
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModulation of aggregation with an electric field; scientific roadmap for a potential non-invasive therapy against tauopathies†
Gaurav Pandeya,
Sudhir Morlaa,
Harshal B. Nemadeb,
Sachin Kumara and
Vibin Ramakrishnan *a
*a
aDepartment of Biosciences and Bioengineering, Indian Institute of Technology Guwahati, Guwahati-781039, India. E-mail: vibin@iitg.ac.in; Fax: +91 361 258 2249; Tel: +91 361 258 2227
bDepartment of Electronics and Electrical Engineering, Indian Institute of Technology Guwahati, Guwahati-781039, India
First published on 6th February 2019
Abstract
Toxic aggregation of tau protein to neurofibrillary tangles (NFTS) is a central pathological event involved in tauopathies. Inhibition of tau protein aggregation can serve as a straightforward therapeutic strategy. However, tau-based therapeutic solutions are not very common. Phenothiazine methylene blue (tau protein inhibitor) is currently the only drug under phase III clinical trials. In this work, a non-invasive strategy is presented for modulating the aggregation of core peptide segments of tau protein (VQIVYK and VQIINK) by using electric fields of varying strengths. We use thioflavin T staining, tyrosine fluorescence assay, electron microscopy, IR, dynamic and static light scattering, and neuronal toxicity estimation, for verifying the effect of electric field on the aggregation kinetics, morphology, conformational state and cellular toxicity of peptide systems. Our observations suggest that electric field arrests the self-assembly of VQIVYK and VQIINK fibrils thereby reducing the neurotoxicity instigated by them. Based on our observations, we propose a prospective scheme for a futuristic non-invasive therapeutic device.
Introduction
Amyloid fibrils are naturally occurring nano-fibrils, associated with a large number of human diseases such as Alzheimer's disease (AD), Parkinson's disease, and type II diabetes mellitus. The phenomenon of amyloidogenesis involves the conversion of normally soluble peptides or proteins into insoluble amyloid plaques which eventually lead to progressive loss of neurons from specific regions of the brain.AD is the most prevalent neurological disorder and affects approximately 50 million people worldwide.1 Although this number is expected to increase to 132 million by 2050, there is no clinically approved therapy to cure or retard its progression.1 Promising therapeutic strategies under research include native state stabilisation or fibrillation inhibition using small molecules,2–6 immunotherapy,7,8 peptide or peptidomimetics,9–18 nanoparticles,19,20 transient metals,21–24 supramolecular inhibition of fibrillation25,26 and sequestering the monomeric form of the peptide.27
Nucleation-dependent aggregation is one of the most accepted models for the formation of amyloid fibrils.28–31 As per this model, the process of fibrillogenesis involves a slow nucleation phase, wherein the monomeric peptide misfolds and undergoes self-assembly to form an oligomer. These oligomers serve as “nuclei” and are rich in β-sheets. The slow (rate-limiting) nucleation phase succeeds a rapid growth phase, which involves self-association of oligomers to yield protofibrils, and they further self-assemble to form mature amyloid fibrils.
The small molecule-based therapeutic strategies to modulate aggregation have shown promise. Some have inhibited fibril formation, while others have arrested fibrillar growth. Nonetheless, they show poor ability to penetrate the blood–brain barrier and are often highly toxic. Non-invasive therapies involving the application of scanning ultrasound,32,33 light,34 transcranial stimulation by magnetism and current,35,36 etc. have also been found effective. Accumulation of misfolded proteins, beta-amyloid (Aβ) and hyperphosphorylated tau, which aggregate and form amyloid plaques and tau tangles are regarded as the putative cause for AD onset. Although Aβ has conventionally been considered as the major malefactor, the investigations in recent years have hinted a shift towards the crucial role, soluble oligomers and neurofibrillary tangles of tau play in the AD disease condition.37,38 The instigation of tau protein aggregation, linked with over 20 neurological disorders known as tauopathies, has been ascribed to the presence of two hexapeptide motifs “Ac-VQIVYK-am (PHF6) and Ac-VQIINK-am (PHF6*)”.16 These motifs are found in R2 and R3 repeat domains of tau protein and have been reported to be critical for conferring the strong aggregation tendency to tau protein.39 Therefore, these peptide segments are regarded as the ideal model for assessing the potential therapies for the AD.40 Here we report an externally applied electric field (EF) induced deceleration of aggregation kinetics of core peptide segments of tau protein VQIVYK and VQIINK.
Several theoretical studies have already reported the potential ability of the electric field, to induce conformational changes in the protein/peptide conformation by modulating the dipole alignments.41–45 Molecular dynamics-based studies in the presence of electric field have predicted that a constant electric field could induce a conformational switch from a β-sheet into an α-helix through a global reordering of the dipole moments at the amide planes.46 The sheet–helix transitions mediated by external electric fields have also been reported to disaggregate amyloid fibrils.47
Several recent experimental findings have also confirmed the possibility of electric field mediated fibrillar disruption.48,49 Porchke et al. reported a cooperative change in the structure of bacteriorhodopsin, induced at a remarkably low field strength of 150 V cm−1.50 We have already tested the effect of the electric field mediated modulation on the core recognition motif of the β-amyloid polypeptide, diphenylalanine (LPhe–LPhe, FF). FF is also the shortest molecule known to self-assemble into ordered nanostructures.51 These findings stimulated our interest towards experimenting further the impact of the electric field upon core peptide segments of tau protein.
