
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnalysis of energies of halogen and hydrogen bonding interactions in the solid state structures of vanadyl Schiff base complexes†
Snehasish Thakura,
Michael G. B. Drewb,
Antonio Franconetti c,
Antonio Frontera*c and
Shouvik Chattopadhyay*a
c,
Antonio Frontera*c and
Shouvik Chattopadhyay*a
aDepartment of Chemistry, Inorganic Section, Jadavpur University, Kolkata 700 032, India. E-mail: shouvik.chem@gmail.com
bSchool of Chemistry, The University of Reading, P. O. Box 224, Whiteknights, Reading RG6 6AD, UK
cDepartament de Química, Universitat de les Illes Balears, Crta. De Valldemossa km 7.5, 07122 Palma, Baleares, Spain. E-mail: toni.frontera@uib.es
First published on 6th February 2019
Abstract
Two mononuclear and two dinuclear vanadium(V) complexes, [VO2L1] (1), [VO2L2] (2), (μ-O)2[V(O)(L3)]2 (3) and (μ-O)2[V(O)(L4)]2·2H2O (4), where HL1 = 4-bromo-6-[(2-phenylaminoethylimino)methyl]phenol, HL2 = 2-((2-(diethylamino)ethylimino)methyl)-4-chlorophenol, HL3 = 2-((2-(ethylamino)ethylimino)methyl)-4-chlorophenol and HL4 = 2-(1-(2-(ethylamino)ethylimino)ethyl)phenol have been synthesized and characterized. Structures of all complexes have been confirmed by single crystal X-ray diffraction studies. Complexes 1, 2, and 3 exhibit significant halogen bonding interactions in their solid state structures. The energies associated to the supramolecular interactions have been explored using Density Functional Theory (DFT) calculations, and further confirmed with non-covalent interaction (NCI) plots.
1. Introduction
Supramolecular chemistry is enriched by the participation of carbon-bound halogen atoms (acting as electron acceptors), forming short contacts with both neutral and negatively charged species, having abilities to act as electron donors.1 This type of supramolecular interaction is commonly known as ‘halogen bonding’ to indicate their similarity with hydrogen bonding interactions.1b,1e These non-covalent interactions have been shown to have significant contribution in controlling the aggregation of different organic molecules in solid,2 liquid,3 and gases.4 Supramolecular chemists are very much interested in analyzing the nature of these halogen bonding interactions.5 Although hydrogen bonding interactions are the most frequently used means to assemble molecules in solid, liquid, and gas phases, and play important roles in stabilizing supramolecular aggregates even in water,6 the literature shows examples where ‘halogen bonding’ interactions may dominate over the hydrogen bonding interactions, depending upon the type of substituents and orientation of the responsible atoms with respect to the electron donor groups.7 However, there is no report in the literature regarding the analysis of halogen bonding interactions in the supramolecular systems generated by vanadyl Schiff base complex building blocks.Recently, we have reported the synthesis and characterization of few vanadyl Schiff base complexes along with the analysis of supramolecular interactions in their solid state structures.8 Supramolecular interactions in those complexes were mainly governed by C–H⋯π, π⋯π (chelate ring) and hydrogen bonding interactions. In the present work, we have designed the Schiff bases in such a way that supramolecular systems could be generated with the help of halogen bonding interactions along with conventional hydrogen bonding interactions. Four tridentate N2O donor Schiff bases, HL1 (4-bromo-6-[(2-phenylaminoethylimino)methyl]phenol), HL2 2-((2-(diethylamino)ethylimino)methyl)-4-chlorophenol, HL3 2-((2-(ethylamino)ethylimino)methyl)-4-chlorophenol and HL4 2-(1-(2-(ethylamino)ethylimino)ethyl)phenol have been used to prepare four complexes, [VO2L1] (1), [VO2L2] (2), (μ-O)2[V(O)(L3)]2 (3) and (μ-O)2[V(O)(L4)]2·2H2O (4). Significant halogen bonding interactions have been observed in the solid state structures of complexes 1, 2 and 3. The energies involved in these interactions have been examined using DFT calculations, and further confirmed with NCI plots. The findings are impressive and hopefully encouraging for coordination as well as supramolecular chemists working in related areas.
2. Experimental section
VOSO4·5H2O was purchased from Loba Chemie Pvt. Ltd. and was of reagent grade. All other starting materials were commercially available, reagent grade, and used as purchased from Sigma-Aldrich without further purification.2.1 Preparation
[VO2L1] (1). Yield: 289 mg (72%); anal. calc. for C15H14BrN2O3V (FW = 401.12) C, 44.91; H, 3.52; N, 6.98%. Found: C, 44.8; H, 3.4; N, 7.1%; FT-IR (KBr, cm−1): 3077(νN–H), 1630(νC
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 925, 825(νVO), UV-Vis, λmax (nm), [εmax (L mol−1 cm−1)] (DMF), 334 (4.1 × 103). Magnetic moment = diamagnetic.
N), 925, 825(νVO), UV-Vis, λmax (nm), [εmax (L mol−1 cm−1)] (DMF), 334 (4.1 × 103). Magnetic moment = diamagnetic.
[VO2L2] (2). Yield: 229 mg (68%); anal. calc. for C13H18ClN2O3V (FW = 336.68): C, 46.37; H, 5.39; N, 8.32%. Found: C, 46.3; H, 5.2; N, 8.4%; FT-IR (KBr, cm−1): 1627 (νC
N), 935, 819 (νVO), UV-Vis, λmax (nm), [εmax (L mol−1 cm−1)] (CH3CN): 331 (3.8 × 103). Magnetic moment = diamagnetic.
(μ-O)2[V(O)(L3)]2 (3). Yield: 219 mg (71%); anal. calc. for C22H28Cl2N4O6V2 (FW = 617.26): C, 42.81; H, 4.57; N, 9.08%; Found: C, 42.7; H, 4.4; N, 9.1%; FT-IR (KBr, cm−1): 1641 (νC
N), 929, 836 (νVO). UV-Vis, λmax (nm), [εmax (L mol−1 cm−1)] (CH3CN): 382 (6.8 × 103). Magnetic moment = diamagnetic.
(μ-O)2[V(O)(L4)]2·2H2O (4). Yield: 211 mg (69%); anal. calc. for C24H38N4O8V2 (FW = 612.46): C, 47.07; H, 6.25; N, 9.15%; Found: C, 47.0; H, 6.1; N, 9.2%; FT-IR (KBr, cm−1): 3212 (νN–H), 1606 (νC
N), 930 (νVO). 464 (νV–O), UV-Vis, λmax (nm), [εmax (L mol−1 cm−1)] (CH3CN): 352 (7.0 × 103). Magnetic moment = diamagnetic.
