
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A novel apoptosis-inducing metabolite isolated from marine sponge symbiont Monascus sp. NMK7 attenuates cell proliferation, migration and ROS stress-mediated apoptosis in breast cancer cells†
Sirpu Natesh Nagabhishek and
Arumugam Madankumar*
and
Arumugam Madankumar*
Cancer Biology Lab, Molecular and Nanomedicine Research Unit, Sathyabama Institute of Science and Technology, Chennai-600119, Tamil Nadu, India. E-mail: madankumarbio@gmail.com; madankumar@sathyabama.ac.in; Tel: +919942110146
First published on 18th February 2019
Abstract
The marine environment has a remarkable source of natural products mainly from marine fungi, which have been a central source of novel pharmacologically bioactive secondary metabolites. In this study, the search for a new potential apoptosis-inducing metabolite is focused on marine sponge-associated symbionts. A total of sixteen different sponges were obtained from the Gulf of Mannar region, India, and twenty-three different marine fungal strains were isolated and tested for antiproliferative activity by the MTT assay. Out of these, Monascus sp. NMK7 associated with the marine sponge Clathria frondifera was found to have a promising antiproliferative property. Furthermore, to isolate the pure active metabolite, the crude material was subjected to column chromatography and HPLC. Structural characterization was conducted by a variety of spectroscopic techniques including UV, IR, MS and NMR. The obtained results from the MS and NMR spectroscopy determined 418.5 Da to be the molecular weight and C24H34O6 to be the molecular formula of the metabolite, indicating the presence of monacolin X (NMKD7). NMKD7 was found to induce dose-dependent cytotoxicity in different human breast cancer cell lines MCF-7, T47D, MDA-MB-231, MDA-MB-468 and MCF-10A normal breast cell after 24 h of exposure. For elucidating the possible mode of cell death, T47D and MDA-MB-468 cells were treated with NMKD7 for 24 h to examine the morphological change of the chromatin (PI & AO/EB). Therefore, it has been suggested as the possible mechanism of apoptosis, and apart from this, it has also exhibited antibacterial and anti-migratory properties as well as induced the ROS stress (DCFH-DA), which causes the mitochondrial membrane potential difference (Rhodamine-123), the loss of cell membrane integrity and eventually cell death. Thus, the present study features a novel promising apoptosis-inducing metabolite (NMKD7) with minimal toxicity, suggesting its potential for biotechnological applications, and substantiates that it should be further considered for the elucidation of molecular targets and signal transduction pathways.
Introduction
The increasing incidences of cancer have led to a global burden to find many diverse therapeutic treatments including surgery, radiation and chemotherapy.1 A large portion of the drugs are targeted for the signaling pathways responsible for cell proliferation and survival.2 Cancer fungotherapy is a promising scientific field that deals with antitumor metabolites derived from fungi,3 the initiation of apoptosis and the limiting of stifling cell proliferation in cancer cells with minimal or no side effects. Sponge-associated fungi metabolites have produced therapeutic compounds that have opened up a new era in marine pharmacology.4Marine fungal metabolites are in the limelight of the drug discovery area as they are very effective therapeutic agents. There are numerous marine fungal metabolites discovered from a vivid source, and they demonstrate a wide range of potent biological activities including anticancer, antibacterial and antiviral.5 Marine fungi, compared to terrestrial fungi, are much less explored and have very good potential for drug discovery in addition to natural product discovery.6,7 Natural products are biologically active substances and are produced in relatively small amounts from rare species of animal or plants whose natural population cannot sustain the extensive collections needed for clinical trials.8 Natural products from the unique environments of oceans and seawater represent new microbes, which are unfamiliar and potent producers of secondary metabolites. Thus, the microorganisms for the screening of bioactive natural products can be considered as an alternative strategy, since it can be an effective approach.9–12 Marine sponges are the hosts for large population including actinomycetes, bacteria and fungi which comprise much as 40% of the sponge tissue's volume and helps in the stabilization of the sponge skeleton, allowing nutrient acquisition, the processing of metabolic waste and secondary metabolite production.13 Marine sponges are sessile soft-bodied organisms and they rely on chemical defenses through the production of secondary metabolites that are often from the associated microorganisms.14 Many sponge-associated fungi are distinguishable from their terrestrial analogues, compounds reported from Aspergillus or Penicillium marine fungi isolates are remarkably different from those of their terrestrial counterparts.15 This suggests the possibility of horizontal gene transfer through evolution. Marine fungi growing in a unique and stressful habitat develop the ability to produce structurally complex and unusual secondary metabolites due to their different transcriptome; proteasome; and finally, their different metabolome, which allows an organism to survive.16 Besides, some of the blockbuster drugs are of fungal origin such as β-lactam, the world's bestselling antibiotic, and cholesterol biosynthesis inhibitors. The anticancer drug paclitaxel was initially isolated from a plant source but was also found in endophytic fungi like Taxomyces andreanae17 and podophyllotoxin, an anticancer drug precursor.18 Hence, there is an increasing recognition for fungal secondary metabolites as a good resource for new drug leads, especially in the area of cancer research.19
Though many natural compounds are widely in use for chemotherapy, the application of such products in folk and traditional medicine has always been an important source of compounds with therapeutic potential3 and has opened the door to look towards several more new promising sources of marine natural products as a potential source of pharmaceuticals. Thus, in this study, we have isolated an apoptotic property-bearing compound produced by a marine sponge-associated fungi and have assessed its anticancer activity in breast cancer cells.
Materials and methods
Reagents and chemicals
All chemicals were purchased from Sigma-Aldrich (USA) and Himedia Laboratories Pvt. Ltd India, unless stated otherwise. Cell culture plastics were purchased from Tarsons Products (P) Ltd, India.Sample collection and sponge identification
Sixteen different marine sponges were collected (Sp1 to Sp16) (data not shown) as entangled specimens from a bottom trawl fish net in the Gulf of Mannar region at 6 AM (latitude-8°41′35.5′′N; longitude-78°08′21.7′′E). The water salinity was 36 ppt, and the temperature was 30.7 °C. The sponge sample was carefully removed from the wires of the trawl net and washed with seawater to remove any sand or adhered debris. Small pieces of samples were cut and transferred into a sterile sample container and transported to the laboratory under the required aseptic conditions, later kept in an icebox, correctly labelled and stored at 4 °C.20A part of the sponge material was preserved in 70% methanol for sponge identification purposes. Based on the color, morphology and spicule pattern of the sponge, the identification was carried out.21
Isolation of sponge-associated fungi and identification
The sponges were washed thoroughly with UV-treated sterile seawater until no visible debris was seen. 1 g of the central core of the sponge tissue was cut and homogenized with 99 mL of phosphate-buffered saline using mortar and pestle. The homogenate was serially diluted and plated on PDA, SDA and MA40S (Osmophilic Agar) to isolate the fungi, using a dilution series of 10−5 by a spread plate technique, and the plates were incubated at 28 °C for 10 days. From this method, 23 different fungi based upon morphology were isolated and checked for their antiproliferative activity. Only the strain showing potent antiproliferative activity was taken further for 18S rRNA sequencing,22,23 and by its morphological characteristics (e.g., the shape of the conidia, colony color), was identified as Monascus sp. NMK7 (GenBank accession no. MG793201). The sequences obtained were aligned using ClustalW and phylogenetically analyzed using MEGA7 with a neighbor-joining tree.24 The fungi surface morphology was analysed using field emission scanning electron microscopy.Inoculum preparation, fermentation and extraction
The isolated culture plate of Monascus sp. NMK7 was taken, and its spores were inoculated into 250 mL Erlenmeyer flasks containing 100 mL of the MA40S (maltose 40 g L−1, sucrose 400 g L−1, and seawater to make up to 1 L) broth. After being cultivated for 72 h on a rotary shaker with 180 rpm at 28 °C, the MA40S (60 mL) was transferred to 54 × 2 L Erlenmeyer flasks each with 600 mL of liquid culture medium and were incubated at 28 °C with shaking at 180 rpm for 8 days. The fermented broth was filtered, and the filtrate was concentrated to one-twentieth of the original volume in a vacuum evaporator at a temperature below 50 °C (rotary vacuum evaporator), which resulted in a dark brown syrup. The crude material was extracted in ethyl acetate and concentrated to dryness in a rotary vacuum evaporator. Further, the soluble fraction was subjected to repeated silica gel column chromatography to purify the compound.Purification of the crude extract by column chromatography and RP-HPLC
The crude extract of Monascus sp. NMK7 was subjected to column chromatography using silica gel (mesh size: 60) using the solvent system hexane and methanol 0–100%. Then different elutions were collected in different test tubes, concentrated and assayed for antiproliferative/cytotoxic activity using MCF7 cell lines. The most active antiproliferative fraction (semi-purified) was taken to HPLC for further purification of the active compound. Further, only the active peak elusion purified cytotoxic fraction was subjected to the reverse-phase high-performance liquid chromatography (RP-HPLC) system using the C18 column (Sunfire-C18, 4.6 × 250 mm), with the photodiode array (PDA) detector (Waters). The mobile phase was applied as the linear gradient of the following: detection – PDA (200–800 nm) with the detection wavelength of 238 nm; mobile phase: A – water, B – acetonitrile; (0–2 min) 70% A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30% B, (2–10 min) 10% A:90% B, (10–20 min) 50% A:50% B, (20–30 min) 70% A:30% B with a flow rate of 1 mL min−1.
