas, E. Ruysbergh, L. Blommaert, B. Vanelslander, K. Sabbe, W. Vyverman, N. De Kimpe and S. Mangelinckx, Mar. Drugs, 2014, 12, 352–367 CrossRef PubMed.
- C. Ma, X. Li, X. liang, K. Jin, J. Cao and W. Xu, Bioorg. Med. Chem. Lett., 2013, 23, 4948–4952 CrossRef CAS PubMed.
- R. M. Forbis and K. L. Rinehart Jr, J. Am. Chem. Soc., 1973, 95, 5003–5013 CrossRef CAS PubMed.
- D. J. am Ende and L. F. Albright, Ind. Eng. Chem. Res., 1994, 33, 840–848 CrossRef CAS.
- Y. Ono, Catal. Today, 2003, 81, 3–16 CrossRef CAS.
- A. K. Roebuck and B. L. Evering, J. Am. Chem. Soc., 1953, 75, 1631–1635 CrossRef CAS.
- S. T. Sie, Ind. Eng. Chem. Res., 1993, 32, 397–402 CrossRef CAS.
- J. Weitkamp, ACS Symposium Series, in Hydrocracking and hydrotreating, ed. J. W. Ward and S. A. Qader, vol. 20, American Chemical Society, Washington, DC, 1975, pp. 1–27, DOI:10.1021/bk-1975-0020.
- A. D. Broadbent and J. M. Stewart, Can. J. Chem., 1983, 61, 1965–1969 CrossRef CAS.
- K. Krohn, G. Carlson, H. Köhle, H. Hussain and I. R. Green, ARKIVOC, 2008, xiv, 216–233 Search PubMed.
- L. Kurc, V. Vopravil and L. Červený, Res. Chem. Intermed., 2001, 27, 249–258 CrossRef CAS.
- Y. Ren, Z. Mi, S. Cheng, L. Wang and Y. Wang, React. Kinet. Catal. Lett., 2004, 83, 71–77 CrossRef CAS.
- P. Shaul, M. Frenkel, E. B. Goldstein, L. Mittelman, A. Grunwald, Y. Ebenstein, I. Tsarfaty and M. Fridman, ACS Med. Chem. Lett., 2013, 4, 323–328 CrossRef CAS PubMed.
- G. Sastre, A. Chica and A. Corma, J. Catal., 2000, 195, 227–236 CrossRef CAS.
- A. Corma and A. V. Orchillés, Microporous Mesoporous Mater., 2000, 35–36, 21–30 CrossRef CAS.
- E. Hedenström, B.-V. Nguyen and L. A. Silks III, Tetrahedron: Asymmetry, 2002, 13, 835–844 CrossRef.
- H. Chiba, H. Agematu, K. Dobashi and T. Yoshioka, J. Antibiot., 1999, 52, 700–709 CrossRef CAS PubMed.
- K. Yamada, N. Wada, H. Onaka, R. Matsubara, R. Isobe, M. Inagaki and R. Higuchi, Chem. Pharm. Bull., 2005, 53, 788–791 CrossRef CAS PubMed.
- F. D. Gunstone, Chem. Phys. Lipids, 1993, 65, 155–163 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR, DEPTQ, HBMC, HSQC, COSY, HRMS, and HPLC of diazaquinomycins H (1) and J (2); 1H NMR, DEPTQ spectra and HPLC chromatogram of isomeric mixture of 1 as well as 2; 1H, 13C NMR spectra and HPLC chromatogram of 3a–c, 4a–b, 5a–b, and 7a–b; 1H, 13C NMR spectra, HRMS, and HPLC chromatogram of 13; synthesis of 14 with a shorter branched side chain and its 1H, 13C NMR spectra; 1H, 13C NMR spectra comparison of 9-methyldecanoic acid and 4-methylpentanoic acid before and after being treated with conc. H2SO4; comparison of NMR spectra for pure diazaquinomycin H (1) with isomeric mixture of 1; and zoomed 1H NMR regions of isomeric mixture of 2 with varying concentrations of CF3CO2D in CDCl3. See DOI: 10.1039/c8ra09792e |
| This journal is © The Royal Society of Chemistry 2019 |