2.2 Physical measurements
Elemental analyses (carbon, hydrogen and nitrogen) were performed using a Perkin Elmer 240C elemental analyzer. IR spectra in KBr (4500–500 cm−1) were recorded with a Perkin Elmer Spectrum Two spectrophotometer. Electronic spectrum of complex 1 (in DMF) and that of complexes 2–4 (in CH3CN) were recorded on a Shimadzu UV-1700 UV-Vis spectrophotometer. The magnetic susceptibility measurements were performed with an EG and PAR vibrating sample magnetometer, model 155 at room temperature (300 K) in a 5000 G magnetic field, and diamagnetic corrections were performed using Pascal's constants.2.3 X-ray crystallography
For each complex, a suitable single crystal was picked and mounted on a glass fiber, and diffraction intensities were measured with an Oxford Diffraction X-Calibur diffractometer, equipped with Mo Kα radiation (λ = 0.71073 Å, 50 kV, 40 mA) at temperature 150 K. Data collection and reduction were performed with the Oxford Diffraction Crystalis system. Structures of the complexes were solved by direct method and refined by full-matrix least squares on F2, using the SHELX-2016/6 package.9 The absorption correction type is empirical with ABSPACK program of Oxford Diffraction Ltd. Non-hydrogen atoms were refined anisotropically. Hydrogen atoms attached to nitrogen and oxygen atoms were located by difference Fourier maps. All other hydrogen atoms were placed in their geometrically idealized positions and constrained to ride on their parent atoms. Programs used: SHELX-2016/6,9 PLATON,10 WINGX,11 ORTEP,12 and MERCURY.13 Significant crystallographic data are summarized in Table 1. Selected bond lengths and bond angles are gathered in Tables 2 and 3, respectively.| Complex | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| Formula | C15H14BrN2O3V | C13H18ClN2O3V | C22H28Cl2N4O6V2 | C24H38N4O8V2 |
| Formula weight | 401.12 | 336.68 | 617.26 | 612.46 |
| Temperature (K) | 150 | 150 | 150 | 150 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Monoclinic |
| Space group | Pc | P21/n | I2/a | P21/n |
| a (Å) | 12.7168(14) | 14.1152(7) | 22.194(2) | 8.9266(4) |
| b (Å) | 6.1962(5) | 6.9353(3) | 7.5879(7) | 6.9648(3) |
| c (Å) | 9.6410(11) | 15.1466(7) | 15.5112(12) | 21.9606(12) |
| α (deg) | 90 | 90 | 90 | 90 |
| β (deg) | 91.74(1) | 106.842(5) | 97.479(10) | 96.547(4) |
| γ (deg) | 90 | 90 | 90 | 90 |
| Z | 2 | 4 | 4 | 2 |
| dcalc (g cm−3) | 1.754 | 1.576 | 1.583 | 1.500 |
| μ (mm−1) | 3.297 | 0.895 | 0.973 | 0.743 |
| F(000) | 400 | 696 | 1264 | 640 |
| Total reflections | 4316 | 9618 | 7844 | 8667 |
| Unique reflections | 3000 | 4096 | 3726 | 3873 |
| Observed data [I > 2σ (I)] | 2678 | 3498 | 3036 | 3256 |
| No. of parameters | 199 | 183 | 167 | 182 |
| R(int) | 0.053 | 0.030 | 0.030 | 0.035 |
| R1, wR2 (all data) | 0.0693, 0.1446 | 0.0464, 0.0947 | 0.0595, 0.1075 | 0.0604, 0.1072 |
| R1, wR2 [I > 2σ (I)] | 0.0609, 0.1345 | 0.0377, 0.0911 | 0.0443, 0.1015 | 0.0476, 0.1091 |
| Complex | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| a Symmetry transformations: a = 1 − x, 2 − y, −z, b = −x, 1 − y, −z. | ||||
| V(1)–O(1) | 1.628(7) | 1.6228(13) | 1.6159(14) | 1.6812(15) |
| V(1)–O(2) | 1.625(8) | 1.6215(14) | 1.6773(15) | 1.6100(15) |
| V(1)–O(11) | 1.903(8) | 1.9160(13) | 1.9150(15) | 1.9033(16) |
| V(1)–N(19) | 2.118(8) | 2.1424(14) | 2.1637(17) | 2.1703(18) |
| V(1)–N(22) | 2.159(9) | 2.2021(14) | 2.1554(18) | 2.1300(18) |
| V(1)–O(2a) | — | — | 2.3096(15) | — |
| V(1)–O(1b) | — | — | — | 2.4008(15) |
| Complex | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| a Symmetry transformations: a = 1 − x, 2 − y, −z, b = −x, 1 − y, −z. | ||||
| O(1)–V(1)–O(2) | 110.4(4) | 110.67(7) | 106.99(7) | 107.49(7) |
| O(1)–V(1)–O(11) | 96.1(3) | 102.95(6) | 101.45(7) | 97.02(7) |
| O(1)–V(1)–N(19) | 137.4(4) | 113.96(6) | 97.03(7) | 152.91(7) |
