
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRetracted Article: MiR-148a agomir based targeting of c-Met and Her-3 is able to attenuate EGFR-T790M mutation driven gefitinib and erlotinib resistance in non-small cell lung cancer cells
Guimin Chena,
Lei Yeb,
Yufei Hana and
Ping Han *c
*c
aDepartment II of Internal Medicine, Lin Yi Cancer Hospital, Linyi, Shandong, China
bDepartment I of Internal Medicine, Lin Yi Cancer Hospital, Linyi, Shandong, China
cDepartment II of Respiration, Linyi People's Hospital, No. 6, East Lingyuan Street, Lanshan District, 276000, Linyi, Shandong, China. E-mail: dmdangmm84@126.com; Tel: +86-0539-8078000
First published on 5th July 2019
Abstract
MiR-148a inhibits NSCLC progression. Whether miR-148a would reduce EGFR tyrosine kinase inhibitor (TKI) resistance of NSCLC cells remains underexplored. In this study, 5 NSCLC patients received surgery and gefitinib treatment but developed pleural metastasis. Patients' NSCLC adopted EGFR T790M mutation. 5 naïve and 5 gefitinib-resisting NSCLC cell lines were derived from patients primary and metastatic tumor tissues, and the 5 gefitinib-resisting NSCLC cell lines were trained with erlotinib to establish the erlotinib-resisting cell lines. MiR-148a levels in cells were analyzed by qRT-PCR. miR-148a overexpression was mimicked by agomir treatment. NSCLC cell malignancy was evaluated by cell proliferation, apoptosis, colony formation and transwell invasion assays. Protein levels of c-Met, Her-3 and IGF-1R were assessed by western blotting. miRNA-mRNA interaction was investigated by luciferase reporter assay and AGO2-RIP. Transient overexpression of MET, ERBB3 or IGF1R gene was achieved by plasmid transfection. Results showed that the MiR-148a level was decreased with the development of gefitinib and erlotinib resistance and that there was an increase in malignancy in NSCLC cells in vitro. Treatment with miR-148a agomir significantly enhanced the cytotoxicity of gefitinib and erlotinib to naïve, gefitinib-resisting and erlotinib-resisting NSCLC cells in vitro while reducing their protein levels of c-Met, Her-3 and IGF-1R, the mRNAs of which were verified as direct targets of miR-148a in NSCLC cells. Restoring c-Met or Her-3 protein levels partially reduced the gefitinib and erlotinib sensitizing effect of miR-148a agomir treatment on NSCLC cells. We concluded that MiR-148a attenuated gefitinib and erlotinib resistance in non-small cell lung cancer cells with EGFR T790M mutation by targeting c-Met and Her-3 expression.
Introduction
As a major breakthrough in non-small cell lung cancer (NSCLC) management, epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) such as gefitinib and erlotinib have been increasingly used to treat NSCLC with EGFR activating mutations. EGFR TKIs function by preferentially binding (reversibly or irreversibly) to mutations on the intracellular region of EGFR that allows the activation of the EGFR kinase domain without EGF ligation, thus inhibiting the overactivation of EGFR signaling and subsequent tumor growth. However, NSCLC cells frequently develop inherited or adaptive resistance to EGFR TKI treatment, either by adopting more mutations on the target site of EGFR TKIs that abrogate the inhibitory effect of these drugs or upregulating the expression of other growth factor receptors, hence bypassing EGFR inhibition and maintaining tumor growth. The T790M mutation occurs in about 60% of patients with EGFR TKI resistance.1 erlotinib is the third generation of EGFR TKI developed to treat NSCLC cells that have developed gefitinib resistance; although this drug hasn't been fully used in the clinic, NSCLC resistance to erlotinib treatment, however, has already been reported.2–4Reduce in miR-148a level has been suggested as possible biomarker for NSCLC diagnosis,5–7 while low miR-148a expression has been found associated with NSCLC progression and patients' worsened outcome,8,9 implying that this microRNA might function as a suppressor in NSCLC development. However, the tumor suppressive mechanism of miR-148a in NSCLC remains underexplored. Previous researches have demonstrated that miR-148a would suppress the gene expression of ERBB3,10 MET11 and IGF1R12 via directly interacting with the target sequences on the 3′ untranslated regions (3′ UTRs) of these genes' mRNAs, thus triggering gene silencing mediated by the RNA-induced silencing complex (RISC). Her-3, Insulin growth factor-1 receptor and c-Met proteins are three growth factor receptors encoded by these three genes, whose upregulation plays pivotal role in the development of EGFR TKI resistance in NSCLC,13–15 and targeting these proteins has been suggested to improve the therapeutic effect of EGFR TKI against NSCLC.16–19
The present research attempted to explore whether and how miR-148a would regulate NSCLC cells' resistance to EGFR TKI treatment. We established patient-derived NSCLC cell lines from naïve and gefitinib-resistant NSCLC tissue samples, and we observed that miR-148a overexpression (mimicked by miR-148a agomir treatment) significantly attenuated the rapid cell proliferation, metastasis and gefitinib resistance of NSCLC cells in vitro. Moreover, after establishing the erlotinib resistant NSCLC cells based on the patient-derived, gefitinib resistant NSCLC cells, we found that miR-148a agomir treatment significantly inhibited growth, metastasis and erlotinib resistance of these cells in vitro. Mechanistically, we found that miR-148a agomir treatment significantly reduced protein level of c-Met and Her-3 in NSCLC cells that were upregulated during the induction of erlotinib resistance, which might be the erlotinib-sensitizing mechanism of miR-148a in NSCLC.
