
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multi-scale instrumental analyses of plasticized polyhydroxyalkanoates (PHA) blended with polycaprolactone (PCL) and the effects of crosslinkers and graft copolymers†
Masakazu Nishida *a,
Tomoko Tanakaa,
Yoshio Hayakawaa,
Takashi Ogurab,
Yoshiaki Itob and
Masahiro Nishidab
*a,
Tomoko Tanakaa,
Yoshio Hayakawaa,
Takashi Ogurab,
Yoshiaki Itob and
Masahiro Nishidab
aNational Institute of Advanced Industrial Science and Technology (AIST), 2266-98 Shimoshidami, Moriyama-ku, Nagoya, 463-8560 Japan. E-mail: m-nishida@aist.go.jp; Fax: +81 52 736 7406; Tel: +81 52 736 7493
bNagoya Institute of Technology, Gokiso-cho, Showa-ku, Nagoya, Aichi 466-8555, Japan
First published on 11th January 2019
Abstract
Details of the mechanism underlying the tensile properties of plasticized polyhydroxyalkanoates (PHA) including poly(butylene succinate) (PBS) were investigated by blending with poly(ε-caprolactone) (PCL) as well as the addition of compatibilizers. Multi-scale instrumental analyses employed micro-focus X-ray CT to provide micro-scale morphology information on the order of ten microns while solid-state NMR spectral and relaxation time analyses contributed knowledge of the environment and molecular mobility of each constituent at the molecular to nano-scale. The blend of plasticized PHA with 50% PCL adopted a sea-island morphology to improve elongation at break in a quasi-static tensile test, which was dominated by the tensile properties of the added PCL. However, impact tensile properties were less improved by PCL addition, because its molecular mobility was suppressed by blending. Meanwhile, peroxy crosslinkers changed the sea-island morphology to homogenous in X-ray CT observations. Although the homogenous morphology sharply lowered the elongation at break in a quasi-static tensile test, the homogenous morphology improved impact tensile properties. Furthermore, graft polymers having acrylonitrile–styrene side-chains did not change the sea-island morphology but increased the molecular mobility of PBS in the plasticized PHA. This weak interaction between the plasticized PHA and PCL improved tensile properties in both quasi-static and impact tensile tests.
Introduction
As part of the solution to global climate problems for a sustainable society, it is desirable to replace petroleum-based polymers with biodegradable equivalents. Polyhydroxyalkanoates (PHA), which are made by a microbial process, have the advantage of consuming non-edible materials as the feedstocks.1 PHA is a family of biopolyesters commonly accumulated by many bacteria, consisting of the following polyesters: poly-3-hydroxybutylate (PHB), poly-3-hydroxyvalerate (PHV), as well as their isomeric polymers and copolymers. The control of chemical structure of the PHA homopolymers and copolymers in both microbial and chemical production is still under development.2 Applications of PHA have extended to various industrial fields with developments in characterization and production. In particular, PHA is useful in medical sectors, such as tissue engineering, bio-implants, drug delivery, and surgical materials, as well as in nanotechnology.3 Other industrial applications include packaging, moulded goods, paper coatings, non-woven fabrics, adhesives, films, and performance additives.4 Considering these applications, the chemomechanical properties of PHA, which are influenced by polymer composition and microstructure, have been investigated.5Chemical modification approaches can alter the characteristics of PHA, such as its mechanical properties, surface structure, amphiphilic character, and rate of degradation.6 Hydroxylation, carboxylation, epoxidation, and chlorination in particular have been considered for use in biomedical applications.7 Blending of PHA with other polymers is the most important method for controlling its thermal and crystallization behaviour and morphology which are closely related to mechanical properties and biodegradability.8 The morphology of poly(3-hydroxybutyrate-co-3-hydroxyvalerate) (PHBV)/polylactide (PLA) blends was studied to assess the correlation to thermal, rheological and barrier properties.9 Depending on the application, bio-based PLA/PHB blend can be manufactured in various shapes. Plasticized PLA/PHB blends with an oligomer of lactic acid, which was extruded for the film formation process, showed a decrease in the glass transition temperature while maintaining the mechanical properties.10 The plasticized PLA/PHB blend could be also produced as flexible fibres by the electrospinning method and the blend ratio affected the biodegradability of the electrospun fibres in composting conditions due to their crystallinities.11 In order to improve melt processability and thermomechanical properties, PHA was blended with poly(butylene adipate-co-terephthalate) (PBAT) by melt extrusion to demonstrate that an acid wash dramatically improved processability.12 Binary blends for PHB and poly(ε-caprolactone) (PCL) blend that were manufactured by a twin screw co-rotating extruder and were subsequently injection moulded showed impact that depended on the PCL content.13
Since PHA is brittle and hydrophilic, it has disadvantages for processability and a small amount of compatibilizer has sometimes been used in blending PHA; the morphology and mechanical properties of the PHA-based blend were changed by compatibilizers, including crosslinkers and graft copolymers. The reactive polymer blending technique changed the physicochemical interactions between the constituent polymers because of in situ formation of the compatibilizing agents, such as a branching/crosslinking copolymer and a graft copolymer.14 An epoxy compatibilizer reacted with the OH group of poly(3-hydroxybutyrate-co-3-hydroxyhexanoate) (PHBH) and the OH/COOH groups of PLA to improve the elongation at break and impact strength of the PHBH/PLA blend.15 Dicumyl peroxide, which acted as a free-radical grafting initiator, compatibilized PHBV/PBS and PHA/PBS blends to ameliorate tensile strength, impact toughness, and elongation at break of injection-moulded blends.16 Grafting maleic anhydride improved the miscibility of a PHBV/PBS blend with incorporation of sepiolite because of a synergistic effect induced by both compatibilizer and filler.17 Starch-based materials can be used as compatibilizer for melt-blending with PHA and PBAT, inhibiting the secondary crystallization of the PHA component in the cast films. Such approaches show promise for flexible packing materials.18
As shown above, the blending of PHA as well as the compatibilizing of PHA-based blends obviously improved the material characteristics of PHA; however, in order to know the details of the mechanism of their functional expression, an analytical method that covers molecular- to nano-scale length scales is necessary. Solid-state NMR is a useful analytical method in particular for both synthetic and natural polymers over these small-scale orders; it has provided significant information about molecular structures and dynamics for amorphous and crystalline phases of PHB19 as well as PHB-based copolymers and blends.20,21 Despite the correlation of microscopic properties to not only the morphology, but also to the mechanical properties of PHA and related materials, solid-state NMR has not yet been applied to analyses of structures and dynamics from the molecular- to nano-scale orders. Using the multi-scale instrumental analyses that range from molecular- to nano-scale (solid-state NMR) to micro-scale (morphology by SEM observation), we have evaluated the material characteristics of biomass polymers, for example, changes of biomass constituents of soft wood22 and other plant materials.23
The present study aims to extend multi-scale instrumental analyses to the multicomponent PHA system to reveal changes at the molecular- to micro-scales that contribute to their mechanical properties. Our preceding paper has revealed the influence of molecular mobility on the compatibility of PHA and PBS for plasticized PHA (commercially available PHA4422P), using solid-state NMR spectral and relaxation time analyses not only at ambient and variable temperatures.24 In a quasi-static tensile test of this plasticized PHA blending with PCL, the addition of 50% PCL dramatically increased the elongation at break and the specimens became pours shape.25 In a dynamic tensile test using a split Hopkinson bar (SHPB) method, a blend consisting of 50% plasticized PHA and 50% PCL gave small elongation at break, which increased with the addition of compatibilizers.26 Here, we focus on plasticized PHA (PHA4422P) blended with various amounts of PCL and with various compatibilizers (crosslinkers and graft polymers). By combining multi-scale instrumental analyses including solid-state NMR spectral and relaxation time analyses and micro-focus X-ray computed tomography (CT) with newly collected systematic tensile test data, the details of the mechanism behind their mechanical properties have been elucidated.
