
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Silver(I) pyridylphosphonates – synthesis, structure, stability and light-insensitivity investigation†
Michaela Rendošováa,
Róbert Gyepes b,
Miroslav Almášia,
Ingrida Bártováa and
Zuzana Vargová*a
b,
Miroslav Almášia,
Ingrida Bártováa and
Zuzana Vargová*a
aDepartment of Inorganic Chemistry, Faculty of Science, P. J. Šafárik University, Moyzesova 11, 041 54 Košice, Slovak Republic. E-mail: zuzana.vargova@upjs.sk
bDepartment of Inorganic Chemistry, Faculty of Science, Charles University, Hlavova 2030, 128 00 Praha, Czech Republic
First published on 11th January 2019
Abstract
Two silver(I) complexes, {[Ag7(2-pypo)3(NO3)]}n (1) and [Ag(3-pypoH)(3-pypoH2)] (2) (pypoH2 – pyridylphosphonic acid) were prepared and characterized by relevant methods in the solid state (elemental, spectral, thermal and structural analysis). Typical argentophilic interactions were observed in complex 1. The synthesized Ag(I) complexes were tested toward reduction to silver(0) using short wavelength UV-radiation and they revealed remarkable light-insensitivity in comparison to silver nitrate. SEM experiments were performed to observe the reduction of silver after the UV-light exposure of the samples. Light-insensitivity with the examined thermal stability and insolubility of the new complexes demonstrates their high potential to be used as light-insensitive materials in medical devices.
Introduction
The chemistry of metal phosphonates has attracted attention in the last few years. Great effort has been devoted to the syntheses of metal complexes and extended networks utilizing pyridylphosphonic acids as promising candidates for functional materials1 with potential applications in catalysis, proton conductivity, ion exchange and magnetic materials, etc.2 Since phosphonic acids can be readily deprotonated, they form anionic ligands that can coordinate to metal centres and compensate for charge3 having several coordination modes. Another advantage of these ligands is their ability to induce polymerization as a result of the strong interaction of the phosphonate group with the metal ion.3In the field of biomedical devices, clinical practice has shown that systemic antibiotics are not able to provide sufficient treatment for implant-associated infections.4 Upon increasing bacterial resistance against current antibiotics the development of new infection-preventing strategies is very important and research on silver and its compounds has regained interest.5 While the number of implanted medical devices is rapidly increasing, the infections associated with biomaterials still represent a significant challenge.6 Bacterial adhesion to implants and biofilm formation causes serious implant-related infections. For example, adhesion of bacteria to dental implant components may lead to the formation of a biofilm that could be responsible for dental diseases like dental caries, periodontal diseases and peri-implantitis.7 One of the possibilities how to prevent the infections is the introduction of an antimicrobial substance, for example, into the glue used in indwelling medical devices introduction or in surface treatment of the devices to prevent pathogenic cell adhesion and subsequent biofilm formation.4 Therefore silver ions which exhibit low cytotoxicity to mammalian cells and broad-spectrum antimicrobial activity have gained attention in a wide range of medicinal applications.5 Experiments with silver salts have led to unsatisfactory results. On the other hand, silver coordination polymeric compounds are promising candidates for surface modification as they exhibit (i) high structural stability during sterilization at 100 °C, (ii) little decomposition upon light exposure, (iii) poor solubility in aqueous media, and (iv) antimicrobial activity.4,7 Pure organic compounds cannot fulfil these specific needs, but some water-insoluble metal complexes (e.g. silver(I) compounds) can.4 For this reason, it is important to study new metal-based compounds that will meet these requirements.4 In this work we present results of our investigation dealing with the synthesis, spectroscopic, structural and photophysical characterization of the new silver(I) pyridylphosphonates {[Ag7(2-pypo)3(NO3)]}n (1) and [Ag(3-pypoH)(3-pypoH2)] (2) as promising materials for implant applications.
Results and discussion
Syntheses of complexes
Complex 1 was synthesized from aqueous solution of 2-pyridylphosphonic acid and silver nitrate at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio. After slow evaporation (one week) at room temperature in the dark, small colourless crystals of compound 1 were formed. The 1:1 molar-ratio reaction of 3-pyridylphosphonic acid and silver nitrate in a mixed solvent H2O:DMSO = 5:1 (DMSO = dimethyl sulfoxide) yielded complex 2. Under the same conditions of evaporation, grey acicular crystals were formed after three weeks. The compounds were stored in the absence of light, they are air-stable and insoluble in water and organic polar solvents.
