
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceApplication of dehydroalanine as a building block for the synthesis of selenocysteine-containing peptides†
Kishorkumar M. Reddy and
Govindasamy Mugesh *
*
Department of Inorganic and Physical Chemistry, Indian Institute of Science, Bangalore 560 012, India. E-mail: mugesh@iisc.ac.in
First published on 20th December 2018
Abstract
Selenocysteine (Sec), the 21st proteinogenic amino acid, is inserted co-translationally into number of natural proteins. It is coded by a dual function stop codon UGA (opal). It is a redox active amino acid found at the active sites of several enzymes that are involved in oxidation–reduction reactions. These enzymes include the three major mammalian selenoproteins glutathione peroxidase (GPx), thioredoxin reductase (TrxR), and iodothyronine deiodinase (Dio). Although Sec is structurally similar to its sulfur analogue cysteine (Cys), the lower pKa of the selenol group in Sec as compared to that of Cys and the interesting redox properties of the selenium atom in peptides and proteins play crucial roles in redox catalysis. However, the chemical synthesis of Sec-containing peptides has been a difficult task. In this paper, we report on a new method for the synthesis of Sec and Sec-containing peptides using dehydroalanine (Dha) as a building block.
Introduction
The genetically encoded selenocysteine (Sec, U)1,2 is incorporated into proteins by suppressing the stop codon UGA.3 The redox active Sec in proteins plays various roles in biological systems, ranging from maintaining the redox homeostasis to hormone activation and protein folding.4 Glutathione peroxidase (GPx), iodothyronine deiodinase (Dio) and thioredoxin reductase (TrxR) are some of the important selenoenzymes that play key roles in mammalian systems.4 The lower pKa of the selenol group in Sec as compared to the thiol group in cysteine (Cys) and the relatively lower reduction potential of the selenium atom in the proteins make Sec a unique moiety for catalysis.5 However, the synthesis of selenoproteins follows a unique pathway. The biosynthesis of Sec involves the use of special mRNA sequence known as selenocysteine inserting sequence (SECIS) adjacent to the UGA codon in the prokaryotes and at the 3′-untranslation region in the eukaryotes. In prokaryotes, it requires a selenophosphate synthetase for the synthesis of selenophosphate, selenocysteine synthase to convert serine to Sec on tRNA, a specialised tRNA and Sec-binding protein to bind with tRNA and SECIS. On the other hand, in eukaryotes, the incorporation of Sec into proteins requires selenophosphate synthetase-2 for the synthesis of selenophosphate, SepSec synthase that can convert the phosphoserine to Sec on tRNA, a specialised tRNA, a Sec-binding protein and an elongation factor to bind with tRNA and SECIS.6Interestingly, the key enzyme SepSec synthase in eukaryotes and selenocysteine synthase in prokaryotes are known to convert phosphoserine and serine, respectively, to selenocysteine on the tRNA using pyridoxal pyrophosphate as a co-factor. Recent studies showed that the mechanism of this pathway involves the formation of a dehydroalanine (Dha) intermediate, which is further converted to Sec by a nucleophilic attack of selenophosphate (Fig. 1).7 The formation of Dha through a spontaneous non-enzymatic elimination of phosphoserine is detected during protein aging in human cells.8 Furthermore, Dha is also produced during lantipeptide natural product biosynthesis in prokaryotes.9
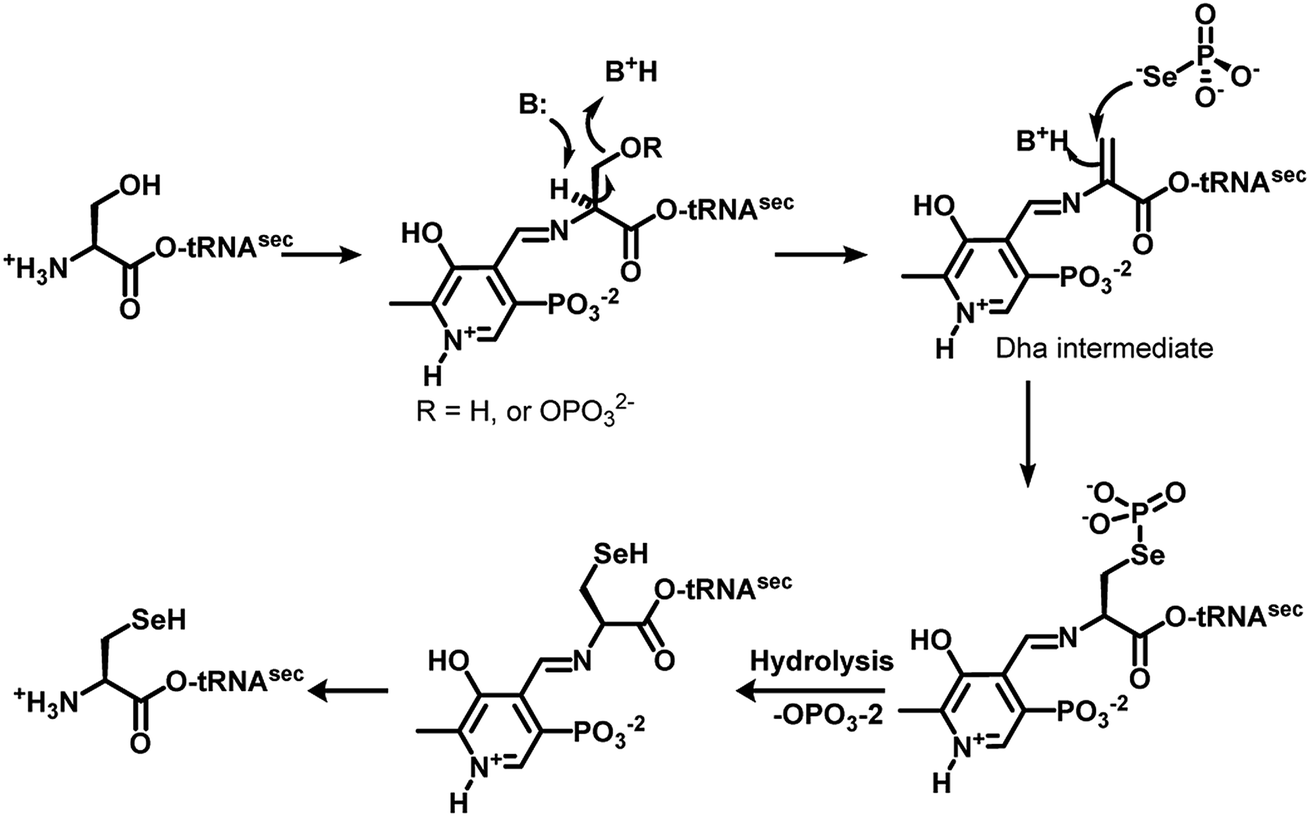 | ||
| Fig. 1 Mechanism of SepSec synthase/selenocysteine synthase, which shows the formation of dehydroalanine before the attack of selenophosphate.7 | ||
Dehydroalanine (Dha) is a widely used precursor for the post-translational modifications in proteins. The amino acid residues serine (Ser), Cys or Sec are first inserted at the position of interest in proteins. These residues are then activated to form good leaving groups, which upon elimination, produces Dha. Synthetically, Dha can be produced by the oxidative elimination of an alkylated derivatives of Cys and Sec. Recently, biosynthetic incorporation of Sec-derivatives followed by an oxidative elimination of the aryl moiety with hydrogen peroxide has been shown to produce Dha moiety on peptides and proteins.10 However, only a few reports are available on the incorporation of selenium into proteins or peptides by chemical modifications. It has been shown that the activation of a serine residue with phenylmethanesulfonyl fluoride (PMSF), followed by displacement of the sulfonate with hydrogen selenide at pH 5.5 can give Sec-containing proteins.11 Another method involving Native Chemical Ligation (NCL) of polypeptides between C-terminal thioester and N-terminal Sec residue has also been reported.12 The solid-phase peptide synthesis (SPPS) of Sec-containing peptides using Fmoc-Sec(pMob)-OH is widely used, but the protected amino acid is known to form Dha via Cα hydrogen atom abstraction mechanism during the elongation step. The Dha residue thus formed can be attacked by piperidine to form β-piperidine adducts.13 As Dha plays a key role in protein modification and SPPS, we have developed a new method to insert Sec into peptides using a synthetic Dha as a building block.