| O(1)–V(1)–N(22) | 90.1(3) | 93.96(6) | 91.50(7) | 92.46(7) |
| O(2)–V(1)–O(11) | 102.8(4) | 96.96(6) | 99.22(7) | 102.64(7) |
| O(2)–V(1)–N(19) | 111.1(3) | 134.29(6) | 154.28(7) | 98.56(7) |
| O(2)–V(1)–N(22) | 96.0(4) | 90.68(6) | 92.65(7) | 93.06(7) |
| O(11)–V(1)–N(19) | 83.8(3) | 82.50(5) | 84.56(7) | 84.16(6) |
| O(11)–V(1)–N(22) | 156.7(3) | 157.52(5) | 158.96(6) | 158.22(6) |
| N(19)–V(1)–N(22) | 76.6(3) | 77.09(5) | 77.38(6) | 78.58(6) |
| O(1)–V(1)–O(1b) | — | — | — | 77.94(6) |
| O(1b)–V(1)–O(2) | — | — | — | 170.29(7) |
| O(1b)–V(1)–O(11) | — | — | — | 84.36(6) |
| O(1b)–V(1)–N(19) | — | — | — | 75.24(5) |
| O(1b)–V(1)–N(22) | — | — | — | 78.49(6) |
| O(1)–V(1)–O(2a) | — | — | 169.79(7) | — |
| O(2)–V(1)–O(2a) | — | — | 78.78(6) | — |
| O(2a)–V(1)–O(11) | — | — | 85.63(6) | — |
| O(2a)–V(1)–N(19) | — | — | 76.15(6) | — |
| O(2a)–V(1)–N(22) | — | — | 79.68(6) | — |
2.4 Theoretical methods
The geometries of the complexes included in this study were computed at the M06-2X/def2-TZVP level of theory using the crystallographic coordinates. Therefore, the interaction energies were calculated using the frozen monomers as they are in the dimers. For all calculations, the GAUSSIAN-09 program has been used.14 The Grimme's dispersion correction15 has also been used as implemented in GAUSSIAN-09 program since it is adequate for the evaluation of non-covalent interactions where dispersion effects are relevant like σ-hole interactions. The basis set superposition error for the calculation of interaction energies has been corrected using the counterpoise method.16 The NCI plot17 isosurfaces have been used to characterize non-covalent interactions. They correspond to both favorable and unfavorable interactions, as differentiated by the sign of the second density Hessian eigenvalue and defined by the isosurface color. The color scheme is a red-yellow-green-blue scale with red for ρcut+ (repulsive) and blue for ρcut− (attractive). Molecular Electrostatic Potential surfaces have been computed at the M06-2X/6-31+G* level using the SPARTAN'10 program version 1.1.0. The SAPT (symmetry adapted perturbation theory)18 has been used to decompose the interaction energy into well-defined components (electrostatic, exchange, induction, and dispersion terms). The DFT-SAPT calculations have been performed using the PBE0/def2-TZVP level of theory and the MOLPRO program.193. Results and discussions
3.1 Synthesis
Four Schiff bases, HL1, HL2, HL3 and HL4, have been prepared by the condensation of appropriate diamines and salicylaldehyde derivatives in CH3CN, following the literature methods.20 The ligands were not isolated and were reacted with VOSO4·5H2O (dissolved either in DMF or CH3CN) in open atmosphere to synthesize two mononuclear vanadium(V) complexes, [VO2L1] (1) and [VO2L2] (2) and two dinuclear vanadium(V) complexes, (μ-O)2[V(O)(L3)]2 (3) and (μ-O)2[V(O)(L4)]2·2H2O (4), where L1, L2, L3 and L4 are the deprotonated mono-anionic forms of the respective Schiff bases. The effort to prepare these complexes under N2 environment was unsuccessful. This is because all the syntheses involves the oxidation of vanadium from +4 (in starting material) to +5 (in complexes 1–4). The oxidation of vanadium centers may therefore be caused by aerial oxygen. Synthetic route to the complexes is outlined in Scheme 1.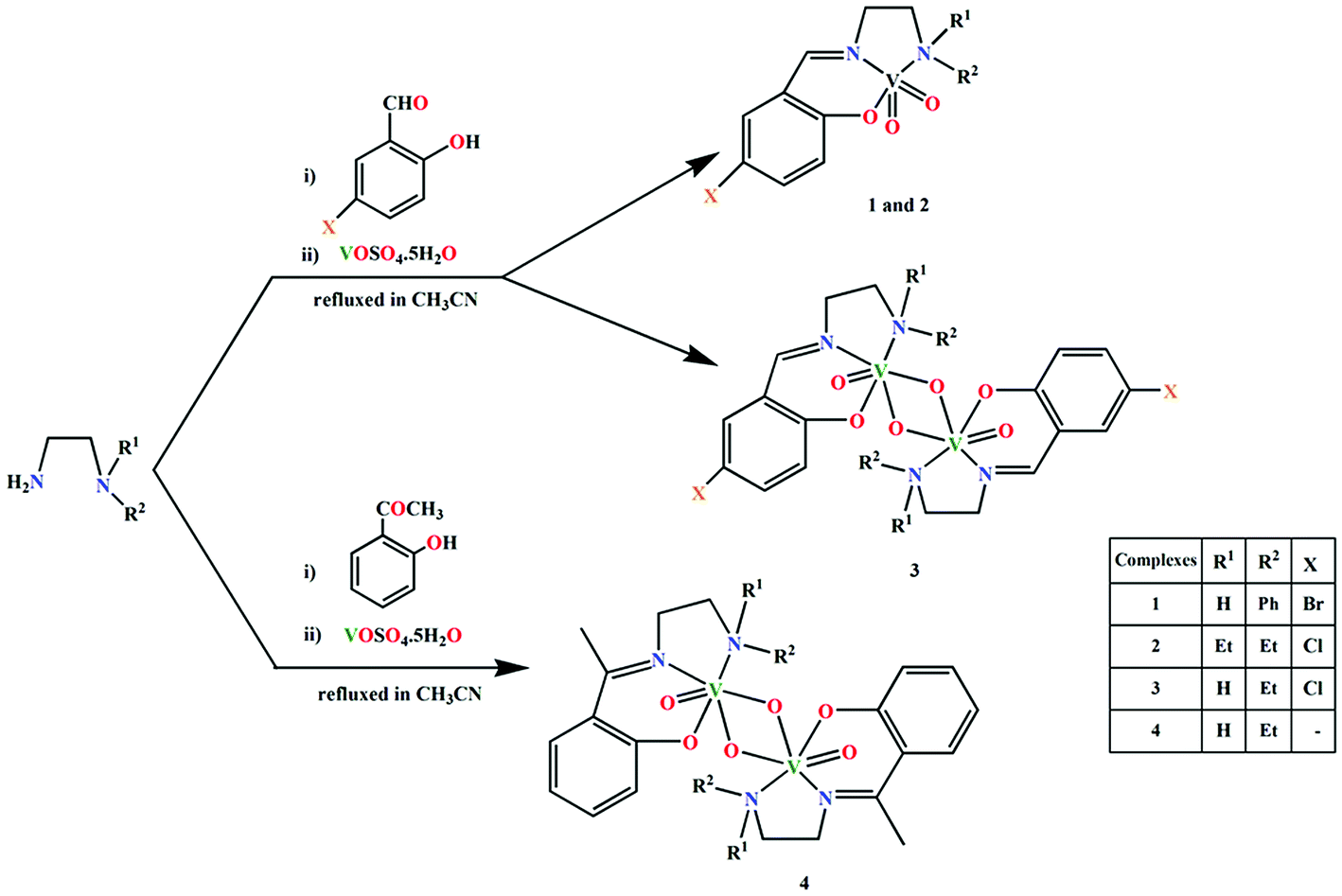 | ||
| Scheme 1 Synthetic route to complexes 1–4. | ||
3.2 Description of structures
O double bonds.23 The bond distances and angles in both complexes are similar to those observed in analogous vanadium(V) complexes with Schiff bases.24 Saturated five membered chelate ring, V(1)–N(19)–C(20)–C(21)–N(22), assumes a half-chair conformation with puckering parameters q = 0.411(12) Å and φ = 282.1(11)° in complex 1, and an envelope conformation with puckering parameters q = 0.4482(17) Å and φ = 104.78(17)° in complex 2.