:1 molar ratio. After slow evaporation (one week) at room temperature in the dark, small colourless crystals of compound 1 were formed. The 1:1 molar-ratio reaction of 3-pyridylphosphonic acid and silver nitrate in a mixed solvent H2O:DMSO = 5:1 (DMSO = dimethyl sulfoxide) yielded complex 2. Under the same conditions of evaporation, grey acicular crystals were formed after three weeks. The compounds were stored in the absence of light, they are air-stable and insoluble in water and organic polar solvents.
Solid-state study
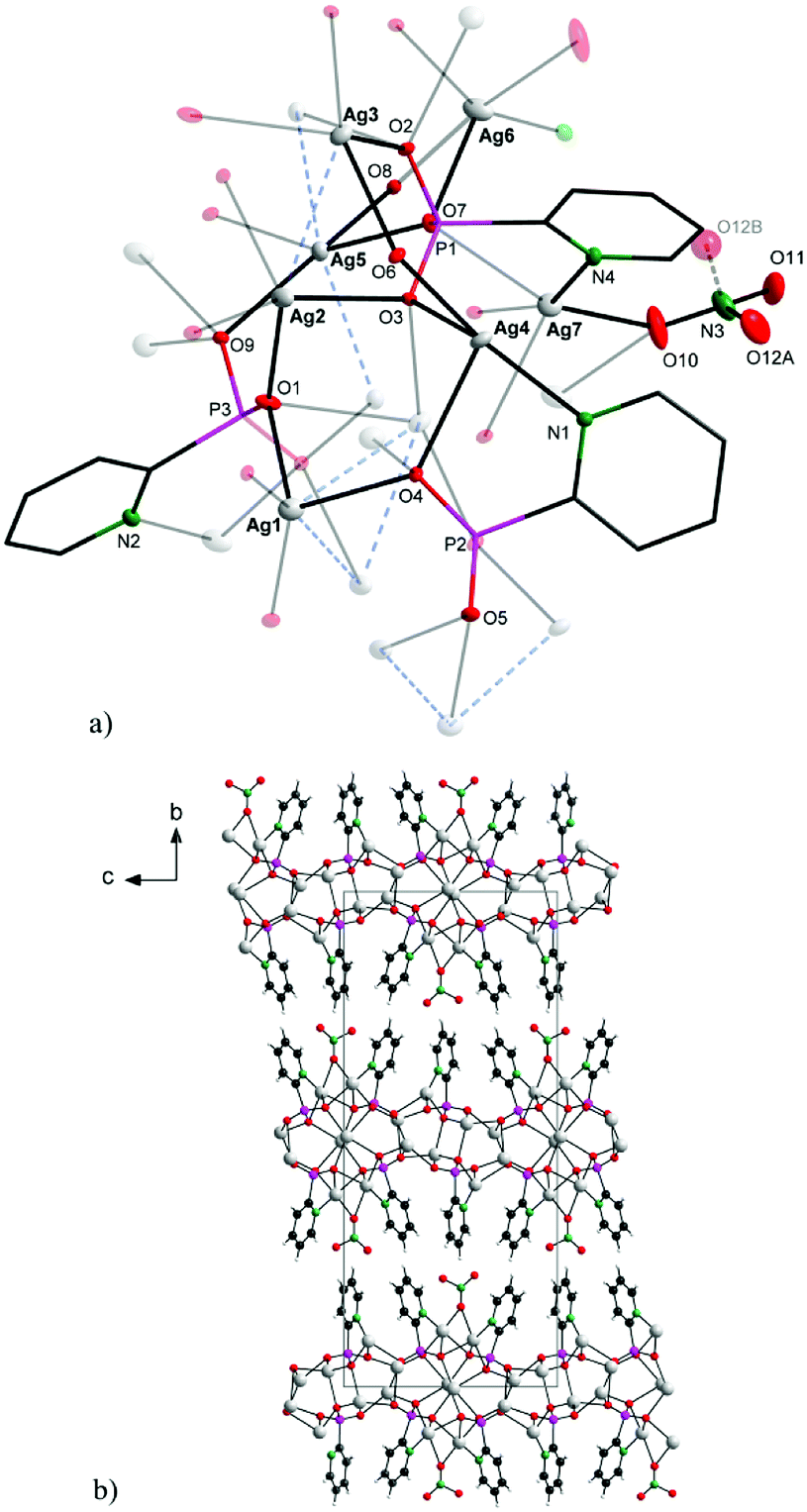 | ||
| Fig. 1 Solid-state structure of the symmetrically independent part (a; upper) and packing diagram viewed along [1 0 0] (b; bottom) for 1. For clarity, labels in the upper picture are given for the most important atoms; atom O12B is omitted on the bottom. | ||
As shown in Fig. 1, the connectivity is quite complicated: the 2-pypo ligand acts as multi-bridging ligand – all of its three oxygen atoms are involved in coordination with silver ions; in addition, the 2-pypo ligand acts as a N,O-chelating ligand, creating a two-dimensional layered structure with layers packed in the ac plane (Fig. 1b).