Results and discussion
Synthesis of peptides having Dha moiety
The early attempts to generate Dha on proteins and peptides relied on sulfonylation of Ser, followed by elimination under experimental conditions that were found to be too harsh for most proteins.14 Recent studies indicate that dehydroalanine (Dha) can be generated under mild conditions via post-translational modifications of Ser or Cys. When these two amino acids are part of peptides or proteins, the elimination of water and hydrogen sulfide, respectively, from these amino acid moieties produce the Dha. In our study, the Dha derivatives 1 and 5a–e were synthesized following the literature procedures with minor modifications.15 The Dha derivative 1 was synthesized starting from L-cysteine. The amine group of cysteine was protected with acid labile Boc and the resulting Boc-L-cysteine was treated with 5 equiv. of K2CO3 in DMF to afford compound 1 in 30% yield, along with the methylated cysteine derivative (2) in about 65% yield (Scheme 1). This observation indicates that it is important to protect the carboxylic group, before treating with methyl iodide for an alkylation–elimination reaction. Davis and co-workers showed that the reaction of BocCysOMe with a series of alkylating agents can produce Dha and the yield of Dha depends on the nature of alkylating agent. They showed that 1,4-dibromo- and 1,4-diiodobutanes are the most efficient of the reagents for the generation of Dha. 1,4-Dichlorobutane, on the other hand, was found to be slow to alkylate, and in this case, they recovered mostly the unreacted BocCysOMe. Other reagents such as 1,5-diiodopentane, 1,4-bis(methansulfonyl)-butandiol and α,α′-dibromo-o-xylene produced BocDhaOMe in only 11–12% yield.16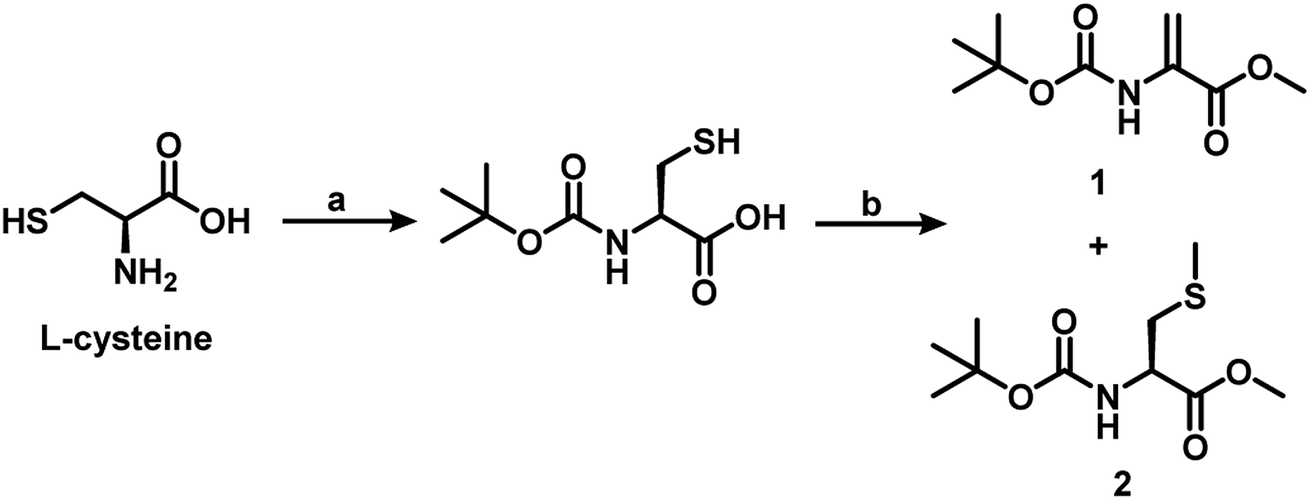 | ||
| Scheme 1 Synthetic route to dehydroalanine 1 from the L-cysteine. (a) 1 M aq. NaOH, Boc anhydride, 1,4 dioxane, 27 °C, 6 h. (b) K2CO3, MeI, DMF, 27 °C, 20 h. | ||
For the synthesis of Dha-based dipeptides 5a–e, we employed the Boc-protected amino acids 3a–e. To achieve this, compounds 3a–e were treated with N-methylmorpholine, ethyl chloroformate and thiophenol in dry THF to form the C-terminal thioesters 4a–e. Peptide C-terminal thioesters are generally used as key intermediates in a variety of applications. Particularly, they have been used recently for the native chemical ligation methods for the total chemical synthesis of proteins.17 Therefore, it was thought worthwhile to explore the use of C-terminal thioesters for the synthesis of compounds 5a–e. The native chemical ligation was performed between compounds 4a–e and L-cysteine to produce the dipeptides bearing free thiol and carboxylic groups. When these dipeptides were treated with K2CO3 in DMF for 24 h, the dipeptides bearing C-terminal dehydroalanine centre (5a–e) were obtained (Scheme 2). Similar to the reaction of Boc-Cys with MeI, the formation of S-methylated peptides was observed as side products in these reactions as well. When we used L-cysteine methyl ester instead of L-cysteine in the native chemical ligation, we obtained dipeptides with a free thiol group. These dipeptides when treated with 1,4-diiodobutane and K2CO3 in DMF for 4 h, afforded C-terminal Dha dipeptides in 90–95% without any other side products.