:30% B, (2–10 min) 10% A:90% B, (10–20 min) 50% A:50% B, (20–30 min) 70% A:30% B with a flow rate of 1 mL min−1.
Structural characterization
500 FWHM. The capillary and cone voltage were set to 3 kV, and the source temperature and desolvation temperature were set to 140 and 400 °C, respectively. The gas flow of the cone was 50 L h−1, and a desolvation gas flow of 1000 L h−1 was set to analyze the mass of the HPLC-purified compound. The ESI data were processed by a Thermo Qual browser, and the collision energy was applied for fragmentation.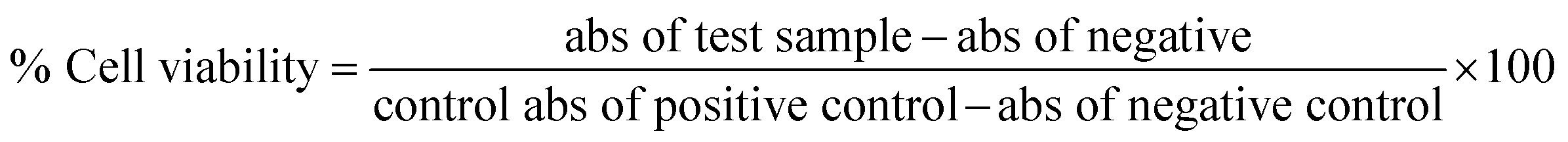 :1 ratio of AO and EB), and it was immediately viewed under an inverted fluorescence microscope (EVOS FL digital inverted fluorescence microscope (AMG)).
:1 ratio of AO and EB), and it was immediately viewed under an inverted fluorescence microscope (EVOS FL digital inverted fluorescence microscope (AMG)).Results
Identification of NMKD7-producing fungi associated with the marine sponge
Initially, sixteen marine sponges (Sp1 to Sp16) were collected, and twenty-three associated fungi (NMK1-23) were isolated based on the colony morphology; each crude was then checked for anticancer activity on the MCF-7 breast cancer cell line (ESI Fig. 1A†). Based on the results, the NMK7 strain associated with the marine sponge (Sp4), showing the highest anticancer activity, was taken for identification.The marine sponge (Sp4) was taxonomically identified based on its morphology and the spicule structure as Clathria frondifera by Dr Siva Leela, a scientist in Zoological Survey of India (ZSI), MBRC, Chennai, Tamil Nadu, India. The fungal strain NMK7 was isolated from the marine sponge Clathria frondifera collected from Gulf of Mannar and was identified using 18S rRNA gene sequencing. The cell surface morphological analysis of the isolate was studied using scanning electron microscopy, which, in support, has shown that the matured, broadly ellipsoid, hyaline, smooth, and 5 μm size (Fig. 1A and B) for the maximum-likelihood phylogenetic analysis revealed that the strain belonged to clade Monascus ruber sp. and was assigned as the Monascus sp. NMK7 strain (Fig. 1C) and submitted into the GenBank with the accession number MG793201.