The P–O interatomic distances are approximately halfway between the P–OH bond and the formal double bond P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O,8 pointing to the negative charge delocalization over the P–O bonds.9 The nitrato ligand is connected with Ag7 and Ag6 having the Ag6–O10 distance 2.658(4) Å and Ag7–O10 2.471(3) Å. The other Ag–O (oxygen atoms of phosphonate groups) distances are in the range 2.234(2)–2.661(3) Å, while the Ag–N distances are 2.212(3) Å, 2.237(3) Å and 2.284(3) Å. These distances together with other bond lengths and angles (see Table S1 in ESI†) are in good accordance with previously published values for other silver complex {[Ag3(triflate)2(2-Hpypo)]}n.3 Moreover, close Ag–Ag contacts had been observed with the metal–metal distance of 2.9298(5)–3.3787(4) Å (Fig. 1a, blue dashed bonds). These contacts are shorter than the sum of their van der Waals radii (3.44 Å), indicating the presence of significant argentophilic interactions.10,11
O,8 pointing to the negative charge delocalization over the P–O bonds.9 The nitrato ligand is connected with Ag7 and Ag6 having the Ag6–O10 distance 2.658(4) Å and Ag7–O10 2.471(3) Å. The other Ag–O (oxygen atoms of phosphonate groups) distances are in the range 2.234(2)–2.661(3) Å, while the Ag–N distances are 2.212(3) Å, 2.237(3) Å and 2.284(3) Å. These distances together with other bond lengths and angles (see Table S1 in ESI†) are in good accordance with previously published values for other silver complex {[Ag3(triflate)2(2-Hpypo)]}n.3 Moreover, close Ag–Ag contacts had been observed with the metal–metal distance of 2.9298(5)–3.3787(4) Å (Fig. 1a, blue dashed bonds). These contacts are shorter than the sum of their van der Waals radii (3.44 Å), indicating the presence of significant argentophilic interactions.10,11
Complex 2 crystallizes in a monoclinic lattice with space group P21/n. The 3-pypo ligand is coordinated to the silver central atom through its pyridine nitrogen. The Ag–N distance is 2.1219(15) Å, which is even shorter than its analogue in 1. Other bond lengths and angles are in good accordance with previously published values for 3-pyridylphosphonic acid8 and other metal complexes with 3-pypo ligand12 and are listed in Table S2 in ESI.† The coordination number of Ag(I) in the complex is two, having a linear arrangement of the nitrogen donor atoms. The 3D arrangement is formed through O3–H3O–O3 hydrogen bonding interactions, where the hydrogen is located in the middle of O–O distance (Fig. 2). Apart from this intermolecular interaction, the structure is stabilized also by O2–H2⋯O1 hydrogen bonds (Fig. 2).