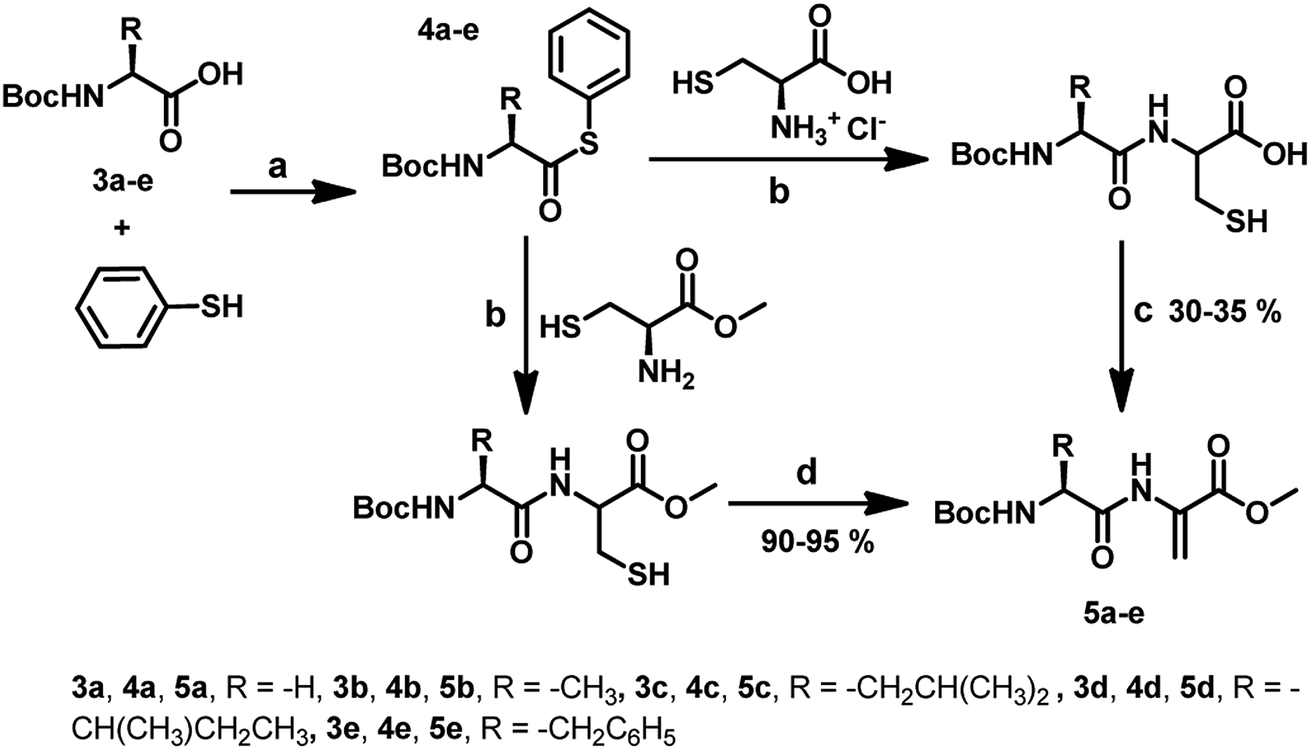 | ||
| Scheme 2 Synthetic route to the dipeptides 5a–e from the protected amino acids 3a–e respectively via the formation of the corresponding thioesters 4a–e. (a) Ethyl chloroformate, N-methylmorpholine, dry THF, molecular sieves, 27 °C, 6 h (b) NaBH4, MeOH, 5 h. (c) K2CO3, MeI, DMF, 27 °C, 20 h. (d) K2CO3, 1,4-diiodobutane, DMF, 27 °C, 4 h. | ||
Reaction of Dha with pMob selenol
Synthetically, Dha is a versatile precursor for various natural and unnatural amino acid derivatives.18 The α,β-unsaturated carbonyl moiety present in Dha can undergo Michael-type conjugate addition reactions with various nucleophiles. These reactions are generally carried out in aqueous media at neutral or slightly basic pH and temperatures below 40 °C, the conditions that are compatible with proteins. Given the higher nucleophilicity of the selenolate as compared to that of a thiolate, it is expected that the Michael addition of aryl selenolates to Dha amides (selena-Michael addition) should be a highly favoured process. It has been shown that Dha is suitable for chemically generating Sec in proteins.19 However, only one example of selena-Michael addition to Dha has been reported in the literature. This involved the formation of a Se-allyl-selenocysteine from a selenium nucleophile derived in situ from the precursor allylselenocyanate.19 However, this methodology may be useful for broader applications, when suitable selenium nucleophiles are available for the Michael addition. For our study, we selected p-methoxybenzyl (pMob) as the protecting group as it is a well-known protecting group for the selenium in Sec.20 For the reactions with Dha, we synthesized pMob diselenide (6), which upon reduction with NaBH4, in situ can generate pMob selenol (7).21 The reduction of compound 6 by NaBH4 was carried out in MeOH, in 50 mM sodium phosphate buffer pH 8.0 to obtain the corresponding selenol 7. When the in situ generated pMob selenol 7 was treated with the Dha derivative 1, the pMob-protected selenocysteine derivative 8 was obtained in good yield. Similarly, the reaction of 7 with the Dha-based dipeptides 5a–e in 50 mM sodium phosphate buffer at pH 8.0 produced the Sec dipeptides 9a–e in quantitative yields (Scheme 3).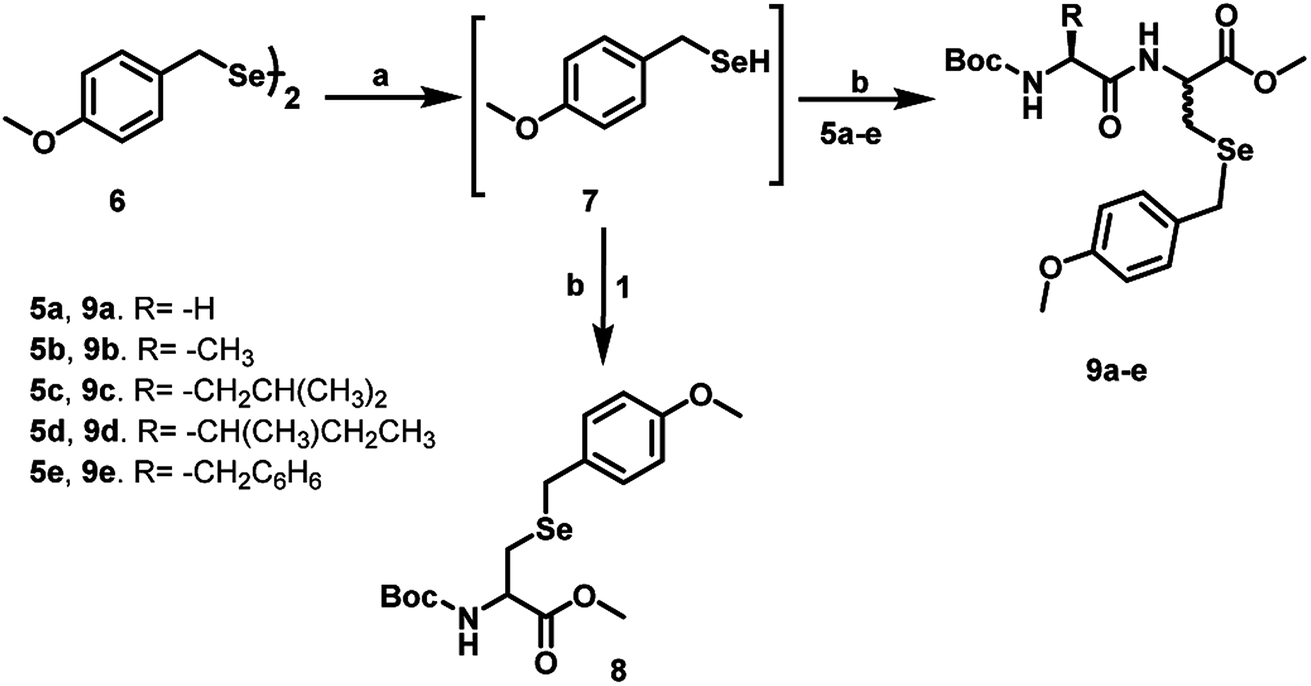 | ||
| Scheme 3 Synthetic route to selenocysteine derivative 8 and dipeptides 9a–e starting from the dipeptides 5a–e. (a) NaBH4, MeOH, 0 °C, 10 min, (b) 50 mM sodium phosphate buffer pH-8, MeOH, 27 °C, 6–8 h. | ||
We have extended this methodology to tripeptides bearing Dha at the C-terminal and central positions. For this purpose, we selected glutathione (GSH, 10) as a model peptide to introduce Dha between two amino acid residues. It is known that GSH is an important antioxidant that prevents damage to cellular components caused by reactive oxygen species (ROS).22 It is a unique tripeptide with a γ-peptide linkage between the carboxyl group of the glutamate side chain and the amine group of cysteine. The carboxyl group of cysteine is attached by