Experimental
Materials
The plasticized PHA pellet, PHA4422P, was purchased from G5 Manufacturing, Singapore. The PHA4422P pellet consisted of 65% PHA, 30% PBS, and 5% crosslinking reagent. The PCL pellet, Capa 6800, was purchased from Perstorp Japan Co. Ltd., Tokyo, Japan. The crosslinking reagents, PERHEXA 25B (liquid, over 90% purity) and PERHEXA 25B-40 (mixed with silica as a white solid, 40% purity), were purchased from NOF CORPORATION, Tokyo, Japan. The graft polymers, Modiper A4400 and Modiper CL430-G, were purchased from NOF CORPORATION, Tokyo, Japan. Modiper A4400 was an ethylene–glycidyl methacrylate copolymer with acrylonitrile–styrene copolymer. Modiper CL430L was polycarbonate with a functional group-modified acrylonitrile–styrene copolymer. All materials were used without further purification. Raw pellets and compatibilizers (1% crosslinkers and 1% graft polymers) were melt-mixed at 175 °C for 20 min at a rotor speed of 50 rpm with a conventional melt-mixer. The mixed sample was moulded into plates of 5 mm thicknesses at 175 °C and 30 MPa for 30 min with a hot press.Static tensile test
Static tensile test specimens with a gauge area of 5 mm × 2 mm and a gauge length of 10 mm were made from the plates using machine processing. Static tensile tests were carried out at a crosshead speed of 0.2 mm min−1 using a universal testing machine. Nominal strain was obtained by images of the gauge length, taken by a digital camera in the elastic region and by the displacement of the crosshead in the plastic region. Nominal stress was calculated using the output of load cell at the crosshead.Dynamic tensile test
Dynamic tensile test specimens with a gauge area of 2 mm × 5 mm and a gauge length of 4 mm were made from the plates using machine processing. At high strain rates, the dynamic properties of the specimens were examined by the tensile split Hopkinson bar test.27 The diameters and lengths of input and output bars were 12 mm and 2000 mm respectively. Strain gauges were applied to both sides of the input and output bars at distances of 1750 mm and 350 mm from the specimen, respectively.Micro-focus X-ray CT observations
Micro-focus X-ray images were recorded with a FLEX-M345CT (Beamsense Co. Ltd., Osaka, Japan) instrument at an acceleration voltage of 40 kV and a tube current of 100 μA. The sample was rotated over 180° at 0.5° steps and two transmission images (the pixel size is 2.12 μm × 2.12 μm to 3.07 μm × 3.07 μm) were acquired using a charge-coupled device (CCD) with the exposure time of 1500 ms. The CT images were obtained by slicing the 3D image reconstructed from the transmission images.Solid-state NMR spectrum measurements
Magic angle spinning (MAS) nuclear magnetic resonance (NMR) spectra were measured on a Varian 400 NMR system spectrometer (Palo Alto, CA) with a Varian 4 mm double-resonance T3 solid probe. The samples were placed in a 4 mm ZrO2 rotor spun at 15 kHz over a temperature range of 20–22 °C. 1H MAS NMR spectra were measured at 399.86 MHz for the 1H nuclei and were collected with a 40 ms acquisition period over a 30.5 kHz spectral width in 16 transients, and a 3 s recycle delay. 13C MAS NMR spectra were collected with a 2.6 μs π/2 pulse at 100.56 MHz for the 13C nuclei and a 40 ms acquisition period over a 30.7 kHz spectral width. Proton decoupling was performed with an 86 kHz 1H decoupling radio frequency with a small phase incremental alteration (SPINAL) decoupling pulse sequence. Cross-polarization/magic angle spinning (CP-MAS) NMR spectra were measured with a 5.0 s recycle and 1024 transients delay, using a ramped-amplitude pulse sequence with a 2 ms contact time and a 2.5 μs π/2 pulse for the 1H nuclei. The amplitude of the 1H pulse was ramped up linearly from 90.5% of its final value during the cross polarization contact time. Pulse saturation transfer/magic angle spinning (PST-MAS) NMR was measured used the single π/2 pulse for the 13C nuclei with a 5 s recycle delay in 2048 transients after saturation of 1H nuclei with 13 consecutive 2.5 μs pulses and a 27.5 μs delay.Nuclei magnetic relaxation analyses
The 1H spin-lattice relaxation time in the laboratory frame (T1H) was indirectly measured via detection of 13C resonance enhanced by cross-polarization, applied after a π pulse to 1H nuclei with the inversion recovery method. The 13C spin-lattice relaxation time in the laboratory frame (T1C) was measured with the conventional Torchia's pulse sequence.28 The T1 analyses were performed with the same solid-state probe with the same contact time and acquisition period used for the 13C CP-MAS NMR spectrum. The 1H spin-lattice relaxation in the rotation frame (T1ρH relaxation) was examined by the observation of signal decay curve with variable contact time over a range of 100–12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 μs in CP-MAS NMR sequence.
000 μs in CP-MAS NMR sequence.
Results and discussion
Effects of PCL ratio and compatibilizers on tensile properties of plasticized PHA/PCL blends
As shown in our previous paper,25 the tensile strength and elongation at break in a quasi-static tensile test were significantly changed by addition of PCL to the plasticized PHA, PHA4422 (PHA/PBS copolymer), depending on the PCL ratio. In particular, over 50% PCL addition increased both the strength and elongation in the quasi-static test. In contrast, the value of the PCL ratio scarcely influenced the impact tensile test of the plasticized PHA/PCL blend. The different effects of the PCL ratio and compatibilizers between the quasi-static and impact tensile tests can be observed in Fig. 1. Comparing the quasi-static (Fig. 1A) and impact (Fig. 1B) tests, the 100% PCL specimen has a smaller elongation at break in the impact test than that in the quasi-static test. The plasticized PHA/PCL blend showed less difference with the PCL content in the impact test than in the quasi-static test, owing to the above tensile properties of PCL.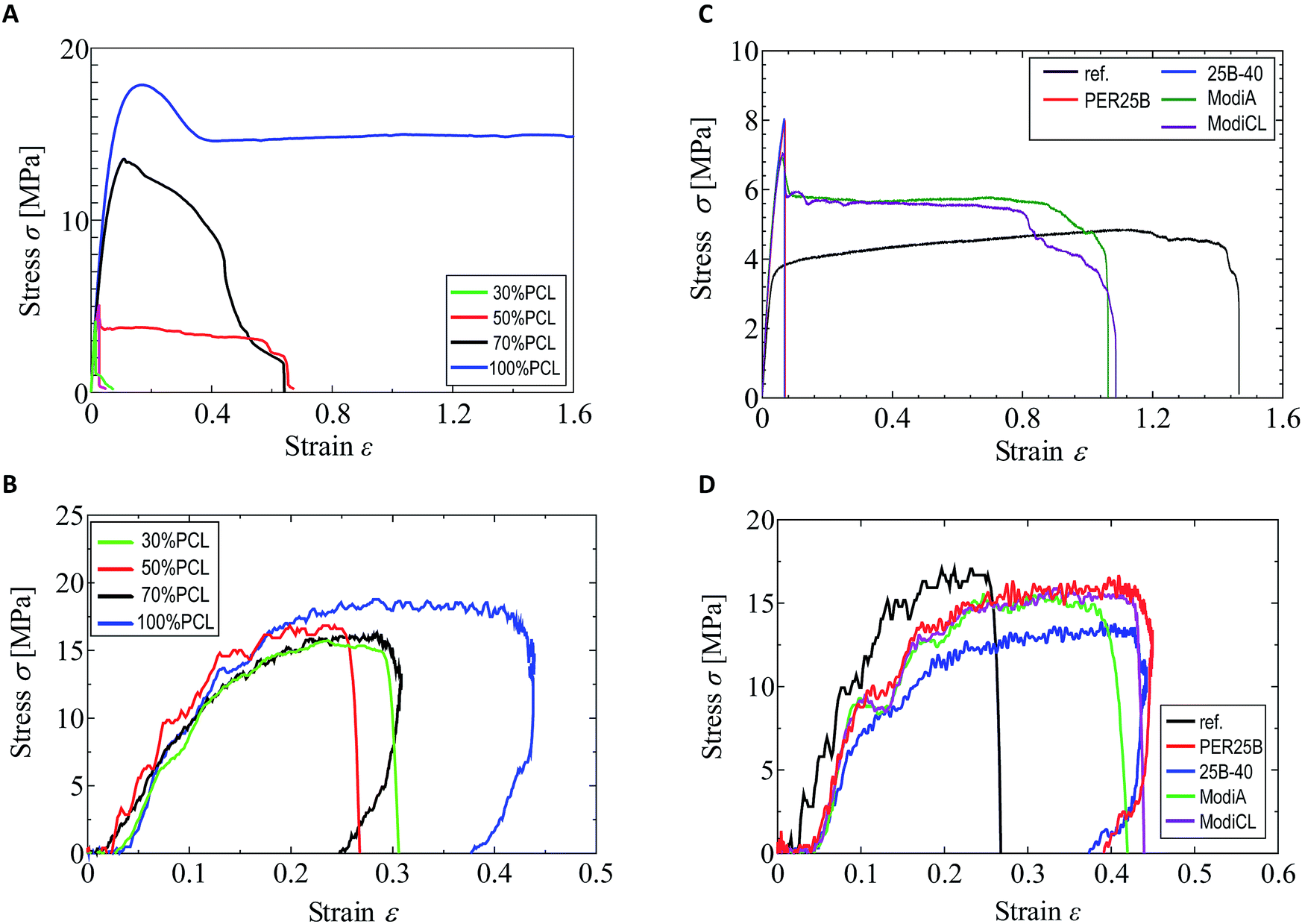 | ||
| Fig. 1 Quasi-static tensile test and impact tensile test of plasticized PHA/PCL blends. (A) Quasi-static (plasticized PHA/PCL blend). (B) Impact (plasticized PHA/PCL blend). (C) Quasi-static (50% PCL with compatibilizers). (D) Impact (50% PCL with compatibilizers). | ||
The tensile strength and elongation at break of the 50% plasticized PHA/50% PCL blend in the impact test noticeably increased with only 1% compatibilizers, such as the crosslinkers (PERHEXA 25B, PERHEXA 25B-40) and graft polymers (Modiper A4400, Modiper CL430L), as previously communicated.26 In the preliminary experiments, the addition amount of the compatibilizer was not greatly different between 1% and 5%. Thus, the addition amount of the compatibilizer was examined for 1% in this study. The additives produced different effects in the quasi-static (Fig. 1C) and impact (Fig. 1D) tests, as well as with changes of the PCL ratio. In the quasi-static test, the crosslinkers significantly decreased elongation at break while they increased tensile strength. The graft polymers in the quasi-static test also increased tensile strength, but they only slightly decreased elongation at break compared with the original blend.