Materials and methods
Establishment of naïve, gefitinib and erlotinib NSCLC cells in vitro
This research was approved by the ethical review committee of Lin Yi Cancer Hospital and all participants' informed consent in written. Primary and metastatic NSCLC tissue specimens were obtained from each of 5 patients who were diagnosed with lung adenocarcinoma and received surgery plus gefitinib treatment but developed pleural metastasis 2–4 years later. Genotyping results indicated the adoption of T790M mutation on EGFR of the patients' metastatic NSCLC cells. 5 naïve and 5 gefitinib resistant NSCLC cell lines were established by transplanting the minced primary and metastatic NSCLC tissue samples subcutaneously on NU/NU nude mice (403, Vital River). The xenografts were excised from the mice when their volumes reached about 1.5 cm3 (calculated by volume = length × width2 × π/6 using a Vernier caliper). The xenografts were then minced and cultured in RPMI-1640 medium supplemented with 10% of FBS and 1% of antibiotic solution in vitro for 20 passages before liquid nitrogen storage. 5 erlotinib resistant NSCLC cell lines were established by training the 5 gefitinib resistant NSCLC cell lines in vitro with erlotinib at 2 μM until tolerance, followed by training with erlotinib at 4 μM until tolerance. Resistance to gefitinib or erlotinib of these NSCLC cells was determined by flow cytometry and clonogenic assays evaluating the cytotoxicity of gefitinib or erlotinib to the putative resistant cells in comparison to sensitive ones.Treatment and transfection
To overexpress MET or ERBB3 gene, cells were transfected with pReceiver-M68 gene overexpressing plasmids (Genecopoeia) with EndoFectin transfection reagent (Genecopoeia). Cells were further assessed 24 hours after transfection. To mimic miR-148a overexpression, cells were treated with miR-148a agomir (synthesized by GenePharma) at 100 nM for 24 hours prior to any other experiment. To induce cell apoptosis, NSCLC cells were challenged by serum deprivation for 12 hours with or without treatment with gefitinib or erlotinib (SML1657 or SML2156, respectively, Sigma-Aldrich) at 10 μM.qRT-PCR
Endogenous miR-148a level in cell culture was evaluated by qRT-PCR. Total RNA in cell culture was extracted using Trizol reagent (R0016, Beyotime), and miR-148a in the total RNA was semi-quantified by 2−ΔΔCt method using miRNA qRT-PCR detection kit (QP015) on a thermo cycler for PCR. snRNA U6 was used as internal reference. Forward primer sequences for miR-148a and U6 are as follows: miR-148a, TCAGTGCACTACAGAACTTTGTAAA; U6, GGGCAGGAAGAGGGCCTAT. Reverse primer was universal and was provided in the miRNA qRT-PCR detection kit. mRNA of MET, ERBB3 or IGF1R in the AGO2-RIP results was detected using BlazeTaq one-step SYBR green RT-qPCR kit (QP070, Genecopoeia) following the product manual. Primers for mRNA detection were as follows: MET forward, AGCAATGGGGAGTGTAAAGAGG; MET reverse, CCCAGTCTTGTACTCAGCAAC; ERBB3 forward, GGTGATGGGGAACCTTGAGAT; ERBB3 reverse, CTGTCACTTCTCGAATCCACTG; IGF1R forward, TCGACATCCGCAACGACTATC; IGF1R reverse, CCAGGGCGTAGTTGTAGAAGAG. 2−ΔCt method was used for semi-quantification.Cell proliferation assay
Cell proliferation was evaluated using Click-iT EdU microplate assay Kit (C10214, Thermo Fisher Scientific). Cells cultured in vitro at log phase (40% confluent) were incubated with 5 μM of EdU for 24 hours, followed by fixation and click labeling following the product manual.Cell apoptosis assay
After induction of cell apoptosis, cells were lifted and collected by incubation with trypsin/EDTA (59418C) for 1 min and centrifugation. Cells were then labeled with apoptosis detection kit (640914, Biolegend) and assessed by low cytometry. Cells with Annexin V+ PI− labeling were considered apoptotic cells.Colony formation assay
Clonogenic survival of NSCLC cells was evaluated by colony formation assay.20 150 cells were inoculated in each well on 6-well-plate and were culture for 2 weeks. Cells were then fixed with acetic acid/methanol at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 (v/v) ratio at room temperature for 5 min, followed by staining with homemade 0.5% crystal violet solution21 at room temperature for 2 hours. Number of colonies with more than 50 cells were counted for evaluation.
:7 (v/v) ratio at room temperature for 5 min, followed by staining with homemade 0.5% crystal violet solution21 at room temperature for 2 hours. Number of colonies with more than 50 cells were counted for evaluation.