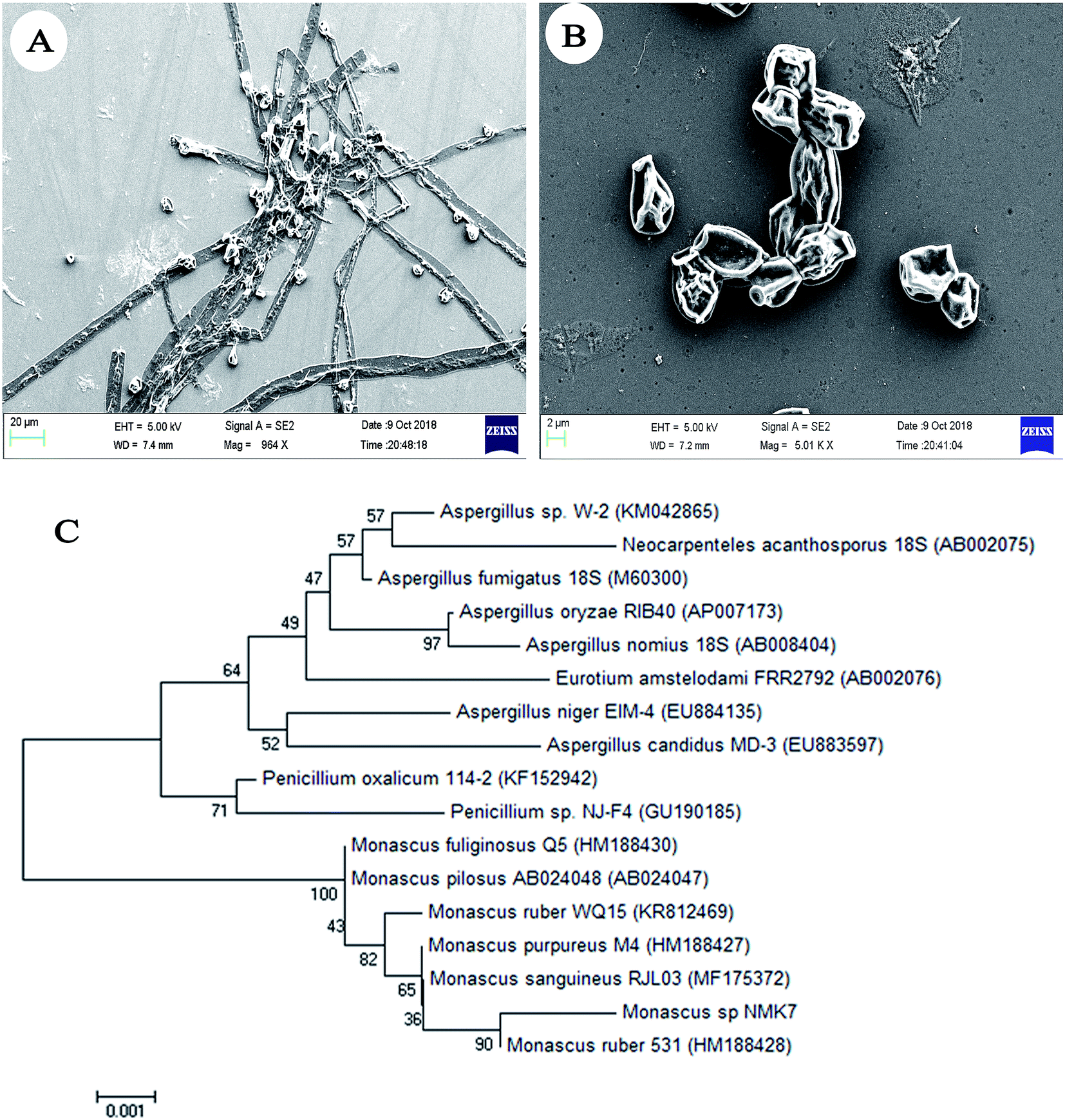 | ||
| Fig. 1 Scanning electron microscopic (SEM) images, (A) and (B) showing the surface morphology of fungi Monascus sp. NMK7 with a broadly ellipsoid body and a hyaline and smooth surface (15000×). (C) Phylogenetic analysis of Monascus sp. NMK7 from the marine sponge Clathria frondifera. Maximum likelihood phylogenetic representation of NMK17 with the neighbor strains by MEGA7 software. | ||
Structural characterization
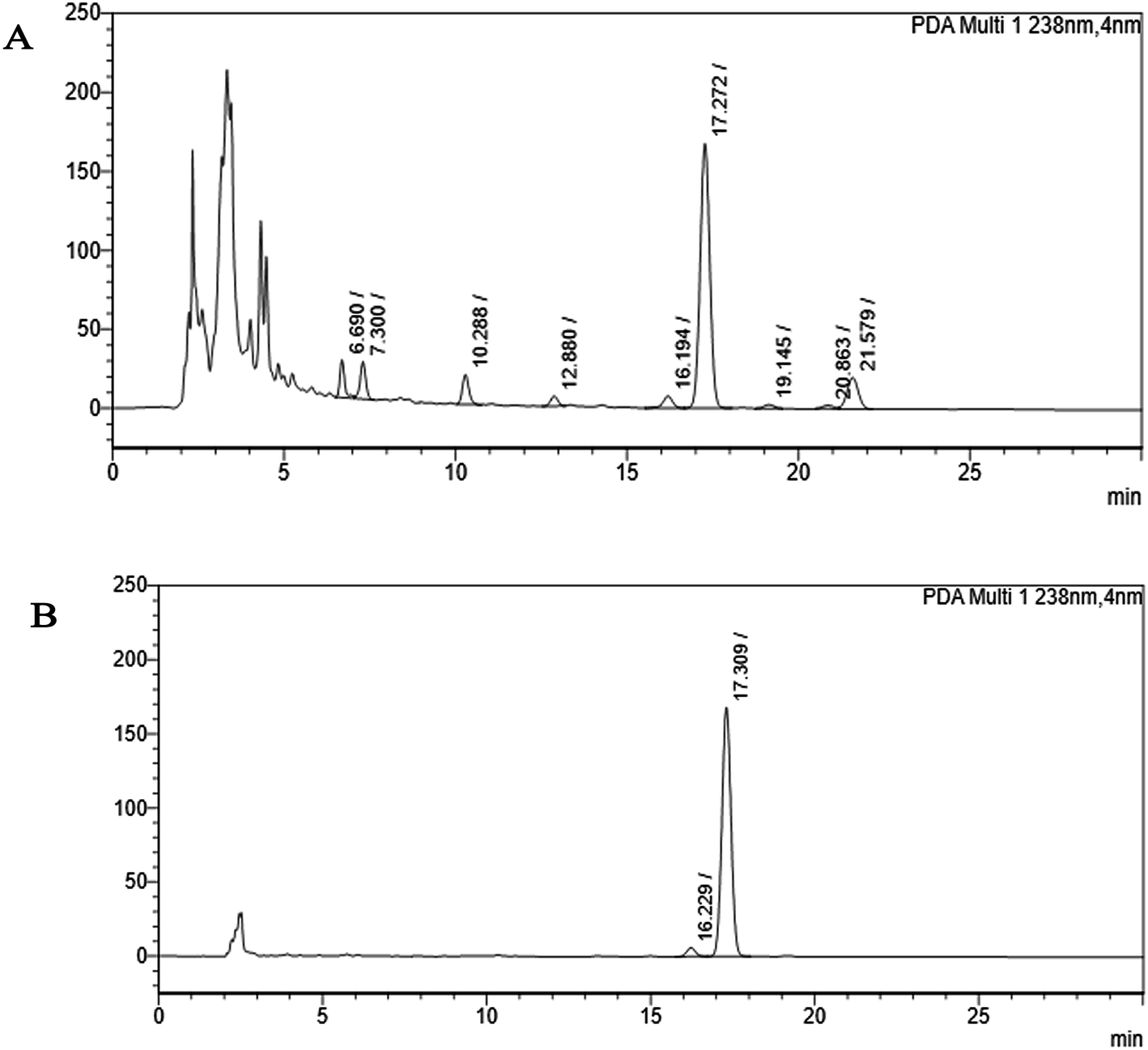 | ||
| Fig. 2 HPLC chromatogram (A) shows the partially purified crude extract of Monascus sp. NMK7 and represents 9 different peaks at 238 nm, indicating various other compounds present apart from the compound of interest. (B) HPLC chromatogram shows a single peak, indicating a pure isolated compound at 17.30 RRT at 238 nm. | ||
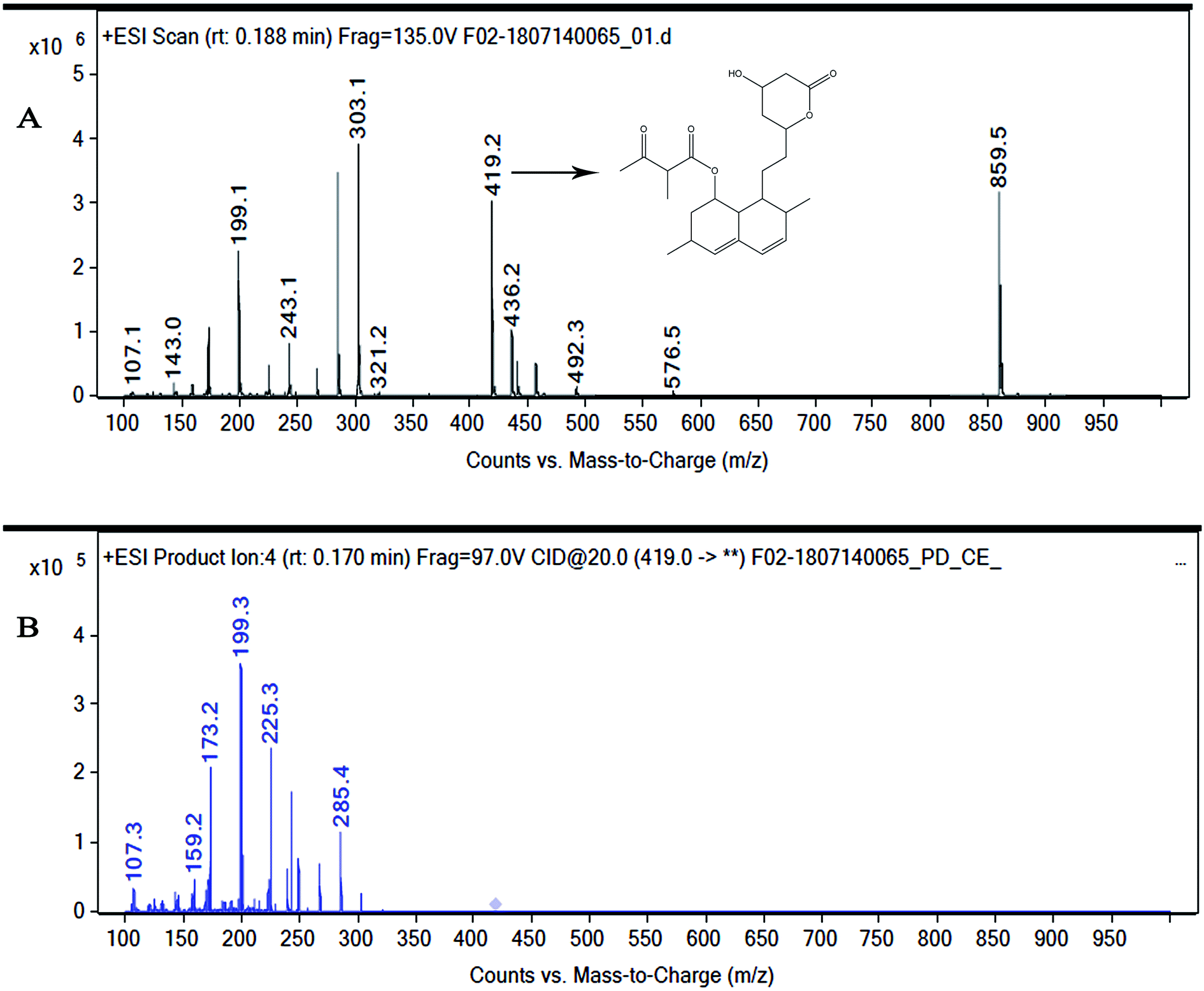 | ||
| Fig. 3 (A) Mass spectrum of NMKD7 (Monacolin X). (MS-ESI) showing a m/z = 419.2 [M + H] and 859.5 [2M + Na+]. The molecular mass was determined to be 418.5 Da. (B) MS fragmentation spectra of the monacolin X peak at m/z = 419.2 at a collision energy of 20, the structure for the peaks at 199.3, 173.2 and 159.2 represents the possible fragmentation form monacolin X. | ||
1H NMR spectroscopy. The isolated compound was dissolved in CDCl3 and characterized by 1H and 13C NMR spectroscopy. The 1H-NMR spectrum of the isolated impurity suggested the presence of the –COCH3 group and one –COCH(CH3)CO– group. Thus, the presence of these groups can be confirmed by locating a singlet of three protons at 2.25 ppm and a multiplet of one proton at 3.50–3.57 ppm. The isolated structure has three alkenes and a proton, which were confirmed by the 1H NMR spectrum. The corresponding [5-alkene–CH] proton corresponded to an observed doublet at 5.97 ppm, and the coupling constant value J = 9.9 Hz. The [4-alkene–CH] group proton was an observed quartet at 5.74–5.79 ppm, with a corresponding coupling constant value of 6.0 Hz, J = 6.3 Hz. The [6-alkene–CH] group proton was an observed triplet at 5.49 ppm, with a corresponding coupling constant value of J = 3.0 Hz (ESI Fig. 2A†).