Morphological observations of plasticized PHA/PLA blends with micro-focus X-ray CT
In order to examine changes of 50% plasticized PHA (PHA/PCL copolymer)/50% PCL blend due to the compatibilizers in terms of micro-scale order, morphological changes of specimens after quasi-static and impact tensile tests were observed using soft-focus X-ray CT for specimens having fractures of different shapes. The morphological difference of 50% plasticized PHA/50% PCL blend after the quasi-static and impact tests is shown in Fig. 2. One can see that a sea-island structure having relatively large island domains (<1000 μm) was observed in both specimens. In the specimen after quasi-static test (Fig. 2A), the island portion was broken to several sections while the sea portion was stretched with minute voids. This morphology after the quasi-static test corresponded to the porous structure previously observed in the visible and low magnification X-ray CT images.25 In contrast, after the impact test (Fig. 2B), both island and sea shapes were not deformed and showed almost no stretching. Since the 100% PCL specimen has a large elongation at break in the quasi-static test while it had a small elongation in the impact test, the sea portion was assigned as PCL and the island portion was assigned as PHA. | ||
| Fig. 2 Micro-focus X-ray CT images of 50% plasticized PHA/50% PCL blends. (A) Without compatibilizers after quasi-static test. (B) Without compatibilizers after impact test. (C) With PERHEXA25B after quasi-static test. (D) With Modiper A4400 after quasi-static test. | ||
Since the effect of compatibilizers appeared in the quasi-static tensile test more clearly, morphological changes of the 50% plasticized PHA/50% PCL blend due to the compatibilizers were examined for the specimens after the quasi-static test using micro-focus X-ray CT. The crosslinker (1% PERHEXA 25B) changed the sea-island structure of plasticized PHA/PCL blend into a homogenous morphology over tens of micron (Fig. 2C). In this case, neither visible cracks nor deformations were observed, being accountable for the very small elongation at break in the quasi-static test. Meanwhile, the graft polymer (1% Modiper A4400) maintained the sea-island structure of the plasticized PHA/PCL blend; however, both the island and sea regions were broken and elongation of sea portion was also observed at the same time (Fig. 2D). Therefore, the graft polymer enhanced the interaction between plasticized PHA and PCL resulting in the fracture of the sea portion consisting of PCL caused by low elongation of PHA.
1H MAS NMR spectra of plasticized PHA/PLA blends
Since 1H nuclei have large dipolar–dipolar interactions with each other, solid-state 1H NMR spectra have wide lines even when using magic-angle spinning (MAS), which can offset the anisotropy of dipoles. The line width of 1H MAS NMR spectra is affected by the spin–spin relaxation (T2 relaxation) which is suppressed by molecular motions. For example, water molecules had a narrower line width because of a longer T2 (greater molecular mobility) while biomass constituents in soft wood showed a wider line shape because of shorter T2 (less molecular mobility).29 Here, the molecular mobility involved in T2 relaxation could be monitored by 1H MAS NMR spectra of plasticized PHA/PCL blends.First, changes of 1H MAS NMR spectra of plasticized PHA/PCL blends with PCL ratio are shown in Fig. 3A. Although both pre-plasticized PHA and PBS copolymer had broad low-intensity signals as shown in Fig. S1 (ESI†), the plasticized PHA (PHA4422P) showed relatively sharp signals not only in the PHA region but also in the PBS region [Fig. 3A(1)]. Therefore, the plasticization of PHA with PBS enhanced molecular motions of both PHA and PBS because of the suppression of T2 relaxation. Like the plasticized PHA, flexible PCL showed sharp high-intensity signals [Fig. 3A(5)]. All plasticized PHA blends including 30%, 50%, and 70% PCL [Fig. 3A(2)–(4)] also showed similar line shapes and intensities to the plasticized PHA and flexible PCL. Thus, molecular mobility concerned with T2 relaxation was little changed by blending with flexible PCL because the copolymerization with PBS has already amplified the molecular mobility of the constituent polymers.
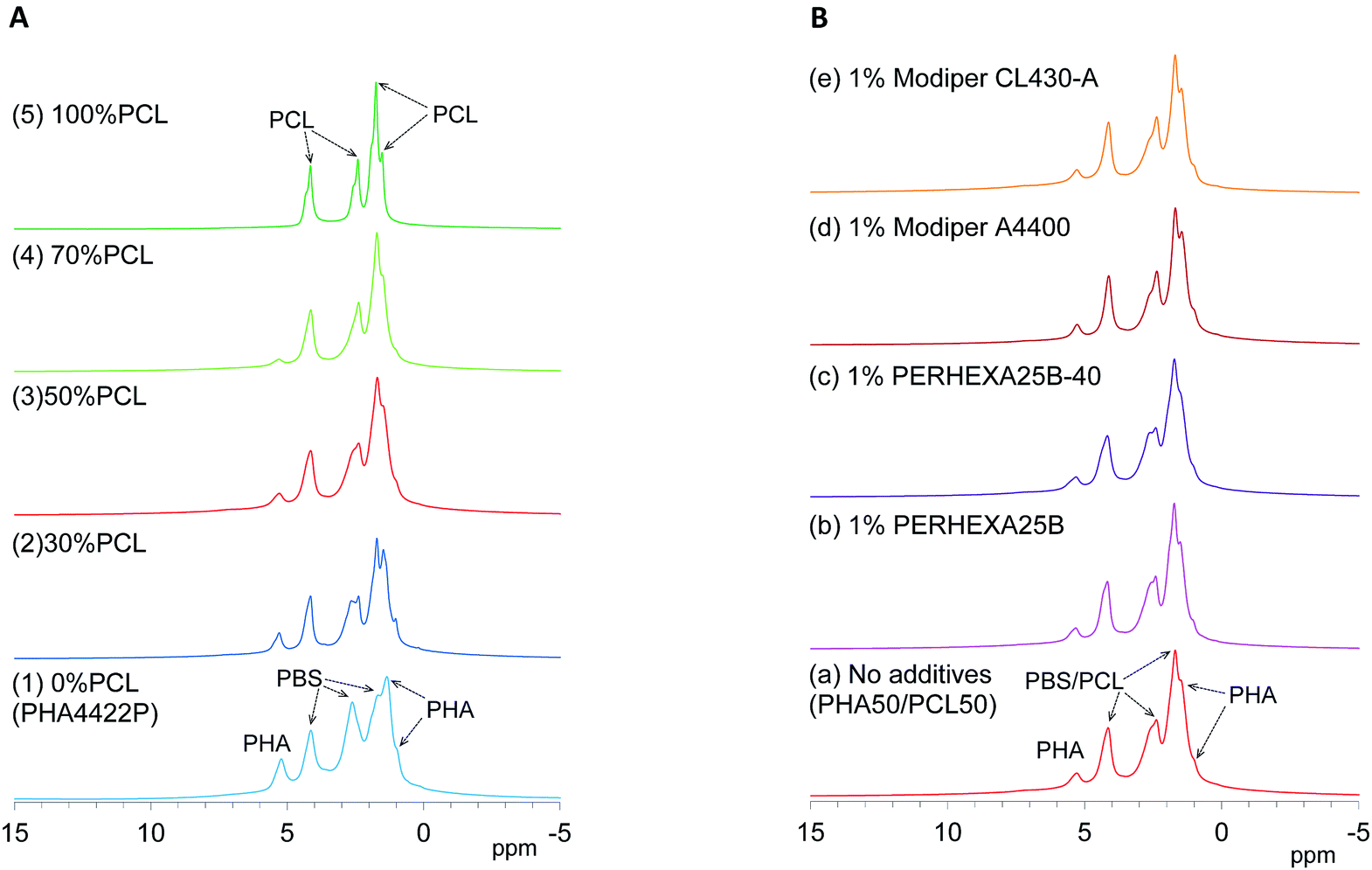 | ||
| Fig. 3 1H MAS NMR spectral changes of plasticized PHA/PCL blends. (A) PCL ratio. (B) Compatibilizers. | ||
Next, changes of 1H MAS NMR spectra of the 50% plasticized PHA/50% PCL blend with the addition of compatibilizers are shown in Fig. 3B. Based on the signal shapes and intensities of 50% plasticized PHA/50% PCL blend without additives [Fig. 3B(a)], the compatibilizers scarcely changed the 1H signal shape except that the addition of PERHEXA25B-40 slightly changed the amount of overlapping 1H signals [Fig. 3B(c)]. Therefore, changing the PCL ratio and the addition of both crosslinkers and graft polymers scarcely changed the molecular mobility involved in T2 relaxation.
