
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRetracted Article: Long non-coding RNA GACAT1 alleviates doxorubicin and vincristine resistance through a PTEN/AKT/mTOR/S6K1 regulatory pathway in gastric cancer†
Hengxuan Ding,
Junfeng Sun,
Ruixin Li and
Guojun Wang *
*
Department of Gastrointestinal Surgery, The First Affiliated Hospital of Zhengzhou University, Jianshe Road 1, Zhengzhou 450052, P. R. China. E-mail: wwangguojun@126.com; Tel: +86-0371-67967137
First published on 11th March 2019
Abstract
Gastric cancer (GC) is a major global health problem. Chemotherapy is a common therapeutic strategy for cancers including GC. However, chemoresistance strikingly limits the clinical applications of chemotherapeutic drugs. Long non-coding RNAs (lncRNAs) have been widely reported to be implicated in the pathogenesis and chemoresistance of cancers including GC. Our work aims to investigate the roles and molecular mechanisms of lncRNA gastric cancer-associated transcript 1 (GACAT1) in regulating doxorubicin (ADR) and vincristine (VCR) resistance in GC. In this text, RT-qPCR assay showed that GACAT1 expression was markedly reduced in ADR- or VCR-resistant GC (SGC7901/ADR or SGC7901/VCR) cells and GC tissues. CCK-8 assay and flow cytometry analysis revealed that GACAT1 overexpression alleviated the resistance of GC cells to ADR and VCR. RT-qPCR and western blot assay disclosed that GACAT1 deactivated the AKT/mTOR/S6K1 signaling pathway and promoted PTEN expression in SGC7901/ADR or SGC7901/VCR cells. Restoration experiments demonstrated that GACAT1 attenuated ADR or VCR resistance by regulating the PTEN/AKT/mTOR/S6K1 pathway in SGC7901/ADR or SGC7901/VCR cells. In vivo experiments demonstrated that GACAT1 overexpression inhibited tumor growth and enhanced ADR- or VCR-mediated anti-tumor effects in GC xenograft tumor models. Taken together, these data revealed that GACAT1 weakened the resistance of GC cells to ADR and VCR by the PTEN/AKT/mTOR/S6K1 regulatory pathway in vitro and in vivo, shedding new light on GACAT1 upregulation as a potential strategy to alleviate chemoresistance in GC.
Introduction
Gastric cancer (GC), a common digestive system malignancy, is responsible for 5.7% of all new cancer cases and 8.2% of all cancer-related deaths in 2018 globally.1 And, an estimated 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 033701 new GC cases and 782685 GC-related deaths occurred in the world in 2018.1 Moreover, GC patients are often diagnosed at the inoperable stages, accompanied by high recurrence rate after surgery resection.2,3 In addition, current therapeutic outcomes for patients with metastatic GC remain dismal with a median survival of less than 1 year.2,4 Chemotherapy is one of prevailing therapeutic approaches for GC.5,6 However, the existence and development of intrinsic or acquired chemoresistance can result in the therapeutic failure of cancers to a large extent.7,8 Hence, it is imperative to seek for potential targets or develop novel strategies to overcome chemoresistance. And, a better understanding on the underlying mechanisms responsible for chemoresistance of anti-tumor drugs may contribute to the reversal of this situation. Doxorubicin (ADR), also termed as adriamycin, has been widely used for the treatment of GC together with 5-fluorouracil, mitomycin C from the late 80's.6,9 Vincristine (VCR) is a microtubule stabilizing chemotherapeutic agent approved by the FDA.10
033701 new GC cases and 782685 GC-related deaths occurred in the world in 2018.1 Moreover, GC patients are often diagnosed at the inoperable stages, accompanied by high recurrence rate after surgery resection.2,3 In addition, current therapeutic outcomes for patients with metastatic GC remain dismal with a median survival of less than 1 year.2,4 Chemotherapy is one of prevailing therapeutic approaches for GC.5,6 However, the existence and development of intrinsic or acquired chemoresistance can result in the therapeutic failure of cancers to a large extent.7,8 Hence, it is imperative to seek for potential targets or develop novel strategies to overcome chemoresistance. And, a better understanding on the underlying mechanisms responsible for chemoresistance of anti-tumor drugs may contribute to the reversal of this situation. Doxorubicin (ADR), also termed as adriamycin, has been widely used for the treatment of GC together with 5-fluorouracil, mitomycin C from the late 80's.6,9 Vincristine (VCR) is a microtubule stabilizing chemotherapeutic agent approved by the FDA.10
Long non-coding RNAs (lncRNAs), a class of non-protein-coding transcripts longer than 200 nucleotides, can regulate a great deal of molecular processes such as gene transcription, protein translation and stability.11,12 Moreover, lncRNAs have emerged as central players in the tumorigenesis and progression of cancers with potential values in the diagnosis, prognosis, screening and treatment of cancers including GC.12–14 And, lncRNAs are involved in mediating chemoresistance in a great variety of cancers.15,16 For example, lncRNA D63785 promoted GC cell proliferation, migration and invasion in vitro and inhibited GC xenograft growth in vivo, as well as enhanced the resistance of GC cells to ADR through regulating microRNA-422a/myocyte enhancer factor-2D (MEF2D) axis.17 lncRNA UCA1 knockdown suppressed cell proliferation and alleviated ADR resistance in GC.18 lncRNA gastric cancer-associated transcript 1 (GACAT1), also named as AC096655.1-002 and LINC00876, has been reported to be abnormally expressed in GC tissues and to be involved in the pathogenesis of GC.19–22 Moreover, our data revealed that GACAT1 was low expressed in GC tissues and GACAT1 expression was associated with TNM stage and tumor distant metastasis in GC. However, the roles and molecular basis of GACAT1 in GC chemoresistance have not been investigated till now. Hence, our study aimed to further explore the roles and molecular mechanisms of GACAT1 in GC tumorigenesis and ADR or VCR resistance.