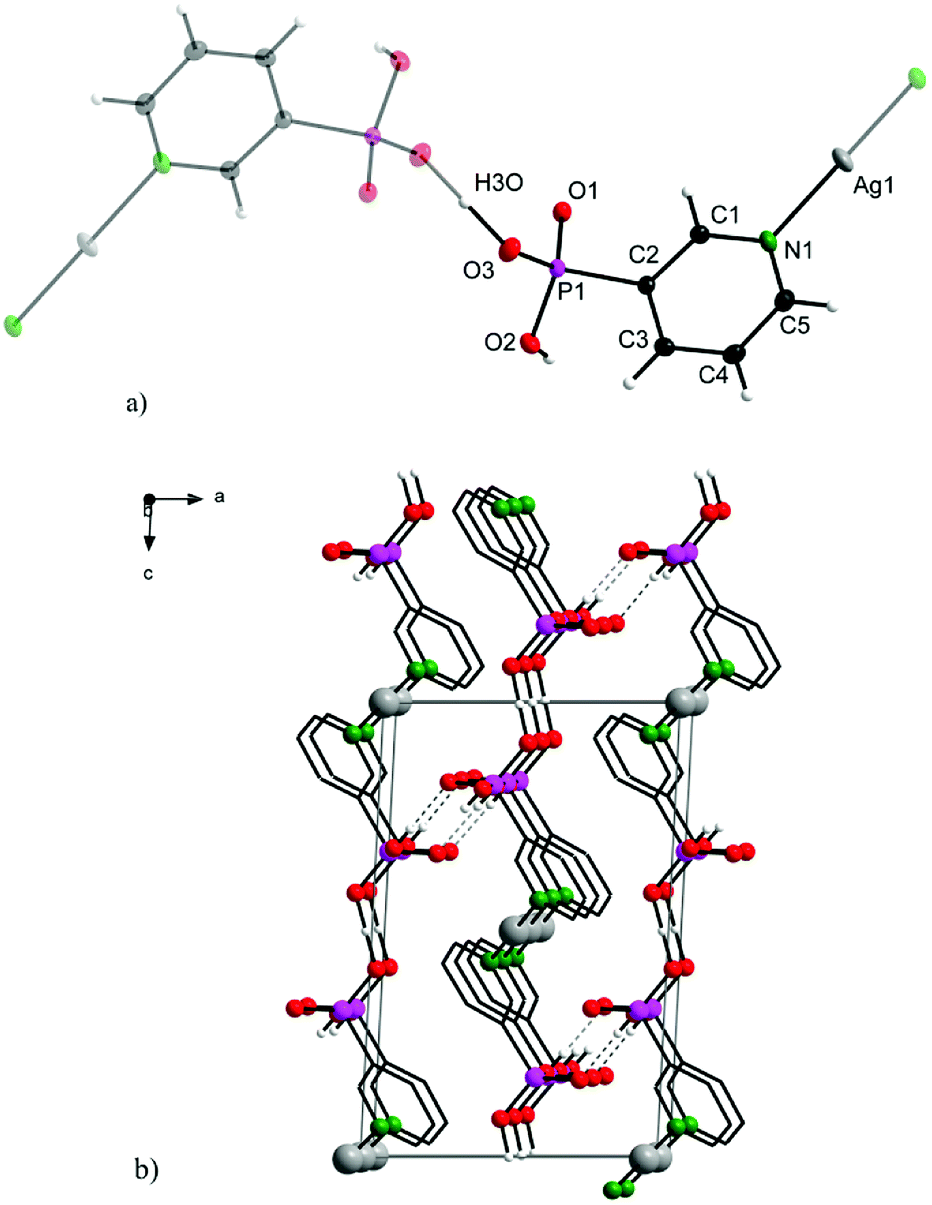 | ||
| Fig. 2 Structure motif (a; upper) and solid-state packing viewed along [0 1 0] (b; bottom) for 2. | ||
Additional π–π stacking interactions are found between the pyridyl rings of the 3-pyridylphosphonato ligands, with an intercentroid distance of 3.7926(11) Å and interplanar spacing 3.4074(7) Å. The ring normal and the vector between the ring centroids form an angle of 26.0°. These values are commonly observed values in complexes with aromatic nitrogen heterocycles.13 The chemical composition of 1 and 2 was in good agreement with results of elemental analyses.
| 2-pypoH2 | Complex 1 | 3-pypoH2 | Complex 2 | |
|---|---|---|---|---|
| ν(O–H) | 3480–3300 | — | 3550–3180 | — |
| ν(CH) | 3108, 3082 | 3054, 3007 | 3118, 3084 | 3110, 3078 |
| ν(C–C), ν(C–N) | 1608, 1520, 1443, 1389 | 1583, 1562, 1459, 1420 | 1626, 1548, 1460, 1368 | 1598, 1566, 1478, 1417 |
| ν(PO3) | 1157,1091, 1030 | 1164, 1154, 1031 | 1178, 1112, 1034 | 1196, 1159, 1118 |
| ν(C–P) | 939 | 948 | 932 | 923 |
| ν(NO3−) | — | 1316 | — | — |
| δ(PO3) | 553, 513, 439 | 570, 528, 460 | 535, 511, 443 | 555, 485, 450 |
| γ(C–H) | 765, 717 | 760, 726 | 727, 682 | 748, 703 |
In summary, observed band shifting in the spectra of complexes may be related to the metal ion coordination as well as to the intermolecular interactions.