13C MAS NMR spectra of plasticized PHA/PCL blends
We have already evaluated the crystallinity and mobility of the plasticized PHA copolymers and their constituent polymers using a combination of 13C CP-MAS NMR and 13C PST-MAS NMR methods.24 The former CP-MAS method enhances signals of rigid portions utilizing magnetization transfer from 1H nuclei to 13C nuclei. The latter PST-MAS method enhances signals of flexible portions near hydrogen atoms using the Nuclear Overhauser Effect (NOE).23 Here, molecular mobility changes due to the blending ratio and compatibilizers were examined by comparison of CP-MAS and PST-MAS signal intensities for each substituent.Fig. 4A shows changes of 13C CP-MAS NMR spectra of plasticized PHA/PCL blends with varying PCL content and Fig. 4B shows their 13C PST-MAS NMR spectra. As described in our previous report, the plasticized PHA (PHA4422P) gave sharp signals for the PHA and PBS constituents, which were readily assigned in the 13C CP-MAS NMR spectrum [Fig. 4A(1)]. Since PHA was more rigid than PBS, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and CH signals of the PHA moiety in the 13C PST-MAS NMR spectrum [Fig. 4B(1)] showed a lower ratio of plasticized PHA to PCL than did the 13C CP-MAS NMR spectrum. However, the CH3 signal of the PHA moiety in the 13C PST-MAS NMR spectrum had a high intensity in a similar manner to the PBS moiety because the CH3 group has a high molecular mobility even in rigid polymer like PHA. For flexible polymer such as PCL, the 13C CP-MAS NMR spectrum [Fig. 4A(5)] showed one CO and four CH2 signals, of which the highest field signal was assigned as an overlapped signal of two CH2 groups. In the 13C PST-MAS NMR spectrum [Fig. 4B(5)], the overlapped signal could be separated and the intensities of signals increased with the result that PCL showed six sharp signals.
O and CH signals of the PHA moiety in the 13C PST-MAS NMR spectrum [Fig. 4B(1)] showed a lower ratio of plasticized PHA to PCL than did the 13C CP-MAS NMR spectrum. However, the CH3 signal of the PHA moiety in the 13C PST-MAS NMR spectrum had a high intensity in a similar manner to the PBS moiety because the CH3 group has a high molecular mobility even in rigid polymer like PHA. For flexible polymer such as PCL, the 13C CP-MAS NMR spectrum [Fig. 4A(5)] showed one CO and four CH2 signals, of which the highest field signal was assigned as an overlapped signal of two CH2 groups. In the 13C PST-MAS NMR spectrum [Fig. 4B(5)], the overlapped signal could be separated and the intensities of signals increased with the result that PCL showed six sharp signals.
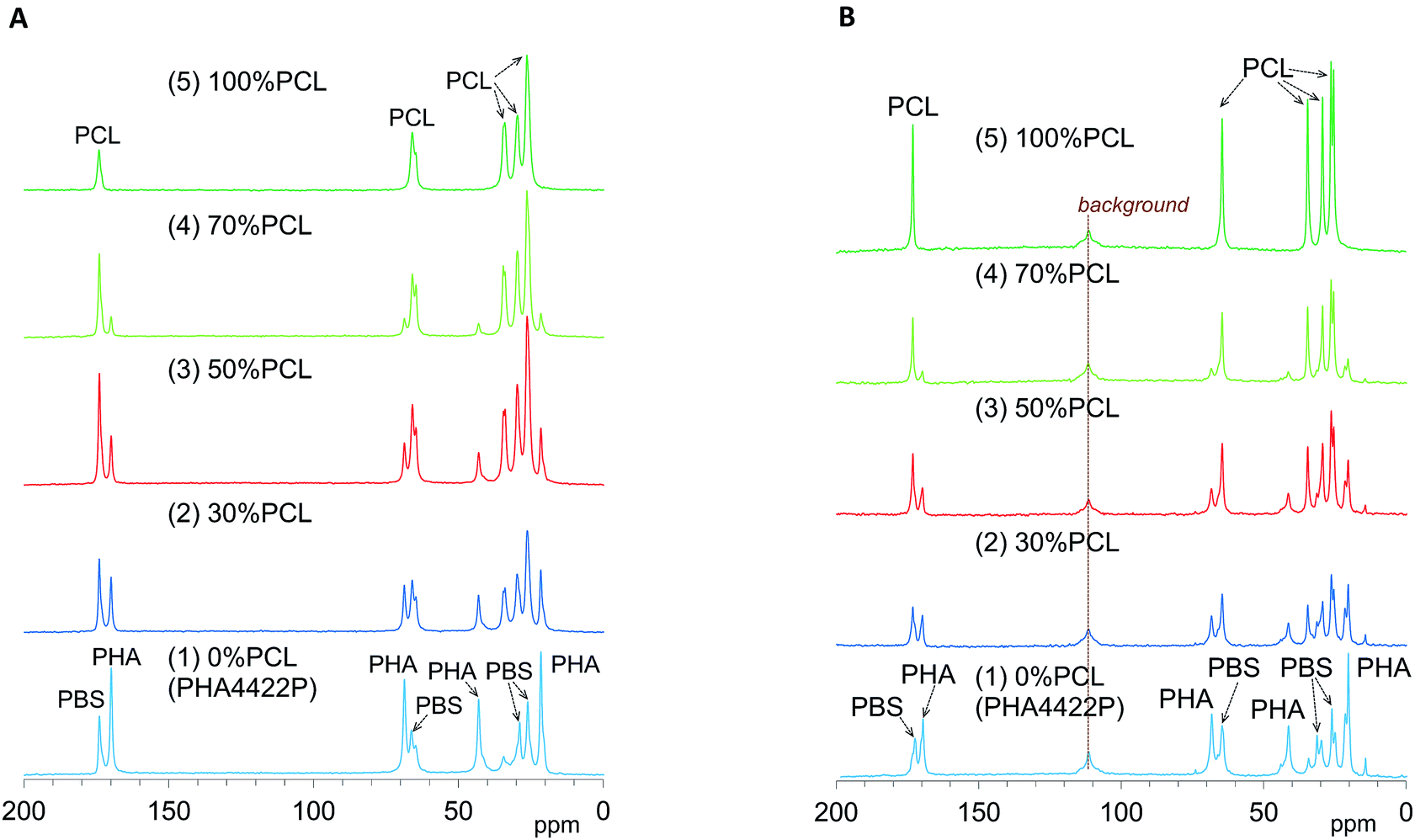 | ||
| Fig. 4 13C MAS NMR spectral changes of plasticized PHA/PCL blends with PCL ratio. (A) 13C CP-MAS NMR. (B) 13C PST-MAS NMR. | ||
In the 13C CP-MAS NMR spectra of the plasticized PHA/PCL blends [Fig. 4A(2)–(4)], the PCL signals except that at 34 ppm (α-CH2) overlapped with the signals derived from PBS in the plasticized PHA. In the 13C PST-MAS NMR spectra of the plasticized PHA/PCL blends [Fig. 4B(2)–(4)], the PCL signals except for α-CH2 still overlapped with the PBS signals. Therefore, the molecular mobility and dynamics of the plasticized PHA/PCL blend is best discussed using the PHA signals and the α-CH2 signal of PCL. At the same time, the molecular mobility and dynamics of PBS in the plasticized PHA/PCL blend can be discussed by comparison of the α-CH2 of PCL and other overlapped PCL/PBS signals. Although the PHA signals in both the 13C CP- and PST-MAS NMR spectra decreased with increasing the PCL content of the plasticized PHA/PCL, the ratio of PST-MAS signal to CP-MAS signal remained almost constant. In contrast, the 13C PST-MAS NMR signal of the α-CH2 of homopolymer (5) had a higher intensity than the 13C CP-MAS NMR signal. This ratio of PST-MAS signal to CP-MAS signal was reduced by blending with the plasticized PHA. Since PST-MAS emphasizes signals of flexible portions, this result indicates that the blending with the plasticized PHA suppressed the molecular mobility of PCL.
The effects of the compatibilizers on molecular mobility were also evaluated by the combination of 13C CP-MAS NMR and 13C PST-MAS NMR methods. Fig. 5A shows changes of the 13C CP-MAS NMR spectra of the 50% plasticized PHA/50% PCL blend with the compatibilizers and Fig. 5B their 13C PST-MAS NMR spectra. The addition of graft polymers (Modiper A4400, Modiper CL430-G) decreased the CO signal intensity of PBS/PCL in the 13C CP-MAS NMR spectra [Fig. 5A(d) and (e)], compared with the 13C PST-MAS NMR spectra [Fig. 5B(d) and (e)], even though the crosslinkers (PERHEXA 25B, PERHEXA 25B-40) exhibited smaller differences between CP-MAS [Fig. 5A(b) and (c)] and PST-MAS [Fig. 5B(b) and (c)]. The combination of the two 13C MAS NMR methods showed that the compatibilizers scarcely changed the molecular mobility involved in NOE except that the graft polymers slightly enhanced the molecular mobility of the CO groups of PBS/PCL.