In the present study, we demonstrated that GACAT1 expression was markedly downregulated in GC tissues and ADR- or VCR-resistant GC cells. Moreover, GACAT1 overexpression mitigated ADR and VCR resistance by regulating PTEN/AKT/mTOR/S6K1 pathway in GC.
Material and methods
Tissue samples
Thirty-five GC patients without any treatment were recruited from the First Affiliated Hospital of Zhengzhou University and all patients signed the written informed consents prior to experiments. GC tissues and adjacent normal tissues were collected from these patients suffered from radical gastrectomy, and then immediately frozen in liquid nitrogen and stored at −80 °C. Our study was approved by Institutional Review Board (IRB) of the First Affiliated Hospital of Zhengzhou University.Cell culture and treatment
GC cell line SGC7901 was ordered from Cell Bank of China Academy of Sciences (Shanghai, China). ADR-resistant SGC7901 cells (SGC7901/ADR) and VCR-resistant SGC7901 cells (SGC7901/VCR) were purchased from Shanghai MEIXUAN Biological Science and Technology Co., ltd (Shanghai, China). SGC7901, SGC7901/ADR and SGC7901/VCR cells were cultured in RPMI-1640 medium (Thermo Scientific, Rockford, IL, USA) supplemented with 10% fetal bovine serum (FBS, Thermo Scientific).Lentivirus
Lentiviruses expressing GACAT1 full length fragment (lenti-GACAT1) and its control lentiviruses lenti-NC, lentiviruses containing GACAT1 or PTEN knockdown fragment (shRNA-GACAT1 or shRNA-PTEN) and their control shRNA-NC were obtained from Hanbio Biotechnology Co., Ltd. (Shanghai, China).Reverse transcription-quantitative PCR (RT-qPCR) assay
Total RNA was extracted from GC tissues and cell lines using Trizol reagent (Thermo Scientific), followed by the removal of DNA using RNase-free DNase I (Thermo Scientific). Then, the purity and concentration of RNA was determined using the NanoDrop ND-2000 spectrophotometer (Thermo Scientific). Next, cDNA first strands were synthesized using M-MLV Reverse Transcriptase (Thermo Scientific) with RNA (1 μg) as the template. Subsequently, real time-quantitative PCR analysis was performed using PowerUp™ SYBR™ Green Master Mix (Thermo Scientific) and specific primers. Data were counted using the 2−ΔΔCT method. GAPDH functioned as the housekeeping gene to normalize the expression of GACAT1 and PTEN. The primers sequences for GACAT1, PTEN and GAPDH were customized from Sangon Biotech Co., ltd. (Shanghai, China). Experiments were repeated more than 3 times.Cell counting kit-8 (CCK-8) assay
Cell viability was estimated through CCK-8 assay using the CCK-8 assay kit (Dojindo Molecular Technologies, Rockville, MD, USA) following the instructions of manufacturer. Briefly, SGC7901/ADR or SGC7901/VCR cells were seeded into 96-well plates in 100 μl of growth medium. Then, these cells were infected with shRNA-NC, shRNA-GACAT1, shRNA-PTEN, lenti-NC, lenti-GACAT1, lenti-GACAT1 + shRNA-NC, or lenti-GACAT1 + shRNA-PTEN for 24 h and then treated with ADR or VCR for another 48 h. At 72 h after infection, 10 μl of CCK-8 solution was added into each well. After 3 h of incubation, cell optical density values were measured at 450 nm. Experiments were repeated more than 3 times.Cell apoptosis detection
Cell apoptotic percentage was determined through Annexin V-FITC Apoptosis Detection Kit (Beyotime, Shanghai, China) referring to the instructions of manufacturer. Generally, cells were collected at 72 h after infection and re-suspended in Annexin V-FITC binding solution (195 μl). Then, cell suspension was treated with Annexin V-FITC (5 μl) and Propidium Iodide (PI, 10 μl) for 20 min at room temperature in the dark. Finally, cell apoptotic percentage was measured using a flow cytometry (BD Bioscience, San Jose, CA, USA). Experiments were repeated more than 3 times.Western blot analysis
Cells were collected at 72 h post-infection and homogenized using RIPA Lysis and Extraction Buffer (Thermo Scientific) containing protease and phosphatase inhibitor (Thermo Scientific). Then, cell supernatant was collected after the centrifugation at the speed of 12000 rpm for 15 min at 4 °C. Following this, protein content was determined using a Pierce BCA Protein Assay Kit (Thermo Scientific). Next, an equal amount of proteins (40 μg per lane) was loaded on 10% SDS-PAGE gels and then transferred to nitrocellulose membranes (Millipore, Bedford, MA, USA). After the blockade of non-specific signals with 5% fat-free milk, the membranes were probed overnight at 4 °C with primary antibodies and sequentially incubated for 1 h at room temperature with secondary antibody conjugated with horseradish peroxidase (HRP). Finally, target bands were visualized using Pierce™ ECL Western Blotting Substrate (Thermo Scientific), and protein signals were quantified using Quantity One Software Version 4.1.1 (BIO-Rad Laboratories, Hercules, CA, USA) by grayscale analysis. Primary antibodies against AKT, phosphorylated-AKT (p-AKT), mTOR, phosphorylated-mTOR (p-mTOR), PTEN, and GAPDH were purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA). And, primary antibodies against S6K1 and phosphorylated-S6K1 (p-S6K1), and goat anti-rabbit secondary antibody was ordered from Abcam, Inc. (Cambridge, UK). Experiments were repeated more than 3 times.