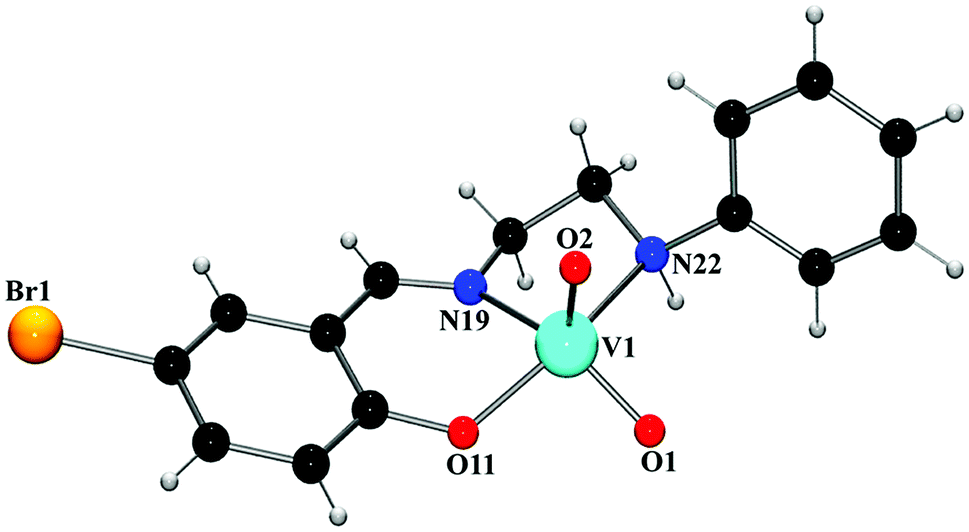 | ||
| Fig. 1 Perspective view of complex 1 with selective atom numbering scheme. | ||
O double bond.25 The V–O and V–N bond lengths in the complexes are comparable to the corresponding values observed in other similar oxovanadium(V) complexes with Schiff bases.24a,26 The chelating angles, N(19)–V(1)–N(22) and N(19)–V(1)–O(22) have the values in the range 78–84°, indicating distortion of the octahedron around vanadium(V) centers.27 The intramolecular V⋯V separations are 3.107(5) Å (complex 3) and 3.206(5) Å (complex 4), which fall within the range of known V⋯V distances in double-bridged vanadium polynuclear systems.28 Depending on the orientation of VO groups with respect to the plane through two vanadium centers and two bridging oxo-groups, the {V2O4}2+ core can have five different configurations (syn and anti-orthogonal, syn- and anti-coplanar, twist) in complexes constructed from two cis-VO2+(pervanadyl) units.29 Both these complexes may be termed as anti-coplanar, as the two terminal VO bonds in both complexes are oriented in an anti arrangement with respect to the {V2O2} plane. The coordination environment may then be best described as two edge-shared vanadium octahedra that are significantly distorted. The assignment of the +5 oxidation state for the vanadium centers in both complexes is confirmed by the bond valence sum calculations,30 which give 4.946 and 4.943 valence units for vanadium centers in complexes 3 and 4, respectively.
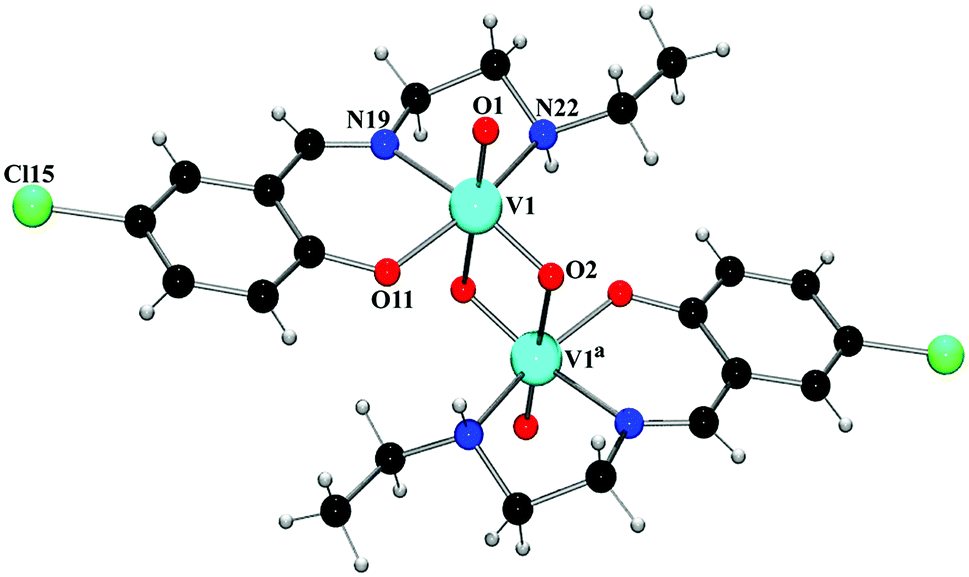 | ||
| Fig. 2 Perspective view of complex 3 with selective atom numbering scheme. Symmetry transformation: a = 1 − x, 2 − y, −z. | ||
3.3 Theoretical studies on supramolecular interactions in complexes 1, 2 and 3
Four new Schiff base complexes of VO2+ have been synthesized and characterized by X-ray crystallography. Some of them contain halogen atoms in the structure (Br or Cl), that participate in halogen bonding or hydrogen bonding interactions. The theoretical study is devoted to analyze the energy associated to the halogen bonding or hydrogen bonding interactions involving the halogen atoms in complexes 1, 2 and 3.In Fig. 3a, a partial view of the X-ray structure of 1 is presented, where an infinite supramolecular chain is formed that is governed by the formation of Br⋯π interactions and where the π-system corresponds to a CC bond of the aromatic ring. First of all, the molecular electrostatic potential (MEP) surface in complex 1 has been computed and it reveals an anisotropic distribution around the Br atom (it is represented only for the Br atom in Fig. 3a, right). The MEP value at the σ-hole of Br is +8 kcal mol−1 and at the belt is −15 kcal mol−1. A dimer, retrieved from the infinite chain, has been computed and the formation energy is moderately strong, ΔE1 = −4.3 kcal mol−1. The DFT-SAPT computed for this dimer shows that the interaction is dominated by induction and dispersion effects that represent the 75% of the sum of the attractive terms followed by the electrostatic term. The ‘non-covalent interaction plot’ (NCI plot) index has also been computed in order to characterize the Br⋯π interaction in the dimer of 1. The NCI plot is an intuitive visualization index that enables the identification of non-covalent interactions easily and efficiently. This plot is convenient to analyze host–guest interactions since it clearly shows which molecular regions interact. The colour scheme is a red-yellow-green-blue scale with red (repulsive) and blue (attractive). Yellow and green surfaces correspond to weak repulsive and weak attractive interactions, respectively. The representation of the NCI plot is shown in Fig. 3c. As noted, the halogen bond is characterized by a green isosurface that covers the aromatic CC bond and thus confirming the existence of the Br⋯π interaction.