1H-NMR spectrum (CDCl3, ppm) 5.97 (d, 1H, J = 9.9 Hz, 5-alkene–CH), 5.74–5.79 (q, 1H, J = 6.0 Hz, J = 6.3 Hz, 4-alkene–CH), 5.49 (t, 1H, J = 3.0 Hz, 6-alkene–CH), 5.35 (d, 1H, J = 3.0 Hz, 1-CH), 4.59–4.65 (m, 1H, 5′-CH), 4.32–4.37 (m, 1H, 3′-CH), 3.50–3.57 (m, 1H, 2′′-CH), 2.33–2.80 (m, 4H, –CH2&–CH), 2.25 (s, 3H, 4′′-CH3), 2.03–2.17 (m, 7H, –CH2&–CH), 1.80–1.99 (m, 4H, –CH2&–CH), 0.86–1.34 (m, 9H, 10,9,5′′-CH3).
13C-NMR spectroscopy. The 13C-NMR spectrum of the isolated compound confirmed the carbonyl signal C-3′′ at 204.88 ppm and the corresponding methyl signal C-2′′ at 53.39 ppm. The 4′′-CH3 group carbon was observed at 27.33 ppm. The 1′′&1′-CO carbon was observed at 170.36 and 169.93 ppm. The [5-alkene–CH] carbon was observed at 129.27 ppm, and the corresponding [4-alkene–CH] group carbon was observed at 128.09 ppm. The [6-alkene–CH] group carbon was observed at 131.59 ppm (ESI Fig. 2B†).
13C-NMR spectrum (CDCl3, ppm): 204.88 (3′′-CO), 170.36 (1′′-CO), 169.93 (1′-CO), 133.34 (-CH), 131.59 (6-alkene–CH), 129.27 (5-alkene–CH), 128.09 (4-alkene–CH), 69.45 (–CH), 62.69 (–CH). 53.39 (2′′-CH), 38.60 (–CH2), 37.34 (–CH), 36.54 (–CH), 36.11 (–CH2), 32.59 (–CH2), 32.50 (–CH2), 30.91 (–CH), 30.60 (–CH), 29.52 (–CH), 24.15 (–CH2), 22.81 (–CH3), 13.78 (–CH3), 12.72 (–CH3).
From the DEPT-135-13C NMR spectrum, the isolated product has five –CH2 carbons in its structure. The five –CH2 carbons were confirmed by the DEPT-135-13C NMR spectrum, and the five –CH2 corresponding values were 24.15 (7′-CH2), 32.49 (6′-CH2), 32.57 (4′-CH2), 36.09 (2-CH2), 38.59 (2′-CH2) (ESI Fig. 3†). The 1H-NMR, 13C and DEPT-135-13C NMR spectra revealed the molecular formula to be C24H34O6 and predicted the structure to be monacolin X (Fig. 4).31
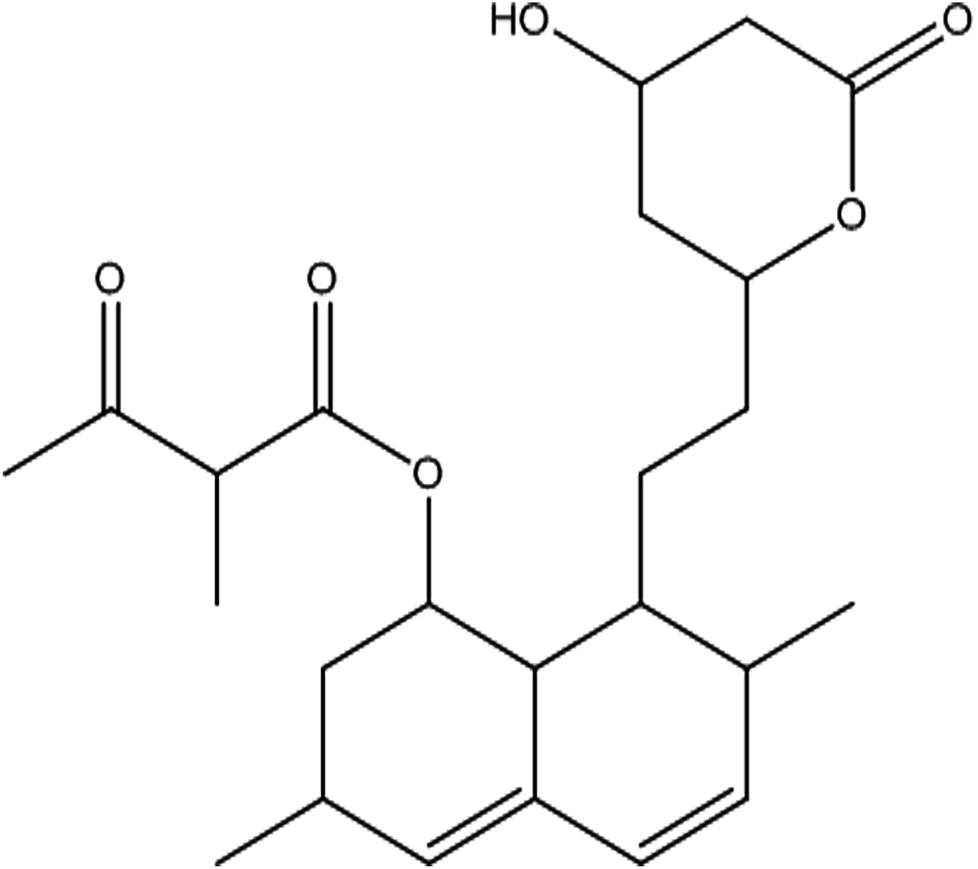 | ||
| Fig. 4 Structure of the isolated molecule monacolin X. | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O carbonyl functional group), and 1612 cm−1 (CC) (ESI Fig. 4A†). Thus, all these functional groups well matched the predicted structure of monacolin X.
O carbonyl functional group), and 1612 cm−1 (CC) (ESI Fig. 4A†). Thus, all these functional groups well matched the predicted structure of monacolin X.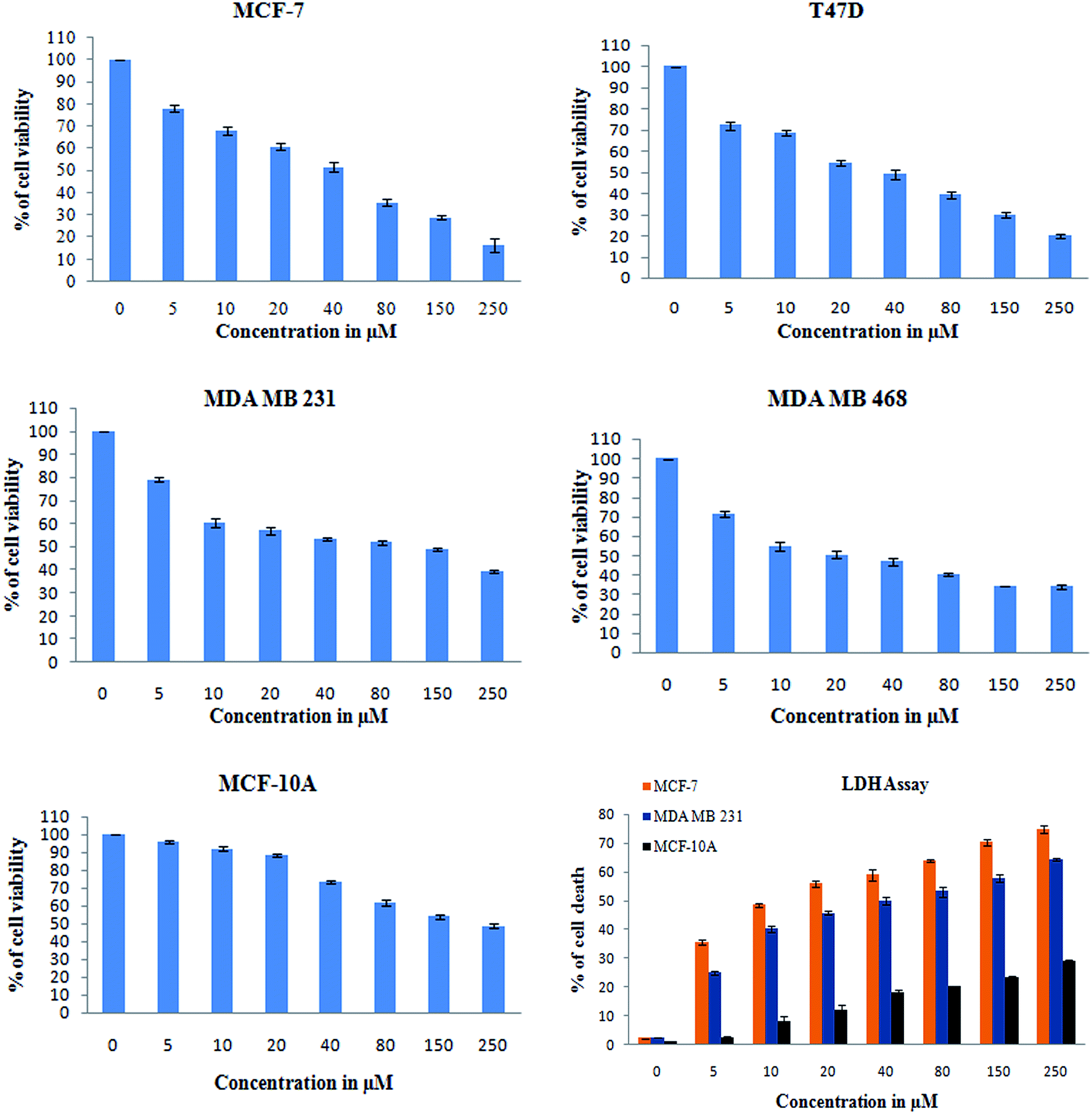 | ||
| Fig. 5 XTT assay was used to check for the antiproliferative and cytotoxic nature of monacolin X on human breast cancer cell lines MCF-7, T47D, MDA-MB-231, MDA-MB-468 and MCF-10A, a non-cancerous breast cell line. The cell viability was measured by XTT assay. All the cells were treated with monacolin X at various concentrations (0–250 μg mL−1) for 24 h. Cell membrane integrity by the release of lactate dehydrogenase (LDH) activity by LDH assay. MCF-7, MDA-MB-231 and MCF-10A cells were treated with monacolin X at various concentrations (0–250 μg mL−1) for 24 h. LDH released into the medium was measured along with blank, untreated cells (0 μM), showing low LDH release in media, whereas treated cells had a dose-dependent release of LDH. | ||
Further, T47D and MDA-MB-468 cells, treated with an IC50 concentration of monacolin X and Dox (1.5 μM), showed significantly increased levels of apoptotic cells in comparison to the untreated cells, where no apoptotic cells were observed. This is evidenced by the acridine orange/ethidium bromide (AO/EB) differential staining method (Fig. 6). The stained cells were characterized to be viable (light green), early apoptotic (yellow fluorescence and condensed chromatin), late apoptotic (orange fluorescence) and nonviable cells (red colored fluorescence).34 Early apoptotic cells with nuclear margination and chromatin condensation, indicated in yellow colour, and late apoptotic cells with fragmented chromatin, indicated in orange colour, were noticed in monacolin X and Dox treated cells. While the control cells have shown intact nuclear architecture, the monacolin X treated cells have shown membrane blebbing, condensed nuclei, and apoptotic bodies.