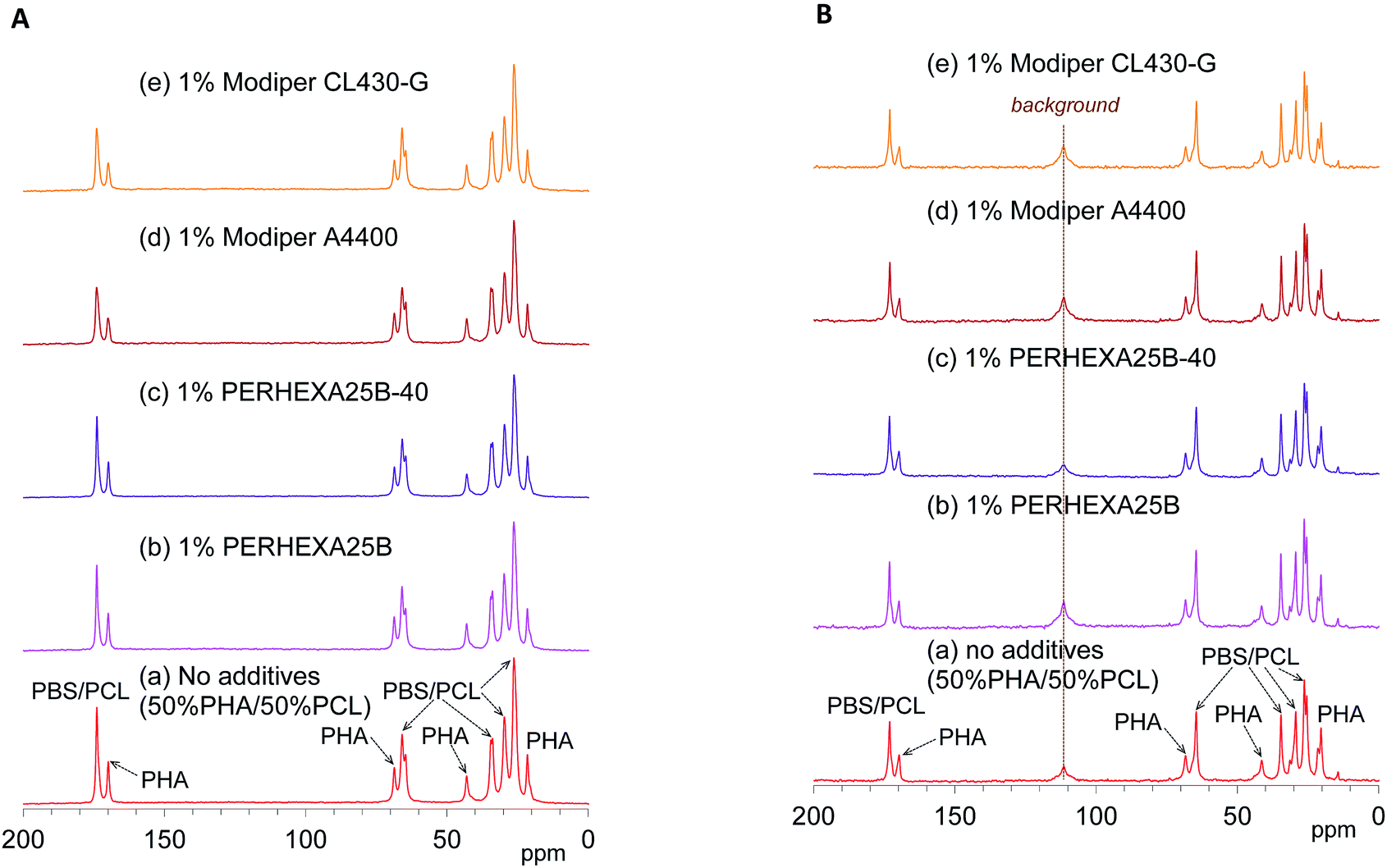 | ||
| Fig. 5 13C MAS NMR spectral changes of 50% plasticized PHA/50% PCL blend with compatibilizers. (A) 13C CP-MAS NMR. (B) 13C PST-MAS NMR. | ||
Spin-lattice relaxation in the laboratory frame of plasticized PHA/PCL blends
As shown in our preceding study, with increasing the measurement temperature, the T1H values of pre-plasticized PHA increased while the T1H values of PBS homopolymer decreased. In the plasticized PHA (PHA/PBS copolymer), however, the T1H values of the PHA moiety were shortened by the copolymerization with PBS at a higher temperature, because the T1H relaxation of PHA moiety was enhanced via PBS, which had shorter T1H value than the PHA moiety.24 On the other hand, the T1H value of PCL homopolymer decreased with increasing temperature as did that of the PBS homopolymer. As shown in Fig. S2 (ESI†), the PCL (5:100% PCL) showed shorter T1H values than the constituent polymers in the plasticized PHA (1:0% PCL).
First we examined the effects of the blending with PCL on the T1H value change for each substituent in the plasticized PHA/PCL (Fig. 6A). Even though PCL had shorter T1H values than the plasticized PHA, the T1H values of the PHA moiety in the plasticized PHA/PCL blends were increased by the addition of PCL. The T1H values of PHA moiety attained a maximum value at 50% PCL addition (3). According to the results of the micro-focus X-ray CT, the increase in the T1H values of the PHA moiety were caused by the change to the sea-island morphology. That is, the T1H relaxation of PHA, accelerated by the addition of PBS, was suppressed by the sea-island morphology. A similar situation applies to the PCL moiety (α-CH2) in the plasticized PHA/PCL blends, where T1H values increased with an increasing fraction of PHA (decreased PCL) because of the inhomogeneous morphology.
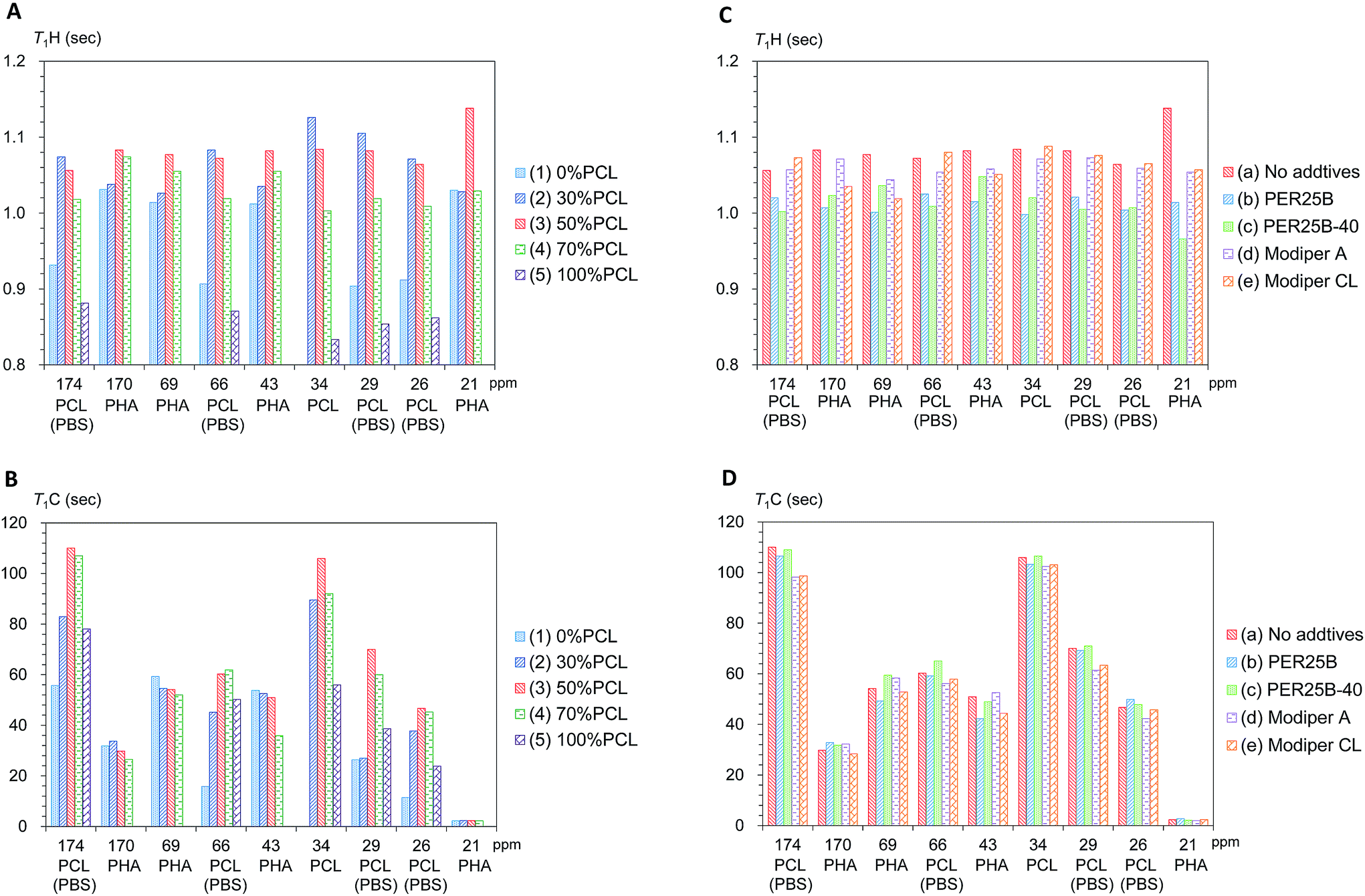 | ||
| Fig. 6 Changes of T1H and T1C values of plasticized PHA/PCL blends. (A) T1H changes with PCL ratio. (B) T1C changes with PCL ratio. (C) T1H changes with compatibilizers. (D) T1C changes with compatibilizers. | ||
The T1C value can provide information on molecular motions for each substituent because its value is less affected by 1H spin diffusion than the T1H value. Actually, the T1C change for each substituent as a function of the PCL content showed a different trend from the T1H change (Fig. 6B). The T1C values of the PHA moiety decreased with increasing PCL content: they attained minimum T1C values at 70% PCL addition (4). In contrast, the α-CH2 of PCL moiety obviously had a maximum T1C value at 50% PCL addition (3). As did the T1C values of pre-plasticized PHA (CH2),24 the T1C values of PCL homopolymer (5) decreased with increasing temperature (Fig. S3, ESI†). The T1C value provides information about rapid molecular motions (MHz order) of polymer constituents in plasticized PHA/PCL blends. Since the T1C values of all substituents except CH3 group decreased with increasing temperature, the molecular motions connected with these T1C values are associated with the long correlation time region (τcω0 ≫ 1).29 Therefore, for the components in plasticized PHA/PCL blends, the increase of T1C equates to a decrease of molecular mobility of a MHz order, which matches with the Larmor frequency of 13C nuclei. The T1C changes associated with blending with 50% PCL show that the sea-island morphology in plasticized PHA/PCL blends considerably decreased the molecular mobility of PCL. In summary, the 13C MAS NMR changes due to blending with PCL revealed that the T1C decrease of PHA in plasticized PHA/PCL blends was caused by the increase of the molecular mobility due to the interaction between PHA and PCL.