Transwell invasion assay
Invasiveness of NSCLC cells was evaluated in vitro by transwell invasion assay using Corning BioCoat Matrigel invasion chamber (08-774-122, Fisher Scientist). 105 cells suspended in serum-free media were added into the chamber, the bottom of which was merged in cell culture media with 5% FBS. Cells were incubated overnight, and those migrated to the underside of the chamber membrane were fixed with 5% glutaraldehyde at room temperature for 10 min, followed by staining with 1% crystal violet solution at room temperature for 30 min. Number of cells migrated through the membrane were counted for evaluation.Western blot
Protein levels of Her-3, c-Met and IGF-1R in NSCLC cells after different treatment were evaluated by Western blot. Cells cultured in vitro were lysed using cell lysis buffer for Western and IP (P0013, Beyotime), and proteins in the cell lysate were separated by SDS-PAGE before transfer onto nitrocellulose membrane. Her-3, c-Met or IGF-1R protein on the membrane were probed with primary and secondary antibodies purchased from Abcam. The antibodies used for Western blot were as follows: anti-Her-3 (ab32121), anti-c-Met (ab216574), anti-IGF-1R (ab131476), anti-GAPDH (ab9485), anti-rabbit IgG H&L (HRP-conjugated, ab6721).MicroRNA target luciferase reporter assay
miR-148a agomir-mediated silencing of MET, ERBB3 or IGF1R gene was determined by microRNA target luciferase reporter assay. The reporter plasmids constructed by Genecopoeia encode Gaussia luciferase gene flanked at 3′ with cDNA of MET, ERBB3 or IGF1R mRNA 3′ UTR. The reporter plasmids also encode secreted alkaline phosphatase as internal reference. Cells were transfected with the reporter plasmids using Endofectin transfection reagent, followed by treatment with vehicle or miR-148a agomir for 24 hours. Activities of Gaussia luciferase and secreted alkaline phosphatase in the cell culture media were determined using secrete-pair Gaussia luciferase assay kit (LF031, Genecopoeia) following the product manual.AGO2-RNA immunoprecipitation (AGO2-RIP)
Association of MET, ERBB3 or IGF1R mRNA with the RISC was evaluated by AGO2-RIP assay. Cell lysate samples (prepared as described above) were incubated with anti-Argonaute-2 antibody (ab32381, Abcam) for 2 hours at 4 °C, followed by incubation with magnetic protein A beads (88845, Thermo Fisher Scientific) for 2 hours at 4 °C. The beads were then enriched by a magnetic field, and mRNAs associated with the beads were extracted using Trizol reagent, followed by detection by qRT-PCR as described above.Statistical analysis
Each dot represents the mean value of three technical replicates using one cell line. Data were presented as fold induction if applicable. Holm-Sidak's multiple comparisons test was performed in Fig. 1A; for the rest of the figures, Student's t test was performed for comparison between two groups, and Tukey's test was performed for multiple comparison. Difference was considered significant when p < 0.05.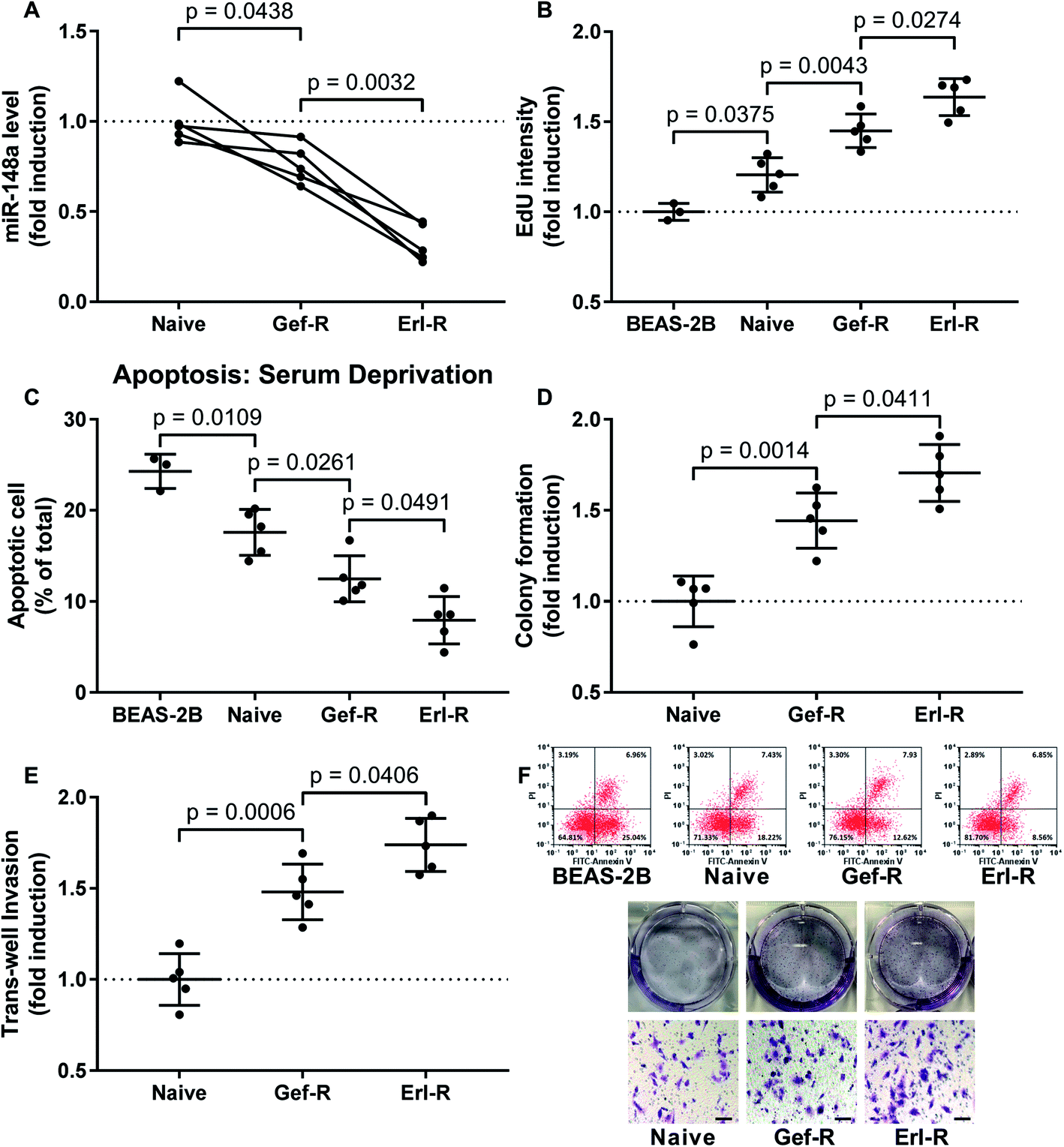 | ||