Materials and methods
Peptide synthesis and characterization
The core tau peptide sequences Ac-VQIVYK-am and Ac-VQIINK-am sequence were synthesized by solid phase peptide synthesis using rink amide resin (Novabiochem) and standard Fmoc chemistry. The synthesized peptide was cleaved from the resin and deprotected using a mixture containing 82.5% trifluoroacetic acid, 5% m-cresol, 5% H2O, 5% thioanisole, and 2.5% ethanedithiol for 12 h at room temperature. The peptide was precipitated in ice-cold diethyl ether. The peptide was dissolved in deionized water and purified on Shimadzu Prominence HPLC system using C-18 reversed phase column. The purified peptide was characterized using MALDI-TOF mass spectrometry in the Laser Desorption positive mode, using α-cyano-4-hydroxycinnamic acid matrix on a MALDI-TOF mass spectrometer (Bruker Autoflex Speed) at the Central Instruments Facility, Indian Institute of Technology Guwahati, India.Monomerization of peptide
The peptide was monomerized by an overnight pretreatment with hexafluoroisopropanol (HFIP), and the solvent was evaporated by air drying/using a speed vac.Pre-aggregation assays
Electric field experiment
The peptide solution was exposed to electric field environment immediately after the addition of Milli-Q water, allowing it to aggregate in the presence of an electric field. No voltage was applied in the case of the control experiment. The following electric field conditions were employed:(i) 150 V cm−1 AC and DC.
(ii) 300 V cm−1 AC and DC.
(iii) 0 V cm−1 (control).
Aggregation kinetics evaluation by thioflavin T fluorescence assay
Thioflavin T (ThT) is a benzothiazole dye obtained from the methylation of dehydrothiotoluidine with sulfonic acid. ThT is used to stain amyloid plaques, or visualize and quantify the presence of fibrillation of misfolded protein aggregates, both in vitro and in vivo. Thioflavin T binds to amyloid fibrils, not to monomer peptides and gives an enhanced fluorescence emission.52 The enhanced fluorescence can be observed by fluorescence microscopy or by fluorescent spectroscopy. To assess the rate of aggregation ThT binding assay was performed by combining 15 μL, 500 μM peptide to a solution of 10 μM ThT in 50 mM sodium phosphate buffer. Fluorescence spectra were recorded on a spectrofluorometer (Jasco FP 8500) at 25 °C using a 1 cm path length quartz cuvette (Helma, Sigma Aldrich). The excitation wavelength was set at 450 nm (slit width = 2.5 nm), and a time scan emission was monitored at 485 nm (slit width = 5 nm).Tyrosine absorbance assay
The tyrosine absorbance was recorded at 280 nm, to determine the soluble concentration of PhF6 peptide in respective samples. Post 16 h, the electric field was turned off, peptide samples were centrifuged at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 20 min, and the supernatant was collected, and its absorbance was measured at 280 nm.
000g for 20 min, and the supernatant was collected, and its absorbance was measured at 280 nm.
Dynamic light scattering (DLS)
To determine the size distribution of aggregates formed in control, as well as electric field, exposed samples, dynamic light scattering analysis, was done, same as described above, after an incubation of 16 hours and 6 hours for PhF6 and PhF6* respectively.Static right angle light scattering assay
To compare the extent of aggregation in the electric field exposed sample to the control sample a right angle scatter was recorded, same as described above, after an incubation of 16 hours and 6 hours for PhF6 and PhF6* respectively.Cell viability assay
MTT assays were performed to assess the cytotoxicity of PhF6 and PhF6* aggregates using neuroblastoma SH-SY5Y, IMR-32 neuronal cell lines and PC-12, rat pheochromocytoma cell lines procured from the repository of the National Centre for Cell Science (NCCS) Pune, India. The SH-SY5Y, IMR-32, and PC-12 cells were maintained in DMEM/F12, DMEM, and RPMI 1640, respectively. All the cells were supplemented with 10% FBS, 100 U mL−1 penicillin and 100 U mL−1 streptomycin at 37 °C under 5% CO2. Prior to the MTT assays, each of the cells (80 μL) were cultured in 96-well plates at a density of 5 × 103 cells per well for 24 h in a CO2 cell culture box (Forma SteriCycle CO2 Incubator 371, Thermo Scientific, USA). Then, the cells were treated with 20 μL control as well as field exposed peptide samples (25 μM) and incubated for another 24 h. After that, 10 μL MTT solutions (5.5 mg mL−1) were added into each well. After 4 h incubation, the medium was replaced with 100 μL DMSO to dissolve the formazan crystals. Then, absorbance at 570 nm was measured by a multifunctional microplate reader (TECAN Austria GmbH, Grödig, Austria). Six replicates were performed, and the data were averaged.Field emission transmission electron microscopy (FETEM) analysis