Next, changes of T1H values with the compatibilizers were examined for each polymer component in the 50% plasticized PHA/50% PCL blend (Fig. 6C). Interestingly, the long T1H values produced by the blending with PCL were shortened again by the addition of crosslinkers (PERHEXA 25B, PERHEXA 25B-40). The T1H values of the 50% plasticized PHA/50% PCL blend containing the crosslinkers had similar T1H values to those of the plasticized PHA without PCL. Meanwhile, the graft polymers (Modiper A4400, Modiper CL430-G) produced almost unchanged T1H values of the PCL and PBS moieties but slightly decreased T1H values of the PHA moiety. The morphology observed by micro-focus X-ray CT with addition of compatibilizers is closely related to the change of the T1H value. The crosslinkers produced a homogenous morphology that enhanced the T1H relaxation of the polymers constituents while the graft polymers maintained the sea-island structure, resulting only in a small change in the T1H relaxation of PHA.
The effect of compatibilizer on the T1C values showed marked changes from the T1H values, as shown in Fig. 6D. The crosslinkers (PERHEXA 25B, PERHEXA 25B-40) scarcely changed the T1C values of the 50% plasticized PHA/50% PCL blend, resulting in only small decreases for PHA (CH and CH2 groups) using PERHEXA 25B. The graft polymers (Modiper A4400, Modiper CL430-G) caused T1C decreases of the overlapped PCL/PBS signals in the 50% plasticized PHA/50% PCL blend. Although both crosslinker and graft polymer changed mechanical properties, the mechanism by which these changes were manifested differed between crosslinker and graft polymer. That is, the crosslinkers changed morphology uniformly while producing little change in the molecular mobility of PHA, while the graft polymers increased the molecular mobility of PBS in the blend without changing the morphology.
Spin-lattice relaxation in the rotational frame of plasticized PHA/PCL blends
Due to the cross-polarization produced by the CP-MAS pulse sequence, the 13C signal increased with the magnetization transfer from 1H nuclei to 13C nuclei and, at the same time, the 13C signal decreased with the 1H spin-lattice relaxation in the rotational frame (T1ρH relaxation). Therefore, the changes of T1ρH relaxation can be monitored by the observation of the arrayed 13C CP-MAS NMR spectra at various contact times.Fig. 7 shows the CH2 signal intensity curves versus contact time for the plasticized PHA/PCL blends for various PCL contents. For the CH2 group, the T1ρH relaxation of the PHA moiety was more enhanced with the addition of PCL, with the result that the 1H spin-lattice relaxation time in the rotational frame (T1ρH) of PHA-CH2 decreased with increasing PCL content (Fig. 7A). Although the T1ρH relaxation of PCL-α-CH2 was suppressed in the 50% PHA/50% PCL blend, it proceeded more rapidly with increasing PCL content over 50% (Fig. 7B). Furthermore, the T1ρH relaxation of the overlapping CH2 features of PBS/PCL was suppressed to various extents depending on the PCL content; that is, the T1ρH of PBS decreased with increasing PCL content (decreasing PHA with PHA content) (Fig. 7C). Therefore, the addition of PCL into the plasticized PHA (the PHA/PBS copolymer) suppressed the T1ρH relaxation of PBS (long T1ρH value), which compensated for the enhanced T1ρH relaxation of PHA (short T1ρH value) resulting from the immiscible morphology as well as the T1H relaxation.
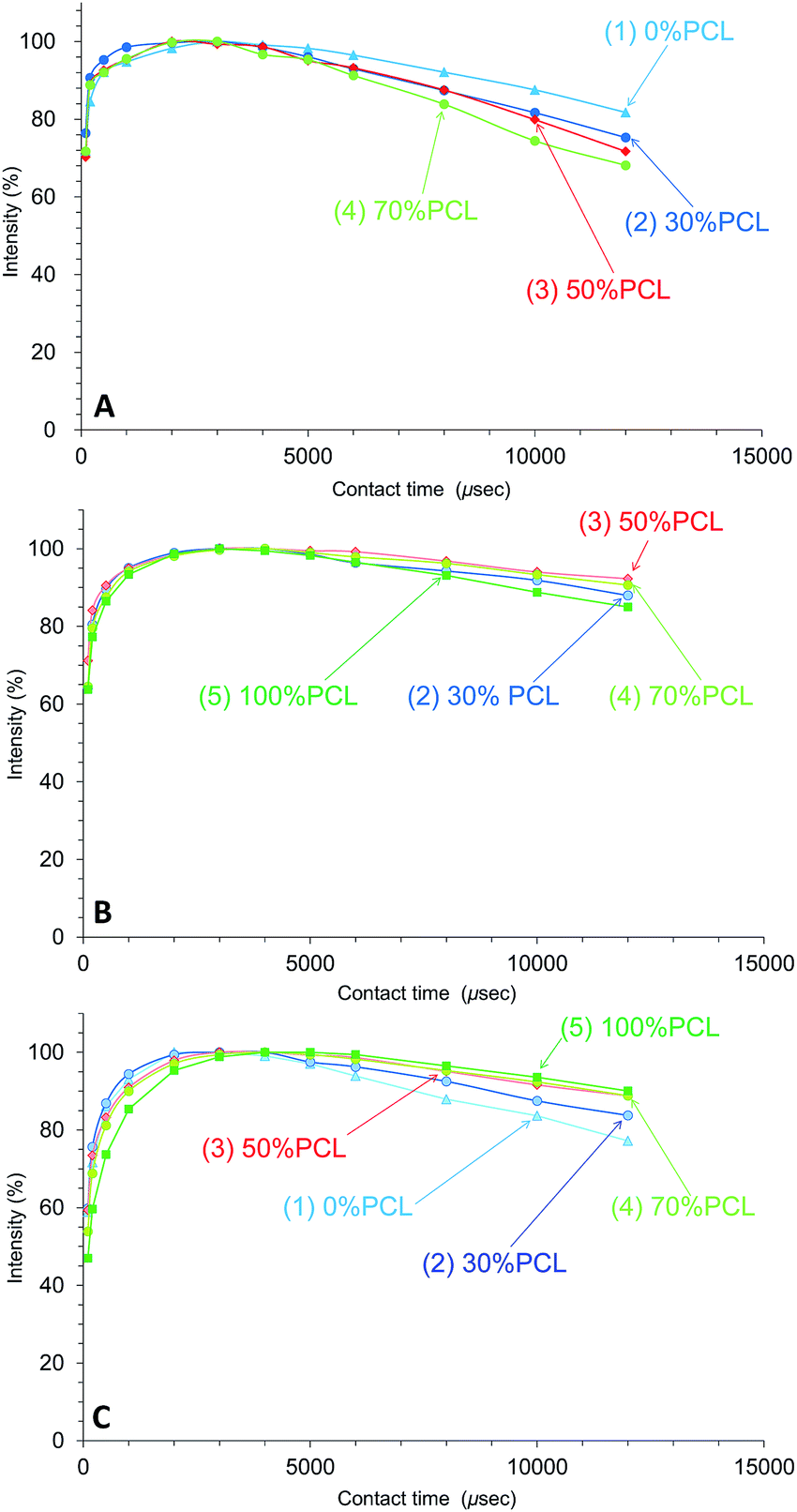 | ||
| Fig. 7 Peak intensity changes of plasticized PHA/PCL blends versus contact time in 13C CP-MAS NMR. (A) PHA, CH2 (43 ppm). (B) PCL, CH2 (34 ppm). (C) PBS-PCL, CH2 (25 ppm). | ||
In contrast to the effect of PCL addition, the T1ρH relaxation changed little with various compatibilizers, as shown in Fig. 8. The T1ρH relaxation of PHA-CH2 was slightly suppressed by the compatibilizers, especially by Modiper CL (Fig. 8A). For CH2 groups in both PCL (Fig. 8B) and overlapping signals from PBS/PCL (Fig. 8C), neither crosslinker or graft polymer produced any significant change in the signal decay curve. That is, the compatibilizers slightly lengthened the T1ρH value of PHA while they scarcely changed the T1ρH values of PCL and PBS. We conclude that T1ρH relaxation is concerned with slow molecular mobility (kHz order) and is not related to the tensile properties but could be an indicator of interactions within constituent polymers of the PHA-based blend.