a normal peptide linkage to glycine. Therefore, the Dha methodology may be useful for the synthesis of the selenium analogue of GSH (GSeSeG). Previously, Tamura et al. synthesized the selenium analogue of glutathione disulphide, γ-L-glutamyl-L-selenocysteinylglycine, by a liquid phase method. In this method, the selenol group of Sec was protected by the pMob group. The overall yield of the final product was ∼9% based on the starting compound, Se-(pMob)-L-selenocysteine.23 Later, Iwaoka and co-workers reported the synthesis and antioxidant activity of GSeSeG. They synthesized GSeSeG from the Sec derivative, Fmoc-Sec(pMob)-OH, using a solid-phase peptide synthesis method.24
To synthesize the GSeSeG via the Dha route, it was necessary to protect the free amino and carboxylic groups. The protected GSH derivative 12 was synthesized by following the literature procedure.25 Briefly, the thiol group of GSH was protected with trityl group by treating the thiol with trityl alcohol in TFA. The resulting trityl-protected GSH was treated with Boc anhydride under basic conditions to afford N-Boc-S-trityl-protected GSH. Esterification of this derivative with MeI in DMF produced N-Boc-S-trityl-glutathione methyl ester 11. The deprotection of the trityl group by treating with triethylsilane in TFA afforded N-Boc-glutathione methyl ester 12. The Dha analogue of glutathione 13 was obtained by treating 12 with 1,4-diiodobutane in DMF (Scheme 4).
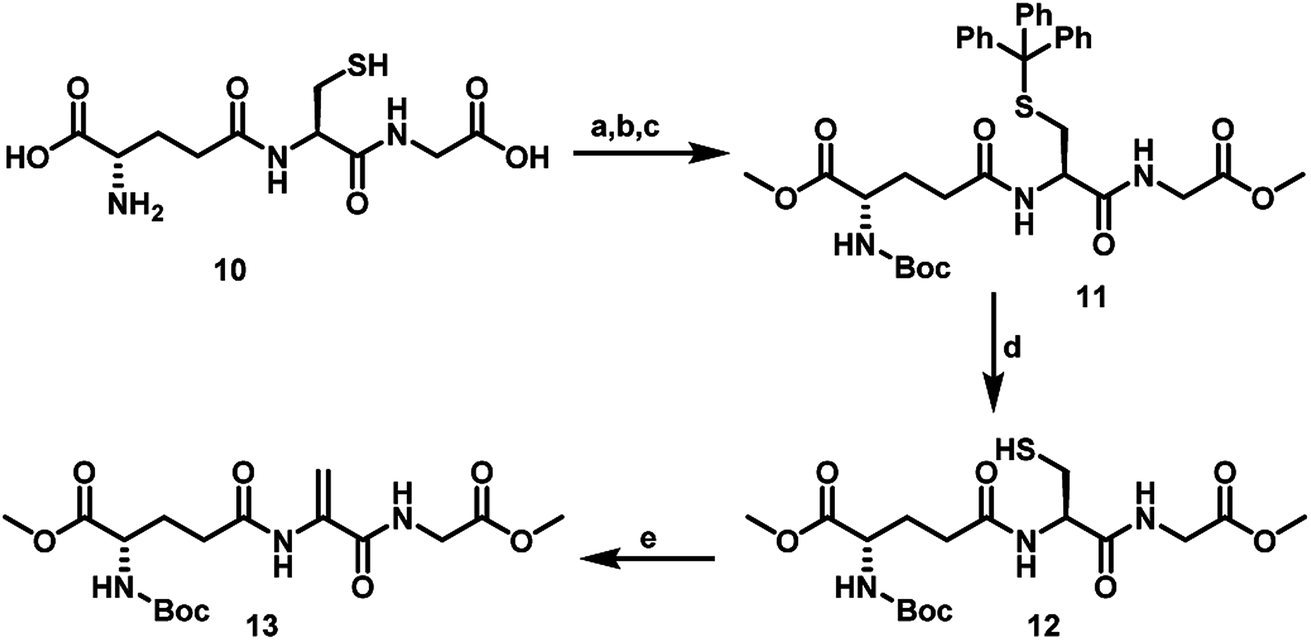 | ||
| Scheme 4 Synthetic route to glutathione analogue 13 starting from the glutathione 10. (a) Trityl alcohol, TFA. (b) Triethylamine, water, Boc anhydride. (c) K2CO3, MeI, DMF. 27 °C, 20 h. (d) Triethylsilane, 4% TFA, DCM, 27 °C, 1 h. (e) K2CO3, 1,4-diiodobutane in DMF, 27 °C, 4 h. | ||
In addition to GSeSeG, several other selenium-containing peptides have been studied as glutathione peroxidase mimetics. Iwaoka and co-workers showed that the change in the amino acid sequence in a series of selenium-containing peptides alters the antioxidant activity.24 For example, they showed that the replacement of the γ-Glu moiety in GSeSeG with a Leu residue significantly decreased the antioxidant activity. Therefore, we aimed at the synthesis of Sec-containing peptides having other amino acids such as Leu and Ala residues. For this, the tripeptide with C-terminal Dha centre (17) was synthesized by following a conventional solution-phase peptide synthesis method. The Boc-protected dipeptide amino acid (Boc-NH-Leu-Ala-COOH, 14) was treated with thiophenol in the presence of N-methylmorpholine and ethyl chloroformate in dry THF to give the dipeptide bearing C-terminal thioester 15, which was used for the native chemical ligation with L-cysteine methyl ester to give the tripeptide 16. Treatment of compound 16 with 1,4 diiodobutane and K2CO3 in DMF for 4 h afforded the tripeptide bearing Dha at C-terminal 17 (Scheme 5). We employed L-cysteine in the chemical ligation and followed by treatment with MeI and K2CO3 in DMF for 24 h afforded the Dha peptide in 30% yield.