Mouse experiment
BALB/c nu/nu mice (male, 6 weeks old, n = 48) were purchased from Experimental Animal Center of Zhengzhou University (Zhengzhou, China), and raised and treated following the National Institutes of Health guidelines for the Care and Use of Laboratory Animals. Also, our animal experiments were approved by the Institutional Committee for Animal Research of the First Affiliated Hospital of Zhengzhou University. Mice were randomly divided into 8 groups with 6 mice in each group. SGC7901/ADR or SGC7901/VCR cells (107 cells per mouse) infected with lenti-NC or lenti-GACAT1 lentiviruses were subcutaneously injected into the right flanks of mice. One weeks later, ADR (1 mg kg−1 body weight, Sigma-Aldrich, St. Louis, MO, USA), VCR (2 mg kg−1 body weight, Sigma-Aldrich) or equal volume sodium chloride injection solution (0.1 ml) were intraperitoneally injected into mice every 4 days for 4 weeks. Tumor volume was monitored every 1 week for a total of 5 weeks using a caliper and calculated with the formula: volume (mm3) = 0.5 × (length × width2). Mice were euthanised at 5 weeks after injection. Then, tumors were resected and weighed.Statistical analysis
Results were obtained from more than triplicate independent experiments and expressed as mean ± standard error of mean (SEM). Differences between groups were estimated by Student's t-test. Differences among groups were analyzed using one-way analysis of variance. Difference was regarded as statistically significant when P value was less than 0.05.Results
GACAT1 expression was markedly reduced in GC tissues and ADR- or VCR-resistant GC cells
At first, RT-qPCR assay revealed that GACAT1 expression was markedly downregulated in 35 cases of GC tissues relative to adjacent normal tissues (Fig. 1A). To further explore the relationship of GACAT1 and ADR or VCR resistance in GC, ADR-resistant SGC7901 cell line (SGC7901/ADR) and VCR-resistant SGC7901 cell line (SGC7901/VCR) were purchased. As presented in ESI Fig. 1,† the IC50 values of ADR increased by 16 fold in SGC7901/ADR cells than that in SGC7901 cells and IC50 values of VCR elevated by 15 fold in SGC7901/VCR cells relative to SGC7901 cells, indicating that these resistant cell lines were successfully established. Moreover, an obvious downregulation of GACAT1 level was observed in SGC7901/ADR and SGC7901/VCR cell lines than that in parental cell line SGC7901 (Fig. 1B), suggesting the link between GACAT1 and drug resistance in GC. | ||
| Fig. 1 GACAT1 expression was markedly reduced in GC tissues and ADR- or VCR-resistant GC cells. (A) RT-qPCR assay was conducted to measure the expression of GACAT1 in 35 pairs of GC tissues and adjacent normal tissues. (B) GACAT1 level was detected by RT-qPCR assay in SGC7901, SGC7901/ADR and SGC7901/VCR cells. *P < 0.05. | ||
GACAT1 overexpression alleviated the resistance of GC cells to ADR and VCR