Peptide sample (20 μL) were drop cast onto a carbon-coated copper mesh grids immediately after taking out from the field after completion of their incubation period and were negatively stained using 2% (w/v) saturated uranyl acetate. After 30 seconds, excess stain was wicked away, and the grids were allowed to air dry. Images were captured with a JEM-2100F field emission transmission electron microscope (JEOL, Tokyo, Japan) at 200 kV.Fourier-transform infrared spectroscopy (FTIR)
Both control as well as field-exposed samples of both PhF6 and PhF6* peptide were cast on potassium bromide (KBr) pellet immediately after taking out from the field. FTIR spectra were recorded on a Perkin-Elmer Model: Spectrum Two IR spectrometer at 1 cm−1 resolution between 1200 cm−1 to 2000 cm−1 (amide 1). The background scan was subtracted from the sample scans, and text files were plotted using OriginPro 8 software.Results and discussion
To test our hypothesis, we synthesised the model peptides (Ac-VQIVYK-am and Ac-VQIINK-am), purified using high-performance liquid chromatography (HPLC) and verified its purity using MALDI-TOF mass spectrometry (ESI, Fig. S1–S4†). We performed an initial aggregation kinetics assay for each of the peptides to determine the time duration of our experiments (ESI, Fig. S5 and S6†). Based on their aggregation kinetics profile, we allowed PhF6 and PhF6* to aggregate for 16 hours and 6 hours respectively, in the presence and absence of alternating and direct current (AC and DC) electric fields of varying strengths by placing them in AC/DC field set-up (Fig. 1).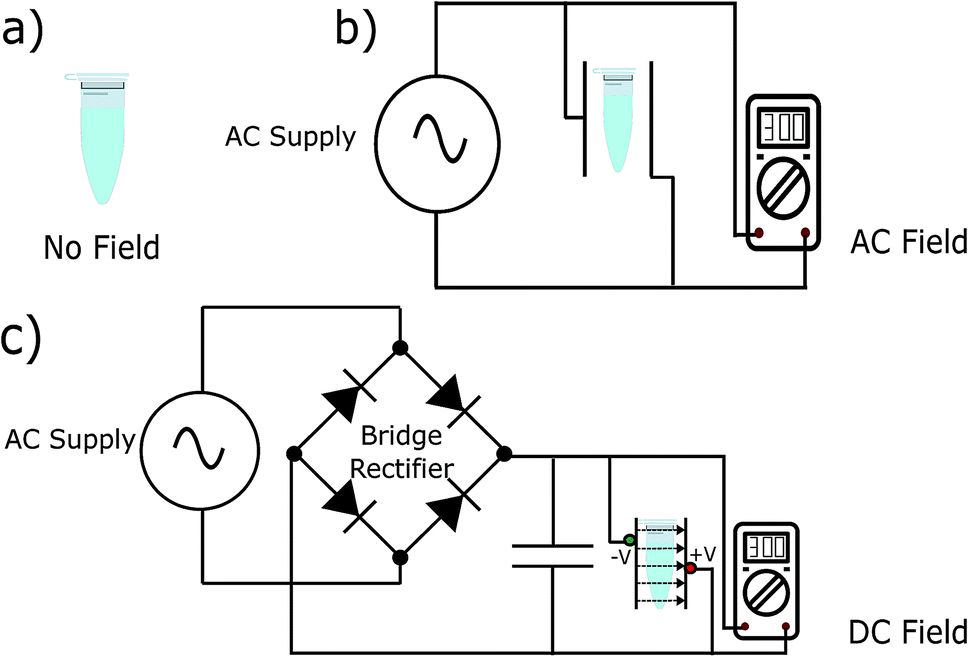 | ||
| Fig. 1 Schematic illustration of the experimental setup. DC electric field setup comprises of horizontal plates connected to a full wave bridge rectifier circuit. Voltage is regulated using an autotransformer. AC field setup is similar to a DC set-up but without a bridge rectifier. Dissolved peptides were allowed to aggregate in (a) ambient conditions (0 V cm−1) and the presence of (b) AC and (c) DC fields. | ||
The kinetics of the amyloid formation was examined by a time-dependent thioflavin T (ThT) fluorescence assay. Thioflavin T is a dye that binds to amyloid fibrils and gives an enhanced fluorescence emission upon binding.48 The fluorescence intensity of PhF6 and PhF6* peptide increased with time, showing a typical sigmoidal curve, in the absence of an electric field. On the contrary, the peptide samples allowed to aggregate in the presence of electric field (both AC and DC) showed 6-fold lesser fluorescence intensity compared to the control, suggesting delayed aggregation kinetics (Fig. 2(a) and 3(a)), ESI, Tables (S1 and S2†). To compare quantitatively, we also examined the extent of aggregation by tyrosine absorbance assay. Each of the respective PhF6 samples was centrifuged at 12000g for 20 minutes, and then the tyrosine absorbance of the supernatant was recorded. The absorbance will be inversely proportional to the magnitude of aggregation. We observed that electric field exposed samples showed higher tyrosine absorbance, and hence less percentage aggregation compared to the control sample (ESI, Fig. S7†). The ThT and tyrosine absorbance data clearly show the retarding effect of the electric field. Several theoretical studies have reported the modulatory effect of electric field on the peptide-based amyloid forming systems.46,47 It has been proposed that electric field forced the peptide dipole to align parallel to its direction, thus modulating the self-assembly. Our experimental observations comply with the earlier theoretical reports, conducted by two different groups headed by Martin E. Garcia and Andrij Baumketner.