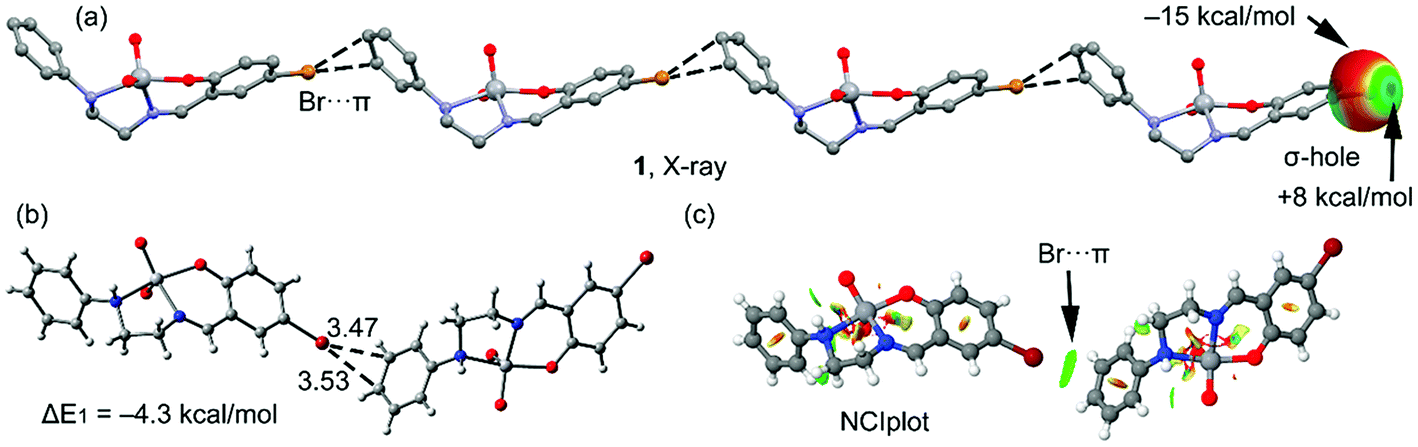 | ||
| Fig. 3 (a) One dimensional supramolecular chain observed in the X-ray solid state structure of 1 and the MEP surface (isodensity = 0.001 a.u.) around the Br atom. Hydrogen atoms have been omitted for clarity. (b) Theoretical model used to evaluate the interaction energy. (c) NCI surface of the dimer of complex 1. The gradient cut-off is s = 0.35 a.u., and the colour scale is −0.04 < ρ < 0.04 a.u. | ||
A partial view of the X-ray structure of 2 is given in Fig. 4a, where an infinite one dimensional supramolecular chain is represented. In this supramolecular assembly, the halogen bonding is established between the Cl atom and one O atom of the VO2+ moiety, which is a better electron donor compared to the π-system. The MEP surface of complex 2 also shows an anisotropic distribution around the Cl atom; however, the MEP value at the σ-hole of Cl is very small, i.e. +2 kcal mol−1 and at the belt is −17 kcal mol−1. A dimer, retrieved from the infinite chain, has been computed and the formation energy is similar to that computed above for complex 1 (ΔE2 = −4.6 kcal mol−1, Fig. 4b), which is likely due to a compensating effect (worse σ-hole donor and better σ-hole acceptor compared to complex 1). The DFT-SAPT in this case shows that the interaction is dominated by electrostatic effects that represent the 55% of the sum of the attractive terms followed by the induction and dispersion terms (45%). The NCI plot index has also been calculated in order to characterize the halogen bonding interaction in the dimer of 2 (Fig. 4c). The halogen bond is characterized by a small green isosurface that is located between the Cl and O atoms. Moreover, an additional green isosurface is located between the aliphatic hydrogen atoms of one methyl group and the negative belt at the Cl atoms, thus also contributing to the stabilization of the dimer and also explaining the larger binding energy obtained for 2 compared to 1.
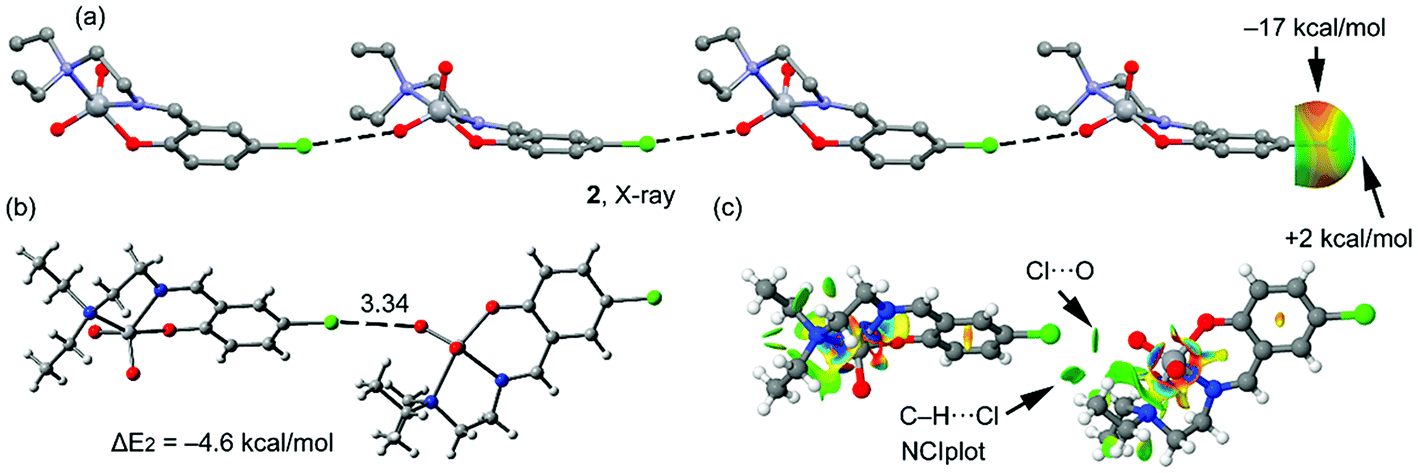 | ||
| Fig. 4 (a) One dimensional supramolecular chain observed in the X-ray solid state structure of 2 and the MEP surface (isodensity = 0.001 a.u.) around the Cl atom. Hydrogen atoms have been omitted for clarity. (b) Dimer retrieved from the crystal structure of complex 2. Distances in Å. (c) NCI surface of the dimer of complex 2. The gradient cut-off is s = 0.35 a.u., and the color scale is −0.04 < ρ < 0.04 a.u. | ||
Complex 3 also has a chlorine atom as substituent; however, it does not participate in halogen bonding interactions. Instead, it establishes several C–H⋯Cl interactions. In fact, its solid state structure (Fig. 5a) shows that each dinuclear complex establishes four sets of C–H⋯Cl interactions: two as donor and two as acceptors, generating a two dimensional sheet. The interaction energy of the hydrogen bonded dimer is modest, ΔE3 = −3.7 kcal mol−1 and smaller than the halogen bonding computed for 1 and 2, thus suggesting that the halogen bond is favoured in these vanadium systems. The DFT-SAPT computed for this interaction shows that it is dominated by electrostatic effects that represent the 75% of the sum of the attractive terms.
