DOI:
10.1039/C8RA10074H
(Paper)
RSC Adv., 2019,
9, 4917-4924
Discovery of trisubstituted nicotinonitrile derivatives as novel human GCN5 inhibitors through AlphaScreen-based high throughput screening
Received
7th December 2018
, Accepted 7th January 2019
First published on 8th February 2019
Abstract
The general control nonrepressed protein 5 (GCN5) is an important target for drug design and drug discovery largely owing to its pathogenic role in malignancies. Chemical probes that target GCN5 have been developed in recent decades, but their potencies are still unsatisfactory. In this study, through an in-house developed AlphaScreen-based high throughput screening platform, radioactive acetylation assays and 2D-similarity based analogue searching, we discovered DC_HG24-01 as the novel hGCN5 inhibitor with the IC50 value of 3.1 ± 0.2 μM. Further docking studies suggested that DC_HG24-01 could occupy the binding pocket of acetyl-CoA cofactor, which laid the foundation for the development of more potent hGCN5 inhibitors in the future. At the cellular level, DC_HG24-01 could retard cell proliferation and block the acetylation of H3K14 leading to cell apoptosis and cell cycle arrest at the G1 phase in MV4-11 cell lines. Taken together, the discovery of DC_HG24-01 may serve as a good starting point to accelerate the development of more potent hGCN5 inhibitors through further structural decoration and provide new insight into the pharmacological treatment of leukemia.
1. Introduction
Post-translation modifications on histones like histone acetylation play the pivotal role in chromatin remodeling and specific gene expression.1,2 In mammalian cells, histone acetylation is stringently regulated by HATs (histone acetyltransferases) and HDACs (histone deacetylases) to maintain homeostasis.3,4 HATs could be further classified into five subfamilies based on sequence similarity and substrate preference: the GNAT (GCN5 related N-acetyltransferase) family,5 MYST (Moz, Ybf2, Sas2, Tip60) family,6 p300/CBP (CREB-binding protein) family,7 nuclear receptor coactivator (NCOA) family and transcription factor-related HATs.8 The general control nonrepressed protein 5 (GCN5), as a member of the HAT superfamily, transfers an acetyl group to the ε-amino group of specific lysine residues on H2B, H3, and H4.9,10 In addition to histones, GCN5 also catalyzes the acetylation of non-histone substrates like transcription factors and other oncoproteins that contributes to several transcription-mediated biological processes including cell-cycle progression and cell metabolism regulation.11,12 Aberrant expression and activation of GCN5 is closely linked with the pathogenesis and progression of several malignant diseases especially cancers.13 GCN5 expression is strongly correlated with the extent of malignancy in non-small lung cancer. Deregulation of GCN5 promotes the acetylation at E2F1, cyclin D1, and cyclin E1 promoters, which accelerates cancer progression.14 In acute lymphoblastic leukemia, GCN5 dynamically regulates the acetylation of chimeric transcription factor E2A-PBX1 facilitating cell transformation in vitro and in vivo.15 In addition, GCN5 could increase the stability of Myc through the acetylation at K323, which is the most frequently deregulated oncoprotein in human cancers. Collectively, these studies provide strong rational for the development of GCN5 inhibitors for anti-cancer therapy.16