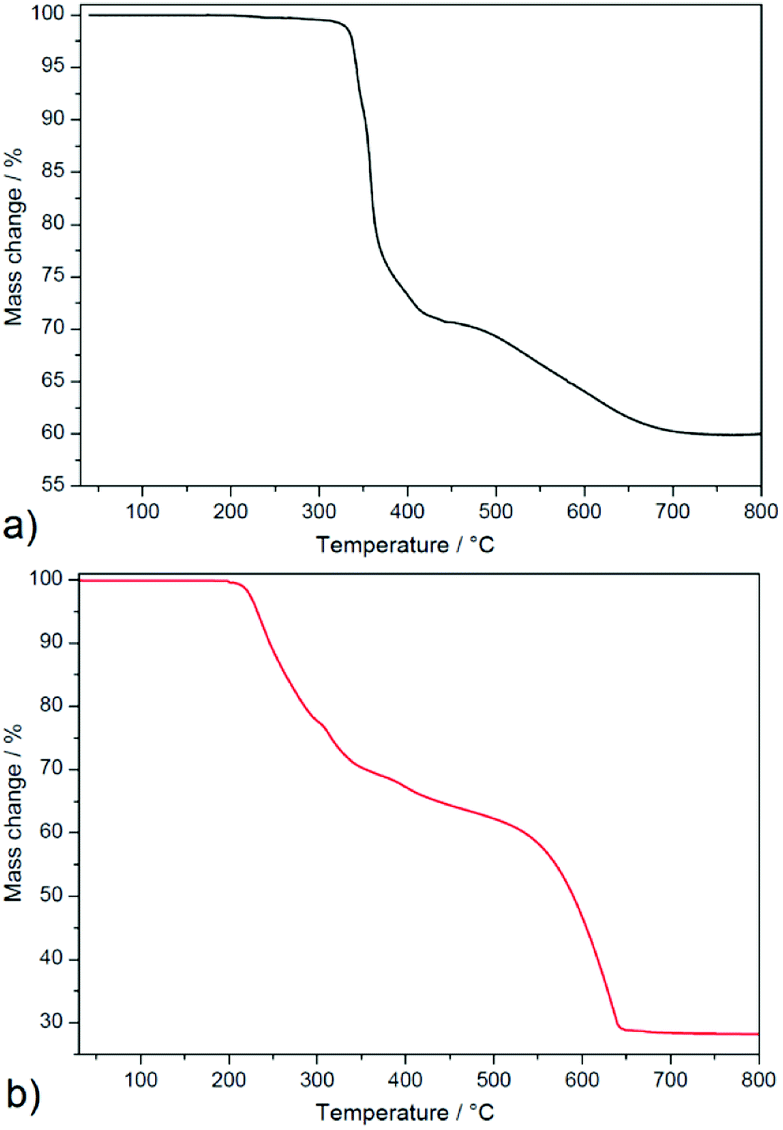 | ||
| Fig. 3 Thermogravimetric curves of 1 (a) and 2 (b) measured in temperature range 30–800 °C and air atmosphere. | ||
Complex 2 (see Fig. 3b) is thermally stable up to 200 °C and thermal decomposition of the organic part occurs in four overlapping decomposition steps. The first three decomposition steps appeared in the temperature range of 200–440 °C in which mass loss of 37.2% was observed and correspond to the thermal decomposition of one 3-pypoH2 molecule (clcd. mass loss 37.4%). The second deprotonated ligand molecule is evolved in the temperature range of 440–650 °C, with mass loss of 36.2% (clcd. mass loss 37.2%). The final product of the thermal decomposition was also silver with residual mass 26.6% (clcd. residual mass 25.4%). Comparison of thermal stability could be performed only for a few compounds, such as {[Cd2(3-pypoH)4]}n·2nDMSO15 and {[Sn2(3-pypo)2]}n,1 which display thermal stability 360 and 325 °C, respectively1,15 (for compound {[Cd2(3-pypoH)4]}n·2nDMSO after removal of the solvent molecules). Higher stability of described compounds could be explained by the polymeric nature of their frameworks compared to the molecular structure of compound 2.