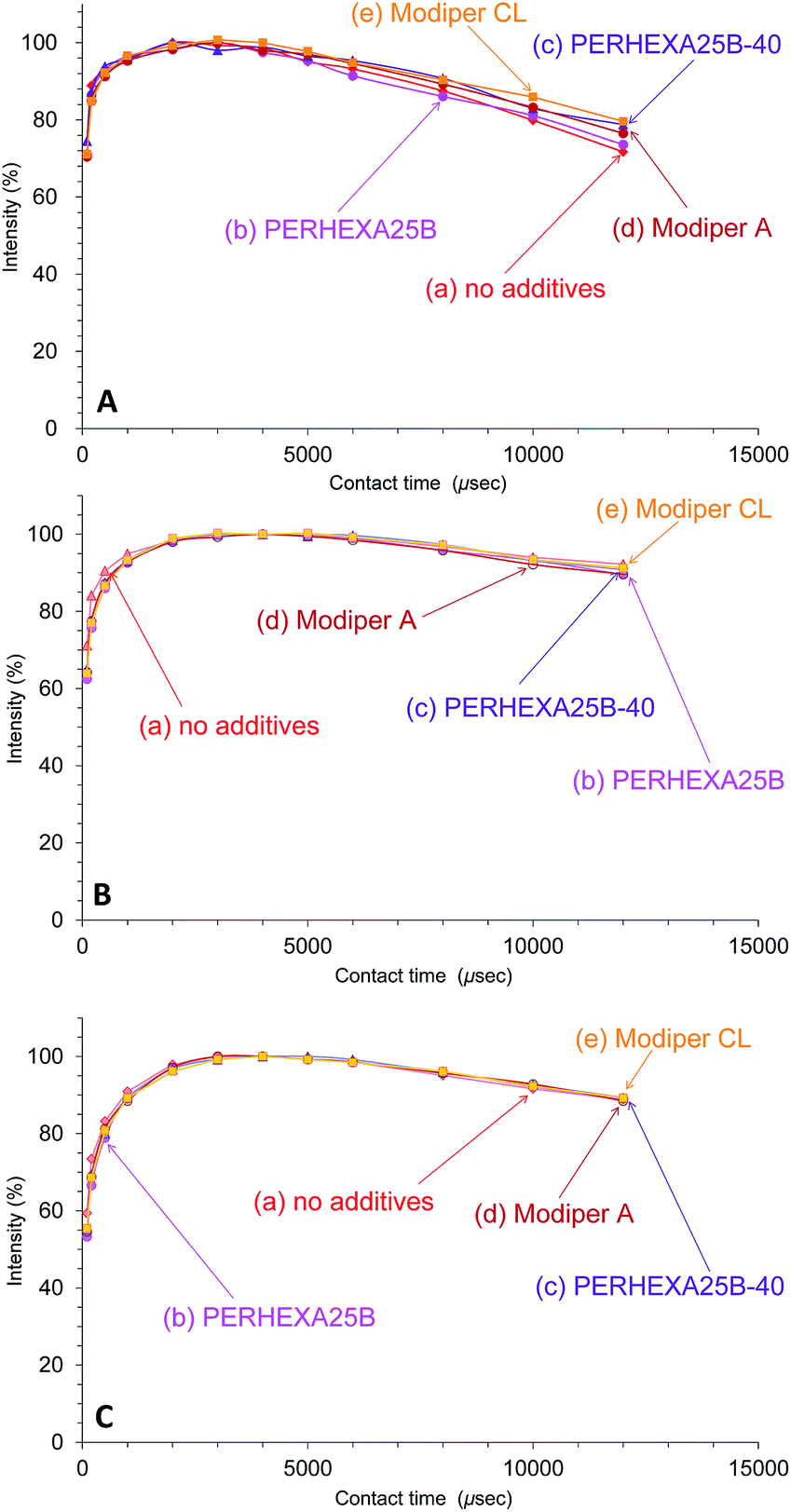 | ||
| Fig. 8 Peak intensity changes of 50% plasticized PHA/50% PCL blends versus contact time in 13C CP-MAS NMR. (A) PHA, CH2 (43 ppm). (B) PCL, CH2 (34 ppm). (C) PBS-PCL, CH2 (25 ppm). | ||
Multi-scale instrumental analyses of PHA/PCL blends for the effects of blend ratio and additives
Based on the micro-focus X-ray CT observation (Fig. 2), Fig. 9 presents schematics of the nanostructural changes of the 50% plasticized PHA/50% PCL blend with/without the compatibilizers after the tensile test. Without compatibilizers [Fig. 9(A)], the blend did not interact with PCL and formed the sea-island morphology seen before tensile tests (a). Although PBS in the blend had a similar chemical structure to PCL, the interaction between the plasticized PHA and PCL was weak in the sea-island morphology. In the quasi-static test (b), the plasticized PHA was difficult to extend, even though PCL readily extended. Because of the weak interaction between the plasticized PHA and PCL, the plasticized PHA in the blend adopted a broken island morphology and exfoliated to form the interface with PCL. In the dynamic tensile test (c), since PCL was also difficult to extend, the 50% plasticized PHA/50% PCL blend was easily broken at a small elongation.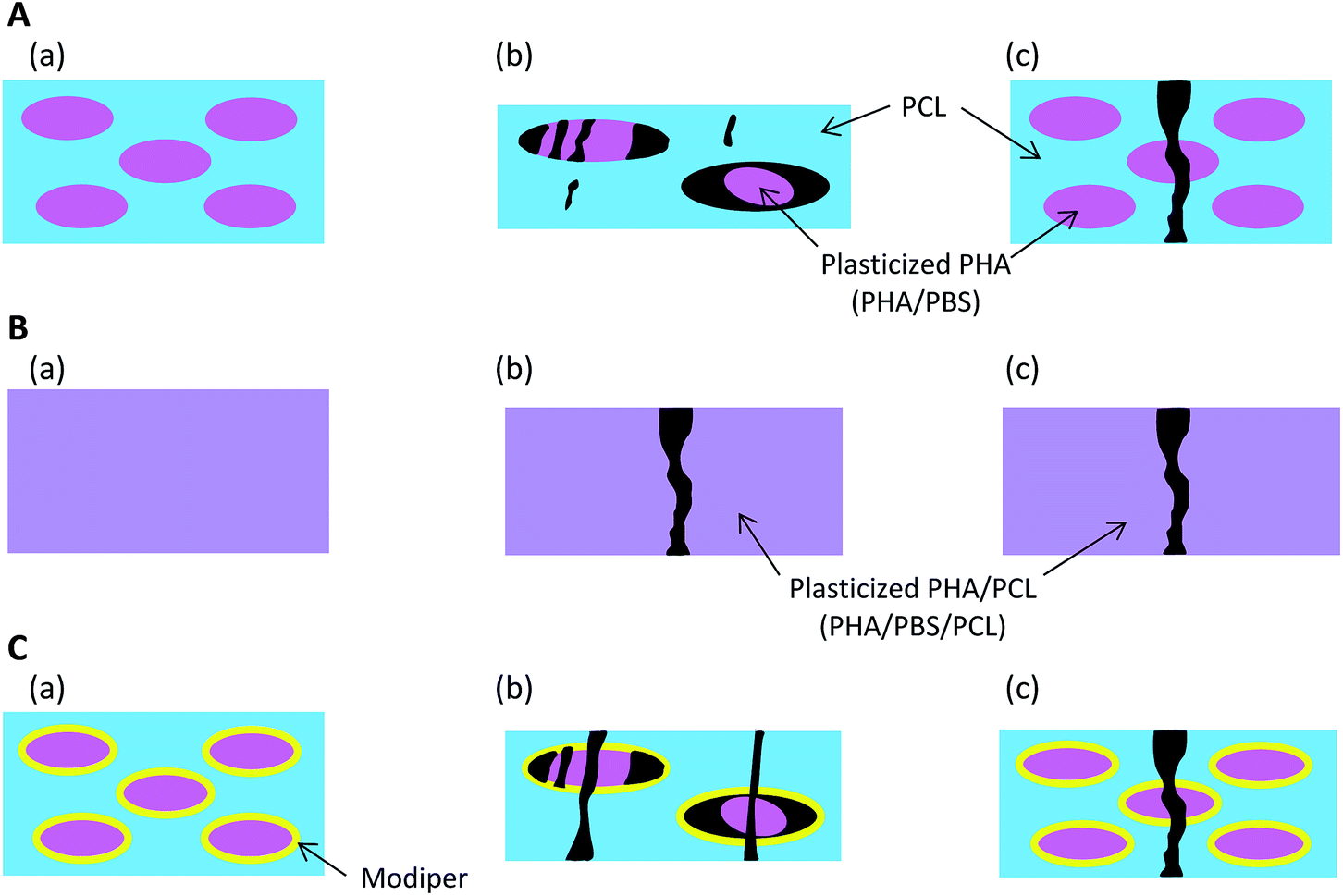 | ||
| Fig. 9 Nanostructural changes of 50% plasticized PHA/50% PCL blend after tensile tests. (A) No additives, (a) before tensile test, (b) after quasi-static test, (c) after dynamic test. (B) Crosslinker, (a) before tensile test, (b) after quasi-static test, (c) after dynamic test. (C) Graft copolymer, (a) before tensile test, (b) after quasi-static test, (c) after dynamic test. | ||
By the addition of a crosslinker into the 50% plasticized PHA/50% PCL blend [Fig. 9(B)], the plasticized PHA became compatible with PCL (a). Both plasticized PHA and PLC integrated to produce a homogenous morphology over tens of micron. In the quasi-static test (b), the unification of the plasticized PHA and PCL restrained stretching of the PCL. The plasticized PHA/PLC blend was easily broken at a small elongation because the quasi-static test was dominated by the rigidity of the PHA moiety. Furthermore, the elongation at break in the dynamic tensile test (c) was improved by the addition of cross-linker because the dynamic test was dominated by the flexibility of the PCL moiety.