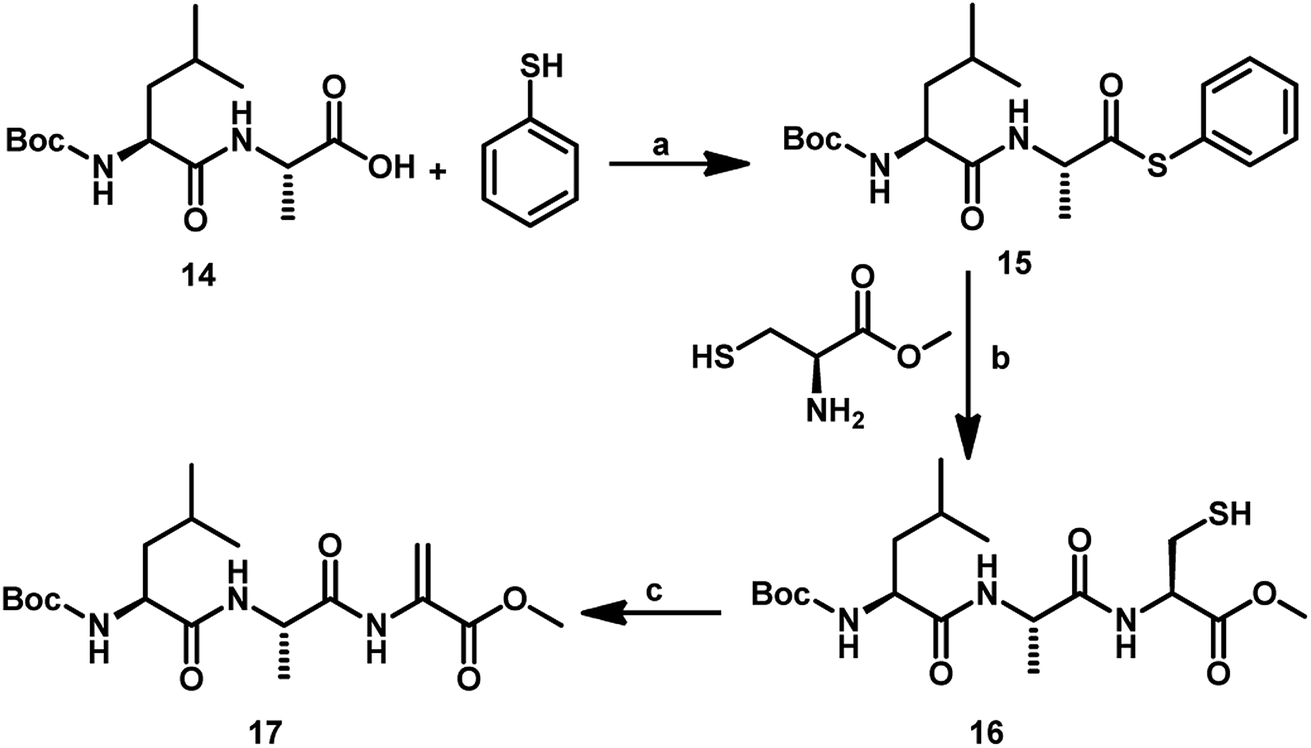 | ||
| Scheme 5 Synthetic route to the tripeptide 17 starting from dipeptide 14. (a) Ethyl chloroformate, N-methylmorpholine, dry THF, molecular sieves, 27 °C, 6 h. (b) NaBH4, MeOH, 5 h. (c) K2CO3, 1,4-diiodobutane, DMF, 27 °C, 4 h. | ||
Although the addition of functionalized thiols to Dha under mild conditions is generally a facile process, the nature of amino acid moiety attached to the Dha appears to modulate its reactivity. In this study, we observed that the nature of solvents plays an important role in the conversions as well as the reaction time. When the coupling of tripeptides bearing Dha (13 and 17) with pMob selenol were performed in MeOH and 50 mM sodium phosphate buffer at pH 8, the selenium analogue of glutathione 18 and selenopeptide 19 were obtained in 80–90% yield. This agrees with an earlier report that the rate of Michael addition of amines and thiols to Dha amides was greatly accelerated in the presence of water (Scheme 6).26
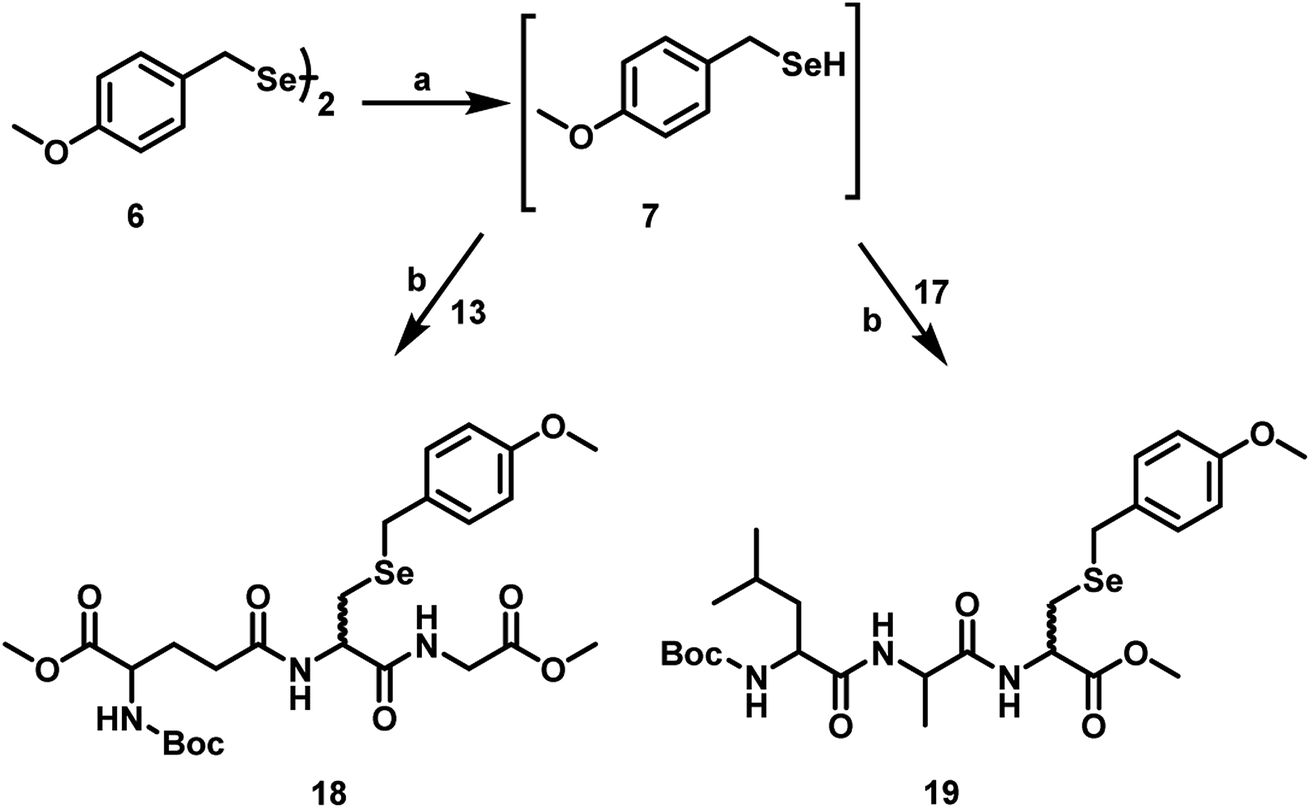 | ||
| Scheme 6 Synthetic route to seleno-tripeptides 18 and 19 starting from the Dha-peptides 13 and 17 respectively. (a) NaBH4, MeOH, 0 °C, 10 min, (b) 50 mM sodium phosphate buffer pH 8, MeOH, 27 °C, 6–8 h. | ||
Formation of epimers
It has been shown earlier that the addition of thiols to Dha lead to epimeric products, and the formation of epimeric (D-/L-) mixtures in these reactions can be viewed as a limitation of the synthetic functionalization of Dha.27 This methodology may not be useful when enantiomerically pure selenium compounds are required. However, for the selenocysteine derivatives, the chirality may not play any key role in certain biological activities such as peroxynitrite scavenging. To understand the number of isomers produced in the selenocysteine derivatives, we carried out 77Se NMR studies in CD3OD at 27 °C (Fig. S1, ESI†). For compound 9d, two signals at 230.4 and 231.4 ppm were observed, indicating the formation of two epimers. As the amino acid attached to the selenocysteine moiety was enantiomerically pure (R-configuration), the two epimers formed in the reactions are expected to have R,R and R,S configurations (Scheme 7).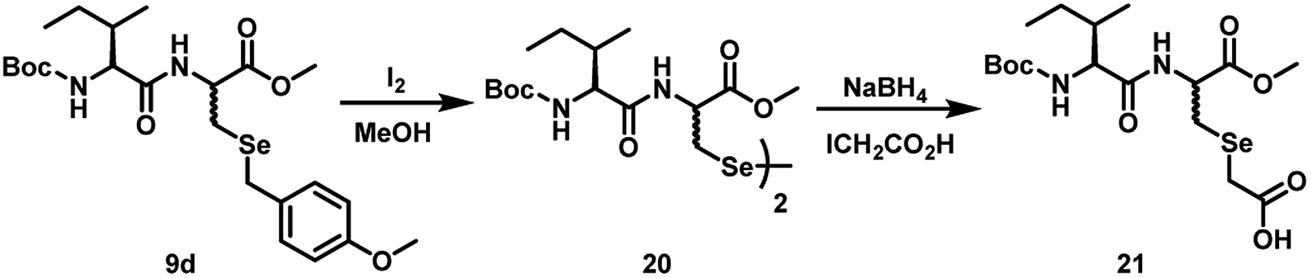 | ||
| Scheme 7 Reaction of the selenocysteine dipeptide 9d with iodine in methanol and a subsequent reaction with sodium borohydride and iodoacetic acid to produce the alkylated derivative 21. | ||
When compound 9d was treated with I2 in MeOH, it produced the corresponding diselenide 20. The 77Se NMR spectrum obtained for 20 showed four peaks at 295.6, 298.1, 298.6, 298.9 ppm, indicating the formation four epimers with possible configurations of R,R,R,S; R,S,R,R; R,R,R,R and R,S,S,R. The reduction of the diselenide 20 by sodium borohydride produced the corresponding selenol, which was trapped by treating the reaction mixture with iodoacetic acid. The alkylated compound 21 also showed two peaks in the 77Se NMR spectrum at 190.4 and 191.1 ppm, indicating the formation of two epimers with R,R and R,S configurations.
Selenocysteine derivatives with a photo-cleavable group
In addition to the use of pMob group, which can be removed from the peptide by treating with iodine in MeOH, it is known that a photo labile group such as o-nitrobenzyl group can also be used as a protecting group for selenium in selenocysteine.28 For the introduction of o-nitrobenzyl group, we synthesized o-nitrobenzyl selenocyanide (o-NO2–C6H4–CH2SeCN) as precursor by following the literature procedure.29 Briefly, the procedure involves treatment of o-nitrobenzyl bromide or chloride with KSeCN in acetone at reflux condition. The product must be recrystallized using EtOH to remove the impurities. Subsequently, o-nitrobenzyl selenocyanide was treated with NaBH4 in MeOH to obtain o-nitrobenzyl selenol, which was added carefully to the Dha derivative 1 in MeOH to obtain compound 22 in around 30% yield (Scheme 8). The irradiation of compound 22 at 366 nm UV light for around 2 h, produced the diselenide 23, which was characterized by ESI-MS (Fig. S2B and S2C, ESI†). The UV-visible spectra of compound 23 exhibited a band (λmax) at around 254, which disappeared upon irradiation (Fig. S2A, ESI†).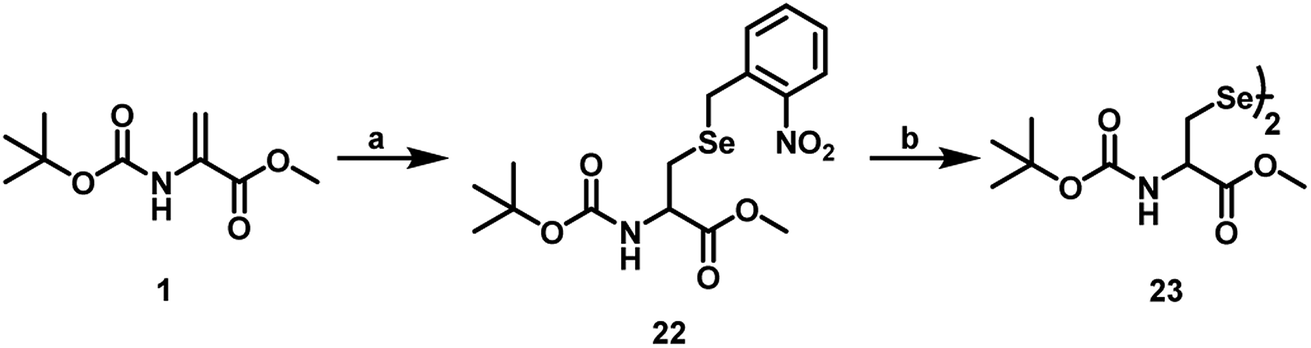 | ||