Next, CCK-8 assay showed that the introduction of ADR or VCR resulted in the dose-dependent reduction of cell viability in SGC7901/ADR or SGC7901/VCR cells infected with lenti-NC or lenti-GACAT1 (Fig. 2A). Moreover, GACAT1-overexpressed SGC7901/ADR or SGC7901/VCR cells presented lower cell viability compared with lenti-NC-infected cells when these cells were stimulated with the same concentration of ADR or VCR (Fig. 2A). Moreover, GACAT1 overexpression induced cell apoptosis in ADR-treated SGC7901/ADR cells or VCR-stimulated SGC7901/VCR cells (Fig. 2B). In a word, these data disclosed that enforced expression of GACAT1 could weaken ADR and VCR resistance in GC cells. Also, our study further demonstrated that GACAT1 knockdown enhanced ADR and VCR resistance in GC cells, as evidenced by increased cell viability and reduced cell apoptotic percentage in SGC7901/ADR or SGC7901/VCR cells infected with shRNA-GACAT1 compared with shRNA-NC-infected cells (ESI Fig. 2†). | ||
| Fig. 2 GACAT1 overexpression alleviated the resistance of GC cells to ADR and VCR. (A) SGC7901/ADR or SGC7901/VCR cells were infected with lenti-NC or lenti-GACAT1 for 24 h. Then, SGC7901/ADR cells were stimulated with different concentrations of ADR (0, 1, 2, 4, 8, 16 μg ml−1) for another 48 h and SGC7901/VCR cells were treated with various doses of VCR (0, 2, 4, 8, 16, 32 μg ml−1) for additional 48 h. Subsequently, cell viability was determined by CCK-8 assay. (B) SGC7901/ADR or SGC7901/VCR cells were infected with lenti-NC or lenti-GACAT1 for 24 h and then treated with ADR (2 μg ml−1) or VCR (4 μg ml−1) for another 48 h. Then, cell apoptotic percentage was determined through flow cytometry. *P < 0.05. | ||
GACAT1 inactivated AKT/mTOR/S6K1 signaling pathway in ADR- or VCR-resistant GC cells
Subsequently, we further demonstrated that the levels of p-AKT, p-mTOR and p-S6K1 were dramatically reduced in SGC7901/ADR or SGC7901/VCR cells following the overexpression of GACAT1 (Fig. 3A and B). Conversely, GACAT1 loss resulted in the notable elevation of p-AKT, p-mTOR and p-S6K1 levels in SGC7901/ADR or SGC7901/VCR cells (Fig. 3A and B). And, GACAT1 upregulation or downregulation had no much influence on the total levels of AKT, mTOR and p-S6K1 in SGC7901/ADR or SGC7901/VCR cells (Fig. 3A and B). That was to say, GACAT1 suppressed the activation of AKT/mTOR/S6K1 signaling pathway in ADR- or VCR-resistant GC cells. | ||
| Fig. 3 GACAT1 inactivated AKT/mTOR/S6K1 signaling pathway in ADR- or VCR-resistant GC cells. (A and B) SGC7901/ADR or SGC7901/VCR cells were infected with lenti-NC, lenti-GACAT1, shRNA-NC, or shRNA-GACAT1 for 24 h and then treated with ADR (2 μg ml−1) or VCR (4 μg ml−1) for another 48 h. Next, protein levels of p-AKT, AKT, p-mTOR, mTOR, p-S6K1 and S6K1 were determined by western blot assay. *P < 0.05. | ||
GACAT1 promoted PTEN expression in SGC7901/ADR or SGC7901/VCR cells
Next, RT-qPCR and western blot assay further presented that PTEN mRNA and protein levels were notably upregulated in SGC7901/ADR or SGC7901/VCR cells following the overexpression of GACAT1 (Fig. 4A–D). Conversely, the depletion of GACAT1 significantly inhibited PTEN expression at mRNA and protein levels in SGC7901/ADR or SGC7901/VCR cells (Fig. 4A–D).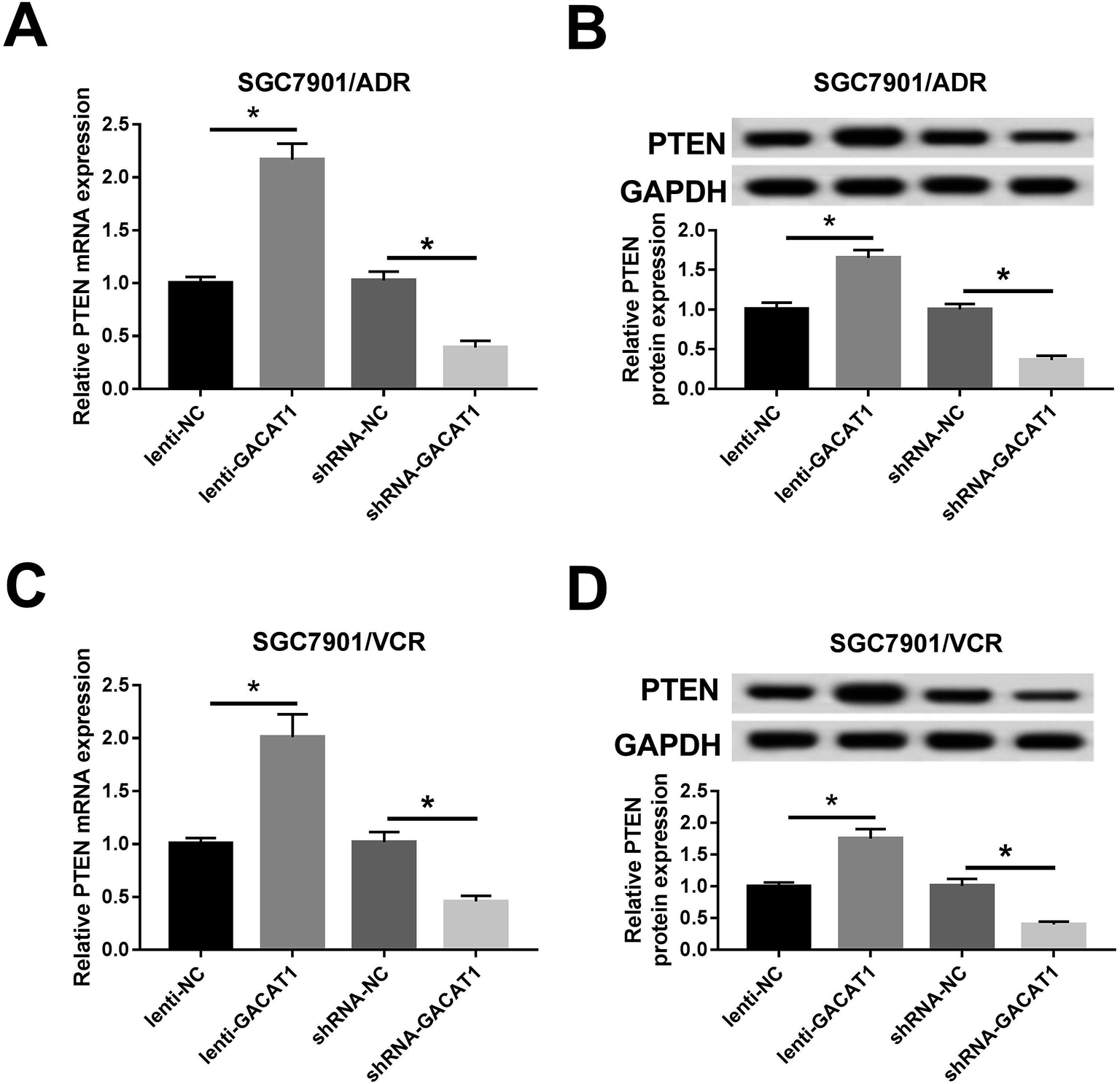 | ||