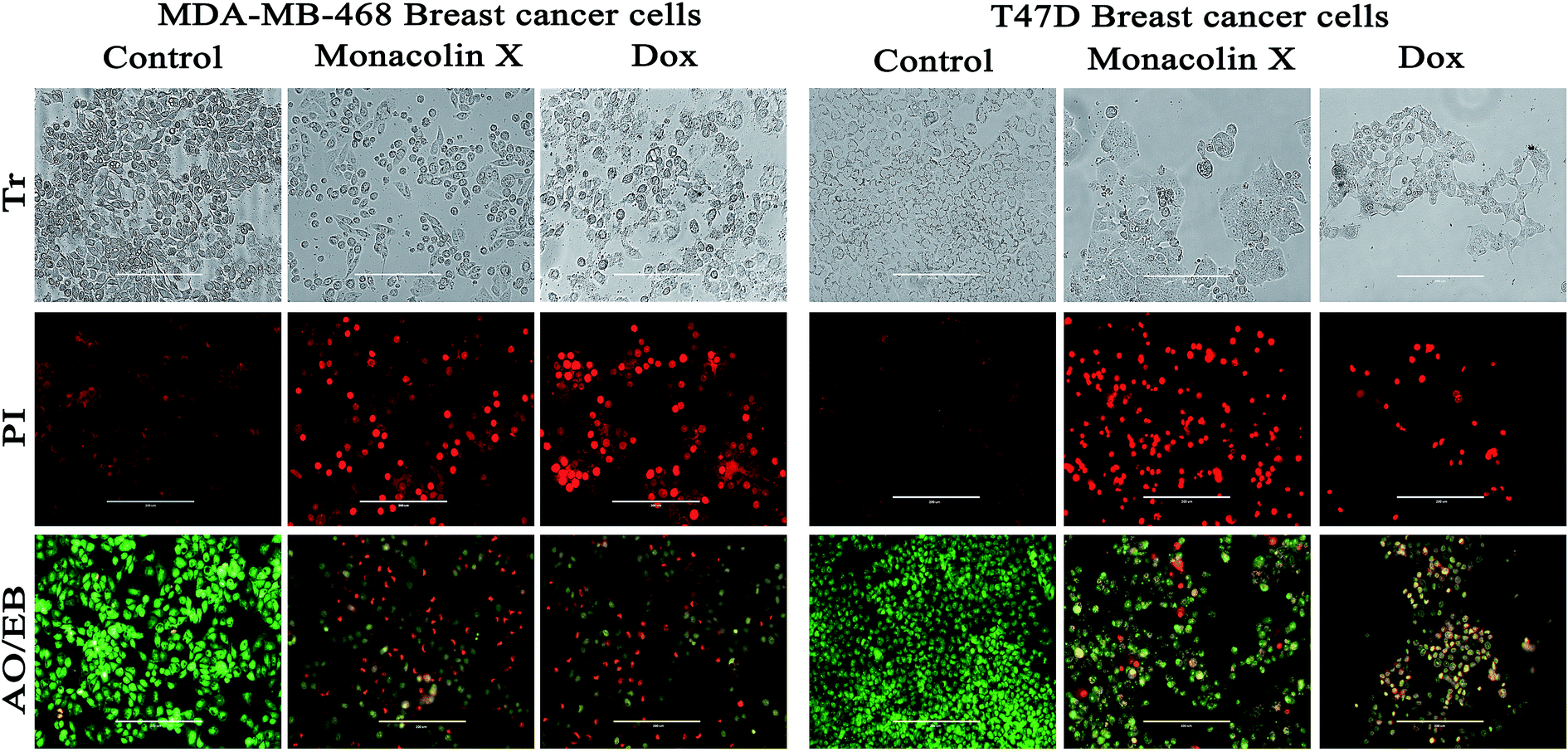 | ||
| Fig. 6 The morphological analysis of MDA-MB-468 and T47D breast cancer cell lines upon treatment with monacolin X (IC50) and Dox (1.5 μM) for 24 h. The morphological changes of treatment group of breast cancer cells showed irregular and shrunken cell morphology, whereas the control group showed normal cell morphology. PI staining by fluorescence microscopy. The percentage of necrotic nuclei after 24 hours of treatment increased enormously when compared to the control, as revealed by nuclear condensation and fragmentation. Apoptotic and nuclear morphological changes in MDA-MB-468 and T47D cells treated with monacolin X and Dox were evaluated with AO/EB dual staining. The stained cells were characterized to viable (light green), early apoptotic (yellow fluorescence and condensed chromatin), late apoptotic (orange fluorescence) and nonviable cells (red-colored fluorescence). In the control, there were only viable cells when compared to the treatment groups which had high numbers of apoptotic cells. Magnification (20×); the experiments were performed in triplicate. | ||
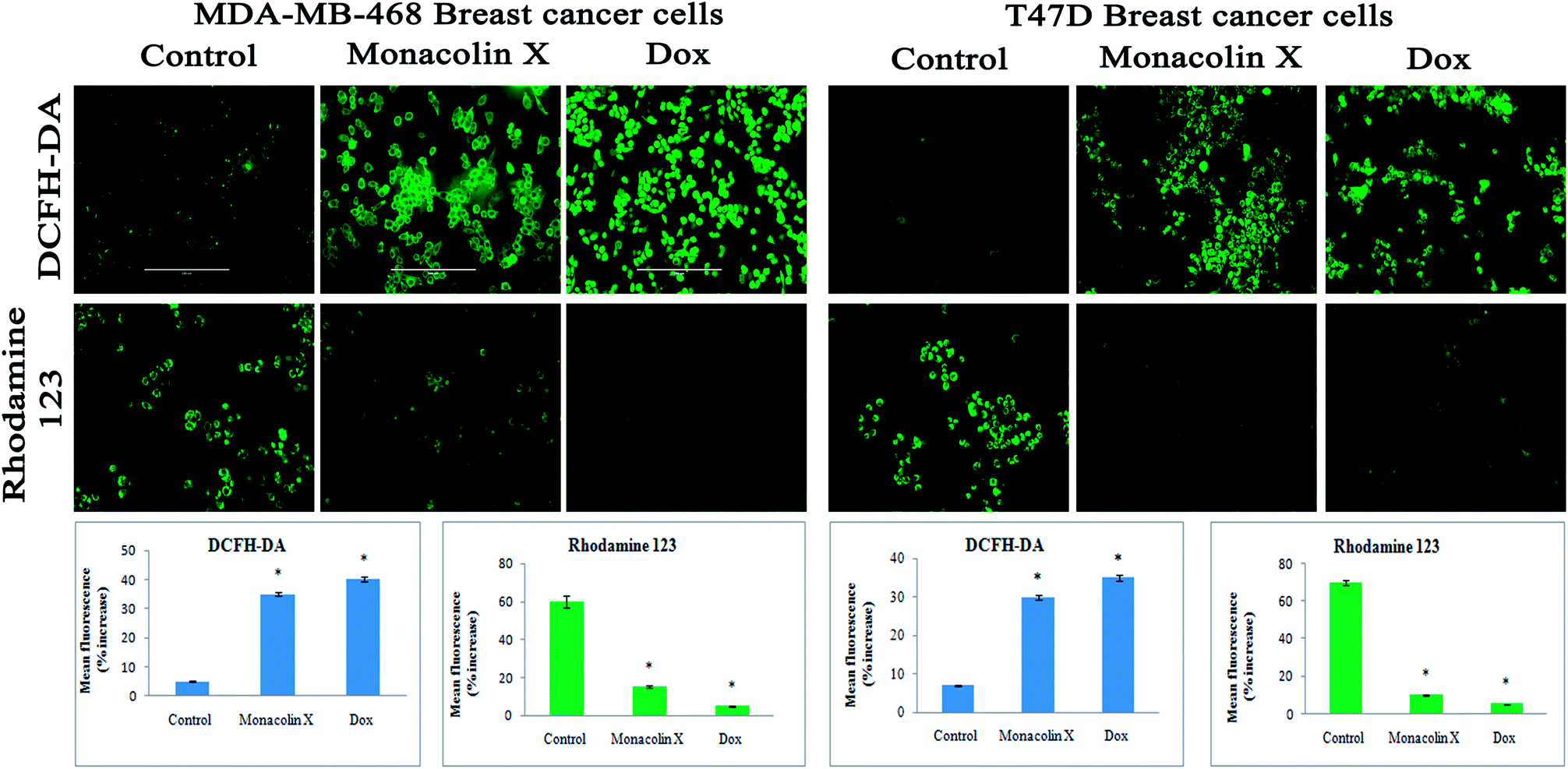 | ||
| Fig. 7 DCFH-DA and Rhodamine-123 staining for ROS and Δψm. The images showed significant ROS generation upon treatment with monacolin X (IC50) and Dox (1.5 μM) treated on MDA-MB-468 and T47D breast cancer cell lines over 24 h. There was a significant difference in the control and treated groups, with high ROS generation in the treated group. Rhodamine-123 staining showed the mitochondrial membrane potential Δψm as decrease fluorescence was observed in monacolin X and Dox, indicating cell death. Magnification (20×); the experiments were performed in triplicate, and the data are expressed as mean ± SD; *p < 0.05, as compared to the control group, was considered as significant. | ||
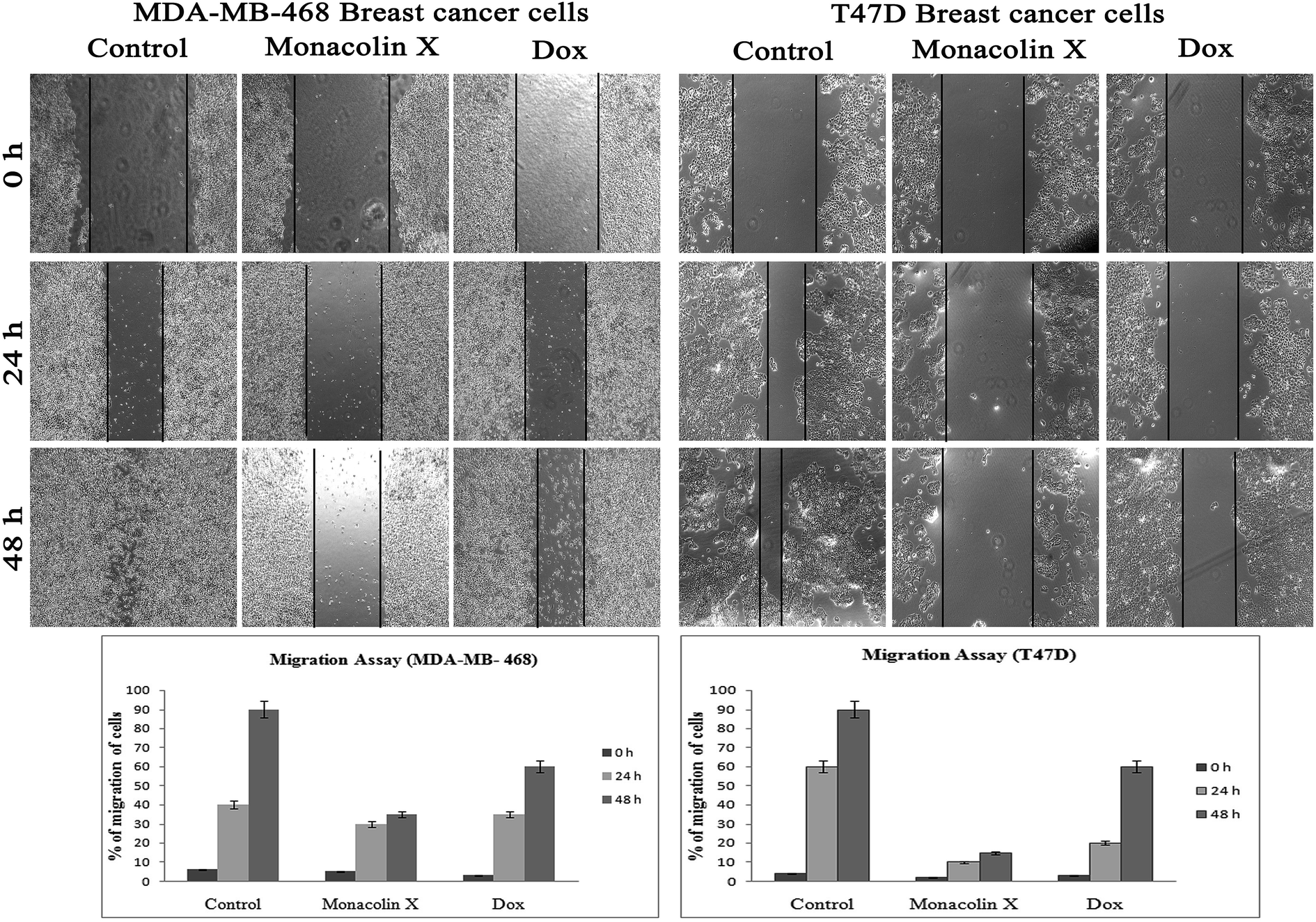 | ||
| Fig. 8 The anti-migratory property of monacolin X (IC50) and Dox (1.5 μM) treated on MDA-MB-468 and T47D breast cancer cell lines for 24 and 48 h. Significant differences between the control groups and treated groups in terms of the number of migrated cells were noted at 24 and 48 h. Treatment I showed fewer migrated cells compared to the control cancer cells. Magnification (4×). Experiments were performed in triplicate and the data were expressed as a mean ± SD; *p < 0.05, as compared to the control group, which was considered as significant. | ||
| Microorganism | MIC (μg mL−1) | |
|---|---|---|
| Monacolin X | Gentamycin | |
| Enterococcus faecalis ATCC 29212 | 38 | 3 |
| Staphylococcus aureus ATCC 25923 | 20 | 2 |
| Pseudomonas aeruginosa ATCC 15442 | 18 | 6 |
| Klebsiella pneumoniae ATCC 13883 | 120 | 3 |
Discussion
Marine-derived fungi synthesize enormous secondary metabolites with complex and unique structures.36 However, very little is known about the biological activity of the extracts and compounds of these fungi. The present work, explored the fungi Monascus sp. NMK7 associated with sponge Clathria frondifera, which secretes the anticancer/antiproliferative compound and polyketide monacolin X. This association of fungi to the host sponge could improve the chemical defense capability. Monascus sp. secretes many secondary metabolites like monascin; ankaflavin; monacolins K, J and L apart from a few other pigments; and polyketides.37 These bioactive compounds have demonstrated their high potential for the development of potent pharmaceutical products. This is the first report on Monascus sp. isolated from the marine sponge in contrast to the previous reports on the terrestrial isolates. Endo et al. reported that Monascus ruber M82121 a mutant strain treated with UV irradiation and N-methyl-N′-nitro-N-nitrosoguanidine, producing monacolin X along with monacolins J and L.31 In this present study, the isolated strain was capable of producing monacolin X without any other treatments or induced mutation. Monacolin X could be produced because of the complex nature of the marine environment.38 Polyketidetrichoharzin is the first metabolite reported from a sponge-associated fungus Trichoderma harzianum Rifai, isolated from the marine sponge Mycale cecilia.39 In particular, sponge-associated fungi have yielded novel metabolites with potent anticancer activities.40 Gymnastatins A, B and C were the first novel cytotoxic metabolites from a strain of Gymnasella dankaliensis, which was isolated from the sponge Halichondria japonica.41In the present study, the purification of the compound was carried out by column chromatography. The active fraction was identified, and further purification was performed by HPLC at 17.2 RRT; the purified compound showed a single peak on the chromatogram fixed PDA at 238 nm. 17.2 RRT elution was concentrated under high vacuum, dried and was subjected to spectral analysis, which revealed the mass of the compound to be 418.2. The peak at m/z = 419 (M + H), the mass spectrum 419.2 [M + H+] and 859.5 [2M + Na+] atomic mass unit (amu), which corresponds to the exact calculated molar mass of Monacolin X, were observed. Thus peaks were observed at m/z = 303.20 (M + H-116), 243.1, 199.1 (M + H-220), 143.0 (M + H-275) and 107.1 (M + H-312). Further, the peak at m/z = 419.2 was further subjected to fragmentation at a collision energy of 20, and peaks were obtained at m/z = 199.3, 173.2, and 159.2. The isolated compound was dissolved in CDCl3 and characterized by 1H and 13C NMR spectroscopy. The 1H-NMR spectrum of the isolated compound suggests the presence of the –COCH3 group and one –COCH (CH3)CO– group; the presence of these groups can be confirmed by locating a singlet of three protons at 2.25 ppm and a multiplet of one proton at 3.50–3.57 ppm. The 13C-NMR spectrum of the isolated compound confirms the carbonyl signal C-3′′ at 204.88 ppm and the corresponding methyne signal C-2′′ at 53.39 ppm. The 4′′-CH3 group carbon was observed at 27.33 ppm. The NMR spectroscopic data determined C24H34O6 to be the molecular formula. Further, the UV spectrum of the compound indicates the peak at 238.2 nm, and FTIR analysis revealed all the functional groups present in the compound. Thus, all this data helped us to predict the structure to be 8-(2-(4-hydroxy-6-oxotetrahydro-2H-pyran-2-yl)ethyl)-3,7-dimethyl-1,2,3,7,8,8a-hexahydronaphthalen-1-yl2-methyl-3-oxobutanoate or monacolin X.31,32
In addition, a parametric approach was used to investigate different hallmarks of the proliferation of cellular components affected by apoptosis and cell viability (AO/EB staining and XTT assay), nuclear activity (PI), the activation of cellular organelles (ROS, Δψm) or the anti-migratory effect. Interestingly, previous reports showed Monascus sp. secondary metabolites and monascorubrin, which inhibits cancer promotion in mice;42 monascin and ankaflavin showed antiproliferation and proapoptosis effects.43,44 It has also been revealed that monacolin K shows a synergistic antitumorigenic activity toward Lewis lung carcinoma cells.45,46 In our study, the cytotoxic activity of monacolin X against MCF-7, T47D, MDA-MB-231, MDA-MB-468 and MCF-10A showed a decrease in the number of viable cells due to an increase of cell death and/or decreased cell proliferation, with an IC50 lower than 50 μM as compared to non-cancerous MCF-10A cells at 170 μM. This indicated decreased cell proliferation in a dose-dependent manner and the induction of cell death through morphological alterations such as cell shrinkage, membrane blebbing, and rounded and detached cells. Apoptosis is an important physiological process for the maintenance of tissue homeostasis and plays a pivotal role in the pathogenesis of various diseases.47 Apoptosis is a programmed cell death, which occurs without eliciting local inflammatory response; in the case of cancer therapy; one of the most important aspects is to induce apoptosis and kill cancer cells via apoptosis induction. Hence, apoptosis was confirmed by AO/EB staining, where the IC50 value was used to evaluate the apoptosis induced by monacolin X. PI staining was used to differentiate necrotic, apoptotic and normal cells. The characteristics of late apoptosis include some loss of membrane integrity and uptake of PI, binding to the nucleotide pair of guanine and cytosine to stain both DNA and RNA and further fluorescence enhancement.48 Monacolin X treatment indicated that more than 40% of cells were dead when compared to the standard control of Dox.
Thus, monacolin X was able to induce nuclear damage and apoptosis, further supporting the role of reactive oxygen species (ROS) and mitochondrial membrane potential (Δψm). Generally, reactive oxygen species (ROS) have a dual role, either beneficial or harmful depending on their levels of accumulation. ROS generally contributes to cell death either by apoptosis or necrosis at the levels beyond the cellular antioxidant defense mechanisms.49 Mitochondria also play a major role in the management of other cellular functions like cell survival and death.50 Hence, for checking for the involvement of mitochondria on treatment with monacolin X – induced cell death, the mitochondrial membrane potential (MMP) was studied using Rhodamine-123, where green fluorescence increased in the treatment group representing the potential difference in the mitochondrial membrane. We concluded from our experimental evidence that nuclear damage leading to apoptosis was possibly mediated by ROS, significantly disrupting the mitochondrial membrane.51 Cell migration in the scratch assay was carried out to demonstrate the anti-migratory property of monacolin X on MDA-MB-468 and T47D cells, showing an anti-migratory effect when compared to standard. Apart from this, monacolin X demonstrated an excellent antibacterial property in both Gram-positive and negative strains, which was confirmed by the MIC method. This indicates monacolin X having a promising range of biological activity.
Conclusion
The present study concludes that this is the first study to isolate monacolin X from wild-type fungi associated with marine sponge collected from the coast of the Gulf of Manner, which has a significant anti-cancer property as it is able to induce apoptosis, increased ROS production and mitochondrial membrane changes. Furthermore, monacolin X exhibits excellent anti-migratory property on both high and low metastatic breast cancer cell lines, also showing antibacterial activity on Gram-positive and Gram-negative bacteria. Thus, the present work emphasizes the role of marine sponge-associated fungi secondary metabolites as an important source of leads for anticancer drug discovery.Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.Authors' contributions
Sirpu Natesh Nagabhishek administered the collection of samples, experiential work, and the interpretation of data, while also carrying out the statistical analysis in addition to studying and drafting the manuscript. Dr A. Madankumar designed the study and drafted the manuscript; both authors read and approved the final manuscript.Conflicts of interest
The authors declare that no conflicts of interest exist.Acknowledgements
The corresponding author is grateful for the grant sanctioned by the Department of Science and Technology, Govt of India under the start-up research grant (young scientist) DST-SERB (SB/YS/LS-101/2014). The authors are very much grateful to the Dr P. Muthuvel, scientist, of Zydus Cadila for helping in predicting the possible structure of the purified compound. The author also thank Dr T. Sasipraba, Pro-vice chancellor, for providing the facility to carry out research work and Mr K. Vishwanathan for the technical help in obtaining SEM images at the Sathyabama Institute of Science and Technology, Chennai. India. The corresponding author is also grateful for the grant sanctioned by Department of Biotechnology, Aquaculture and Marine Biotechnology, Govt of India under the taskforce (BT/PR11664/AAQ/3/682/2014) research grant.References
- S. S. Hasson, H. A.-S. AS, J. Z. Al-Busaidi, M. S. Al-Balushi, F. L. Hakkim, L. Rashan, G. M. Aleemallah and A. A. Al-Jabri, Asian Pac. J. Cancer Prev., 2018, 19, 1917–1925 CAS.
- K. Domvri, P. Zarogoulidis, K. Darwiche, R. F. Browning, Q. Li, J. F. Turner, I. Kioumis, D. Spyratos, K. Porpodis, A. Papaiwannou, T. Tsiouda, L. Freitag and K. Zarogoulidis, J. Cancer, 2013, 4, 736–754 CrossRef CAS PubMed.
- A. Blagodatski, M. Yatsunskaya, V. Mikhailova, V. Tiasto, A. Kagansky and V. L. Katanaev, Oncotarget, 2018, 9, 29259–29274 CrossRef PubMed.
- T. R. Thomas, D. P. Kavlekar and P. A. LokaBharathi, Mar. Drugs, 2010, 8, 1417–1468 CrossRef CAS PubMed.
- S. K. Deshmukh, V. Prakash and N. Ranjan, Front. Microbiol., 2017, 8, 2536 CrossRef PubMed.
- M. S. Butler, A. A. Robertson and M. A. Cooper, Nat. Prod. Rep., 2014, 31, 1612–1661 RSC.
- T. F. Molinski, D. S. Dalisay, S. L. Lievens and J. P. Saludes, Nat. Rev. Drug Discovery, 2009, 8, 69–85 CrossRef CAS PubMed.