In summary, complex 1 displayed higher thermal stability compared to 2. Its higher thermal stability compared to complex 2 could be attributed to the higher crystal packing and complicated covalent bonding formations between building blocks in the crystal structure of 1. Moreover, the conjunction between Ag–Ag interactions helps to stabilize the polymeric network, which resulted in higher thermal robustness of 1.
 | ||
| Fig. 4 Light-insensitivity study of new silver(I) pyridylphosphonates toward UV-radiation at 254 nm in comparison to silver nitrate. | ||
The light-insensitive characteristics of silver(I) compounds as well as their thermal stability and poor solubility are important for their potential applications in medical devices as additives to curable photopolymers in dental implants.4,17 Practically, for the intention of desired application of synthesized compounds only minutes of light stability are needed.4
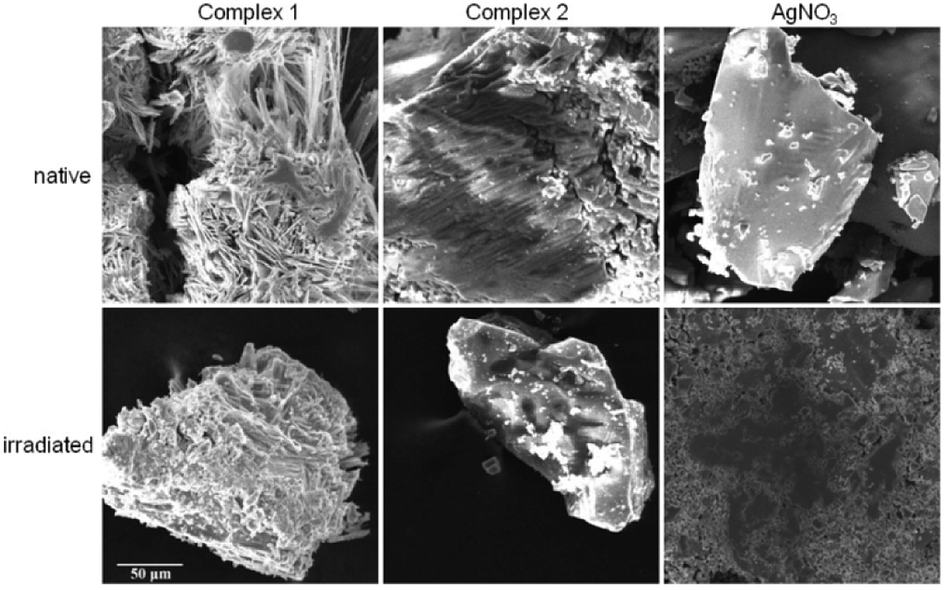 | ||
| Fig. 5 SEM images of native and irradiated silver(I) complexes and silver nitrate at voltage of 20 kV. The scale applies to all images. | ||
Conclusions
Two silver(I) pyridylphosphonates were prepared and fully characterized by IR, CHN, TG and X-ray analysis. Typical argentophilic interactions were observed in complex 1. The IR spectra of both complexes are in a good correlation with structural observations. As was demonstrated by thermal analysis, complexes are thermally stable up to 200 °C, while complex 1 exhibits even higher thermal stability than complex 2. The synthesized Ag(I) complexes have demonstrated remarkable light-insensitivity toward UV-radiation in comparison to silver nitrate. SEM observations revealed dark spots on the most UV-sensitive sample, silver nitrate, that may represent reduced elemental silver on the surface of the irradiated sample.Thermal stability, light-insensitivity and poor solubility are important properties of potential materials for dental implants and indwelling medical devices. A high thermal stability of used materials is required for equipment sterilization and water insolubility is needed for no leaching them out from the surface of the indwelling devices. In conclusion, new silver(I) pyridylphosphonates fulfil these requirements for their potential applications in medical devices and demonstrate high potential to be used as light-insensitive materials and therefore they are suitable candidates for further biocompatibility studies.