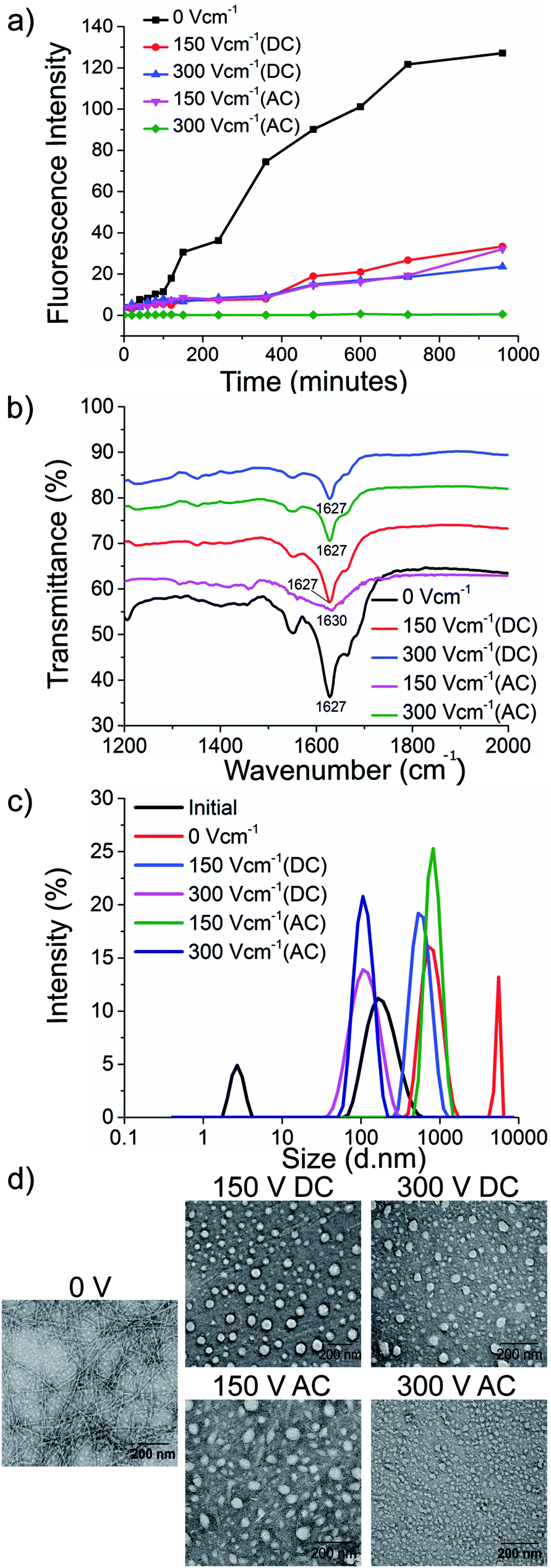 | ||
| Fig. 2 Retardation of PhF6 aggregation by the electric field. Fibrillation kinetics of PhF6 was examined by (a) ThT fluorescence emission, (b) secondary structure determined by FTIR spectroscopy, (c) size estimation using dynamic light scattering (DLS) (d) TEM micrographs. All samples were allowed to aggregate in the presence and absence of DC and AC electric field of varying strengths. | ||
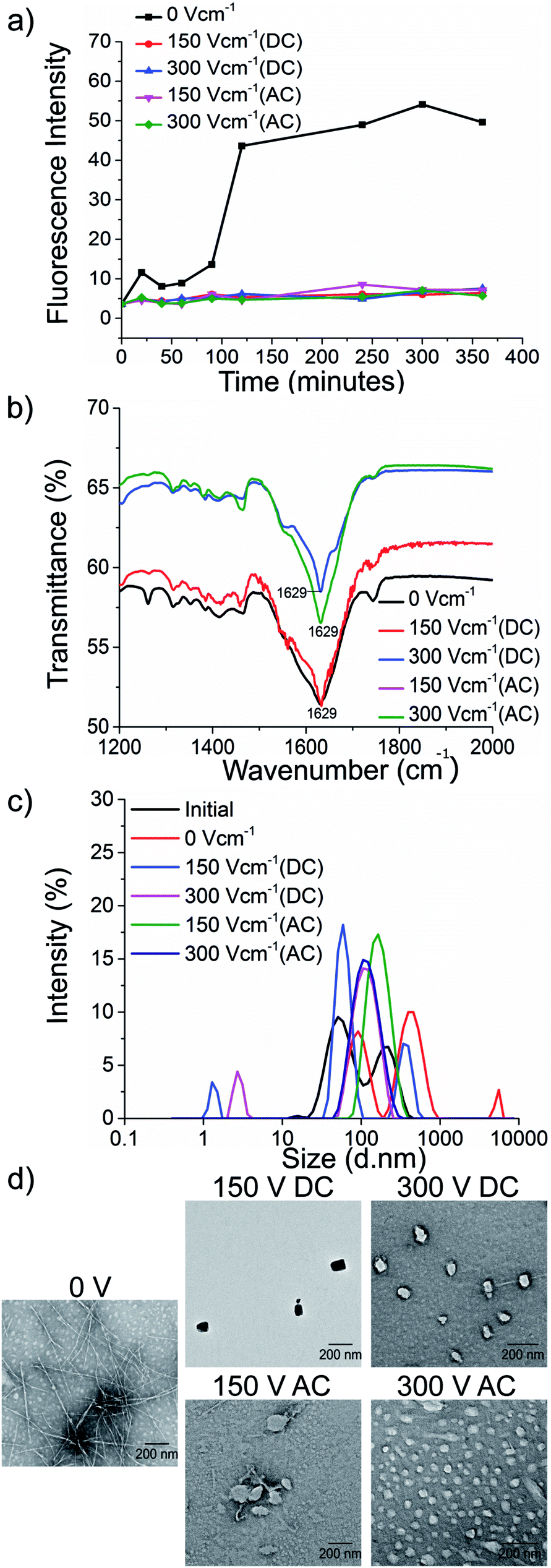 | ||
| Fig. 3 Retardation of PhF6* aggregation by the electric field. Fibrillation kinetics of PhF6* was examined by (a) ThT fluorescence emission, (b) secondary structure determined by FTIR spectroscopy, (c) size estimation using dynamic light scattering (DLS) (d) TEM micrographs. All samples were allowed to aggregate in the presence and absence of DC and AC electric field of varying strengths. | ||
Field-emission transmission electron microscope (FETEM) analysis has been performed to investigate the effects of electric field on the morphology of PhF6 and PhF6* aggregates. The control sample shows the presence of dense fibrils. Contrastingly, electric field samples show spherical structures and no fibrillar assemblies were observed. (Fig. 2(d), and 3(d)).
The secondary structure conformation of each sample was determined by Fourier-transform infrared spectroscopy (FTIR) analysis. Both electric filed treated as well as untreated PhF6 and PhF6* samples showed the amide I band, a characteristic signature of β-sheet (Fig. 2(b), and 3(b)).53–55 FTIR results indicate that though electric field and control sample differ in their aggregation kinetics and morphology, they have same β-sheet conformation. This result suggests that the morphological difference induced by EF is manifested in the supramolecular assembly levels and not at the molecular level.
The size of aggregates has been reported to increase with time.56 To compare the size distribution of self-assemblies formed by electric field exposed and control sample, dynamic and static light scattering measurements were recorded. The DLS plot of control samples shows an increase in size, with time. However, in the case of electric field samples, this increase is markedly reduced (Fig. 2(c), and 3(c)). Static light scattering experiment further supported this observation, wherein, the control samples having large-sized aggregates, showed significantly high scattering intensities compared to the electric field exposed samples (ESI, Fig. S8, and S9†).
The cytotoxic self-assemblies of amyloid and tau protein aggregates are regarded as the hallmark of AD pathology and have been attributed to causing neuronal loss and synaptic dysfunction. Hence, intervening in the self-assembly phenomenon may serve as a complementary therapeutic option. The retardation of aggregation kinetics of our model peptides (PhF6 and PhF6*), in the presence of an electric field, confirms the efficacy of the electric field for therapeutic applications in amyloidogenic diseases. Therefore, we assessed the improvement in the cell viability as a result of electric field treatment using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction assay. Human neuroblastoma SH-SY5Y and IMR-32 neuronal cell lines and PC-12, rat pheochromocytoma cell lines were treated with 25 μM PhF6 and PhF6* peptides solutions. The peptide samples were initially allowed to aggregate in the presence (treated) or absence (untreated) of an electric field.
The assay results show that the untreated aggregated PhF6 peptide solution reduced cell viability of SH-SY5Y, IMR-32 and PC-12 cells to 55%, 68%, and 50% respectively. Field treated samples, on the other hand, rescued the cell viability from 18% to 45%, depending on the field strength and cell type (Fig. 4(a), ESI, Tables S3–S5†). Similarly, in the case of PhF6*, peptide samples were showing cell viability estimates at 60%, 91%, and 76%, for SH-SY5Y, IMR-32 and PC-12 cell lines respectively.
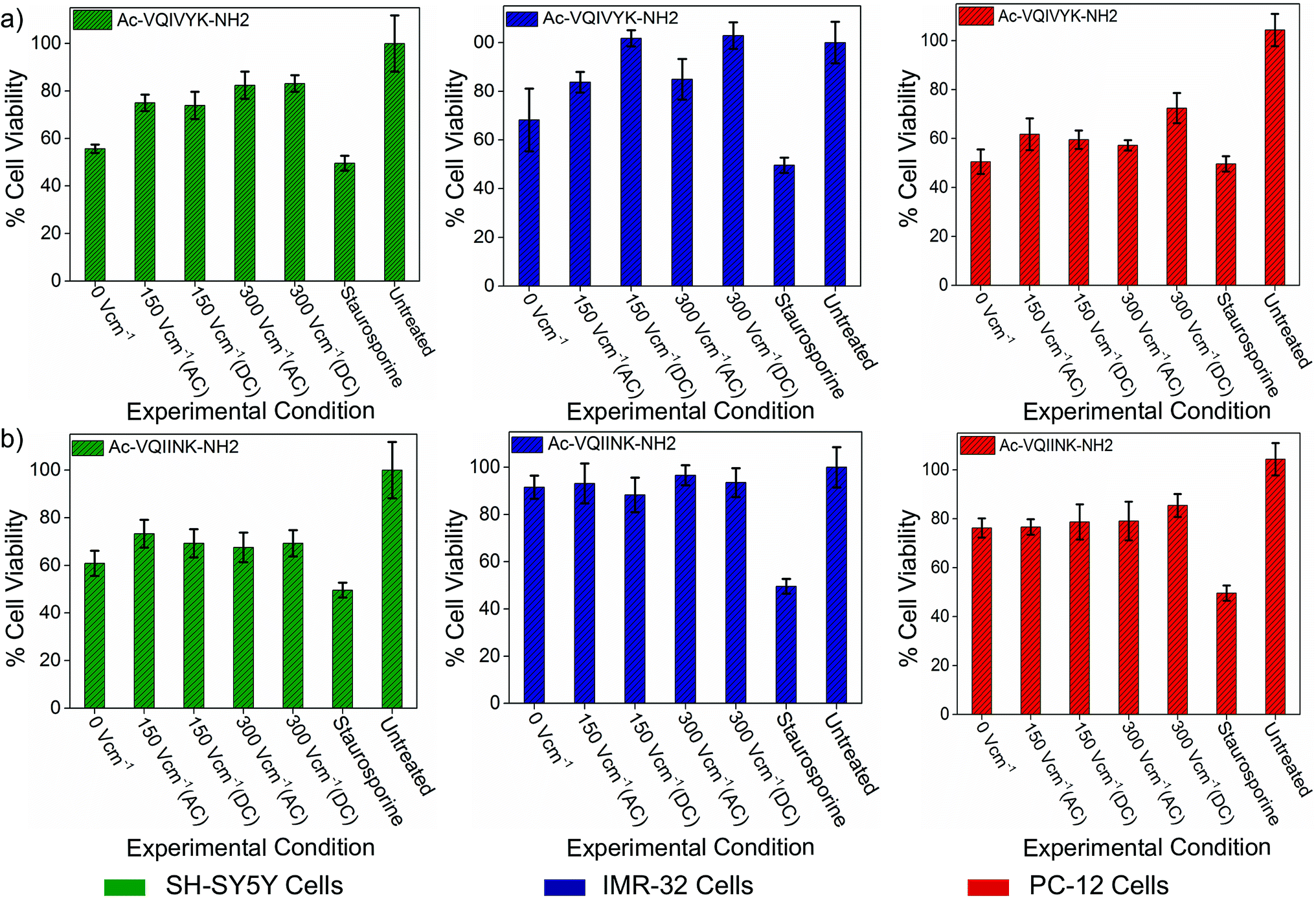 | ||
| Fig. 4 Effect of DC and AC electric field on the cellular toxicity of PhF6 and PhF6* peptide. Post incubation, peptide samples allowed to aggregate in the presence and absence of AC/DC electric field were added to SH-SY5Y, IMR-32 and PC-12 cells. The cytotoxicity effect was determined using an MTT assay. The electric field exposure reduced the toxicity of PhF6 and PhF6* peptide evident from the increased cell viability compared to the control. Additional data for the cell viability is available in Table S3 of ESI.† | ||
Here also, the electric field treatment resulted in cell viability improvements up to 36% (Fig. 4(b), ESI, Tables S3–S5†). These cell viability assay results support the neuroprotective behaviour of the electric field.
Conclusions
Aggregation of the protein chain to the ordered and patterned functional or dysfunctional forms is principally guided by physical forces such as electrostatics, van der Waals and hydrogen bonding interactions. Formation of biological forms is ‘rooted in its physics’, is an observation by D'Arcy Wentworth Thompson in his book on growth and form at the beginning of the last century, while attempting to quantitatively assess structural patterns and formations in biological systems.57 Inspiration to modulate peptide assembly using a physical perturbant like the electric field is derived from such and similar observations. Our experimental results convincingly prove that the electric field exposure decelerated the aggregation kinetics of PhF6 and PhF6* peptides. The structures formed by both the peptides, allowed to aggregate under field conditions were spherical and lacked the usual fibrillar morphology formed by the control peptide. The size of the aggregates formed by EF treated samples was also smaller compared to the untreated ones. Both EF treated as well as untreated samples had β-sheet conformation, indicating that the EF-induced modulatory effect is at the assembly, and not at the molecular level. Both PhF6 and PhF6* aggregates are toxic to neuronal cells. A comparative experiment with EF treated and untreated samples with two neuronal cell lines and one normal cell, showed that EF treated samples were 18 to 45% less toxic, under various experimental conditions. Pathological aggregation of tau protein has been reported to have links with more than 20 neurodegenerative diseases, collectively referred to as tauopathies. Despite having a causative association with diseases affecting millions of people worldwide, limited tau-based therapeutic options are available. We infer that electric field modulation can effectively retard tau protein aggregation and supramolecular assembly of the core peptide segment of tau. Mechanistic investigations on the possible effects of the electric field using molecular dynamics simulations have been published in two earlier studies by three different groups.46,47,58 Our experimental observations qualitatively align more with the mechanistic predictions of Martin E. Garcia46 and Andrij Baumketner.47Future work
Through this paper, we prefer to go further by designing an experimental set-up to assess the electric field based modulation of peptide aggregation and amyloid formation in disease model systems. We are presenting the outlines of a prospective device as a future therapeutic option, subject to a series of in vivo studies and clinical trials (ESI, Fig. S10†). However, toxicity estimation of EF has to be more rigorously tested, but recent positive reports on the efficacy of similar non-invasive therapies are very optimistic.32–36 The electric field can, therefore, be referred to advanced phases of clinical examination, for potential use as a therapeutic option against tauopathies.Author contribution
VR designed the experiment. HB designed electric field set-up. GP fabricated the electric field set-up. GP and SM performed the experiments. VR, GP analyzed, interpreted data, and contributed to the editing of the manuscript. SK and SM analyzed and interpreted cell viability assay data. GP and VR analyzed the results and wrote the paper in close collaboration with all the authors. VR conceived & directed the ideas, planning and overall execution.Conflicts of interest
There are no conflicts to declare. However, a patent has been filed on the core theme of this manuscript (WO/2019/012556).Acknowledgements
We acknowledge Department of Biotechnology (grant No. BT/565/NE/U-Excel/2016) and Board of Research in Nuclear Sciences (grant no. 35/14/07/2017-BRNS/35090), Govt. of India, for funding. We thank Central Instruments Facility, IIT Guwahati for instrument support. Patent on EF induced non-invasive device, No. WO/2019/012556.References
- M. Prince, A. Comas-Herrera, M. Knapp, M. Guerchet and M. Karagiannidou, World Alzheimer Report 2016 Improving healthcare for people living with dementia. Coverage, Quality and costs now and in the future, 2016, pp. 1–140, https://www.alz.co.uk/research/world-report-2016 Search PubMed.
- S. E. Chastain and M. Moss, Biophys. J., 2015, 108, 357a CrossRef.
- A. Abelein, L. Lang, C. Lendel, A. Gräslund and J. Danielsson, FEBS Lett., 2012, 586, 3991–3995 CrossRef CAS PubMed.
- J. G. Moe, P. K. Krishnamurthy, P. Lopez, G. Papiani, D. Romero, H. Bian, M. E. McDonnel, A. B. Reitz, C. Gluchowski and E. J. Davidowitz, Alzheimers Dement, 2016, 12, P434 CrossRef.
- M. Pickhardt, T. Neumann, D. Schwizer, K. Callaway, M. Vendruscolo, D. Schenk, P. St George-Hyslop, E. M. Mandelkow, C. M Dobson and L. McConlogue, Curr. Alzheimer. Res., 2015, 12, 814–828 CrossRef CAS PubMed.
- B. Cheng, H. Gong, H. Xiao, R. B. Petersen, L. Zheng and K. Huang, Biochim. Biophys. Acta, 2013, 1830, 4860–4871 CrossRef CAS PubMed.
- D. L. Brody and D. M. Holtzman, Annu. Rev. Neurosci., 2008, 31, 175–193 CrossRef CAS PubMed.
- D. L. Castillo-Carranza, M. J. Guerrero-Muñoz, U. Sengupta, C. Hernandez, A. D. Barrett, K. Dineley and R. Kayed, J. Neurosci., 2015, 35, 4857–4868 CrossRef CAS PubMed.
- M. H. Viet, S. T. Ngo, N. S. Lam and M. S. Li, J. Phys. Chem. B, 2011, 115, 7433–7446 CrossRef CAS PubMed.
- S. Ribarič, Molecules, 2018, 23, 283 CrossRef PubMed.
- P. Ryan, B. Patel, V. Makwana, H. R. Jadhav, M. Kiefel, A. Davey, T. A. Reekie, S. Rudrawar and M. Kassiou, ACS Chem. Neurosci., 2018, 9, 1530–1551 CrossRef CAS PubMed.
- D. Goyal, S. Shuaib, S. Mann and B. Goyal, ACS Comb. Sci., 2017, 19, 55–80 CrossRef CAS PubMed.
- H. Li, F. Rahimi and G. Bitan, ACS Chem. Neurosci., 2016, 7, 845–856 CrossRef CAS PubMed.
- Y. X. Zhang, S. W. Wang, S. Lu, L. X. Zhang, D. Q. Liu, M. Ji, W. Y. Wang and R. T. Liu, FEBS Lett., 2017, 591, 3615–3624 CrossRef CAS PubMed.
- C. Soto, E. M. Sigurdsson, L. Morelli, R. A. Kumar, E. M. Castaño and B. Frangione, Nat. Med., 1998, 4, 822–826 CrossRef CAS PubMed.
- M. Chemerovski-Glikman, M. Frenkel-Pinter, R. Mdah, A. Abu-Mokh, E. Gazit and D. Segal, Chem. Eur. J., 2017, 23, 9618–9624 CrossRef CAS PubMed.
- E. Andreetto, E. Malideli, L. M. Yan, M. Kracklauer, K. Farbiarz, M. Tatarek-Nossol, G. Rammes, E. Prade, T. Neumüller and A. Caporale, Angew. Chem., Int. Ed. Engl., 2015, 54, 13095–13100 CrossRef CAS PubMed.
- J. A. Plumley, J. Ali-Torres, G. Pohl and J. J. Dannenberg, J. Phys. Chem. B, 2014, 118, 3326–3334 CrossRef CAS PubMed.
- C. Cabaleiro-Lago, F. Quinlan-Pluck, I. Lynch, S. Lindman, A. M. Minogue, E. Thulin, D. M. Walsh, K. A. Dawson and S. Linse, J. Am. Chem. Soc., 2008, 130, 15437–15443 CrossRef CAS PubMed.
- L. Xiao, D. Zhao, W.-H. Chan, M. M. Choi and H.-W. Li, Biomaterials, 2010, 31, 91–98 CrossRef CAS PubMed.
- K. J. Barnham, V. B. Kenche, G. D. Ciccotosto, D. P. Smith, D. J. Tew, X. Liu, K. Perez, G. A. Cranston, T. J. Johanssen and I. Volitakis, Proc. Natl. Acad. Sci., 2008, 105, 6813–6818 CrossRef CAS PubMed.
- A. Kumar, L. Moody, J. F. Olaivar, N. A. Lewis, R. L. Khade, A. A. Holder, Y. Zhang and V. Rangachari, ACS Chem. Neurosci., 2010, 1, 691–701 CrossRef CAS PubMed.
- B. Y.-W. Man, H.-M. Chan, C.-H. Leung, D. S.-H. Chan, L.-P. Bai, Z.-H. Jiang, H.-W. Li and D.-L. Ma, Chem. Sci., 2011, 2, 917–921 RSC.
- Y. Jiao and P. Yang, J. Phys. Chem. B, 2007, 111, 7646–7655 CrossRef CAS PubMed.
- B. Ojha, H. Liu, S. Dutta, P. P. Rao, E. P. Wojcikiewicz and D. Du, J. Phys. Chem. B, 2013, 117, 13975–13984 CrossRef CAS PubMed.
- H. H. Lee, T. S. Choi, S. J. C. Lee, J. W. Lee, J. Park, Y. H. Ko, W. J. Kim, K. Kim and H. I. Kim, Angew. Chem., Int. Ed. Engl., 2014, 53, 7461–7465 CrossRef CAS PubMed.
- L. M. Luheshi, W. Hoyer, T. P. de Barros, I. van Dijk Härd, A.-C. Brorsson, B. Macao, C. Persson, D. C. Crowther, D. A. Lomas and S. Ståhl, PLoS Biol., 2010, 8, e1000334 CrossRef PubMed.
- J. T. Jarrett and P. T. Lansbury Jr, Cell, 1993, 73, 1055–1058 CrossRef CAS PubMed.
- M. Wogulis, S. Wright, D. Cunningham, T. Chilcote, K. Powell and R. E. Rydel, J. Neurosci., 2005, 25, 1071–1080 CrossRef CAS PubMed.
- W.-F. Xue, S. W. Homans and S. E. Radford, Proc. Natl. Acad. Sci., 2008, 105, 8926–8931 CrossRef CAS PubMed.
- S. Kumar and J. Walter, Aging, 2011, 3, 803 Search PubMed.
- G. Leinenga and J. Götz, Sci. Transl. Med., 2015, 7, 278ra233 Search PubMed.
- R. J. Hatch, G. Leinenga and J. Götz, PLoS One, 2016, 11, e0164278 CrossRef PubMed.
- D. M. Johnstone, C. Moro, J. Stone, A.-L. Benabid and J. Mitrofanis, Front. Neurosci., 2016, 9, 500 CrossRef PubMed.
- I. Gonsalvez, R. Baror, P. Fried, E. Santarnecchi and A. Pascual-Leone, Curr. Alzheimer. Res., 2017, 14, 362–376 CAS.
- S. Mondragón-Rodríguez, G. Perry, F. Pena-Ortega and S. Williams, Curr. Alzheimer. Res., 2017, 14, 40–46 CrossRef.
- W. Noble, D. P. Hanger, C. C. Miller and S. Lovestone, Front. Neurol., 2013, 4, 83 Search PubMed.
- M. G. Spillantini and M. Goedert, Lancet Neurol., 2013, 12, 609–622 CrossRef CAS PubMed.
- P. Ganguly, T. D. Do, L. Larini, N. E. LaPointe, A. J. Sercel, M. F. Shade, S. C. Feinstein, M. T. Bowers and J.-E. Shea, J. Phys. Chem. B, 2015, 119, 4582–4593 CrossRef CAS PubMed.
- C. Schirmer, E. Lepvrier, L. Duchesne, O. Decaux, D. Thomas, C. Delamarche and C. Garnier, Biochim. Biophys. Acta, 2016, 1860, 2598–2609 CrossRef CAS PubMed.
- Y. Lu, X.-F. Shi, F. R. Salsbury Jr and P. Derreumaux, J. Chem. Phys., 2018, 148, 045105 CrossRef PubMed.
- X. Wang, Y. Li, X. He, S. Chen and J. Z. Zhang, J. Phys. Chem. A, 2014, 118, 8942–8952 CrossRef CAS PubMed.
- W. Zhao and R. Yang, J. Phys. Chem. B, 2009, 114, 503–510 CrossRef PubMed.
- A. Budi, F. S. Legge, H. Treutlein and I. Yarovsky, J. Phys. Chem. B, 2008, 112, 7916–7924 CrossRef CAS PubMed.
- C. M. Kelly, T. Northey, K. Ryan, B. R. Brooks, A. L. Kholkin, B. J. Rodriguez and N.-V. Buchete, Biophys. Chem., 2015, 196, 16–24 CrossRef CAS PubMed.
- P. Ojeda-May and M. E. Garcia, Biophys. J., 2010, 99, 595–599 CrossRef CAS PubMed.
- A. Baumketner, J. Phys. Chem. B, 2014, 118, 14578–14589 CrossRef CAS PubMed.
- N. K. Pandey, S. Mitra, M. Chakraborty, S. Ghosh, S. Sen, S. Dasgupta and S. Dasgupta, J. Phys. D: Appl. Phys., 2014, 47, 305401 CrossRef.
- S. Sen, M. Chakraborty, S. Goley, S. Dasgupta and S. Dasgupta, Biophys. Chem., 2017, 226, 23–33 CrossRef CAS PubMed.
- D. Porschke, Biophys. J., 1996, 71, 3381–3391 CrossRef CAS.
- G. Pandey, J. Saikia, S. Sasidharan, D. C. Joshi, S. Thota, H. B. Nemade, N. Chaudhary and V. Ramakrishnan, Sci. Rep., 2017, 7, 2726 CrossRef PubMed.
- H. Levine III, Protein Sci., 1993, 2, 404–410 CrossRef PubMed.
- D. Suhas, H. Jeong, T. Aminabhavi and A. Raghu, Polym. Eng. Sci., 2014, 54, 24–32 CrossRef CAS.
- A. Raghu, G. Anita, Y. Barigaddi, G. Gadaginamath and T. Aminabhavi, J. Appl. Polym. Sci., 2007, 104, 81–88 CrossRef CAS.
- A. Raghu, G. Gadaginamath, N. Mathew, S. Halligudi and T. Aminabhavi, J. Appl. Polym. Sci., 2007, 106, 299–308 CrossRef CAS.
- S. Banerjee, M. Hashemi, Z. Lv, S. Maity, J.-C. Rochet and Y. L. Lyubchenko, Sci. Rep., 2017, 7, 45592 CrossRef PubMed.
- D. A. Thompson, On Growth and Form, Cambridge University Press, 1942 Search PubMed.
- F. Toschi, F. Lugli, F. Biscarini and F. Zerbetto, J. Phys. Chem. B, 2008, 113, 369–376 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Static right angle scatter depicting FF self-assembly formation (Section 1), aggregation kinetics profiles of PhF6 and PhF6* peptides (Section 2), aggregation assay plots for PhF6 peptide (Section 3), aggregation assay plots for PhF6* peptide (Section 4), illustration of a potential electric field based therapeutic device, comparative thioflavin T fluorescence data of PhF6 and PhF6* peptides (Section 6), and cell viability assay data (Section 7), which are not explicitly detailed in the main text. See DOI: 10.1039/c8ra09993f. |
| This journal is © The Royal Society of Chemistry 2019 |