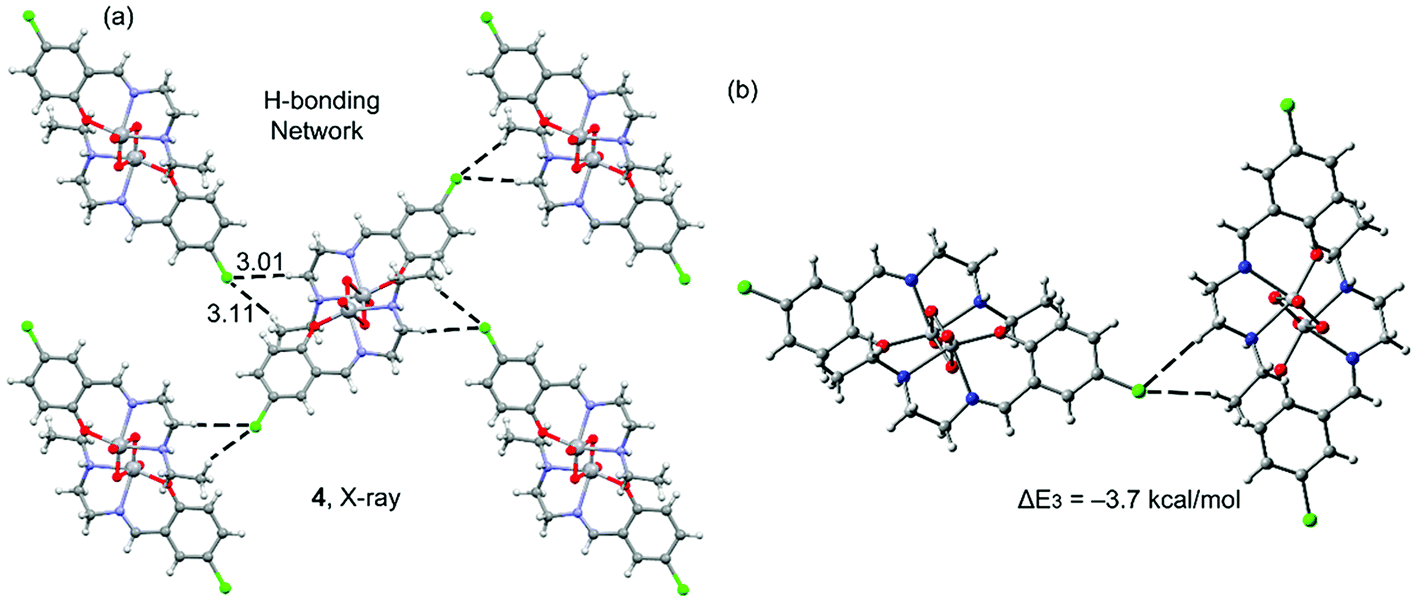 | ||
| Fig. 5 Partial view of the X-ray solid state structure of 3 (a) and the theoretical model (b). Distances in Å. | ||
3.4 IR, electronic spectra and magnetic moments
Strong and sharp bands due to azomethine (CN) groups in the range 1600–1640 cm−1 have been customarily noticed in the IR spectra of these complexes.31 Strong bands around 930 cm−1 and 825 cm−1 in all complexes may be assigned to asymmetric and symmetric ν(OVO) vibrations of cis-VO2 groups present.32 The ν(V–O) bands can be monitored around 460 cm−1 for all complexes.33 These values are in good agreement with similar vanadium(V) Schiff base complexes. Sharp bands around 3310 cm−1 may be assigned to ν(N–H) stretching vibrations in complexes 3 and 4.34 Bands in the range of 2998–2806 cm−1 due to alkyl C–H bond stretching vibrations are customarily noticed in the IR spectra of all the complexes.35
Electronic spectrum of each complex shows an absorption band around 350 nm. This band may be assigned as a ligand-to-metal charge transfer (LMCT) transition, originating from the pπ orbital on the phenolate oxygen to the empty d orbitals of the vanadium(V).36 As all these vanadium(V) complexes has a 3d0 configuration, d–d bands are not expected to appear in their electronic spectra.37
All these complexes are diamagnetic as expected for mononuclear and dinuclear vanadium(V) complexes with d0 configuration, as also observed for other similar vanadium(V) complexes.38
4. Conclusion
Four new VO2+ complexes with Schiff base ligands have been prepared and structurally characterized. Supramolecular interactions were investigated in details. An infinite one dimensional supramolecular chain is formed in complex 1, governed by the formation of Br⋯π interactions (where the π-system corresponds to a CC bond of the aromatic ring). A dimer, retrieved from the infinite chain, has been computed and the formation energy is moderately strong, ΔE1 = −4.3 kcal mol−1. In the supramolecular assembly of complex 2, the halogen bonding is established between the Cl atom and one O atom of the VO2+ moiety, which is a better electron donor compared to the π-system. A dimer, retrieved from the infinite chain, has been computed and the formation energy is similar to that computed for complex 1 (ΔE2 = −4.6 kcal mol−1), which is likely due to a compensating effect (worse σ-hole donor and better σ-hole acceptor compared to complex 1). Complex 3 has a chlorine atom in the ligand part; however, it does not participate in halogen bonding interactions. Instead, it establishes several C–H⋯Cl interactions. In fact, its solid state structure shows that each dinuclear complex establishes four sets of C–H⋯Cl interactions: two as donor and two as acceptors, generating a two dimensional sheet. The interaction energy of the hydrogen bonded dimer is modest, ΔE3 = −3.7 kcal mol−1 and smaller than the halogen bonding computed for 1 and 2, thus suggesting that the halogen bond is favoured in these vanadium systems. All these results were further corroborated with the molecular electrostatic potential (MEP) surface calculation and NCI plot index computational tool.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
S. T. thanks the DST, India, for awarding a Junior Research Fellowship (IF160359). A. Frontera and A. Franconetti gratefully acknowledge the financial support of this work by the MINECO of Spain (project CTQ2017-85821-R, FEDER funds). In addition, AF thanks the MINECO/AEI from Spain for a “Juan de la Cierva” contract. They thank the CTI (UIB) for free allocation of computer time. M. G. B. D. thanks the University of Reading and the EPSRC (U. K.) for funds for the diffractometer.Notes and references
- (a) J. P. M. Lommerse, A. J. Stone, R. Taylor and F. H. Allen, J. Am. Chem. Soc., 1996, 118, 3108–3116 CrossRef CAS; (b) P. Politzer, P. Lane, M. C. Concha, Y. Ma and J. S. Murray, J. Mol. Model., 2007, 13, 305–311 CrossRef CAS PubMed; (c) P. Metrangolo, F. Meyer, T. Pilati, G. Resnati and G. Terraneo, Angew. Chem., Int. Ed., 2008, 47, 6114–6127 CrossRef CAS PubMed; (d) P. Politzer, J. S. Murray and M. C. Concha, J. Mol. Model., 2007, 13, 643–650 CrossRef CAS PubMed; (e) P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2010, 12, 7748–7757 RSC.