| Fig. 4 GACAT1 induced PTEN expression in SGC7901/ADR or SGC7901/VCR cells. (A–D) SGC7901/ADR or SGC7901/VCR cells were infected with lenti-NC, lenti-GACAT1, shRNA-NC or shRNA-GACAT1 for 24 h and then treated with ADR (2 μg ml−1) or VCR (4 μg ml−1) for another 48 h. Next, PTEN mRNA (A and C) and protein (B and D) levels were measured by RT-qPCR assay and western blot assay, respectively. *P < 0.05. | ||
GACAT1 attenuated ADR and VCR resistance by regulating PTEN/AKT/mTOR/S6K1 pathway in GC cells
Subsequently, we further demonstrated that cell viability was restored in GACAT1-overexpressed SGC7901/ADR or SGC7901/VCR cells following the depletion of PTEN when these cells were stimulated with same doses of ADR or VCR (Fig. 5A). Moreover, PTEN loss inhibited the increase of cell apoptotic percentage induced by GACAT in SGC7901/ADR and SGC7901/VCR cells (Fig. 5B). In a word, these outcomes manifested that PTEN knockdown abrogated the palliative effect of GACAT1 on ADR and VCR resistance in GC cells. Next, western blot assay further unveiled that PTEN loss abated the inhibitory effect of GACAT1 on AKT/mTOR/S6K1 pathway in SGC7901/ADR and SGC7901/VCR cells, as evidenced by the increase of p-AKT, p-mTOR and p-S6K1 levels in lenti-GACAT1-infected cells after the depletion of PTEN (Fig. 5C and D). Also, our study demonstrated that PTEN knockdown triggered the conspicuous increase of cell viability and the noticeable decrease of cell apoptotic percentage in SGC7901/ADR and SGC7901/VCR cells, suggesting that PTEN depletion potentiated ADR and VCR resistance in GC cells (ESI Fig. 3†). Moreover, PTEN silence induced the activation of AKT/mTOR/S6K1 signaling pathway in SGC7901/ADR and SGC7901/VCR cells (ESI Fig. 3†). In a word, these data disclosed that GACAT1 weakened ADR and VCR resistance by regulating PTEN/AKT/mTOR/S6K1 pathway in GC cells.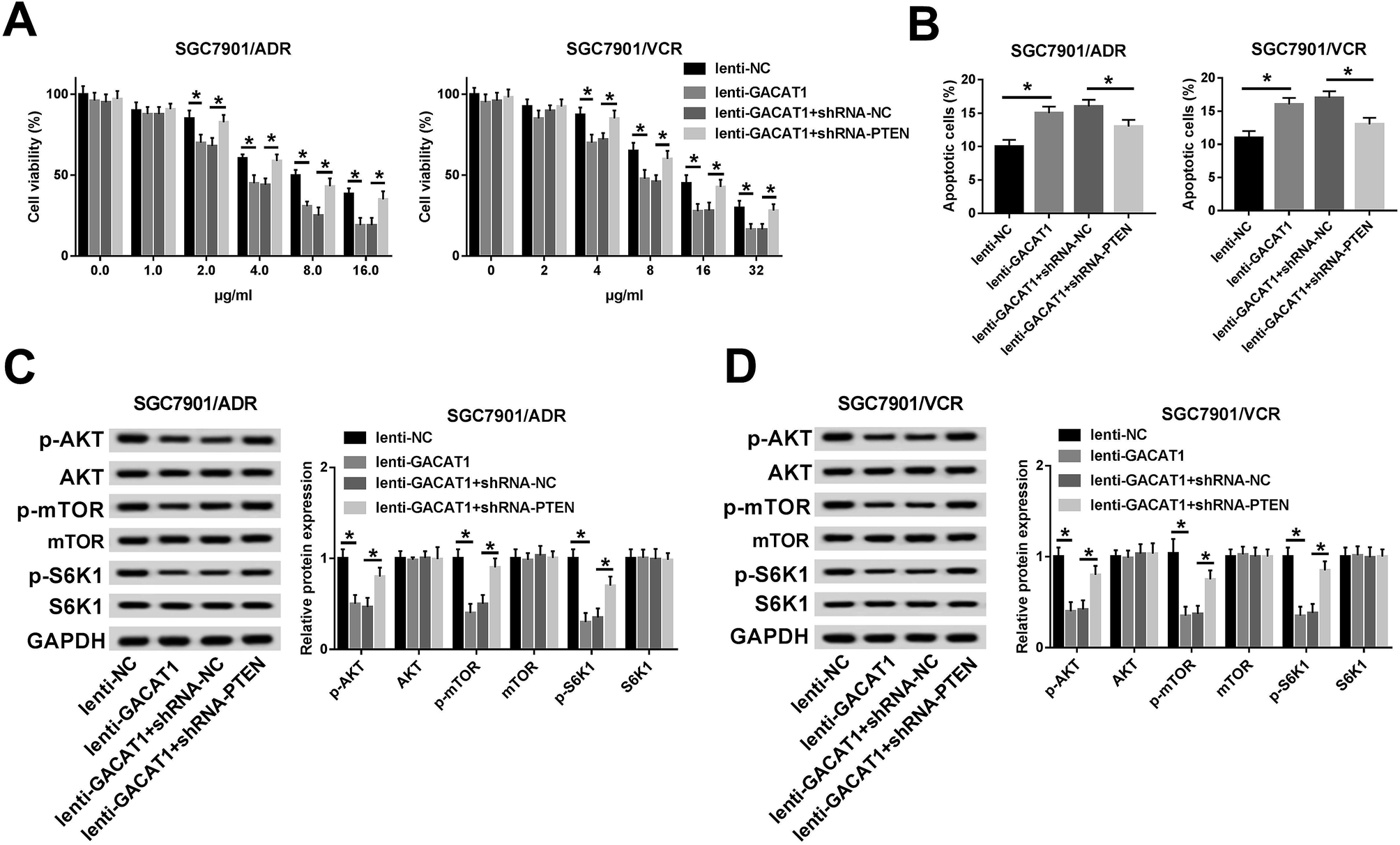 | ||
| Fig. 5 GACAT1 attenuated ADR and VCR resistance by regulating PTEN/AKT/mTOR/S6K1 signaling pathway in GC cells. (A) SGC7901/ADR or SGC7901/VCR cells were infected with lenti-NC, lenti-GACAT1, lenti-GACAT1 + shRNA-NC, or lenti-GACAT1 + shRNA-PTEN for 24 h and then treated with various doses of ADR or VCR for another 48 h, followed by the measurement of cell viability through CCK-8 assay. (B–D) SGC7901/ADR or SGC7901/VCR cells were infected with lenti-NC, lenti-GACAT1, lenti-GACAT1 + shRNA-NC, or lenti-GACAT1 + shRNA-PTEN for 24 h and then treated with ADR (2 μg ml−1) or VCR (4 μg ml−1) for another 48 h. Following this, cell apoptotic percentage was examined through flow cytometry (B), protein levels of AKT, mTOR, p-AKT, p-mTOR, p-S6K1 and S6K1 were determined by western blot assay (C and D). *P < 0.05. | ||
GACAT1 overexpression inhibited tumor growth and mitigated ADR and VCR resistance in GC mouse xenograft models