| Fig. 1 Downregulation of miR-148a in NSCLC cells associated with the development of EGFR TKI resistance and increase in NSCLC cell malignancy in vitro. (A) miR-148a level in the 15 NSCLC cell lines were evaluated by qRT-PCR. (B) Cell proliferations of the 15 NSCLC cell lines and non-malignant lung epithelial cell line BEAS-2B were evaluated by EdU assay. (C) Cell apoptosis of the 15 NSCLC cell lines and BEAS-2B cells induced by serum deprivation was evaluated by flow cytometry after FITC-Annexin V/PI labeling. (D) The overall cell growth of the 15 NSCLC cell lines was evaluated by colony formation assay. (E) Invasiveness of the 15 NSCLC cell lines was evaluated by transwell invasion assay. (F) are representative results of data in (C), (D) and (E). Bar indicates 100 μM. Data in BEAS-2A group come from experiments using 3 vials of BEAS-2A cells revived from preservation in liquid nitrogen. Naïve, NSCLC cells derived from primary tumor tissue. Gef-R, NSCLC cells with gefitinib resistance. Erl-R, NSCLC cells with erlotinib resistance. Mean value of the naïve group or the BEAS-2A group was used for data normalization. | ||
Results
Downregulation of miR-148a associated with increased malignancy of NSCLC cells in vitro
To investigate whether miR-148a would regulate the sensitivity of NSCLC cells to EGFR TKIs, we established 15 NSCLC cell lines including 5 naïve, 5 gefitinib-resistant and 5 erlotinib-resistant NSCLC cell lines derived from patients' tissue specimens, and first compared miR-148a expression levels in these cells. Our data suggested that, comparing to naïve cells derived from primary NSCLC tissue specimens, gefitinib and erlotinib-resisting NSCLC cells showed decreased miR-148a expression, and erlotinib-resisting NSCLC cells showed even lower miR-148a level than gefitinib-resisting cells (Fig. 1A). To characterize the malignancy of these naïve, gefitinib-resistant and erlotinib-resistant NSCLC cells, we performed cell proliferation, apoptosis, colony formation and transwell invasion assays. Our data showed that the cell proliferation, resistance to serum deprivation, colony formation and transwell invasion capacity of gefitinib and erlotinib-resisting NSCLC cells were higher than that of naïve NSCLC cells in vitro, and the erlotinib-resisting NSCLC cells showed higher malignancy that the gefitinib-resisting ones (Fig. 1). These results suggested that reduce in miR-148a level and increase in malignancy associated with the acquisition of gefitinib and erlotinib resistance in NSCLC cells.MiR-148a agomir pre-treatment significantly increased the sensitivity of NSCLC cells to gefitinib and erlotinib in vitro
To investigate whether increasing miR-148a level would restore the EGFR TKI sensitivity of NSCLC cells, we utilized miR-148a agomir treatment to mimic miR-148a overexpression. Our data showed that pre-treatment with miR-148a agomir significantly increased gefitinib and erlotinib induced cell apoptosis in the naïve, gefitinib- and erlotinib-resisting NSCLC cells in vitro, in which miR-148a agomir pre-treatment showed more potent impact on erlotinib-resisting NSCLC cells than the naïve and gefitinib-resisting ones (Fig. 2). These results suggested that miR-148a would attenuate gefitinib and erlotinib resistance of NSCLC cells in vitro.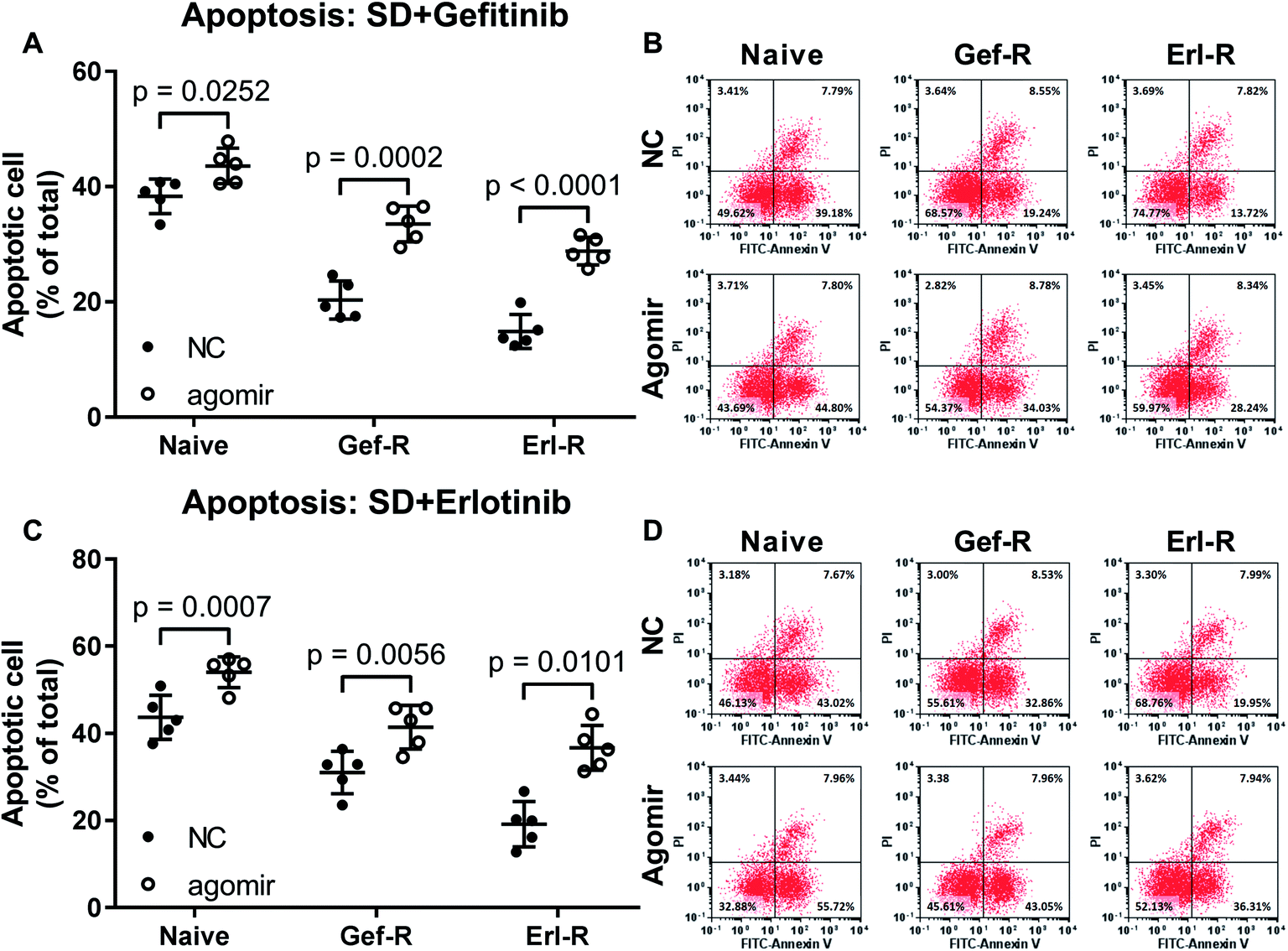 | ||