- (a) A. Farina, S. V. Meille, M. T. Messina, P. Metrangolo, G. Resnati and G. Vecchio, Angew. Chem., Int. Ed., 1999, 38, 2433–2436 CrossRef CAS; (b) T. Caronna, R. Liantonio, T. A. Logothetis, P. Metrangolo, T. Pilati and G. Resnati, J. Am. Chem. Soc., 2004, 126, 4500–4501 CrossRef CAS PubMed.
- (a) M. T. Messina, P. Metrangolo, W. Panzeri, E. Ragg and G. Resnati, Tetrahedron Lett., 1998, 39, 9069–9072 CrossRef CAS; (b) H. L. Nguyen, P. N. Horton, M. B. Hursthouse, A. C. Legon and D. W. Bruce, J. Am. Chem. Soc., 2004, 126, 16–17 CrossRef CAS PubMed.
- (a) O. Dumele, B. Schreib, U. Warzok, N. Trapp, C. A. Schalley and F. Diederich, Angew. Chem., Int. Ed., 2017, 56, 1152–1157 CrossRef CAS PubMed; (b) A. C. Legon, Angew. Chem., 1999, 111, 2850–2880 CrossRef; (c) A. C. Legon, Chem.–Eur. J., 1998, 4, 1890–1897 CrossRef CAS.
- (a) A. M. Maharramova, N. Q. Shikhaliyeva, G. T. Suleymanovaa, A. V. Gurbanova, G. V. Babayevaa, G. Z. Mammadovaa, F. I. Zubkovc, V. G. Nenajdenkod, K. T. Mahmudov and A. J. L. Pombeiro, Dyes Pigm., 2018, 159, 135–141 CrossRef; (b) D. Franchinia, F. Dapiaggia, S. Pieraccinia, A. Fornib and M. Sironi, Chem. Phys. Lett., 2018, 712, 89–94 CrossRef; (c) J. M. Guevara-Vela, D. Ochoa-Resendiz, A. Costales, R. Hernández-Lamoneda and A. M. Pendás, ChemPhysChem, 2018, 19, 2512–2517 CrossRef CAS PubMed.
- (a) L. Brunsveld, J. A. J. M. Vekemans, J. H. K. K. Hirschberg, R. P. Sijbesma and E. W. Meijer, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4977–4982 CrossRef CAS PubMed; (b) C. M. A. Leenders, L. Albertazzi, T. Mes, M. M. E. Koenigs, A. R. A. Palmans and E. W. Meijer, Chem. Commun., 2013, 49, 1963–1965 RSC; (c) M. Henry, ChemPhysChem, 2002, 3, 607–616 CrossRef CAS PubMed; (d) C. Schmuck and W. Wienand, Angew. Chem., Int. Ed., 2001, 40, 4363–4369 CrossRef CAS; (e) F. Wang, M. A. J. Gillissen, P. J. M. Stals, A. R. A. Palmans and E. W. Meijer, Chem.–Eur. J., 2012, 18, 11761–11770 CrossRef CAS PubMed; (f) T. Kato, N. Mizoshita and K. Kanie, Macromol. Rapid Commun., 2001, 22, 797–814 CrossRef CAS; (g) C. M. Paleos and D. Tsiourvas, Liq. Cryst., 2001, 28, 1127–1161 CrossRef CAS; (h) F. C. Gozzo, L. S. Santos, R. Augusti, C. S. Consorti, J. Dupont and M. N. Eberlin, Chem.–Eur. J., 2004, 10, 6187–6193 CrossRef CAS PubMed.
- (a) P. Metrangolo and G. Resnati, Chem.–Eur. J., 2001, 7, 2511–2519 CrossRef CAS PubMed; (b) E. Corradi, S. V. Meille, M. T. Messina and G. Resnati, Angew. Chem., Int. Ed., 2000, 39, 1782–1786 CrossRef CAS; (c) C. B. Aakeröy, M. Fasulo, N. Schultheiss, J. Desper and C. Moore, J. Am. Chem. Soc., 2007, 129, 13772–13773 CrossRef PubMed; (d) G. C. Cole, A. C. Legon and P. Ottaviani, J. Chem. Phys., 2002, 117, 2790 CrossRef CAS.
- (a) S. Thakur, S. Roy, A. Bauzá, A. Frontera and S. Chattopadhyay, Inorg. Chim. Acta, 2017, 467, 212–220 CrossRef CAS; (b) S. Thakur, N. Sarkar, M. G. B. Drew, A. Bauzá, A. Frontera and S. Chattopadhyay, Polyhedron, 2018, 142, 83–92 CrossRef CAS.
- (a) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed; (b) G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- A. L. Spek, Acta Crystallogr., 2009, D65, 148–155 CrossRef PubMed.
- J. L. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef.
- M. N. Burnett and C. K. Johnson, ORTEP-3: Oak Ridge Thermal Ellipsoid Plot Program for Crystal Structure Illustrations, Report ORNL-6895, Oak Ridge National Laboratory, Oak Ridge, TN, USA, 1996 Search PubMed.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. R. Monge, R. Taylor, J. V. D. Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef PubMed.