In recent decades, tremendous efforts have been devoted to the drug discovery of HAT inhibitors, including bi-substrate inhibitors, natural products and small molecule inhibitors17–23 (Fig. 1). However, bi-substrate inhibitors show poor physicochemical properties that restrict their further applications in cellular studies. Natural products like anacardic acid, garcinol and curcumin suffer from structure instability and synthetic complexity making them difficult for medicinal chemistry optimization. In 2017, Lasko et al. reported the identification of selective p300/CBP inhibitor, namely A-485, which could selectively inhibit proliferation in lineage-specific tumor types.24 Shortly after the discovery of A-485, Jonathan and co-workers identified the first-in-class KAT6A/B inhibitor.25 Despite the continuing success of the development of the p300/CBP and KAT6A/B inhibitors, less progress has been made towards the development of GCN5 inhibitors. In our previous work, we reported 1,8-acridinedione derivatives as yeast GCN5 inhibitors with the IC50 value of 6.8 μM,26 but there is still a lack of effective inhibitors against human GCN5. In this study, we reported a novel hGCN5 inhibitor DC_HG24-01 that was discovered and validated by the amplified luminescent proximity homogeneous assay based high throughput screening assay and radioactive acetylation assay. The SPR study demonstrated the direct binding between hGCN5 and DC_HG24-01. Molecular docking studies proposed that DC_HG24-01 occupied the pocket of acetyl-CoA substrate, which may account for its inhibitory activity in vitro. In MV4-11 leukemia cells, DC_HG24-01 retarded cell growth, induced cell apoptosis and cell cycle arrest. Taken together, our study proves that DC_HG24-01 is a novel, potent hGCN5 inhibitor, which can provide novel structural clues to develop potent hGCN5 inhibitors for cancer therapy.
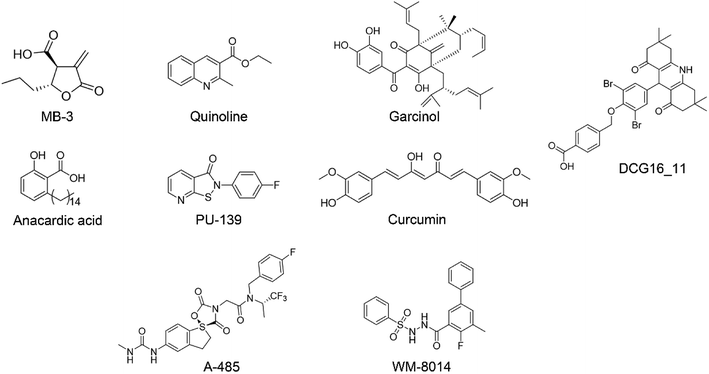 |
| | Fig. 1 The representative HAT inhibitors reported in the literature. | |
2. Results and discussions
2.1 The establishment of AlphaScreen-based high throughput screening platform
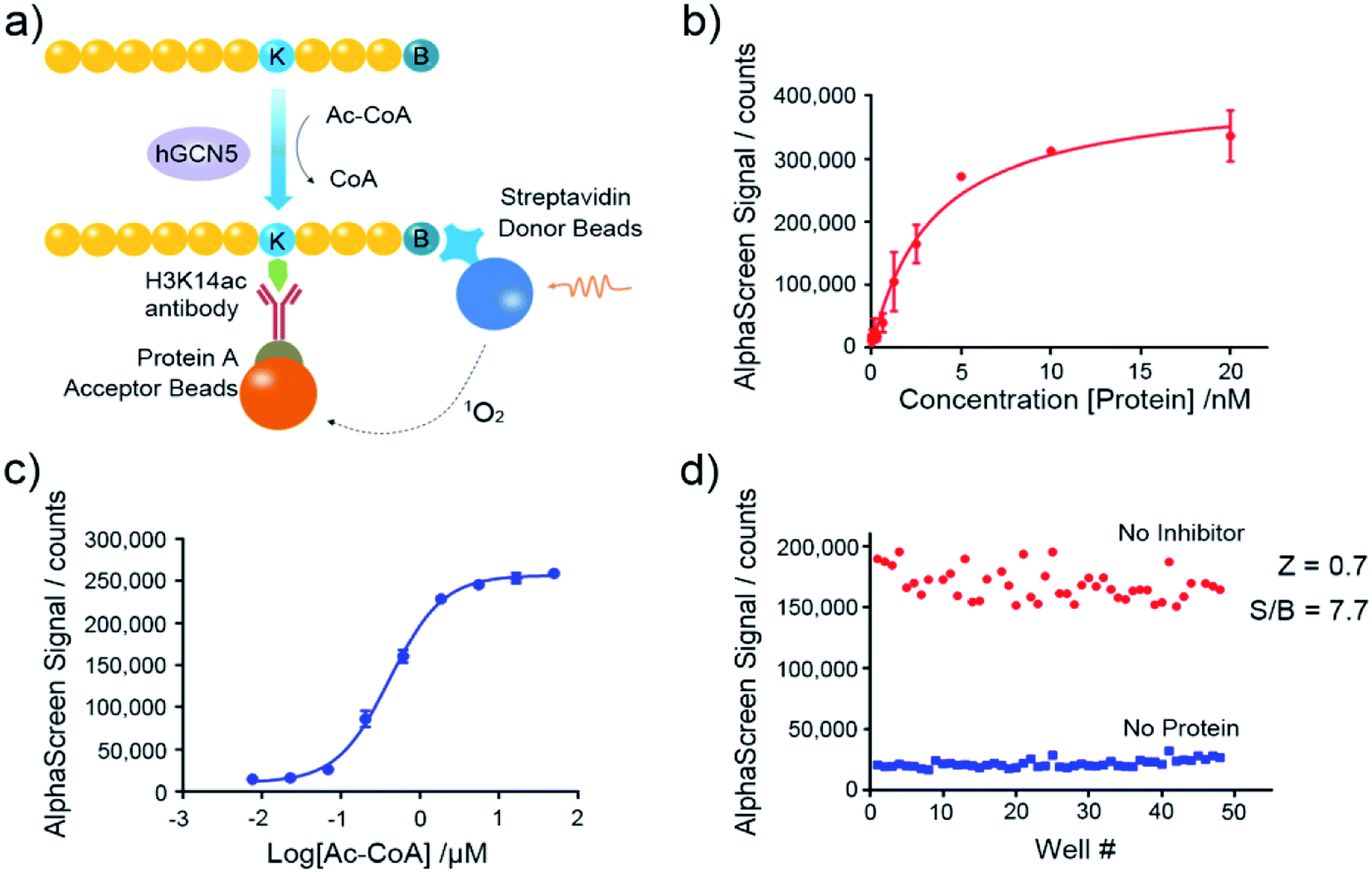
Nowadays, the AlphaScreen technology, as one of most useful, front line biophysical methods for primary high-throughput screening (HTS), has been widely applied in the target-based drug screening campaign.27,28 Herein, we utilized AlphaScreen-based screening assays with the aim to develop potent hGCN5 inhibitors (Fig. 2a). Firstly, different concentrations of hGCN5 were titrated ranging from 0.002 nM to 20 nM with excess H3 substrate and acetyl-CoA to determine the optimum concentration for the following screening experiments (Fig. 2b). The titration was then repeated with different concentrations of the cofactor in the presence of a fixed amount of hGCN5 (Fig. 2c). Considering the assay sensitivity and robustness, 5 nM hGCN5 and 500 nM acetyl-CoA were selected as the suitable assay conditions for the subsequent experiments. Finally, we determined Z-factor to quantify the assay quality (Fig. 2d). The Z-factor in our assay was 0.7, which is considered to yield qualified assay format.29 Through a series of assay optimization processes, we finally established a robust and highly sensitive immunoassay platform which enables high throughput screening and quick activity determination of hGCN5 inhibitors.