| Fig. 2 Pre-treatment with miR-148a agomir sensitized NSCLC cells to gefitinib and erlotinib. Cell apoptosis was induced by serum deprivation (SD) plus gefitinib or erlotinib treatment. Cells were treated with miR-148a agomir before apoptosis induction. Cell lines used for these experiments were as those described in Fig. 1. NC, cells treated with vehicle (water). Agomir, cells were treated with miR-148a agomir. | ||
MiR-148a agomir treatment significantly inhibited c-Met, Her-3 and IGF-1r expression in NSCLC cells via mRNA targeting
Previous reports have suggested MET, ERBB2 and IGF1R as target genes of miR-148a, and proteins encoded by these genes participate in the EGFR TKI resistance mechanism in NSCLC cells. To investigate whether miR-148a would sensitize NSCLC cells to EGFR TKI by targeting these genes, we performed western blotting and found that the protein level of c-Met, Her-3 and IGF-1R were significantly reduced in all 15 patient-derived NSCLC cell lines in vitro by miR-148a agomir treatment (Fig. 3A–D). We further performed luciferase reporter assay that confirmed the 3′ UTR of MET, ERBB3 and IGF1R mRNAs as direct targets of miR-148a (Fig. 3E), and our AGO2-RNA RIP results showed that treatment with miR-148a agomir would significantly increase the association of MET, ERBB3 or IGF1R mRNA with the RISC complex in NSCLC cells (Fig. 3F). These results suggested that miR-148a would reduce protein expression of c-Met, Her-3 and IGF-1R by targeting their mRNAs in NSCLC cells.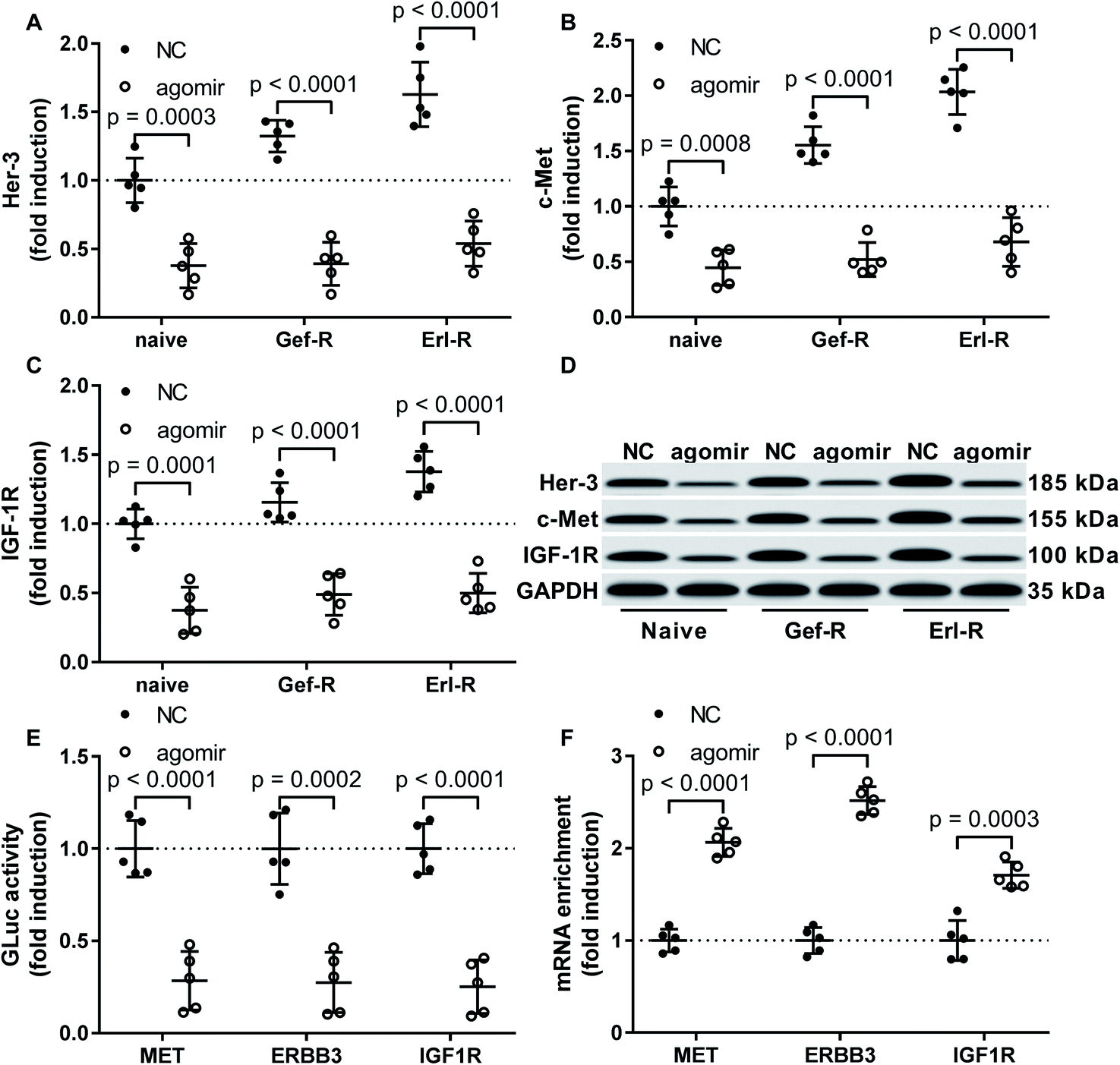 | ||
| Fig. 3 Treatment with miR-148a agomir downregulated Her-3, c-Met and IGF-1R protein expression in NSCLC cells. (A–D) Protein levels of Her-3, c-Met and IGF-1R in the 15 NSCLC cell lines with or without miR-148a agomir treatment were compared by Western blot. (E) Interaction of miR-148a agomir with the 3′ UTR of MET, ERBB3 or IGF1R gene was verified by luciferase reporter assay. (F) miR-148a agomir mediated silencing of MET, ERBB3 or IGF1R gene was verified by AGO2-RIP assay. Cell lines and treatment were as described in Fig. 2. | ||
Restoring c-Met and Her-3 protein expression partially abrogated the gefitinib and erlotinib-sensitizing effect of MiR-148a agomir co-treatment on NSCLC cells in vitro
To verify whether miR-148a sensitize NSCLC cells to gefitinib and erlotinib by reducing c-Met, Her-3 and IGF-1R protein expression, we co-transfected MET, ERBB3 or IGF1R gene overexpressing plasmid into gefitinib and erlotinib resisting NSCLC cells to restore their c-Met, Her-3 or IGF-1R protein level that was downregulated by miR-148a agomir treatment. Cell apoptosis assay results showed that restoring c-Met or Her-3 protein level in gefitinib or erlotinib resisting NSCLC cells partially recovered their resistance to gefitinib or erlotinib induced apoptosis, while the rescue effect of IGF-1R protein restoration seemed minimal (Fig. 4A–F). These results suggested that miR-148a would sensitize NSCLC cells to EGFR TKIs at least in part by inhibiting c-Met and Her-3 protein expression.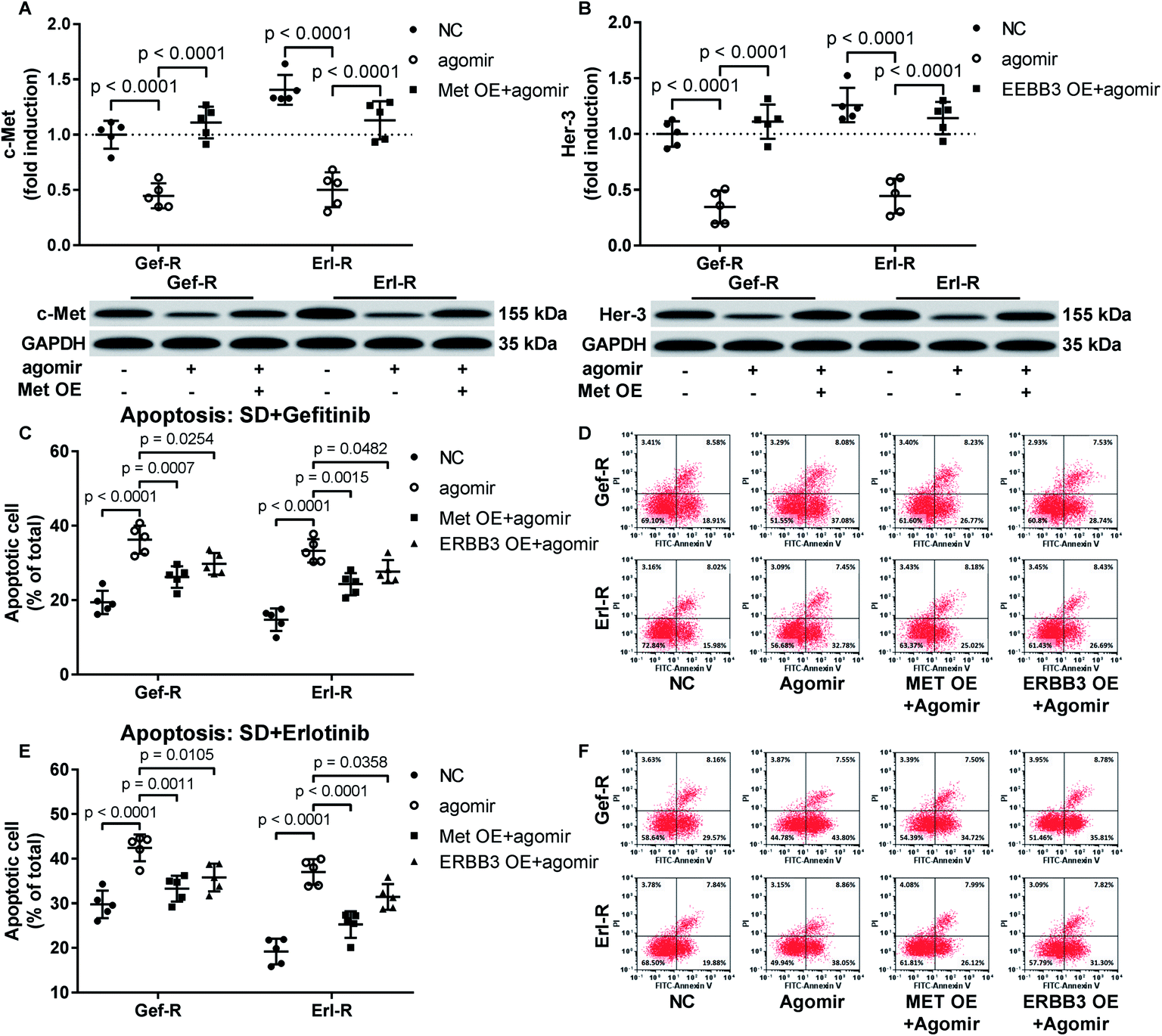 | ||