Scheme 8 Coupling of o-nitrobenzyl selenol with the Dha derivative 1 to afford the selenocysteine derivative 22. (a) o-NO2–C6H4–CH2SeCN, NaBH4, MeOH, 0 °C for 30 min; 27 °C, 8 h (b) 366 nm UV light, ACN and phosphate buffer pH 7 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 2 h. :1), 2 h. | ||
Conclusions
In this paper, we reported the incorporation of selenocysteine into peptides via dehydroalanine intermediate (Dha). Using this methodology, several di- tri- and penta-peptides having Dha moiety were synthesized. A selenium moiety has been introduced into these peptides using p-methoxybenzyl (pMob) selenol, derived from the corresponding diselenide. The pMob group can be cleaved easily by treating the peptides with iodine in methanol. The Dha methodology was found to be useful, particularly, for the synthesis of the selenium analogue of glutathione. The Dha route can also be used to synthesize selenocysteine derivatives having photo-cleavable o-nitrobenzyl group, which can be readily cleaved under UV irradiation. Although the Dha methodology yields the selenium compounds as mixture of epimers, the compounds produced by this method should be useful for studies that do not depend on the chirality of the selenocysteine moiety.Experimental section
General procedure
All the amino acids were purchased from GL Biochem (Shanghai) Ltd. Methanol was obtained from Merck. All other chemicals were of the highest purity available. Most reactions were carried out in a well-ventilated fume hood to avoid the unpleasant odour and toxic nature of the reaction mixtures involved. THF was dried over sodium metal with benzophenone. Thin-layer chromatography analyses were carried out on pre-coated silica gel plates (Merck), and spots were visualized under UV radiation. Column chromatography was performed on glass columns loaded with silica gel. 1H (400 MHz), 13C (100.56 MHz), and 77Se (76.29 MHz) NMR spectra were obtained on a Bruker 400 MHz NMR spectrometer. Chemical shift values are cited with respect to SiMe4 as internal (1H and 13C) and Me2Se as external (77Se) standard. Mass spectral studies were carried out on a Bruker Daltonics Esquire 6000 plus mass spectrometer with ESI-MS mode analysis.Synthesis of 1
General procedure for the synthesis of thioesters 4a–e, 15
The Boc-protected amino acid (3a–e, 14) (1.0 equiv., 5.71 mmol), N-methylmorpholine (1.2 equiv., 6.85 mmol) and dry THF (25 mL) were mixed in a dry flask containing 4 Å molecular sieves and the solution was cooled to 0 °C. To this, ethyl chloroformate (1.5 equiv., 8.56 mmol) was added and the reaction mixture was stirred for 15 min. Thiophenol (1.2 equiv., 6.85 mmol) and N-methylmorpholine (1.2 equiv., 6.85 mmol) were added and the reaction mixture was slowly warmed to 27 °C. The stirring was continued for 12 h at this temperature. The completion of the reaction was monitored by TLC. The reaction mixture was filtered to remove molecular sieves, and the solvent was removed under reduced pressure. The resulting residue was dissolved in dichloromethane (100 mL) and washed sequentially with NaHCO3 (3 × 50 mL), H2O (3 × 50 mL), and brine (3 × 50 mL). The organic phase was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The product was purified by column chromatography to afford the products (4a–e, 15) (80–90%) as white solids. The spectral values obtained were matching with the literature values.15,30General procedure for the synthesis of Dha peptides 5a–e, 17
Cysteine·HCl (1.0 equiv., 3.74 mmol) was added to MeOH (15 mL). NaBH4 (2.0 equiv., 7.48 mmol) was added slowly, and the mixture was stirred for 1 h under N2 atm. Thioesters (4a–e, 15) (1.0 equiv., 3.74 mmol) was added in MeOH (10 mL), and the reaction mixture was stirred till the starting materials were fully consumed (∼4 h, TLC analysis). After the removal of MeOH under reduced pressure, the residue was treated with water (20 mL). The reaction mixture was acidified with 1 N HCl, extracted with ethylacetate (3 × 50 mL) and washed with water (2 × 25 mL), followed by brine (3 × 25 mL). The organic layer was dried over anhydrous Na2SO4. After reducing the volume of the solvent to ∼10 mL, TECP (0.5 equiv., 1.87 mmol) was added to reduce any disulphide bonds formed during the progress of the reaction. The reaction mixture was diluted with water (50 mL) and the aqueous layer was extracted once again with ethylacetate (3 × 50 mL) and washed with