- B. Jeziorski, R. Moszynski and K. Szalewicz, Chem. Rev., 1994, 94, 1887–1930 CrossRef CAS.
- MOLPRO, A package of ab initio programs, version 2010.1; Molpro, Cardiff, U.K., 2010 Search PubMed.
- (a) L. Sacconi and I. Bertini, Inorg. Chem., 1966, 5, 1520–1522 CrossRef CAS; (b) M. S. Shongwe, U. A. Al-Zaabi, F. Al-Mjeni, C. S. Eribal, E. Sinn, I. A. Al-Omari, H. H. Hamdeh, D. Matoga, H. Adams, M. J. Morris, A. L. Rheingold, E. Bill and D. J. Sellmyer, Inorg. Chem., 2012, 51, 8241–8253 CrossRef CAS PubMed; (c) Y.-Q. Cui, Z.-L. You, X.-F. Li, X.-L. Wang, Y.-P. Ma, C. Wang and C.-L. Zhang, Transition Met. Chem., 2010, 35, 159–163 CrossRef CAS.
- A. W. Addison, T. N. Rao, J. Reedijk, J. V. Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349 RSC.
- C. R. Cornman, K. M. Geiser-Bush, S. P. Rowley and P. D. Boyle, Inorg. Chem., 1997, 36, 6401–6408 CrossRef CAS.
- G. Asgedom, A. Sreedhara, J. Kivikoski, E. Kolehmainen and C. P. Rao, J. Chem. Soc., Dalton Trans., 1996, 93–97 RSC.
- (a) L. M. Mokry and C. J. Carrano, Inorg. Chem., 1993, 32, 6119–6121 CrossRef CAS; (b) E. Kwiatkowski, G. Romanowski, W. Nowicki, M. Kwiatkowski and K. Suwińska, Polyhedron, 2003, 22, 1009–1018 CrossRef CAS.
- (a) G. Romanowski, E. Kwiatkowski, W. Nowicki, M. Kwiatkowski and T. Lis, Polyhedron, 2008, 27, 1601–1609 CrossRef CAS; (b) A. Sarkar and S. Pal, Polyhedron, 2007, 26, 1205–1210 CrossRef CAS.
- (a) C. A. Duncan, E. P. Copeland, I. A. Kahwa, A. Quick and D. J. Williams, J. Chem. Soc., Dalton Trans., 1997, 6, 917–919 RSC; (b) S. Mondal, M. Mukherjee, K. Dhara, S. Ghosh, J. Ratha, P. Banerjee and A. K. Mukherjee, Cryst. Growth Des., 2007, 7, 1716–1721 CrossRef CAS.
- Z.-L. You, D.-H. Shi, J.-C. Zhang, Y.-P. Maa, C. Wang and K. Li, Inorg. Chim. Acta, 2012, 384, 54–61 CrossRef CAS.
- (a) C. A. Root, J. D. Hoeschele, C. R. Cornman, J. W. Kampf and V. L. Pecoraro, Inorg. Chem., 1993, 32, 3855–3861 CrossRef CAS; (b) S. Pal and S. Pal, J. Chem. Crystallogr., 2000, 30, 329–333 CrossRef CAS.
- (a) Y. Zhang, R. C. Haushalter and J. Zubieta, Inorg. Chim. Acta, 1997, 260, 105–110 CrossRef CAS; (b) W. Plass, Angew. Chem., Int. Ed. Engl., 1996, 35, 627–631 CrossRef CAS.
- (a) R. Codd, T. W. Hambley and P. A. Lay, Inorg. Chem., 1995, 34, 877–882 CrossRef CAS; (b) H. H. Thorp, Inorg. Chem., 1992, 31, 1585–1588 CrossRef CAS.
- (a) P. Bhowmik, S. Chattopadhyay, M. G. B. Drew, C. Diaz and A. Ghosh, Polyhedron, 2010, 29, 2637–2642 CrossRef CAS; (b) P. Bhowmik, H. P. Nayek, M. Corbella, N. Aliaga-Alcalde and S. Chattopadhyay, Dalton Trans., 2011, 40, 7916–7926 RSC; (c) F. H. Suydam, Anal. Chem., 1963, 35, 193–195 CrossRef CAS; (d) S. Chattopadhyay, M. S. Ray, S. Chaudhuri, G. Mukhopadhyay, G. Bocelli, A. Cantoni and A. Ghosh, Inorg. Chim. Acta, 2006, 359, 1367–1375 CrossRef CAS.
- (a) M. R. Maurya, A. Kumar, M. Abid and A. Azam, Inorg. Chim. Acta, 2006, 359, 2439–2447 CrossRef CAS; (b) E. Kwiatkowski, G. Romanowski, W. Nowicki, M. Kwiatkowski and K. Suwińska, Polyhedron, 2007, 26, 2559–2568 CrossRef CAS.
- X. Wang, X. M. Zhang and H. X. Liu, Inorg. Chim. Acta, 1994, 223, 193–197 CrossRef CAS.
- (a) S. Rayati, N. Sadeghzadeh and H. R. Khavasi, Inorg. Chem. Commun., 2007, 10, 1545–1548 CrossRef CAS; (b) G. F. Svatos, C. Curran and J. V. Quagliano, J. Am. Chem. Soc., 1955, 77, 6159–6163 CrossRef CAS.
- G. Grivani, A. D. Khalaji, V. Tahmasebi, K. Gotoh and H. Ishida, Polyhedron, 2012, 31, 265–271 CrossRef CAS.
- (a) R. Dinda, P. Sengupta, S. Ghosh and T. C. W. Mak, Inorg. Chem., 2002, 41, 1684–1688 CrossRef CAS PubMed; (b) G. Asgedom, A. Sreedhara, J. Kivikoski, E. Kolehmainen and C. P. Rao, J. Chem. Soc., Dalton Trans., 1996, 93–97 RSC.
- M. R. Maurya, A. Kumar, A. R. Bhat, A. Azam, C. Bader and D. Rehder, Inorg. Chem., 2006, 45, 1260–1269 CrossRef CAS PubMed.
- (a) J. Dai, S. Akiyama, M. Munakata and M. Mikuriya, Polyhedron, 1994, 13, 2495–2499 CrossRef CAS; (b) T. Ghosh, B. Mondal, T. Ghosh, M. Sutradhar, G. Mukherjee and M. G. B. Drew, Inorg. Chim. Acta, 2007, 360, 1753–1761 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1879191–1879194 contain the supplementary crystallographic data for complexes 1–4 respectively. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra09947b |
| This journal is © The Royal Society of Chemistry 2019 |