| Fig. 4 Restoring c-Met or Her-3 protein level by transient overexpression attenuated the EGFR TKI sensitizing effect of miR-148a agomir treatment in gefitinib or erlotinib resistant NSCLC cells. (A and B) c-Met or Her-3 protein level was evaluated by Western blot. (C–F) Cell apoptosis induced by serum deprivation (SD) plus gefitinib or erlotinib treatment was evaluated by flow cytometry after FITC-Annexin V/PI labeling. Cells with or without gene overexpression (OE) plasmid transfection were treated with vehicle (NC) or miR-148a agomir before apoptosis induction. | ||
Discussions
Development of resistance to EGFR TKI treatment has been increasingly becoming a severe threat to NSCLC patients, and investigation on the molecular mechanism underlying the adoption of EGFR TKI resistance of NSCLC cells as well as the development of novel interventions against EGFR TKI resistance might significantly improve the treatment outcome and patient's prognosis.22 In this research we investigated how miR-148a would regulate EGFR TKI sensitivity of NSCLC cells, and whether the treatment of miR-148a agomir would re-sensitize NSCLC cells to EGFR TKI-induced cell death. Our data suggested that endogenous miR-148a level was decreased with the development of EGFR TKI resistance and increase in malignancy in NSCLC cells, and treatment with miR-148a agomir would significantly augment gefitinib and erlotinib induced cell apoptosis in EGFR TKI resistant NSCLC cells in vitro, presumably by downregulating the protein expression of c-Met and Her-3.Activation of c-Met upon ligation with its ligand hepatocyte growth factor bypasses EGFR inhibition by activating the JAK-STAT3, PI3K-Akt-mTOR and MAPK pathways,23,24 while Her-3 might activate c-Met or EGFR without ligand binding by dimerizing with these growth factor receptors.13,25 Moreover, previous study suggested that the coexistence of Met ad T790M mutation contributed to EGFR TKI resistance in advanced NSCLC.26 These works indicated c-Met and Her-3 might serve as possible therapeutic target for the treatment of EGFR TKI resistant, metastatic or advanced-stage NSCLC. Our data demonstrated that c-Met and Her-3 protein levels in NSCLCs derived from metastatic tumor tissue samples were significantly higher than that in those from primary tissue samples, and erlotinib resistance training further increased c-Met and Her-3 protein level in the NSCLC cells, suggesting that the upregulation of these two proteins might participate in the gefitinib and erlotinib resisting mechanisms in NSCLC cells. IGF-1R protein level in the three patient-derived NSCLC cells we constructed showed no statistically significant difference, possibly suggesting that this growth factor receptor was not involved in the EGFR TKIs resisting mechanisms in these cells.