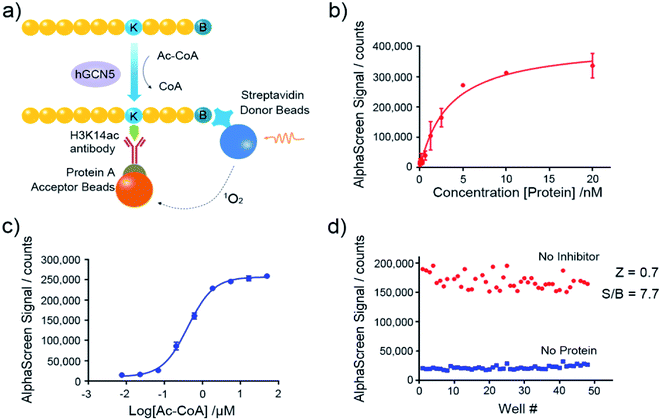 |
| | Fig. 2 Assay development of AlphaScreen-based HTS platform. (a) Schematic representation of AlphaScreen assay for the detection of hGCN5 enzymatic activity. (b) The enzyme titration curve in the presence of 100 nM biotinylated H3 (1–23) peptide substrate and 15 μM acetyl-CoA. (c) The cofactor titration curve with the concentration of acetyl-CoA ranging from 8 nM to 50 μM. (d) Z-factor determination of established AlphaScreen assays. Data in panels (b and c) is presented as mean ± SD, n = 3. | |
2.2 The discovery of hit compound DC_HG24 as a novel hGCN5 inhibitor through established AlphaScreen assay
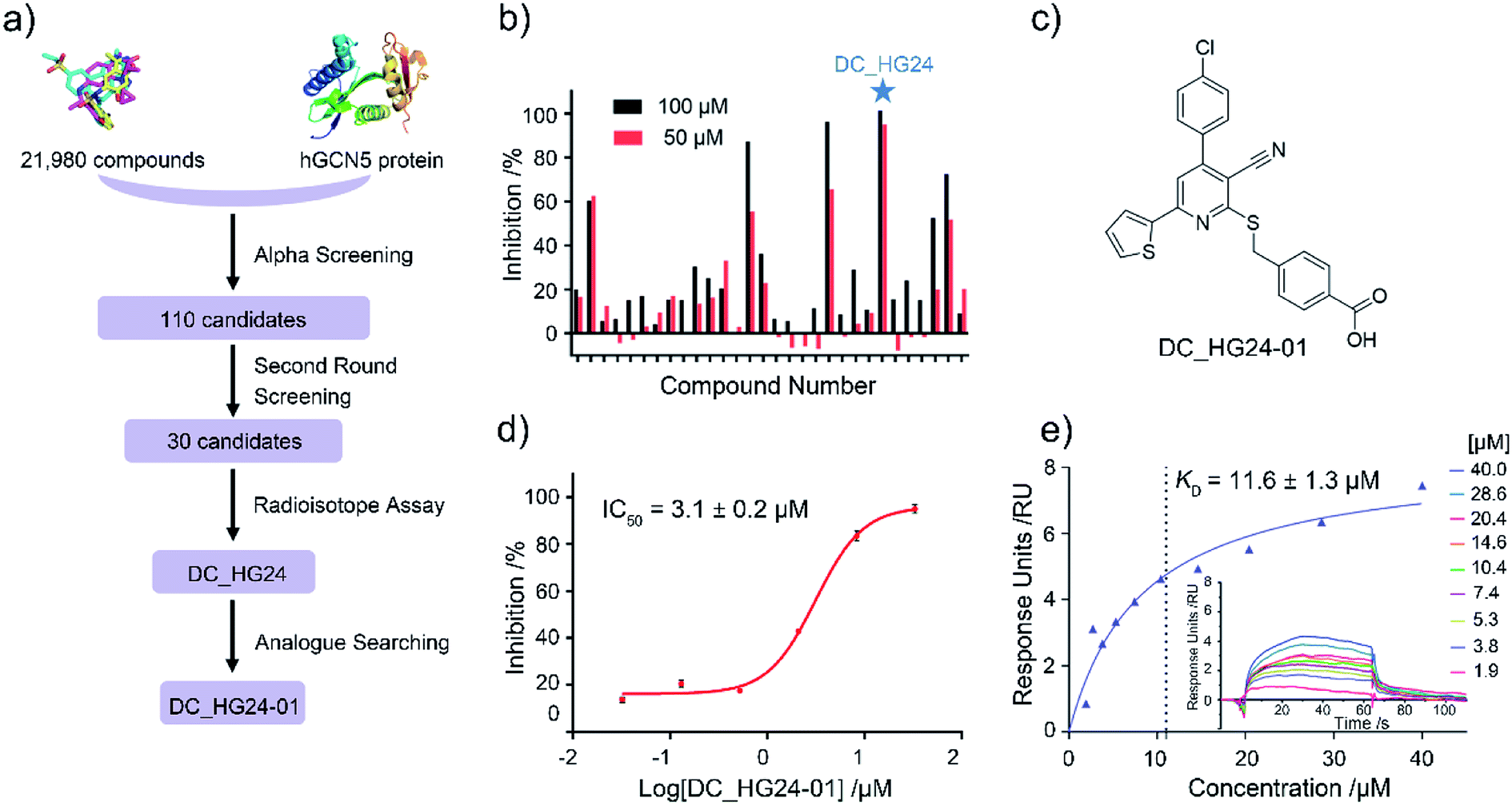
Based on established AlphaScreen assay, we carried out HTS screening against in-house library containing 21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 980 diverse small-molecule compounds in a 384-well assay format (Fig. 3a). 110 compounds that showed over 70% inhibitory rate at 100 μM were selected and retested in duplicate at 100 μM and 50 μM, respectively. After two rounds of screening based on AlphaScreen assay, top 30 compounds with moderate inhibitory activities were chosen with a manageable number for post-HTS hit validation.
980 diverse small-molecule compounds in a 384-well assay format (Fig. 3a). 110 compounds that showed over 70% inhibitory rate at 100 μM were selected and retested in duplicate at 100 μM and 50 μM, respectively. After two rounds of screening based on AlphaScreen assay, top 30 compounds with moderate inhibitory activities were chosen with a manageable number for post-HTS hit validation.
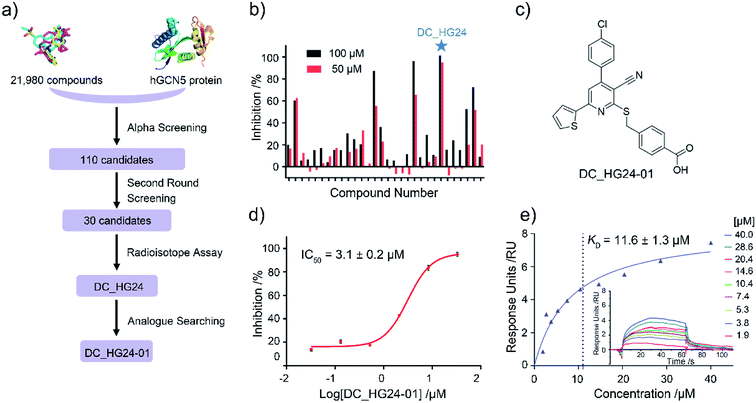 |
| | Fig. 3 The results of preliminary high throughput screening and hit validation. (a) Flowchart of AlphaScreen-based high throughput screening for the discovery of hGCN5 inhibitors. (b) Inhibitory activities of top 30 compounds from high throughput screening determined by radioisotope acetylation assays. (c) The chemical structure of DC_HG24-01. (d) The IC50 determination of DC_HG24-01 against hGCN5 HAT domain measured by radioisotope acetylation assays. (e) SPR binding data for the interactions of DC_HG24-01 with hGCN5. The compound was prepared at the concentrations of 40 μM, 28.6 μM, 20.4 μM, 14.6 μM, 10.4 μM, 7.4 μM, 5.3 μM, 3.8 μM and 1.9 μM, respectively. Data in panels (d and e) is presented as mean ± SD, n = 3. | |
2.3 Radioactive acetylation assay
Considering HTS assays are usually prone to false positives and artificial effects, the inhibitory activity of 30 candidates were tested using the 3H-labeled radioactive acetylation assay at the concentrations of 100 μM and 50 μM, respectively, which was regarded as the gold standard assay format for epi-enzyme activity detection30,31 (Fig. 3b). Among them, DC_HG24 showed the most potent inhibitory activity towards hGCN5 with the IC50 value of 12.7 ± 0.9 μM (Table 1). To explore hGCN5 inhibitors with better potency, we employed the 2D-similarity search in Pipeline Pilot program to obtain the derivatives based on the structure of DC_HG24 and then validated the inhibitory activities of this series of compounds by the radioactive acetylation assay (Table 1). Among them, DC_HG24-01 exhibited the best potency with an IC50 value of 3.1 ± 0.2 μM (Fig. 3c and d). Thus, we chose DC_HG24-01 for follow-up biophysical and biochemical evaluation.