On the addition of the graft polymer [Fig. 9(C)], although the 50% plasticized PHA/50% PCL blend still adopted the sea-island morphology, the interaction between the plasticized PHA and PCL became strengthened (a), as confirmed by the relaxation time analyses. However, the unifying effect of the graft polymer on the plasticized PHA/PLC blend was weaker than with the crosslinker. Therefore, both plasticized PHA and PCL were broken with a larger elongation at break in the quasi-static test (b). Since the unification of the plasticized PHA/PLC blend mobilized the plasticized PHA and PCL together, the elongation at break was also increased by the addition of graft polymer in the dynamic test (c).
Conclusions
With the aim of extending application of multi-scale instrumental analyses to biodegradable polymers, plasticized polyhydroxyalkanoates (PHA), containing 65% PHA, 30% poly(butylene succinate) (PBS), and 5% crosslinker, were examined we respect to blending with poly(ε-caprolactone) (PCL) as well as the addition of compatibilizers, such as crosslinkers and graft polymers. Blending PHA with more than 50% PCL increased the tensile strength and elongation at break in the quasi-static tensile test, although the tensile properties were independent of PCL content in the impact tensile test. The crosslinkers markedly lowered the elongation at break in the quasi-static tensile test, while the graft polymers only produced a slight decrease. Both crosslinkers and graft polymers increased the tensile strength and elongation at break in the impact tensile test. Micro-focus X-ray CT observations revealed a sea-island morphology in the 50% plasticized PHA/50% PCL blend. Although the morphology was slightly changed by the addition of graft polymers having acrylonitrile–styrene copolymers as a side-chain, it became homogenous over distances of more than ten microns with the addition of peroxy crosslinkers. The combination of 13C CP- and PST-MAS NMR spectra showed that the molecular mobility of PCL was suppressed by blending with the plasticized PHA while the compatibilizers left the molecular motions almost unchanged, with the exception of the CO groups of PBS/PCL. The 1H spin-lattice relaxation time (T1H) values of the plasticized PHA/PCL blend increased in the sea-island morphology at 50% PCL addition. The lengthened T1H value of the 50% plasticized PHA/50% PCL blend was shortened again by the crosslinkers, which also changed the sea-island morphology into a homogenous mixture. The T1H value of blend was shortened to a smaller degree by the graft polymers, which also produced less change in the sea-island morphology. Blending with the plasticized PHA increased the 13C spin-lattice relaxation time (T1C) value of PCL, indicating that the sea-island morphology decreased the molecular mobility of the added PCL. Meanwhile, the T1C values of the PHA moiety decreased with increasing PCL content to indicate that the added PCL amplified the molecular mobility of PHA. Concerning the effects of compatibilizer on the T1C values, the graft polymers noticeably decreased the T1C value of the PBS constituent. The 1H spin-lattice relaxation in the rotational frame (T1ρH relaxation) of PHA was enhanced by suppression of the T1ρH relaxation of PBS because of the immiscible morphology, while the T1ρH relaxation was little changed by the addition of compatibilizers. The molecular mobility of the polymers constituents in the plasticized PHA was changed by blending with PCL and the change of molecular mobility caused the increase of tensile strength and elongation at break in the quasi-static tensile test. In contrast, the molecular mobility of the blend was not changed by the crosslinker but was by the graft polymer, while the morphology was not changed by the graft polymer but was by the crosslinker. For these reasons, a different trend for the tensile properties in quasi-static and impact tests was observed when using either crosslinkers or graft polymers. In conclusion, to improve the tensile properties of biodegradable polymers, it is necessary not only to add a component having superior tensile properties but also to match the interaction and molecular mobility of the new component with the original polymer. Considering the tensile properties from the interactions between the constituent polymers, the graft polymer was suitable for polymer blends having different molecular mobility, such as plasticized PHA/PCL blends. In contrast, the crosslinker would be more suitable for polymer blends having similar molecular mobility to each other. NMR is a good indicator of success as the T1H and T1ρH values, which are the indexes involved in the tensile properties, decrease with an increasing interaction while the T1C value decrease with increasing the molecular mobility. Our present study showed that the multi-scale instrumental analyses gave information about molecular mobility and morphology concerned with the tensile properties of synthetic polymers. Studies of materials characteristics are currently in progress for not only synthetic polymers, but also for natural polymers, using multi-scale instrumental analyses.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to express special thanks to Kohei Yasuda, Nagoya Institute of Technology, for help with the tensile test data analyses.References
- S. Chanprateep, J. Biosci. Bioeng., 2010, 111, 621 CrossRef PubMed.
- X. Gao, J.-C. Chen, Q. Wu and G.-Q. Chen, Curr. Opin. Biotechnol., 2011, 22, 768 CrossRef CAS PubMed.
- Z. A. Raza, S. Abid and I. M. Banat, Int. Biodeterior. Biodegrad., 2018, 126, 45 CrossRef CAS.
- E. Bugnicourt, P. Cinelli, A. Lazzeri and V. Alvarez, eXPRESS Polym. Lett., 2014, 8, 791 CrossRef.
- B. Laycock, P. Halley, S. Pratt, A. Werker and P. Lant, Prog. Polym. Sci., 2013, 38, 536 CrossRef CAS.
- Z. A. Raza, S. Riaz and I. M. Banat, Biotechnol. Prog., 2018, 34, 29 CrossRef CAS PubMed.
- D. Kai and X. J. Loh, ACS Sustainable Chem. Eng., 2014, 2, 106 CrossRef CAS.
- H. Verhoogt, B. A. Ramsay and B. D. Favis, Polymer, 1994, 35, 5155 CrossRef CAS.
- I. Zembouai, M. Kaci, S. Bruzaud, A. Benhamida, Y.-M. Corre and Y. Grohens, Polym. Test., 2013, 32, 842 CrossRef CAS.
- I. Armentano, E. Fortunati, N. Burgos, F. Dominici, F. Luzi, S. Fiori, A. Jiménez, K. Yoon, J. Ahn, S. Kang and J. M. Kenny, LWT--Food Sci. Technol., 2015, 64, 980 CrossRef CAS.
- M. P. Arrieta, J. López, D. López, J. M. Kenny and L. Peponi, Eur. Polym. J., 2015, 73, 433 CrossRef CAS.
- M. Larsson, O. Markbo and P. Jannasch, RSC Adv., 2016, 6, 44354 RSC.
- D. Garcia-Garcia, J. M. Ferri, T. Boronat, J. Lopez-Martinez and R. Balart, Polym. Bull., 2016, 73, 3333 CrossRef CAS.
- Y. Ke, X. Y. Zhang, S. Ramakrishna, L. M. He and G. Wu, Mater. Sci. Eng., C, 2017, 70, 1107 CrossRef CAS PubMed.
- Y.-X. Zhou, Z.-G. Huang, X.-Q. Diao, Y.-X. Weng and Y.-Z. Wang, Polym. Test., 2015, 42, 17 CrossRef CAS.
- P. Ma, D. G. Hristova-Bogaerds, P. J. Lemstra, Y. Zhang and S. Wang, Macromol. Mater. Eng., 2012, 297, 402 CrossRef CAS.
- A. Chikh, A. Benhamida, M. Kaci, I. Pillin and S. Bruzaud, Polym. Test., 2016, 53, 19 CrossRef CAS.
- Y. Parulekar and A. K. Mohanty, Macromol. Mater. Eng., 2007, 292, 1218 CrossRef CAS.
- F. G. Morin and R. H. Marchessault, Macromolecules, 1992, 25, 576 CrossRef CAS.
- A. Cao, N. Asakawa, N. Yoshie and Y. Inoue, Polymer, 1999, 40, 3309 CrossRef CAS.
- L. Zhang, H. Tang, G. Hou, Y. Shen and F. Deng, Polymer, 2007, 48, 2928 CrossRef CAS.
- M. Nishida, T. Tanaka, T. Miki, Y. Hayakawa and K. Kanayama, Cellulose, 2017, 24, 5295 CrossRef CAS.
- M. Nishida, T. Tanaka, T. Miki, T. Ito and K. Kanayama, Ind. Crops Prod., 2018, 114, 1 CrossRef CAS.
- M. Nishida, T. Tanaka, Y. Hayakawa and M. Nishida, Polymers, 2018, 10, 506 CrossRef.
- M. Nishida, T. Ogura, H. Shinzawa, M. Nishida and W. Kanematsu, J. Mol. Struct., 2016, 1124, 92 CrossRef CAS.
- M. Nishida, Y. Ito, H. Shinzawa, M. Nishida and Y. Hayakawa, Key Eng. Mater., 2016, 715, 39 Search PubMed.
- M. Nishida, H. Ichihara, H. Watanabe, N. Fukuda and H. Ito, in Dynamic Behavior of Materials, ed. V. Chalivendra, B. Song and D. Casem, Springer New York, 2012, vol. 1, p. 329 Search PubMed.
- D. A. Torchia, J. Magn. Reson., 1978, 30, 613 CAS.
- M. Nishida, T. Tanaka, T. Miki, Y. Hayakawa and K. Kanayama, RSC Adv., 2017, 7, 54532 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra10045d |
| This journal is © The Royal Society of Chemistry 2019 |