Experimental
Materials
Silver(I) nitrate was purchased from Lachema (Czech Republic), dimethyl sulfoxide (DMSO), palladium(II) acetate (Pd(OAc)2) and 3-bromopyridine from Sigma-Aldrich Chemie, 2-bromopyridine, diisopropyl phosphite and N,N-diisopropylethylamine from Fluka, 1,1′-bis(diphenylphosphino)ferrocene (dppf) from Alfa Aesar, bromotrimethylsilane from Acros, dichloromethane, methanol and acetonitrile from VWR Chemicals.Synthesis of 2- and 3-pyridylphosphonic acids
Acids were prepared according to Belabassi:18 diisopropyl phosphite (4.8 mmol, 1.2 equiv.), 2- or 3-bromopyridine (4.0 mmol, 1.0 equiv.), N,N-diisopropylethylamine (5.2 mmol, 0.9 mL, 1.3 equiv.), Pd(OAc)2 (0.04 mmol, 1 mol%), and 1,1′-bis(diphenylphosphino)ferrocene (dppf) (0.044 mmol, 1.1 mol%) were added to acetonitrile (dry, 15 mL) and the reaction mixture was then heated at reflux for 24 h under argon atmosphere. After reaction, the solution was concentrated under reduced pressure (LC separation, TLC detection). The resulting oil was dissolved in CH2Cl2 and treated with bromotrimethylsilane (2.2 equiv.) at room temperature under N2. When silylation was complete (24–48 h, monitored by 31P NMR), the solvent was removed in vacuo and MeOH was added. The phosphonic acids were obtained as solids either directly, or by precipitation from water.Synthesis of complex {[Ag7(2-pypo)3(NO3)]}n (1)
Silver(I) nitrate (100 mg, 0.59 mmol) was dissolved in water (5 mL) and added dropwise to the 5 mL of 2-pyridylphosphonic acid aqueous solution (93.6 mg; 0.59 mmol). The reaction mixture was stirred constantly for 10 min and then allowed to stand at room temperature. Crystals formed by slow evaporation (after five days) were filtered off, washed with diethyl ether and dried under vacuum for 24 h. Yield: 27%. Elemental analysis – found: C, 13.80; H, 0.76; N 4.51. Calc. for C15H12N4O12P3Ag7: C, 13.98; H, 0.94; N, 4.35%.Synthesis of complex [Ag(3-pypoH)(3-pypoH2)] (2)
Silver(I) nitrate (100 mg, 0.59 mmol) was dissolved in water (5 mL) and added dropwise to the 18 mL of 3-pyridylphosphonic acid H2O:DMSO (5:1 v/v) solution (93.6 mg; 0.59 mmol). The reaction mixture was stirred constantly for 10 min and then allowed to stand at room temperature. Crystals formed by slow evaporation (after three weeks) were filtered off, washed with diethyl ether and dried under vacuum for 24 h. Yield: 58%. Elemental analysis – found: C, 28.16; H, 2.45; N, 6.61. Calc. for C10H11N2O6P2Ag: C, 28.26; H, 2.61; N, 6.59%.
Physical measurements
Diffraction data for 1 were measured on a Bruker D8 Venture Kappa Duo diffractometer equipped with a PHOTON 100 detector and IμS 3.0 microfocus sources; data for 2 were obtained on a Nonius Kappa CCD diffractometer equipped with a Bruker APEX II detector and graphite monochromator. For both measurements, Mo Kα (λ = 0.71073 Å) was used as the primary radiation. The phase problem was solved by intrinsic phasing and structure models were refined against F2 using the SHELX program suite.19 All non-hydrogen atoms (excepting O12B in 1) were refined anisotropically. All hydrogen atoms were refined isotropically; those on aromatic rings were included in idealized positions. Important crystallographic data are listed in Table S3 in ESI.† Structure figures were drawn using the DIAMOND software.20Infrared spectra were recorded on Avatar FT-IR 6700 spectrometer from 4000 to 400 cm−1 using ATR (attenuated total reflectance) technique.
Elemental analysis was performed with a CHNOS Elemental Analyzer Vario MICRO from Elementar Analysensysteme GmbH.
Thermal behaviour of compounds 1 and 2 was studied by thermogravimetry (TG) using TA instrument TGA Q-500 in atmosphere of air. Samples were heated with heating rate 10 °C min−1 in temperature range from 30 to 800 °C and air flow rate 60 cm3 min−1. Before thermal measurements, gentle grinding of the sample and careful packing into the platinum crucibles were performed. The mass of samples used in the analyses was within 10–15 mg. Obtained thermoanalytical curves were analysed using Origin computational program (version 6.1052, Origin Lab Northampton, MA, USA).