Next, in vivo experiments further disclosed that GACAT1 overexpression, ADR introduction or VCR treatment resulted in the reduction of tumor volume and weight in GC mouse xenograft models (Fig. 6A and B). Moreover, GACAT1 upregulation potentiated ADR- or VCR-mediated anti-tumor effect in GC mouse xenograft models (Fig. 6A and B).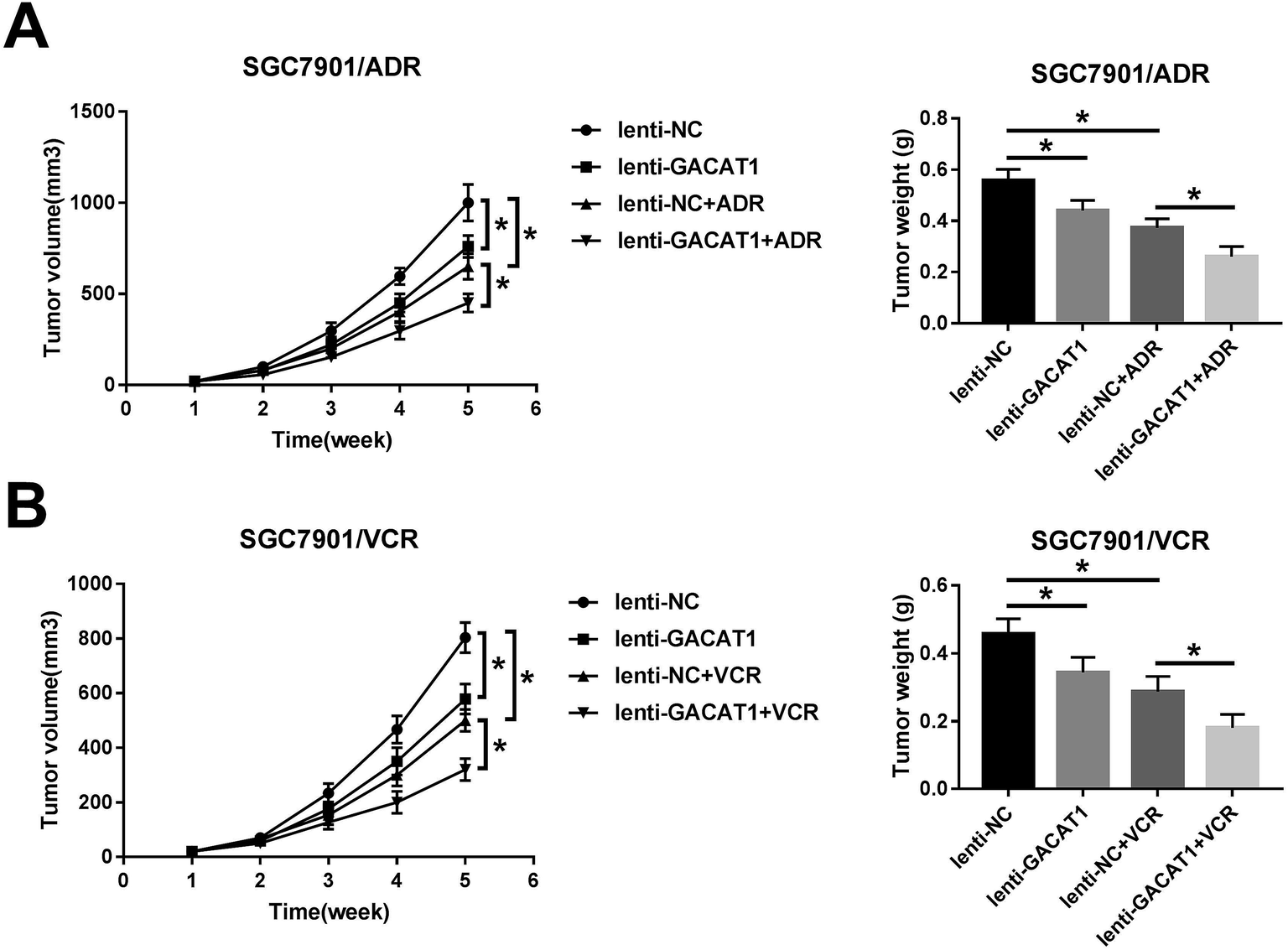 | ||
| Fig. 6 GACAT1 overexpression inhibited tumor growth and mitigated ADR and VCR resistance in GC mouse xenograft models. (A and B) SGC7901/ADR or SGC7901/VCR cells infected with lenti-NC or lenti-GACAT1 lentiviruses were subcutaneously injected into the right flanks of mice. At 1 weeks after cell injection, ADR (1 mg kg−1 body weight) or VCR (2 mg kg−1 body weight) or equal volume sodium chloride injection solution were intraperitoneally injected into mice every 4 days for 4 weeks. Then, tumor volume was measured every 1 week for a total of 5 weeks. Tumors were weighed at 5 weeks after cell injection. *P < 0.05. | ||
Discussion
GC is a huge threat for human health and life worldwide with the morbidity and mortality respectively ranking the fifth and third in all cancers.1 Chemotherapy is an effective approach to improve survival time and life quality of GC patients, while side effects and resistance of chemotherapeutic agents greatly limit their usage in the treatment for cancers.5,23 Recently, accumulating non-coding RNAs and genes have been found as central regulators in cancer tumorigenesis and chemoresistance.24,25In the present study, we demonstrated that GACAT1 was lower expressed in GC tissues and GACAT1 overexpression inhibited the growth of GC xenograft tumors in vivo. Similarly, Sun et al. disclosed that GACAT1 expression was significantly downregulated in GC tissues and was associated with GC metastasis and differentiation.21 However, some documents pointed out that GACAT1 expression was strikingly upregulated in GC tissues compared with adjacent normal tissues.20,22 And, GACAT1 overexpression promoted cell proliferation, migration and invasion by downregulating miR-378 and regulating miR-149/zinc finger and BTB domain containing 2 (ZBTB2) and miR-149/SP1 pathways in GC.22,26 In this text, the main aim of our study was to investigate whether GACAT1 could regulate ADR or VCR resistance in GC, as well as related molecular mechanisms.
Our data firstly revealed that GACAT1 level was strikingly reduced in ADR- or VCR-resistant GC cells (SGC7901/ADR or SGC7901/VCR) than that in parental cells (SGC7901), suggesting the link of GACAT1 and ADR or VCR resistance in GC. Moreover, our study further demonstrated that GACAT1 overexpression could weaken the resistance of GC cells to ADR and VCR. Inversely, GACAT1 knockdown potentiated ADR or VCR resistance in GC cells. Moreover, in vivo experiments disclosed that ectopic expression of GACAT1 enhanced ADR- or VCR-mediated anti-tumor effect in GC xenograft models. That was to day, GACAT1 upregulation relieved the resistance of GC cells to ADR and VCR in vitro and in vivo.