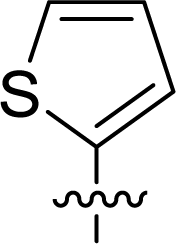
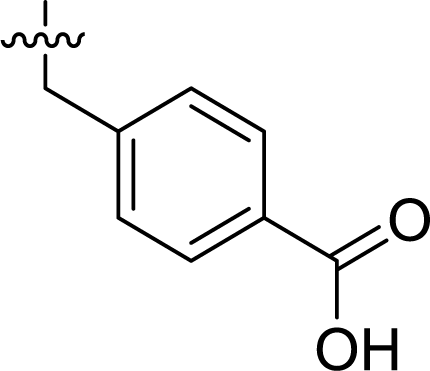
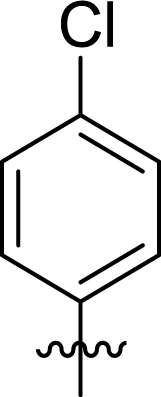
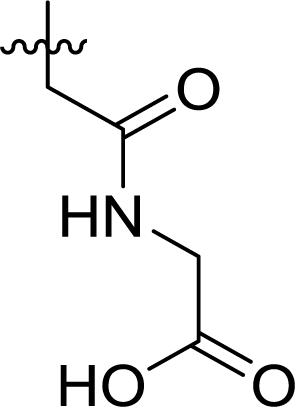
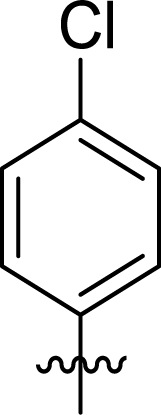
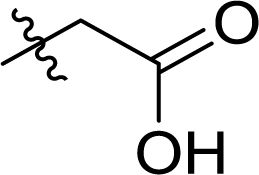
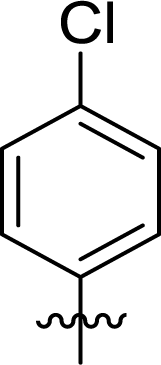
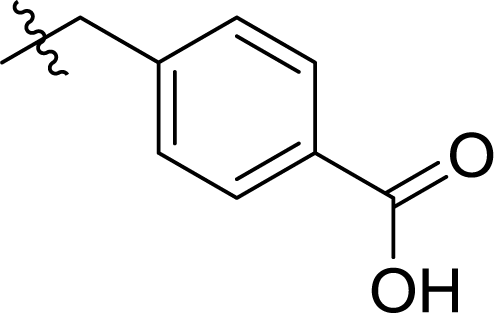
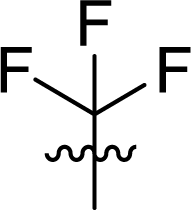
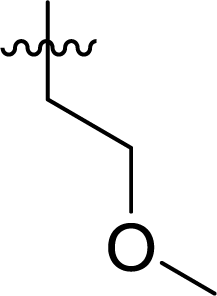
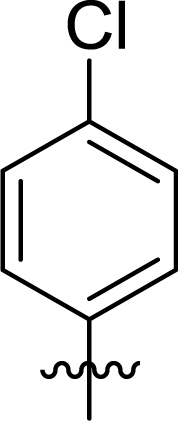
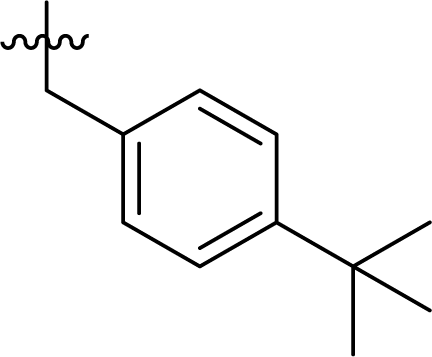
Table 1 Structures and their relative activity of DC_HG24-01 and its analoguesa
2.4 Surface plasmon resonance-based binding assay
To determine whether DC_HG24-01 could directly bind to the hGCN5 enzyme, surface plasmon resonance (SPR) based binding assay was performed to assess the binding affinity. SPR binding curves demonstrated the non-covalent interaction patterns of DC_HG24-01. The result proved that DC_HG24-01 bound directly to the immobilized hGCN5 HAT domain with the equilibrium dissociation constant (KD) value of 11.6 ± 1.3 μM, which is consistent with the inhibitory activity measured by radioisotope acetylation results (Fig. 3e).
2.5 Structure-activity relationship (SAR) analysis and molecular docking analysis
In order to further explore the SAR of DC_HG24-01, we further evaluated the SAR of DC_HG24-01 and its derivatives based on the results from radioisotope acetylation assays. The results revealed that the replacement of carboxyl group in DC_HG24-01 to alkyl groups like alkoxy group (DC_HG24-05) and tert-butyl group (DC_HG24-06) in R1 led to dramatically decreased inhibitory activity against hGCN5. Another finding is that the activity gradually decreased when the chlorobenzene group in R2 was replaced with less hydrophobic groups like thiophene group (DC_HG24-01) and trifluoromethyl group (DC_HG24-04). Shortening the distance between sulfur atom and carboxyl group in R1 led to DC_HG24-03 with weaker potency, which prompted us that the geometry and length of this linker were essential to retain the inhibitory activity.
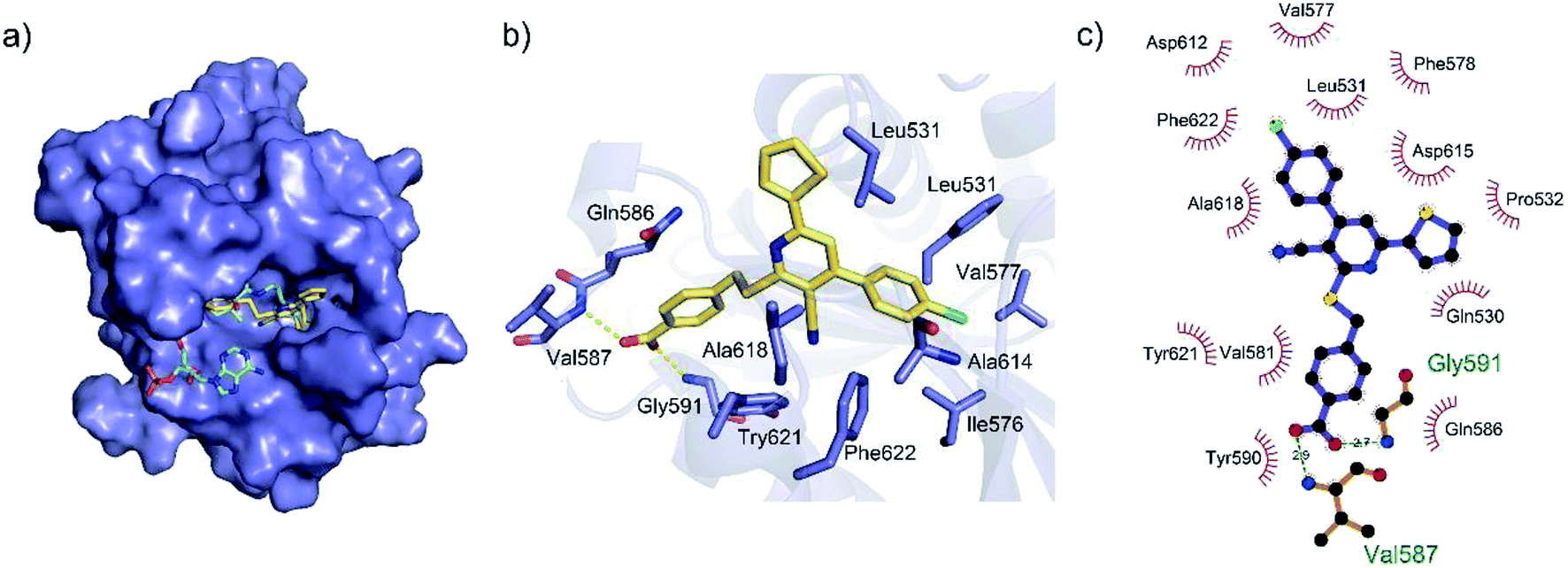
To gain a better understanding of the molecular basis for its inhibitory activity, molecular docking studies were performed to investigate the binding mode of DC_HG24-01 to hGCN5. The docking results revealed that DC_HG24-01 occupied the pocket of Ac-CoA cofactor and the carboxyl group formed the hydrogen bond with V587 and G591, which clarified the molecular mechanism for better inhibitory activity of DC_HG24-01 compared with its analogues (Fig. 4a and b). Meanwhile, the chlorobenzene group formed extensive hydrophobic contact with neighboring residues F622, A618, V577, L531, F578 (Fig. 4c). Hydrophobic contact was also observed for benzene group in R1 with W621, V581 in hGCN5. The replacement of benzene group to alkoxy group (DC_HG24-02) showed slightly weaker activity (Table 1).
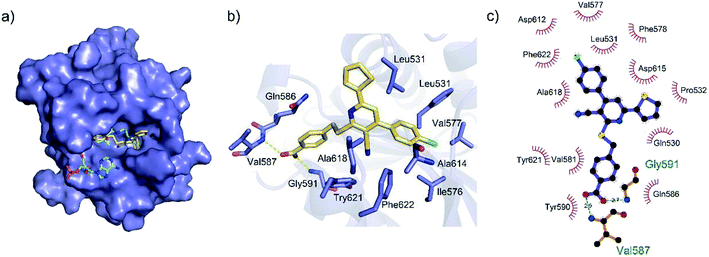 |
| | Fig. 4 Predicted binding mode between DC_HG24-01 and human GCN5. (a) Superimposition of the binding modes showing DC_HG24-01 in the acetyl CoA-binding pocket of human GCN5 (PDB ID 1Z4R). (b) Close-up view of the key interactions stabilizing DC_HG24-01 in acetyl CoA binding pocket. DC_HG24-01 is depicted as yellow sticks, the surrounding key residues are shown in purple sticks and labelled. Hydrogen bonds are shown as a yellow dashed line. (c) Schematic diagram showing putative interactions between hGCN5 and DC_HG24-01. Hydrogen-bonding interactions are denoted by dotted green lines and residues involved in the hydrophobic interactions are shown as starbursts. | |
2.6 Cell-based assays
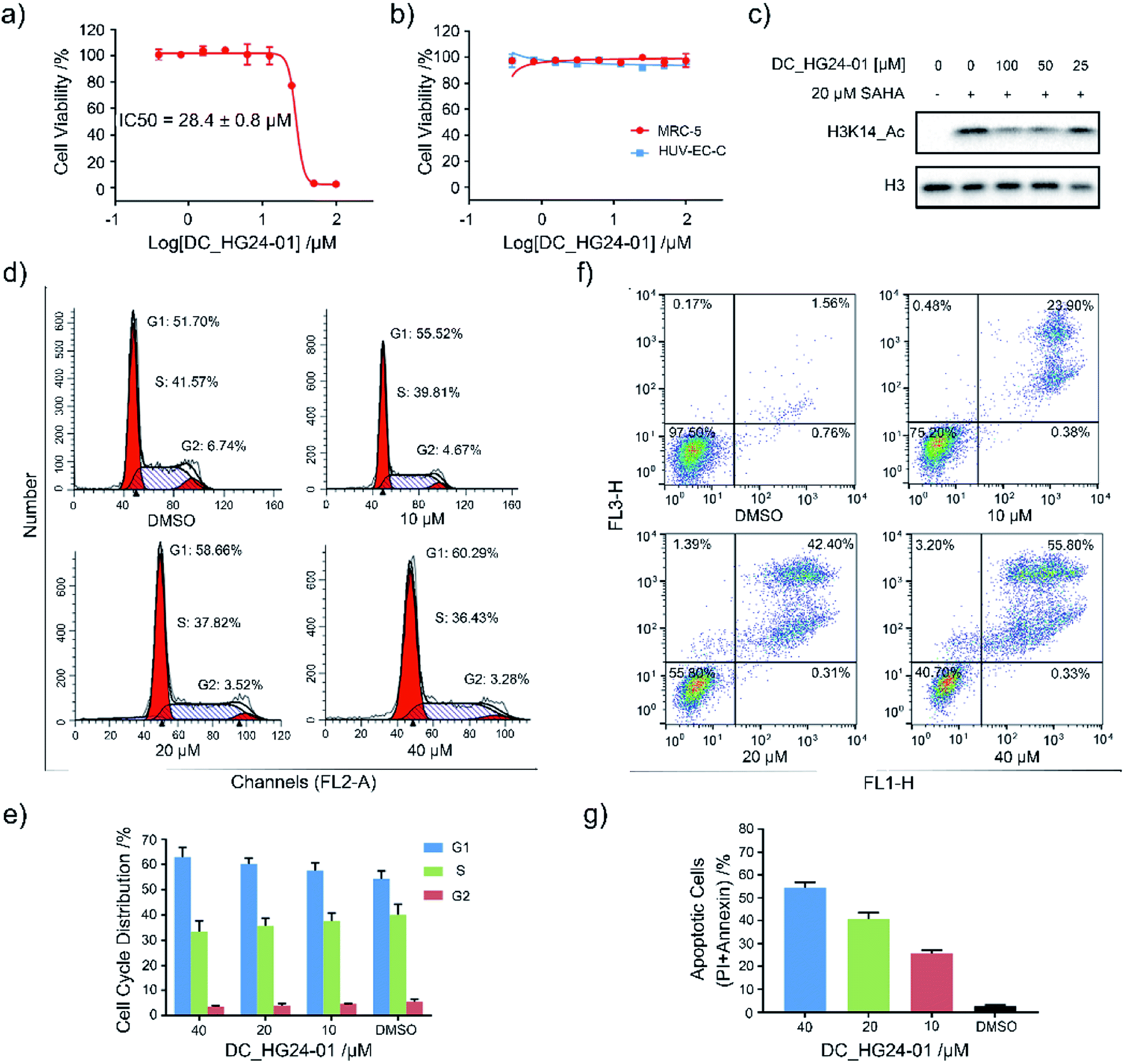
Considering the pathological function of GCN5 in acute lymphoblastic leukemia,15 we evaluated the efficacy of DC_HG24-01 in leukemia cell models. The cell viability assays showed that after 72 h treatment, DC_HG24-01 inhibited the leukemia cell proliferation in a dose-dependent manner with the IC50 value of 28.4 ± 0.8 μM (Fig. 5a). Meanwhile, DC_HG24-01 showed minimal effect on the proliferation of normal cells including the umbilical vein/vascular endothelium cells HUV-EC-C and MRC5 cell lines from normal lung tissue (Fig. 5b). In addition, DC_HG24-01 could block the acetylation of H3K14 in cellular context (Fig. 5c).