Light-sensitivity of synthesized Ag(I) pyridylphosphonates was studied at 298 K using an ultraviolet germicidal lamp PROLUX G with a hot cathode (30 W, 254 nm). Complexes were gently ground into a powder and placed on a microscope slide. The radiation source was positioned 10 cm from the samples. Silver nitrate was used as a control substance to access the light stability of silver(I) complexes. An optical microscope with a 10 times microscope objective lens was used to observe the changes and digital pictures of the random part of the samples were taken in intervals 0, 15, 30, 60 and 120 min of radiation exposure time using a digital ocular camera (Dino Eye, 5 Mpx). Images were used without any further contrast or brightness editing. SEM images were taken with Scanning Electron Microscope VEGA (TESCAN) at 20 kV using secondary electron detector and back scatter electron detector.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by Slovak grant agency KEGA 008UPJŠ-4/2018, Pavol Jozef Safarik University in Kosice VVGS-PF-2018-799 and Slovak Research and Development Agency under the contract No. APVV-15-0520. The authors are very grateful to Dr Vladimír Komanický for SEM measurements. Authors thank the Ministry of Education, Science, Research and Sport of the Slovak Republic for the financial support of the TRIANGEL team in the frame of the scheme “Top Research Teams in Slovakia”.References
- H. Perry, J. Zoń, J. Law and A. Clearfield, J. Solid State Chem., 2010, 183, 1165–1173 CrossRef CAS.
- Y. S. Ma, Y. F. Yang, S. Gao, Y. Z. Li and L. M. Zheng, J. Solid State Chem., 2006, 179, 3017–3023 CrossRef CAS.
- J. A. Fry, C. R. Samanamu, J. L. Montchamp and A. F. Richards, Eur. J. Inorg. Chem., 2008, 463–470 CrossRef CAS.
- N. Gerasimchuk, A. Gamian, G. Glover and B. Szponar, Inorg. Chem., 2010, 49, 9863–9874 CrossRef CAS PubMed.
- S. Eckhardt, P. S. Brunetto, J. Gagnon, M. Priebe, B. Giese and K. M. Fromm, Chem. Rev., 2013, 113, 4708–4754 CrossRef CAS PubMed.
- E. R. Kenawy, S. D. Worley and R. Broughton, Biomacromolecules, 2007, 8, 1359–1384 CrossRef CAS PubMed.
- P. S. Brunetto, T. V. Slenters and K. M. Fromm, Materials, 2011, 4, 355–367 CrossRef CAS PubMed.
- J. Zon, V. Videnova-Adrabinska, J. Janczak, M. Wilk, A. Samoc, R. Gancarz and M. Samoc, CrystEngComm, 2011, 13, 3474–3484 RSC.
- K. D. Demadis, S. D. Katarachia and M. Koutmos, Inorg. Chem. Commun., 2005, 8, 254–258 CrossRef CAS.
- M. Rendošová, Z. Vargová, J. Kuchár, D. Sabolová, Š. Levoča, J. Kudláčová, H. Paulíková, D. Hudecová, V. Helebrandtová, M. Almáši, M. Vilková, M. Dušek and D. Bobáľová, J. Inorg. Biochem., 2017, 168, 1–12 CrossRef.
- H. Schmidbaur and A. Schier, Angew. Chem., Int. Ed., 2015, 54, 746–784 CrossRef CAS.
- J. L. Zhou, X. Q. He, B. Liu, Y. Huo, S. C. Zhang and X. G. Chun, Transition Met. Chem., 2010, 35, 795–800 CrossRef CAS.
- C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 21, 3885–3896 RSC.
- M. Rendošová, Z. Vargová, D. Sabolová, N. Imrichová, D. Hudecová, R. Gyepes, B. Lakatoš and K. Elefantová, J. Inorg. Biochem., 2018, 186, 206–216 CrossRef PubMed.
- P. Ayyappan, O. R. Evans, B. M. Foxman, K. A. Wheeler, T. H. Warren and W. Lin, Inorg. Chem., 2001, 40, 5954–5961 CrossRef CAS.
- M. Wilk, K. N. Jarzembska, J. Janczak, M. Duczmal, J. Hoffmann and V. Videnova-Adrabinska, RSC Adv., 2014, 4, 58858–58866 RSC.
- M. I. Azócar, G. Gómez, C. Velásquez, R. Abarca, M. J. Kogan and M. Páez, Mater. Sci. Eng., C, 2014, 37, 356–362 CrossRef PubMed.
- Y. Belabassi, S. Alzghari and J. L. Montchamp, J. Organomet. Chem., 2008, 693, 3171–3178 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- K. Brandenburg and H. Putz, DIAMOND–Crystal and Molecular Structure Visualization, v4.5, Crystal Impact GbR, Bonn, 2017 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1883488 and 1883489. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra10136a |
| This journal is © The Royal Society of Chemistry 2019 |