- P. Krishnan, M. Balasubramaniam, D. Roy, K. Sarma, R. Hairun and J. Sunde, Advances in Animal and Veterinary Sciences, 2013, 2(1), 20–25 Search PubMed.
- J. W. Blunt, B. R. Copp, R. A. Keyzers, M. H. G. Munro and M. R. Prinsep, Nat. Prod. Rep., 2017, 34, 235–294 RSC.
- J. Bao, Y. L. Sun, X. Y. Zhang, Z. Han, H. C. Gao, F. He, P. Y. Qian and S. H. Qi, J. Antibiot., 2013, 66, 219–223 CrossRef CAS PubMed.
- J. Peng, J. Jiao, J. Li, W. Wang, Q. Gu, T. Zhu and D. Li, Bioorg. Med. Chem. Lett., 2012, 22, 3188–3190 CrossRef CAS PubMed.
- Z. H. Xin, Y. Fang, L. Du, T. Zhu, L. Duan, J. Chen, Q. Q. Gu and W. M. Zhu, J. Nat. Prod., 2007, 70, 853–855 CrossRef CAS PubMed.
- M. W. Taylor, R. Radax, D. Steger and M. Wagner, Microbiol. Mol. Biol. Rev., 2007, 71, 295–347 CrossRef CAS PubMed.
- M. Adnan, E. Alshammari, M. Patel, S. Amir Ashraf, S. Khan and S. Hadi, PeerJ, 2018, 6, 5049 CrossRef PubMed.
- A. H. Aly, A. Debbab, J. Kjer and P. Proksch, Microb. Biotechnol., 2010, 41, 1–16 Search PubMed.
- R. D. Firn and C. G. Jones, Mol. Microbiol., 2000, 37, 989–994 CrossRef CAS PubMed.
- A. Stierle, G. Strobel and D. Stierle, Science, 1993, 260, 214–216 CrossRef CAS PubMed.
- T. Amna, S. C. Puri, V. Verma, J. P. Sharma, R. K. Khajuria, J. Musarrat, M. Spiteller and G. N. Qazi, Can. J. Microbiol., 2006, 52, 189–196 CrossRef CAS PubMed.
- G. Hendrik, E. Ietidal and P. Alexander, Phytochem. Rev., 2010, 9(4), 537–545 CrossRef.
- S. N. Nagabhishek, A. Madankumar and K. Gayathri, Mol. Biol. Rep., 2018, 45(6), 2641–2651 CrossRef PubMed.
- N. A. Hooper, Sponguide guide to sponge collection and identification, Australia, 2000 Search PubMed.
- T. J. White, T. Bruns, S. Lee and J. Talor, Amplification and Direct Sequencing of Fungal Ribosomal RNA Genes for Phylogenetics, Academic Press Inc., New York, 1990, pp. 315–322 Search PubMed.
- Z. Wu, X. R. Wang and G. Blomquist, J. Environ. Monit., 2002, 4, 377–382 RSC.
- S. Kumar, G. Stecher and K. Tamura, Mol. Biol. Evol., 2016, 33, 1870–1874 CrossRef CAS PubMed.
- A. A. Alshatwi, P. Subash-Babu and P. Antonisamy, Exp. Toxicol. Pathol., 2016, 68, 89–97 CrossRef CAS PubMed.
- M. Leite, M. Quinta-Costa, P. S. Leite and J. E. Guimaraes, Anal. Cell. Pathol., 1999, 19, 139–151 CrossRef CAS PubMed.
- F. Warleta, C. S. Quesada, M. Campos, Y. Allouche, G. Beltran and J. J. Gaforio, Nutrients, 2011, 3, 839–857 CrossRef CAS PubMed.
- S. M. Chacko, K. G. Nevin, R. Dhanyakrishnan and B. P. Kumar, Toxicol. Rep., 2015, 2, 1213–1221 CrossRef CAS PubMed.
- M. Karlstetter, E. Lippe, Y. Walczak, C. Moehle, A. Aslanidis, M. Mirza and T. Langmann, J. Neuroinflammation, 2011, 8, 125 CrossRef CAS PubMed.
- J. N. Eloff, J. Ethnopharmacol., 1998, 60, 1–8 CrossRef CAS PubMed.
- A. Endo, K. Hasumi, T. Nakamura, M. Kunishima and M. Masuda, J. Antibiot., 1985, 38, 321–327 CrossRef CAS PubMed.
- C. Belwal, P. K. Goyal, A. Balte, S. Kolhe, K. Chauhan, A. S. Rawat and A. Vardhan, Sci. Pharm., 2014, 82, 43–52 CrossRef CAS PubMed.
- B. N. Prashanth, R. Shashi, D. Kaushik, S. Aditya, D. ubhasis, M. Abhijit and M. Mahitosh, BMC Cancer, 2013, 13, 273 CrossRef PubMed.
- T. Afsar, J. H. Trembley, C. E. Salomon, S. Razak, M. R. Khan and K. Ahmed, Sci. Rep., 2016, 6, 23077 CrossRef CAS PubMed.
- J. E. Stine, H. Guo, X. Sheng, X. Han, M. N. Schointuch, T. P. Gilliam, P. A. Gehrig, C. Zhou and V. L. Bae-Jump, Oncotarget, 2016, 7, 946–960 CrossRef PubMed.
- A. Debbab, A. H. Aly, W. H. Lin and P. Proksch, Microb. Biotechnol., 2010, 3, 544–563 CrossRef CAS PubMed.
- L. Martinkova, P. Patakova-Juzlova, V. Krent, Z. Kucerova, V. Havlicek, P. Olsovsky, O. Hovorka, B. Rihova, D. Vesely, D. Vesela, J. Ulrichova and V. Prikrylova, Food Addit. Contam., 1999, 16, 15–24 CrossRef CAS PubMed.
- T. Y. Lung, L. Y. Liao, J. J. Wang, B. L. Wei, P. Y. Huang and C. L. Lee, Mar. Drugs, 2016, 14(6), 106 CrossRef PubMed.
- K. Motomasa, U. Hideto, M. Katsuyoshi and K. isao, Tetrahedron Lett., 1993, 34(49), 7925–7928 CrossRef.
- P. R. Jensen and W. Fenical, Drugs Sea, 2000, 6–29 CAS.
- T. Amagata, M. Tanaka, T. Yamada, K. Minoura and A. Numata, J. Nat. Prod., 2008, 71, 340–345 CrossRef CAS PubMed.
- K. Yasukawa, M. Takahashi, S. Natori, K. Kawai, M. Yamazaki, M. Takeuchi and M. Takido, Oncology, 1994, 51, 108–112 CrossRef CAS PubMed.
- T. Akihisa, H. Tokuda, M. Ukiya, A. Kiyota, K. Yasukawa, N. Sakamoto, Y. Kimura, T. Suzuki, J. Takayasu and H. Nishino, Chem. Biodiversity, 2005, 2, 1305–1309 CrossRef CAS PubMed.
- N. W. Su, Y. L. Lin, M. H. Lee and C. Y. Ho, J. Agric. Food Chem., 2005, 53, 1949–1954 CrossRef CAS PubMed.
- C. I. Lee, C. L. Lee, J. F. Hwang, Y. H. Lee and J. J. Wang, Appl. Microbiol. Biotechnol., 2013, 97, 1269–1278 CrossRef CAS PubMed.
- B. Y. Ho and T. M. Pan, J. Agric. Food Chem., 2009, 57, 8258–8265 CrossRef CAS PubMed.
- M. Agostini, P. Tucci and G. Melino, Biochem. Biophys. Res. Commun., 2011, 414, 451–455 CrossRef CAS PubMed.
- S. L. Fink and B. T. Cookson, Infect. Immun., 2005, 73, 1907–1916 CrossRef CAS PubMed.
- W. Fiers, R. Beyaert, W. Declercq and P. Vandenabeele, Oncogene, 1999, 18, 7719–7730 CrossRef CAS PubMed.
- L. Galluzzi, I. Vitale, J. M. Abrams, E. S. Alnemri, E. H. Baehrecke, M. V. Blagosklonny, T. M. Dawson, V. L. Dawson, W. S. El-Deiry, S. Fulda, E. Gottlieb, D. R. Green, M. O. Hengartner, O. Kepp, R. A. Knight, S. Kumar, S. A. Lipton, X. Lu, F. Madeo, W. Malorni, P. Mehlen, G. Nunez, M. E. Peter, M. Piacentini, D. C. Rubinsztein, Y. Shi, H. U. Simon, P. Vandenabeele, E. White, J. Yuan, B. Zhivotovsky, G. Melino and G. Kroemer, Cell Death Differ., 2012, 19, 107–120 CrossRef CAS PubMed.
- M. L. Circu and T. Y. Aw, Free Radical Biol. Med., 2010, 48, 749–762 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra09886g |
| This journal is © The Royal Society of Chemistry 2019 |