AKT/mTOR pathway has been well documented as a key signaling platform in orchestrating a wide range of cellular and biological processes such as proliferation, metabolism and survival.27,28 Moreover, AKT/mTOR pathway is frequently dysregulated in many malignancies and its activation can promote tumor initiation and progression as well as enhance the resistance of chemotherapeutic agents.29–31 Additionally, previous studies showed that AKT, p-AKT, p-mTOR, and their downstream targets were highly expressed in GC tissues, indicating that AKT/mTOR pathway was activated in GC.32,33 And, AKT/mTOR pathway has been proposed as potential therapeutic targets for GC due to their crucial functions in GC tumorigenesis and chemoresistance.34,35
PTEN, a negative regulator of AKT/mTOR pathway, has been well known as a tumor suppressor in numerous malignances including GC.36–38 Moreover, PTEN expression and function was closely associated with chemoresistance,38,39 and loss of PTEN function was a potential mechanism of AKT-mediated chemoresistance in GC.40 Additionally, previous studies revealed that the suppression of PI3K/AKT pathway ameliorated ADR and VCR resistance in GC.41–43 Hence, the link of GACAT1, PTEN/AKT/mTOR pathway and ADR or VCR resistance was further explored in GC cells.
Our outcomes showed that GACAT1 overexpression inactivated AKT/mTOR/S6K1 signaling pathway in SGC7901/ADR or SGC7901/VCR cells. And, GACAT1 knockdown or PTEN loss activated AKT/mTOR/S6K1 signaling pathway in SGC7901/ADR or SGC7901/VCR cells. Moreover, PTEN expression was markedly upregulated in GACAT1-overexpressed SGC7901/ADR or SGC7901/VCR cells, but was remarkably downregulated in GACAT1-depleted cells. And, PTEN knockdown improved ADR and VCR resistance by activating AKT/mTOR/S6K1 signaling pathway in GC cells. Furthermore, we further demonstrated that GACAT1 mitigated the resistance of GC cells to ADR and VCR by PTEN/AKT/mTOR/S6K1 regulatory pathway in vitro.
Taken together, our data revealed that GACAT1 suppressed GC tumorigenesis and alleviated the resistance of GC cells to ADR and VCR by regulating PTEN/AKT/mTOR/S6K1 signaling pathway, proving a deep insight into the roles and molecular basis of GACAT1 in modulating ADR and VCR resistance and hinting the potential values of GACAT1 in the mitigation of multidrug resistance in GC. Also, our outcomes might contribute to the further development of treatment for GC due to the inhibitory effects of GACAT1 on GC tumorigenesis and chemoresistance. However, the downstream targets and other regulatory pathways of GACAT1 need to be further delved. Moreover, it is indispensable to perform a more thorough investigation on the direct or indirect interaction between GACAT1 and PTEN. Also, it is requisite to further explore the functions and regulatory mechanisms of GACAT1 in GC tumorigenesis and progression.
Ethics approval and consent to participate
Thirty-five GC patients without any therapy were recruited in the First Affiliated Hospital of Zhengzhou University with written informed consents from all subjects. Our study was approved by Institutional Review Board (IRB) of the First Affiliated Hospital of Zhengzhou University. Our animal experiments were approved by the Institutional Committee for Animal Research of the First Affiliated Hospital of Zhengzhou University. All experiments were performed in compliance with the guidelines of the First Affiliated Hospital of Zhengzhou University.Conflicts of interest
All author declare that they have no financial and non-financial conflicts of interest in this research.Acknowledgements
This study was approved by the Science and Technology Research Key Project of Henan Provincial Education Department (Grant no 17A320045).References
- F. Bray, J. Ferlay, I. Soerjomataram, R. L. Siegel, L. A. Torre and A. Jemal, Ca-Cancer J. Clin., 2018, 68, 394–424 CrossRef PubMed.
- E. Van Cutsem, X. Sagaert, B. Topal, K. Haustermans and H. Prenen, Lancet, 2016, 388, 2654–2664 CrossRef CAS.
- J. A. Ajani, T. A. D'Amico, K. Almhanna, D. J. Bentrem, J. Chao, P. Das, C. S. Denlinger, P. Fanta, F. Farjah and C. S. Fuchs, J. Natl. Compr. Cancer Network, 2016, 14, 1286–1312 CrossRef.
- M. Riihimäki, A. Hemminki, K. Sundquist, J. Sundquist and K. Hemminki, Oncotarget, 2016, 7, 52307–52316 CrossRef PubMed.
- K. Harada, D. Mizrak Kaya, Y. Shimodaira and J. A. Ajani, Gastric Cancer, 2017, 20, 92–101 CrossRef CAS PubMed.
- A. D. Wagner, N. L. Syn, M. Moehler, W. Grothe, W. P. Yong, B. C. Tai, J. Ho and S. Unverzagt, Cochrane Database Syst. Rev., 2017, 8, Cd004064 Search PubMed.
- S. T. Pan, Z. L. Li, Z. X. He, J. X. Qiu and S. F. Zhou, Clin. Exp. Pharmacol. Physiol., 2016, 43, 723–737 CrossRef CAS PubMed.
- J. J. Marin, R. Al-Abdulla, E. Lozano, O. Briz, L. Bujanda, J. M. Banales and R. I. Macias, Anti-Cancer Agents Med. Chem., 2016, 16, 318–334 CrossRef CAS PubMed.
- M. Orditura, G. Galizia, V. Sforza, V. Gambardella, A. Fabozzi, M. M. Laterza, F. Andreozzi, J. Ventriglia, B. Savastano, A. Mabilia, E. Lieto, F. Ciardiello and F. De Vita, World J. Gastroenterol., 2014, 20, 1635–1649 CrossRef CAS PubMed.
- R. O. Carlson, Expert Opin. Invest. Drugs, 2008, 17, 707–722 CrossRef CAS PubMed.
- S. U. Schmitz, P. Grote and B. G. Herrmann, Cell. Mol. Life Sci., 2016, 73, 2491–2509 CrossRef CAS PubMed.
- Y. Tang, B. B. Cheung, B. Atmadibrata, G. M. Marshall, M. E. Dinger, P. Y. Liu and T. Liu, Cancer Lett., 2017, 391, 12–19 CrossRef CAS PubMed.
- A. Bhan, M. Soleimani and S. S. Mandal, Cancer Res., 2017, 77, 3965–3981 CrossRef CAS PubMed.
- M. Nasrollahzadeh-Khakiani, M. Emadi-Baygi, W. A. Schulz and P. Nikpour, Briefings Funct. Genomics, 2017, 16, 129–145 CAS.
- M. Majidinia and B. Yousefi, DNA Repair, 2016, 45, 25–33 CrossRef CAS PubMed.
- M. E. Askarian-Amiri, E. Leung, G. Finlay and B. C. Baguley, Methods Mol. Biol., 2016, 1395, 207–227 CrossRef CAS PubMed.
- Z. Zhou, Z. Lin, Y. He, X. Pang, Y. Wang, M. Ponnusamy, X. Ao, P. Shan, M. A. Tariq and P. Li, Mol. Ther.--Nucleic Acids, 2018, 12, 405–419 CrossRef CAS PubMed.
- C. Shang, Y. Guo, J. Zhang and B. Huang, Cancer Chemother. Pharmacol., 2016, 77, 1061–1067 CrossRef CAS PubMed.
- B. Xiao and J. Guo, Tumor Biol., 2013, 34, 3271 CrossRef PubMed.
- T. Xia, Q. Liao, X. Jiang, Y. Shao, B. Xiao, Y. Xi and J. Guo, Sci. Rep., 2014, 46088, DOI:10.1038/srep06088.
- W. Sun, Y. Wu, X. Yu, Y. Liu, H. Song, T. Xia, B. Xiao and J. Guo, Tumor Biol., 2013, 34, 2697–2701 CrossRef CAS PubMed.
- D. Wang, Y. Ji, W. Li, Y. Yu and C. Liu, Int. J. Clin. Exp. Pathol., 2017, 10, 6364–6374 Search PubMed.
- S. M. Kim and S. H. Park, World J. Gastroenterol., 2015, 21, 8811–8816 CrossRef PubMed.
- D. Ayers and J. Vandesompele, Genes, 2017, 8, 95 CrossRef PubMed.
- H. Xia and K. M. Hui, Curr. Med. Chem., 2014, 21, 3029–3041 CrossRef CAS PubMed.
- X. Shi, X. Wang and Y. Hua, J. Cancer, 2018, 9, 3715–3722, DOI:10.7150/jca.27546.
- A. Ghigo, F. Morello, A. Perino and E. Hirsch, Subcell. Biochem., 2012, 58, 183–213 CAS.
- T. Ersahin, N. Tuncbag and R. Cetin-Atalay, Mol. BioSyst., 2015, 11, 1946–1954 RSC.
- P. M. LoRusso, J. Clin. Oncol., 2016, 34, 3803–3815 CrossRef PubMed.
- H. J. Lim, P. Crowe and J. L. Yang, J. Cancer Res. Clin. Oncol., 2015, 141, 671–689 CrossRef CAS PubMed.
- H. A. Burris 3rd, Cancer Chemother. Pharmacol., 2013, 71, 829–842 CrossRef PubMed.
- O. Tapia, I. Riquelme, P. Leal, A. Sandoval, S. Aedo, H. Weber, P. Letelier, E. Bellolio, M. Villaseca, P. Garcia and J. C. Roa, Virchows Arch., 2014, 465, 25–33 CrossRef CAS PubMed.
- D. S. Byun, K. Cho, B. K. Ryu, M. G. Lee, J. I. Park, K. S. Chae, H. J. Kim and S. G. Chi, Int. J. Cancer, 2003, 104, 318–327 CrossRef CAS PubMed.
- K. Almhanna, J. Strosberg and M. Malafa, Anticancer Res., 2011, 31, 4387–4392 CAS.
- S. E. Al-Batran, M. Ducreux and A. Ohtsu, Int. J. Cancer, 2012, 130, 491–496 CrossRef CAS PubMed.
- M. S. Song, L. Salmena and P. P. Pandolfi, Nat. Rev. Mol. Cell Biol., 2012, 13, 283–296 CrossRef CAS PubMed.
- L. L. Zhang, J. Liu, S. Lei, J. Zhang, W. Zhou and H. G. Yu, Cell. Signalling, 2014, 26, 1011–1020 CrossRef CAS PubMed.
- W. T. Xu, Z. Yang and N. H. Lu, Asian Pac. J. Cancer Prev., 2014, 15, 17–24 CrossRef PubMed.
- S. Zhang and D. Yu, Clin. Cancer Res., 2010, 16, 4325–4330 CrossRef CAS PubMed.
- E. Oki, H. Baba, E. Tokunaga, T. Nakamura, N. Ueda, M. Futatsugi, K. Mashino, M. Yamamoto, M. Ikebe, Y. Kakeji and Y. Maehara, Int. J. Cancer, 2005, 117, 376–380 CrossRef CAS PubMed.
- X. Xie, B. Tang, J. Zhou, Q. Gao and P. Zhang, Oncol. Rep., 2013, 30, 773–782 CrossRef CAS PubMed.
- W. Zhou, X. Fu and H. Yu, Xibao Yu Fenzi Mianyixue Zazhi, 2017, 33, 1354–1359 Search PubMed.
- H. G. Yu, Y. W. Ai, L. L. Yu, X. D. Zhou, J. Liu, J. H. Li, X. M. Xu, S. Liu, J. Chen and F. Liu, Int. J. Cancer, 2008, 122, 433–443 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra10030f |
| This journal is © The Royal Society of Chemistry 2019 |