water(2 × 25 mL), followed by brine (3 × 25 mL). The organic layer was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The intermediates formed in these reactions were used directly for the next step without further purification. These intermediate products were dissolved in DMF (25 mL) and added MeI (5 equiv., 18.7 mmol) and K2CO3 (5 equiv., 18.7 mmol) at 0 °C. After 15 min the ice bath was removed, and the reaction mixture was stirred at 27 °C for about 12 h. After completion of the reaction, which was followed by TLC, dil. HCl was added to neutralize the reaction mixture and the compound was extracted with ethylacetate (3 × 50 mL). The organic layer was washed with brine solution (5 × 25 mL) to remove DMF. It was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The product was purified by column chromatography using 100–200 mesh silica and 20% petroleum ether and ethylacetate as eluent to afford the Dha-containing peptide 5a–e, 17 as viscous liquids which becomes solids on standing for long time.General procedure for the synthesis of Sec derivatives
NaBH4 (4 equiv., 6.0 mmol) was added portion-wise to a solution of p-methoxy benzyldiselenide or o-NO2–C6H4–CH2SeCN (2 equiv., 3.0 mmol) in degassed MeOH (1 mL per 0.03 mmol) at 0 °C under N2 atmosphere. An immediate evolution of hydrogen gas was observed. After 5 min, this solution was added dropwise to Dha containing aminoacid (1)/dipeptides (5a–e)/tripeptides (13, 17) (1 equiv., 1.50 mmol) in 0.5 M sodium phosphate (pH-8) (1 mL per 0.003 mmol) and stirred at 27 °C for 4–6 h. The reaction mixture was treated with EtOAc and washed subsequently with 1 M aq. HCl, saturated NaHCO3, water and brine. The organic layers were combined and dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude residue was purified by column chromatography (100–200 mesh silica gel for compounds 8, 9a–9e, and 230–400 mesh silica gel for compound 18, 19 & 22) to afford the protected selenocysteine derivatives, seleno dipeptides 8, 9a–e, 18, 19, in 80–90% yields, and 22 in 35% yield.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This study was supported by the Science and Engineering Research Board (SERB), New Delhi, India. K. M. R. thanks the CSIR for a research fellowship. G. M. acknowledges the SERB for the award of J. C. Bose National fellowship (SB/S2/JCB-067/2015).Notes and references
- (a) T. C. Stadtman, Science, 1974, 183, 915–922 CrossRef CAS; (b) J. E. Cone, R. M. Del Río, J. N. Davis and T. C. Stadtman, Proc. Natl. Acad. Sci. U. S. A., 1976, 73, 2659–2663 CrossRef CAS.
- (a) A. Böck, K. Forchhammer, J. Heider and C. Baron, Trends Biochem. Sci., 1991, 16, 463–467 CrossRef; (b) A. Bock, K. Forchhammer, J. Heider, W. Leinfelder, G. Sawers, B. Veprek and F. Zinoni, Mol. Microbiol., 1991, 5, 515–520 CrossRef CAS.
- (a) F. Zinoni, A. Birkmann, T. C. Stadtmant and A. Böck, Proc. Natl. Acad. Sci. U. S. A., 1986, 83, 4650–4654 CrossRef CAS; (b) I. Chambers, J. Frampton, P. Goldfarb, N. Affara, W. McBain and P. R. Harrison, EMBO J., 1986, 5, 1221–1227 CrossRef CAS; (c) B. J. Lee, P. J. Worland, J. N. Davis, T. C. Stadtman and D. L. Hatfield, J. Biol. Chem., 1989, 264, 9724–9727 CAS.
- (a) L. Flohé, E. A. Günzler and H. H. Schock, FEBS Lett., 1973, 32, 132–134 CrossRef; (b) J. T. Rotruck, A. L. Pope, H. E. Ganther, A. B. Swanson, D. G. Hafeman and W. G. Hoekstra, Science, 1973, 179, 588–590 CrossRef CAS; (c) H. Sies, Angew. Chem., Int. Ed., 1986, 25, 1058–1071 CrossRef; (d) D. Behne, A. Kyriakopoulos, H. Meinhold and J. Köhrle, Biochem. Biophys. Res. Commun., 1990, 173, 1143–1149 CrossRef CAS PubMed; (e) T. Tamura and T. C. Stadtman, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 1006–1011 CrossRef CAS PubMed; (f) G. Mugesh, W.-W. du Mont and H. Sies, Chem. Rev., 2001, 101, 2125–2180 CrossRef CAS; (g) J. Köhrle, Methods Enzymol., 2002, 347, 125–167 Search PubMed; (h) G. Roy, B. K. Sarma, P. P. Phadnis and G. Mugesh, J. Chem. Sci., 2005, 117, 287–303 CrossRef CAS; (i) M. Conrad, M. Schneider, A. Seiler and G. W. Bornkamm, Biol. Chem., 2007, 388, 1019–1025 CAS; (j) K. P. Bhabak and G. Mugesh, Acc. Chem. Res., 2010, 43, 1408–1419 CrossRef CAS; (k) T. Wirth, Angew. Chem., Int. Ed., 2015, 55, 10074–10076 CrossRef PubMed; (l) S. Mondal, K. Raja, U. Schweizer and G. Mugesh, Angew. Chem., Int. Ed., 2016, 55, 7606–7630 CrossRef CAS PubMed.