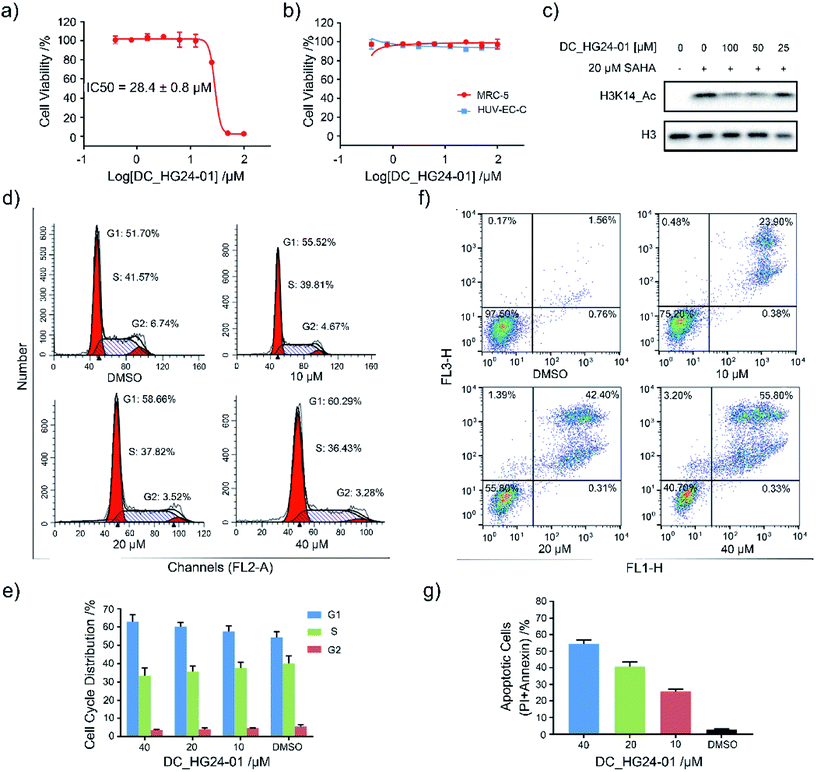 |
| | Fig. 5 Evaluation of the cellular activity of compound DC_HG24-01. (a) The IC50 curve of DC_HG24-01 against MV4-11 leukemia cell proliferation. (b) Cell proliferation inhibition assay of DC_HG24-01 on MRC5 and HUV-EC-C normal cells. (c) Western blot analysis of H3K14ac in MV4-11 leukemia cells treated with the 0 μM, 25 μM, 50 μM, 100 μM for 48 h. (d and e) Cell cycle analysis. MV4-11 cells were treated with 0 μM, 10 μM, 20 μM and 40 μM DC_HG24-01 for 24 h. (f and g) Cell apoptosis analysis. MV4-11 cells were treated with 0 μM, 10 μM, 20 μM and 40 μM DC_HG24-01 for 72 h. Data in panels (a and b) and (e–g) is represented as mean ± SD, n = 3. | |
To further elaborate the antiproliferation mechanism of DC_HG24-01, we examined the effects of DC_HG24-01 on cell apoptosis and cell cycle via flow cytometric analysis. We found that DC_HG24-01 caused cell cycle arrest at G1 phase in a concentration-dependent manner after 24 h treatment (Fig. 5d–e). Then cell apoptosis analysis was conducted, which showed increased cell apoptosis upon the treatment with DC_HG24-01 for 72 h (Fig. 5f and g). Collectively, the above results demonstrated that DC_HG24-01 was a bioactive, low-toxic inhibitor, which hold great potential for leukemia treatment.
3. Conclusions
In the recent decades, it has been well established that histone acetyltransferase GCN5 is involved in a wide range of physiologies and is a contributor in pathogenesis and progression of several diseases including cancers. Although several inhibitors toward GCN5 have been development, there is still shortage of effective small molecules targeting human GCN5. In this study, through two rounds of AlphaScreen screening together with the radioactive acetylation assay, we discovered DC_HG24-01 as a potent small molecule inhibitor, which effectively inhibited hGCN5 enzyme activity with the IC50 value of 3.1 ± 0.2 μM. SPR convinced that DC_HG24-01 could bind to hGCN5 with the KD value of 11.6 ± 1.3 μM. Further molecular docking studies proposed a substrate acetyl-CoA-dependent inhibitory mode, which provided the interpretation of structure activity relationship in atomic level. In cellular context, DC_HG24-01 restricted MV4-11 growth and could modulate the cellular H3K14 acetylation pattern. At the same time, DC_HG24-01 induced MV4-11 leukemia cells apoptosis and cell cycle arrest at G1 phase. Taken together, the discovery of DC_HG24-01 represents an advance on the road to develop more potent hGCN5 inhibitors and could assist in exploring the elusive role of hGCN5 in physiological and pathological processes.
4. Experimental
4.1 Protein expression and purification
The cDNA encoding HAT domain of hGCN5 (residues 497–662) was cloned into a modified pET-28a vector, which included an N-terminal His6-tag. The recombinant protein was overexpressed as previously described.32 The cells were harvested by centrifugation at 2655 × g for 8 min at 4 °C and sonicated in lysis buffer (50 mM Hepes pH 8.0, 500 mM NaCl, 20 mM imidazole, 5% Glycerol and 1 mM TCEP). The lysate was centrifuged at 39191 × g for 45 min and the cleared supernatant was loaded onto HisTrap HP column (GE Healthcare). The column was washed with wash buffer (50 mM Hepes pH 8.0, 500 mM NaCl, 100 mM imidazole, 5% glycerol and 1 mM TCEP). Bound protein was eluted with elution buffer containing 300 mM imidazole. The eluted protein was further purified by gel-filtration chromatography on a Superdex 75 column (GE Healthcare) equilibrated in SEC buffer (20 mM Hepes pH 8.0, 250 mM NaCl, 5% glycerol, 1 mM TCEP). The protein was snap-frozen in liquid nitrogen and stored at −80 °C.
4.2 Amplified luminescent proximity homogeneous (AlphaScreen) assay
AlphaScreen assays were performed following the protocol previously described.33 Compounds from the in-house library were diluted in dilution buffer (20 mM Hepes pH 8.0, 150 mM NaCl) and incubated with 10 nM hGCN5 in 384-well plate at 25 °C. 100 nM H3 peptide (ARTKQTARKSTGGKAPRKQLATK-Biotin, synthesized by Shanghai ChinaPeptide Corporation) mixed with 2 μM Ac-CoA (Sigma, cat. no. A2056) in modified assay buffer was added to start the reaction with the incubation time of 30 min. For signal detection, 5 μL streptavidin coated donor beads (6760002, PerkinElmer Life Science) and 5 μL protein A coated acceptor beads (6760137M, PerkinElmer Life Science)/anti-H3K14-ac antibody (Cell Signaling Technology, #7627) mixture were added to the whole system and incubated for another 60 min. Finally, the data was collected using EnVision (PerkinElmer Life Science) and analyzed in Graphpad Prism 7.0. The determination of Z-factor in AlphaScreen assays was conducted as previously described.28
4.3 Radioactive acetylation assay
Inhibitors were diluted in DMSO and pretreated with hGCN5 enzyme for 15 min at room temperature. Substrate solution in optimized assay buffer containing H3 peptide and [3H] Ac-CoA (PerkinElmer, cat. no. NET290) was added into each well to start the reaction. After 60 min of incubation at 37 °C, reaction was stopped with cold Ac-CoA (Sigma, cat. no. A2056). Then 25 μL mix solution was transferred to 384-well Flashplate (Perkin Elmer, cat. no. SMP410A001PK) from the assay plate. After several washes with ddH2O containing 0.1% Tween-20, the signal was collected on Microbeta. IC50 values were derived by fitting the data using nonlinear regression in GraphPad Prism 7.0.
4.4 Surface plasmon resonance (SPR) based binding assay
The SPR binding assay was performed on the Biacore T200 instrument (GE Healthcare). The purified protein was immobilized on a CM5 sensor chip by amine coupling at a concentration of 20 μg mL−1. The SPR system was equilibrated with HBS buffer (25 mM Hepes pH 8.0, 150 mM NaCl, 0.05% (v/v) P20 surfactant) containing 0.2% (v/v) DMSO. Compounds were diluted using HBS buffer with the concentrations ranging from 40 μM to 1.9 μM. The association and dissociation time was set as 50 s and 60 s with the flow rate at 30 μL min−1, respectively. Finally, data analysis was conducted via the state and binding model of T200 evaluation software (GE Healthcare). Data was fitted globally to a 1:1 binding model using Biacore T200 Evaluation software to determine the binding kinetic rate constants, ka (M−1 s−1) and kd (s−1), and the equilibrium dissociation constant KD (M) or steady-state binding levels were fitted to determine the equilibrium dissociation constant KD (M) alone. The errors were determined in GraphPad Prism 7.0 and were represented as mean ± SD of three replicates.
4.5 2D-similarity based analogue searching and molecular docking
The 2D-similarity based analogue searching was performed using Pipeline Pilot (version 7.5; Accelrys Inc., San Diego, CA, USA) as previously described.34 Molecular docking studies were performed in order to analyze the binding modes of DC_HG24-01 and its derivatives. Firstly, the crystal structure of hGCN5 was fetched from the protein data bank (PDB code: 1Z4R). The ligands were prepared by LigPrep module to generate of all stereoisomers and different protonation states and the protein states were optimized by the Protein preparation Wizard module in Maestro (Schrödinger, LLC: New York, NY, 2009). A 30 Å × 30 Å × 30 Å receptor grids centered on Ac-CoA were generated in the Receptor Grid Generation Module. Then docking study was performed using Glide using extra-precision (XP) mode with enhanced planarity of conjugated pi groups, and strain correction terms applied.
4.6 Cell proliferation assays
MV4-11 and HUV-EC-C, MRC-5 cell lines were purchased from ATCC. All cell lines were cultured at 37 °C with 5% CO2 in RPMI1640 medium or DMEM medium (Life Technologies) with 10% FBS and 1% PS (Life Technologies). For cell proliferation assay, the cells were plated in 96-well plates (Corning) in total volume of 100 μL and treated with DMSO or indicated concentration of compounds for 72 h. CellTiter-Glo luminescent assays (Promega) were used to determine the fraction of viable cells according to the manufacturer's instruction. Briefly, 100 μL of reconstituted CellTiter-Glo reagent was added to each well and incubated at room temperature for 10 min. Then, 60 μL of the mixture was transferred to the white 96-well luminometer plate, and plates were measured on EnVision (PerkinElmer). All treatments were performed in triplicate. IC50 values were derived by fitting the data using nonlinear regression in GraphPad Prism 7.0.
4.7 Western blot
The western blot assay was conducted as previously described.26 20 μM SAHA was used (CSNpharm, catalog no. CSN11991) to elevate basal acetylation levels. After 48 h treatment of DC_HG24-01, the cells were lysed in 1 × SDS loading buffer. Total cell lysates were separated by 12% SDS–PAGE gel and transferred to nitrocellulose membranes. The membranes were blocked in 5% (w/v) nonfat milk for 1 h and incubated with primary antibodies overnight at 4 °C. Then the membrane was incubated with diluted HRP-linked anti-rabbit IgG or HRP-linked anti-mouse IgG for 1 h. Following three washes with TBST, bands were detected in a ChemiScope3400 imaging system using ECL substrate.
4.8 Cell cycle and apoptosis assay
For cell cycle analysis, MV4-11 cells were cultured under the conditions described above, and 1 × 106 cells were seeded in 6-well plate (Corning). After 24 h treatment with indicated concentration of compound, cells were harvested and resuspended in 70% enthanol overnight at 4 °C for fixation. Then samples were washed with PBS and incubated with PI/RNase Staining buffer (BD Pharmingen) at room temperature for flow cytometry analysis. For the apoptosis assay, the cells were collected at 72 h and cell apoptosis was measured using Annexin V-FITC Apoptosis Detection Kit (Vazyme Biotech) based on manufacturer's instructions. All samples were measured by FACSCalibur (BD Pharmingen) and data was analyzed by Modfit and FlowJo V7.6.1.
Authors' contributions
Conception and design: X. X. and C. L.; development of methodology: H. T., J. W., W. L. R. Z. Y. X., L. Y. and C. L.; acquisition of data: H. T., J. W., W. L, Y. L. and R. L.; analysis and interpretation of data: W. L. J. H., T. L., J. W., Y. L., C. Z., H. X., Y. C. and C. W. Writing, review, and/or revision of the manuscript: H. T., J. W., W. L. and H. X. Administrative, technical, or material support: K. C., H. J., Y. Z., X. X., and C. L. Study supervision: X. X. and C. L.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
We thank the staff members of the Large-Scale Protein Preparation System at the National Facility for Protein Science in Shanghai (NFPS), Zhangjiang Lab, China for providing technical support and assistance in data collection and analysis. We gratefully acknowledge financial support from the National Natural Science Foundation of China (21472208, 81625022, 81430084 to C. L. and 81803554 to Y. Z); China Postdoctoral Science Foundation (2017M621571 to L. Y.); K. C. Wong Education Foundation to C. L. and Chinese Academy of Sciences (XDA12020353 and XDA12050401).
References
- P. Cheung, C. D. Allis and P. Sassone-Corsi, Cell, 2000, 103, 263–271 CrossRef CAS.
- G. J. Narlikar, H.-Y. Fan and R. E. Kingston, Cell, 2002, 108, 475–487 CrossRef CAS.
- C. Choudhary, C. Kumar, F. Gnad, M. L. Nielsen, M. Rehman, T. C. Walther, J. V. Olsen and M. Mann, Science, 2009, 325, 834–840 CrossRef CAS.
- K. Balasubramanyam, M. Altaf, R. A. Varier, V. Swaminathan, A. Ravindran, P. P. Sadhale and T. K. Kundu, J. Biol. Chem., 2004, 279, 33716–33726 CrossRef CAS.
- F. Dyda, D. C. Klein and A. B. Hickman, Annu. Rev. Biophys. Biomol. Struct., 2000, 29, 81–103 CrossRef CAS.
- V. Sapountzi and J. Cote, Cell. Mol. Life Sci., 2011, 68, 1147–1156 CrossRef CAS.
- T. Jenuwein and C. D. Allis, Science, 2001, 293, 1074–1080 CrossRef CAS.
- S. Vernarecci, F. Tosi and P. Filetici, Epigenetics, 2014, 5, 105–111 CrossRef.
- M. H. Kuo, J. E. Brownell, R. E. Sobel, T. A. Ranalli, R. G. Cook, D. G. Edmondson, S. Y. Roth and C. D. Allis, Nature, 1996, 383, 269–272 CrossRef CAS.
- M. Grunstein, Nature, 1997, 389, 349–352 CrossRef CAS.
- L. Howe, D. Auston, P. Grant, S. John, R. G. Cook, J. L. Workman and L. Pillus, Genes Dev., 2001, 15, 3144–3154 CrossRef CAS.
- M. Sakai, T. Tujimura-Hayakawa, T. Yagi, H. Yano, M. Mitsushima, H. Unoki-Kubota, Y. Kaburagi, H. Inoue, Y. Kido, M. Kasuga and M. Matsumoto, Nat. Commun., 2016, 7, 13147 CrossRef CAS.
- K. K. Lee and J. L. Workman, Nat. Rev. Mol. Cell Biol., 2007, 8, 284–295 CrossRef CAS PubMed.
- L. Chen, T. Wei, X. Si, Q. Wang, Y. Li, Y. Leng, A. Deng, J. Chen, G. Wang, S. Zhu and J. Kang, J. Biol. Chem., 2013, 288, 14510–14521 CrossRef CAS.
- T. Holmlund, M. J. Lindberg, D. Grander and A. E. Wallberg, Leukemia, 2012, 27, 578–585 CrossRef.
- A. Farria, W. Li and S. Y. Dent, Oncogene, 2015, 34, 4901–4913 CrossRef CAS.
- F. Manzo, F. P. Tambaro, A. Mai and L. Altucci, Expert Opin. Ther. Pat., 2009, 19, 761–774 CrossRef CAS.
- A. N. Poux, M. Cebrat, C. M. Kim, P. A. Cole and R. Marmorstein, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 14065–14070 CrossRef CAS.
- F. J. Dekker and H. J. Haisma, Drug Discovery Today, 2009, 14, 942–948 CrossRef CAS.
- M. Biel, A. Kretsovali, E. Karatzali, J. Papamatheakis and A. Giannis, Angew. Chem., Int. Ed. Engl., 2004, 43, 3974–3976 CrossRef CAS.
- J. M. Gajer, S. D. Furdas, A. Gründer, M. Gothwal, U. Heinicke, K. Keller, F. Colland, S. Fulda, H. L. Pahl, I. Fichtner, W. Sippl and M. Jung, Oncogenesis, 2015, 4, e137 CrossRef CAS.
- P. Ornaghi, D. Rotili, G. Sbardella, A. Mai and P. Filetici, Biochem. Pharmacol., 2005, 70, 911–917 CrossRef CAS.
- W. Lu, H. Xiong, Y. Chen, C. Wang, H. Zhang, P. Xu, J. Han, S. Xiao, H. Ding, Z. Chen, T. Lu, J. Wang, Y. Zhang, L. Yue, Y. C. Liu, C. Zhang, Y. Yang, H. Jiang, K. Chen, B. Zhou and C. Luo, Bioorg. Med. Chem., 2018, 26(20), 5397–5407 CrossRef CAS.
- L. M. Lasko, C. G. Jakob, R. P. Edalji, W. Qiu, D. Montgomery, E. L. Digiammarino, T. M. Hansen, R. M. Risi, R. Frey, V. Manaves, B. Shaw, M. Algire, P. Hessler, L. T. Lam, T. Uziel, E. Faivre, D. Ferguson, F. G. Buchanan, R. L. Martin, M. Torrent, G. G. Chiang, K. Karukurichi, J. W. Langston, B. T. Weinert, C. Choudhary, P. de Vries, J. H. Van Drie, D. McElligott, E. Kesicki, R. Marmorstein, C. Sun, P. A. Cole, S. H. Rosenberg, M. R. Michaelides, A. Lai and K. D. Bromberg, Nature, 2017, 550, 128–132 CrossRef CAS.
- J. B. Baell, D. J. Leaver, S. J. Hermans, G. L. Kelly, M. S. Brennan, N. L. Downer, N. Nguyen, J. Wichmann, H. M. McRae, Y. Yang, B. Cleary, H. R. Lagiakos, S. Mieruszynski, G. Pacini, H. K. Vanyai, M. I. Bergamasco, R. E. May, B. K. Davey, K. J. Morgan, A. J. Sealey, B. Wang, N. Zamudio, S. Wilcox, A. L. Garnham, B. N. Sheikh, B. J. Aubrey, K. Doggett, M. C. Chung, M. de Silva, J. Bentley, P. Pilling, M. Hattarki, O. Dolezal, M. L. Dennis, H. Falk, B. Ren, S. A. Charman, K. L. White, J. Rautela, A. Newbold, E. D. Hawkins, R. W. Johnstone, N. D. Huntington, T. S. Peat, J. K. Heath, A. Strasser, M. W. Parker, G. K. Smyth, I. P. Street, B. J. Monahan, A. K. Voss and T. Thomas, Nature, 2018, 560, 253–257 CrossRef CAS.
- H. Xiong, J. Han, J. Wang, W. Lu, C. Wang, Y. Chen, L. Fulin, N. Zhang, Y. C. Liu, C. Zhang, H. Ding, H. Jiang, W. Lu, C. Luo and B. Zhou, Eur. J. Med. Chem., 2018, 151, 740–751 CrossRef CAS.
- J. Xing, W. Lu, R. Liu, Y. Wang, Y. Xie, H. Zhang, Z. Shi, H. Jiang, Y. C. Liu, K. Chen, H. Jiang, C. Luo and M. Zheng, J. Chem. Inf. Model., 2017, 57, 1677–1690 CrossRef CAS.
- T. Lu, J. C. Hu, W. C. Lu, J. Han, H. Ding, H. Jiang, Y. Y. Zhang, L. Y. Yue, S. J. Chen, H. L. Jiang, K. X. Chen, H. F. Chai and C. Luo, Acta Pharmacol. Sin., 2018, 39, 1544–1552 CrossRef CAS.
- J. H. Zhang, T. D. Y. Chung and K. R. Oldenburg, J. Biomol. Screening, 1999, 4, 67–73 CrossRef.
- A. Ciulli, Methods Mol. Biol., 2013, 1008, 357–388 CrossRef CAS.
- Y. L. Wang, L. J. Li, B. D. Zhang, J. Xing, S. J. Chen, W. Wan, Y. K. Song, H. Jiang, H. L. Jiang, C. Luo and M. Y. Zheng, J. Med. Chem., 2017, 60, 2026–2036 CrossRef CAS.
- A. Schuetz, G. Bernstein, A. Dong, T. Antoshenko, H. Wu, P. Loppnau, A. Bochkarev and A. N. Plotnikov, Proteins, 2007, 68, 403–407 CrossRef CAS.
- A. Schneider, S. Chatterjee, O. Bousiges, B. R. Selvi, A. Swaminathan, R. Cassel, F. Blanc, T. K. Kundu and A. L. Boutillier, Neurotherapeutics, 2013, 10, 568–588 CrossRef CAS.
- F. Meng, S. Cheng, H. Ding, S. Liu, Y. Liu, K. Zhu, S. Chen, J. Lu, Y. Xie, L. Li, R. Liu, Z. Shi, Y. Zhou, Y. C. Liu, M. Zheng, H. Jiang, W. Lu, H. Liu and C. Luo, J. Med. Chem., 2015, 58, 8166–8181 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence cd,
Rukang
Zhang
cd,
Yiqian
Xie
c,
Yu-Chih
Liu
f,
Rongfeng
Liu
f,
Liyan
Yue
c,
Kaixian
Chen
cdg,
Hualiang
Jiang
cd,
Yuanyuan
Zhang
cd,
Rukang
Zhang
cd,
Yiqian
Xie
c,
Yu-Chih
Liu
f,
Rongfeng
Liu
f,
Liyan
Yue
c,
Kaixian
Chen
cdg,
Hualiang
Jiang
cd,
Yuanyuan
Zhang