On this basis, we found that pre-treatment with miR-148a agomir would significantly reduce the protein levels of c-Met, Her-3 and IGF-1R in all the NSCLC cell lines we constructed, presumably by mediating their mRNA degradation via interacting with the 3′ UTR of their mRNAs. We further confirmed that the EGFR TKI sensitizing effect of miR-148a agomir treatment on NSCLC cells was at least in part mediated by the downregulation of c-Met and Her-3 protein levels. MiRNA agomir are mimics to endogenous miRNAs with the same RNA sequences and biological functions, while these artificially engineered oligonucleotides are chemically modified, allowing them to penetrate cell membrane and resist RNA degradation mechanism in the cytosol (according to product description). In this research we observed that treating the cells with miR-148a agomir in the absence of transfection reagent could reduce MET, ERBB3 and IGF1R gene expressions via mRNA targeting, and previous researches also described the application of agomir in mice for cancer treatment.27,28 These results imply that co-treatment with miR-148a agomir might be a possible approach to overcome EGFR TKI resistance in NSCLC, although this idea as well as the tolerability of miR-148 agomir application need to be verified by in vivo experiments. Furthermore, to better understanding the mechanism, the effect of miR-148a on NSCLC cells with or without T790 mutations and exogenous EGFR-T790M overexpression system should be analyzed in further study.
Besides MET, ERBB3 and IGF1R gene expressions that have been investigated in this research, our bioinformatic analysis results alto suggested ERBB2 mRNA as a possible target of miR-148a. Upregulation of Her-2 protein encoded by ERBB2 gene also participates in EGFR TKI resistance and tumor progression mechanism in NSCLC,25,29,30 and targeting this tumor promoting protein might reduce EGFR TKI resistance in NSCLC and improve treatment outcome.31,32 Whether ERBB2 mRNA is a target of miR-148a in its regulation of EGFR TKI resistance in NSCLC will be investigated in our future research.
Conclusion
Our results for the first time demonstrated that miR-148a would reduce the EGFR TKI resistance of NSCLC cells in vitro by downregulating MET and ERBB3 gene expressions, thus re-sensitizing the NSCLC cells to gefitinib and erlotinib treatment. Employment of miR-148a agomir might be a possible approach to overcome EGFR TKI resistance in NSCLC treatment.Ethical statement
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of “Lin Yi Cancer Hospital” and approved by the Animal Ethics Committee of “Lin Yi Cancer Hospital”.Funding
None.Conflicts of interest
All authors declare that there are no conflicts of interest in this research.References
- H. A. Yu, M. E. Arcila, N. Rekhtman, C. S. Sima, M. F. Zakowski, W. Pao, M. G. Kris, V. A. Miller, M. Ladanyi and G. J. Riely, Clin. Cancer Res., 2013, 19, 2240–2247 CrossRef CAS.
- K. R. Jakobsen, C. Demuth, A. T. Madsen, D. Hussmann, J. Vad-Nielsen, A. L. Nielsen and B. S. Sorensen, Oncogenesis, 2017, 6, e307 CrossRef CAS.
- Y. Kuang, A. Rogers, B. Y. Yeap, L. Wang, M. Makrigiorgos, K. Vetrand, S. Thiede, R. J. Distel and P. A. Janne, Clin. Cancer Res., 2009, 15, 2630–2636 CrossRef CAS.
- T. Li, Y. H. Ling and R. Perez-Soler, J. Thorac. Oncol., 2008, 3, 643–647 CrossRef.
- J. S. Yang, B. J. Li, H. W. Lu, Y. Chen, C. Lu, R. X. Zhu, S. H. Liu, Q. T. Yi, J. Li and C. H. Song, Tumour Biol., 2015, 36, 3035–3042 CrossRef CAS.