- (a) A. P. Arnold, K. S. Tan and D. L. Rabenstein, Inorg. Chem., 1986, 25, 2433–2437 CrossRef CAS; (b) B. J. Byun and Y. K. Kang, Biopolymers, 2011, 95, 345–353 CrossRef CAS PubMed.
- (a) W. Leinfelder, E. Zehelein, M. A. MandrandBerthelot and A. Böck, Nature, 1988, 331, 723–725 CrossRef CAS PubMed; (b) A. A. Turanov, X. M. Xu, B. A. Carlson, M. H. Yoo, V. N. Gladyshev and D. L. Hatfield, Adv. Nutr., 2011, 2, 122–128 CrossRef CAS PubMed; (c) E. G. Varlamova, M. V. Goltyaev, S. V. Novoselov, V. I. Novoselov and E. E. Fesenko, Mol. Biol., 2013, 47, 558–567 CAS.
- (a) S. Palioura, R. L. Sherrer, T. A. Steitz, D. Söll and M. Simonovic, Science, 2009, 325, 321–325 CrossRef CAS PubMed; (b) K. Forchhammer and A. Böck, J. Biol. Chem., 1991, 266, 6324–6328 CAS.
- (a) P. M. Masters, Calcif. Tissue Int., 1985, 37, 236–241 CrossRef CAS PubMed; (b) Z. Wang, B. Lyons, R. J. W. Truscott and K. L. Schey, Aging Cell, 2014, 13, 226–234 CrossRef CAS PubMed.
- (a) P. J. Knerr and W. A. van der Donk, Annu. Rev. Biochem., 2012, 81, 479–505 CrossRef CAS PubMed; (b) Y. Shi, X. Yang, N. Garg and W. A. van der Donk, J. Am. Chem. Soc., 2011, 133, 2338–2341 CrossRef CAS PubMed.
- J. Wang, S. M. Schiller and P. G. Schultz, Angew. Chem., Int. Ed., 2007, 46, 6849–6851 CrossRef CAS PubMed.
- Z. P. Wu and D. Hilvert, J. Am. Chem. Soc., 1989, 111, 4513–4514 CrossRef CAS.
- (a) R. J. Hondal, B. L. Nilsson and R. T. Raines, J. Am. Chem. Soc., 2001, 123, 5140–5141 CrossRef CAS PubMed; (b) R. Quaderer, A. Sewing and D. Hilvert, Helv. Chim. Acta, 2001, 84, 1197–1206 CrossRef CAS.
- (a) D. Besse and L. Moroder, J. Pept. Sci., 1997, 3, 442–453 CrossRef CAS PubMed; (b) M. Rooseboom, N. P. E. Vermeulen, L. Andreadou and J. N. M. Commandeur, J. Pharmacol. Exp. Ther., 2000, 294, 762–769 CAS.
- D. H. Strumeyer, W. N. White and D. E. Koshland Jr, Proc. Natl. Acad. Sci. U. S. A., 1963, 50, 931–935 CrossRef CAS.
- L. Markey, S. Giordani and E. M. Scanlan, J. Org. Chem., 2013, 78, 4270–4277 CrossRef CAS PubMed.
- (a) D. H. Rich, J. Tam, P. Mathiaparanam, J. A. Grant and C. Mabuni, J. Chem. Soc., Chem. Commun., 1974, 897–898 RSC; (b) J. M. Chalker, S. B. Gunnoo, O. Boutureira, S. C. Gerstberger, M. Fernández-González, G. J. L. Bernardes, L. Griffin, H. Hailu, C. J. Schofield and B. G. Davis, Chem. Sci., 2011, 2, 1666–1676 RSC.
- (a) P. E. Dawson and S. B. H. Kent, Annu. Rev. Biochem., 2000, 69, 923–960 CrossRef CAS PubMed; (b) P. Thapa, R. Y. Zhang, V. Menon and J. P. Bingham, Molecules, 2014, 19, 14461–14483 CrossRef PubMed.
- (a) B. G. Davis, R. C. Lloyd and J. B. Jones, Chem.–Asian J., 2009, 4, 630–640 CrossRef PubMed; (b) J. Dadová, S. R. G. Galam and B. G. Davis, Curr. Opin. Chem. Biol., 2018, 46, 71–81 CrossRef PubMed.
- Y. A. Lin, O. Boutureira, L. Lercher, B. Bhushan, R. S. Paton and B. G. Davis, J. Am. Chem. Soc., 2013, 135, 12156–12159 CrossRef CAS PubMed.
- (a) M. D. Gieselman, L. Xie and V. A. Wilfred A, Org. Lett., 2001, 3, 1331–1334 CrossRef CAS PubMed; (b) R. J. Hondal, Protein Pept. Lett., 2005, 12, 757–764 CrossRef CAS PubMed.
- O. Flögel, G. Casi, D. Hilvert and D. Seebach, Helv. Chim. Acta, 2007, 90, 1651–1666 CrossRef.
- A. Pompella, A. Visvikis, A. Paolicchi, V. De Tata and A. F. Casini, Biochem. Pharmacol., 2003, 66, 1499–1503 CrossRef CAS PubMed.
- T. Tamura, T. Oikawa, A. Ohtaka, N. Fujii, N. Esaki and K. Soda, Anal. Biochem., 1993, 208, 151–154 CrossRef CAS PubMed.
- S. Yoshida, F. Kumakura, I. Komatsu, K. Arai, Y. Onuma, H. Hojo, B. G. Singh, K. I. Priyadarsini and M. Iwaoka, Angew. Chem., Int. Ed., 2011, 50, 2125–2128 CrossRef CAS PubMed.
- D. Crich, V. Krishnamurthy and T. K. Hutton, J. Am. Chem. Soc., 2006, 128, 2544–2545 CrossRef CAS PubMed.
- B. N. Naidu, M. E. Sorenson, T. P. Connolly and Y. Ueda, J. Org. Chem., 2003, 68, 10098–10102 CrossRef CAS PubMed.
- C. D. Spicer and B. G. Davis, Nat. Commun., 2014, 5, 4740 CrossRef CAS PubMed.
- M. Abbas and L. A. Wessjohann, Org. Biomol. Chem., 2012, 10, 9330–9333 RSC.
- D. Plano, Y. Baquedano, D. Moreno-Mateos, M. Font, A. Jiménez-Ruiz, J. A. Palop and C. Sanmartín, Eur. J. Med. Chem., 2011, 46, 3315–3323 CrossRef CAS PubMed.
- (a) X. Guan, M. Drake and Z. Tan, Org. Lett., 2013, 15, 6128–6131 CrossRef CAS PubMed; (b) Q. Wan, J. Chen, Y. Yuan and S. J. Danishefsky, J. Am. Chem. Soc., 2008, 130, 15814–15816 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra09880h |
| This journal is © The Royal Society of Chemistry 2019 |