- M. X. Huang, J. Cancer Res. Ther., 2016, 12, 671–675 CrossRef CAS.
- Y. He, S. Ren, Y. Wang, X. Li, C. Zhou and F. R. Hirsch, J. Thorac. Dis., 2018, 10, 5080–5085 CrossRef.
- F. Duan, W. Liu, X. Fu, Y. Feng, L. Dai, S. Cui and Z. Yang, Oncotarget, 2017, 8, 77999–78010 Search PubMed.
- L. Li, Y. Y. Chen, S. Q. Li, C. Huang and Y. Z. Qin, Med. Sci. Monit., 2015, 21, 1155–1161 CrossRef CAS.
- J. Yu, Q. Li, Q. Xu, L. Liu and B. Jiang, J. Biomed. Res., 2011, 25, 170–177 CrossRef CAS.
- J. P. Zhang, C. Zeng, L. Xu, J. Gong, J. H. Fang and S. M. Zhuang, Oncogene, 2014, 33, 4069–4076 CrossRef CAS.
- Q. Xu, Y. Jiang, Y. Yin, Q. Li, J. He, Y. Jing, Y. T. Qi, Q. Xu, W. Li, B. Lu, S. S. Peiper, B. H. Jiang and L. Z. Liu, J. Mol. Cell Biol., 2013, 5, 3–13 CrossRef CAS PubMed.
- H. Lyu, A. Han, E. Polsdofer, S. Liu and B. Liu, Acta Pharm. Sin. B, 2018, 8, 503–510 CrossRef.
- D. Westover, J. Zugazagoitia, B. C. Cho, C. M. Lovly and L. Paz-Ares, Ann. Oncol., 2018, 29, i10–i19 CrossRef CAS.
- A. B. Cortot, C. E. Repellin, T. Shimamura, M. Capelletti, K. Zejnullahu, D. Ercan, J. G. Christensen, K. K. Wong, N. S. Gray and P. A. Janne, Cancer Res., 2013, 73, 834–843 CrossRef CAS.
- M. Nishio, A. Horiike, H. Murakami, N. Yamamoto, H. Kaneda, K. Nakagawa, H. Horinouchi, M. Nagashima, M. Sekiguchi and T. Tamura, Lung Cancer, 2015, 88, 275–281 CrossRef.
- Y. L. Wu, L. Zhang, D. W. Kim, X. Liu, D. H. Lee, J. C. Yang, M. J. Ahn, J. F. Vansteenkiste, W. C. Su, E. Felip, V. Chia, S. Glaser, P. Pultar, S. Zhao and B. Peng, et al., J. Clin. Oncol., 2018, JCO2018777326 Search PubMed.
- A. Hurbin, M. Wislez, B. Busser, M. Antoine, C. Tenaud, N. Rabbe, S. Dufort, F. de Fraipont, D. Moro-Sibilot, J. Cadranel, J. L. Coll and E. Brambilla, J. Pathol., 2011, 225, 83–95 CrossRef CAS.
- Y. Lee, Y. Wang, M. James, J. H. Jeong and M. You, Mol. Carcinog., 2016, 55, 991–1001 CrossRef CAS.
- N. A. Franken, H. M. Rodermond, J. Stap, J. Haveman and C. van Bree, Nat. Protoc., 2006, 1, 2315–2319 CrossRef CAS.
- M. Feoktistova, P. Geserick and M. Leverkus, Cold Spring Harb. Protoc., 2016, 2016, pdb.prot087379 CrossRef.
- D. H. Lee, Pharmacol. Ther., 2017, 174, 1–21 CrossRef CAS.
- A. Drilon, F. Cappuzzo, S. I. Ou and D. R. Camidge, J. Thorac. Oncol., 2017, 12, 15–26 CrossRef PubMed.
- J. A. Engelman and P. A. Janne, Clin. Cancer Res., 2008, 14, 2895–2899 CrossRef.
- R. Roskoski Jr, Pharmacol. Res., 2014, 79, 34–74 CrossRef PubMed.
- L. Y. Gou, A. N. Li, J. J. Yang, X. C. Zhang, J. Su, H. H. Yan, Z. Xie, N. N. Lou, S. Y. Liu, Z. Y. Dong, H. F. Gao, Q. Zhou, W. Z. Zhong, C. R. Xu and Y. L. Wu, Oncotarget, 2016, 7, 51311–51319 Search PubMed.
- Y. Tang, J. Pan, S. Huang, X. Peng, X. Zou, Y. Luo, D. Ren, X. Zhang, R. Li, P. He and Q. Wa, J. Exp. Clin. Cancer Res., 2018, 37, 160 CrossRef.
- F. Fang, B. Huang, S. Sun, M. Xiao, J. Guo, X. Yi, J. Cai and Z. Wang, Cell Death Dis., 2018, 9, 395 CrossRef.
- S. Wang, Y. Song, F. Yan and D. Liu, Front. Med., 2016, 10, 383–388 CrossRef.
- G. R. Ricciardi, A. Russo, T. Franchina, G. Ferraro, M. Zanghi, A. Picone, A. Scimone and V. Adamo, J. Thorac. Oncol., 2014, 9, 1750–1762 CrossRef.
- S. Liu, S. Li, J. Hai, X. Wang, T. Chen, M. M. Quinn, P. Gao, Y. Zhang, H. Ji, D. A. E. Cross and K. K. Wong, Clin. Cancer Res., 2018, 24, 2594–2604 CrossRef CAS.
- S. S. Ramalingam, J. C. Yang, C. K. Lee, T. Kurata, D. W. Kim, T. John, N. Nogami, Y. Ohe, H. Mann, Y. Rukazenkov, S. Ghiorghiu, D. Stetson, A. Markovets, J. C. Barrett and K. S. Thress, et al., J. Clin. Oncol., 2018, 36, 841–849 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |