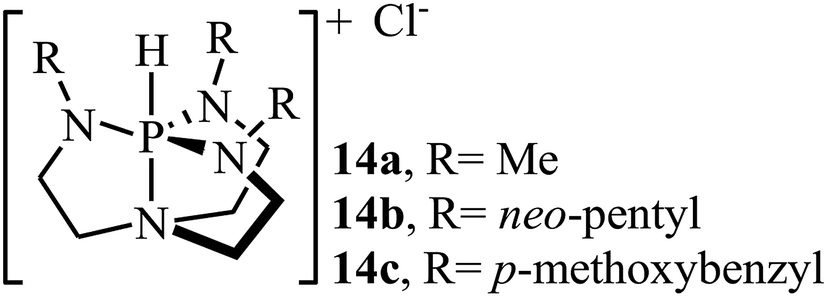DOI:
10.1039/C8RA10233C
(Review Article)
RSC Adv., 2019,
9, 3884-3899
Cycloaddition of atmospheric CO2 to epoxides under solvent-free conditions: a straightforward route to carbonates by green chemistry metrics
Received
13th December 2018
, Accepted 13th January 2019
First published on 29th January 2019
Abstract
The conversion of carbon dioxide (CO2) into value-added organic compounds has received more and more attention over recent years, not only because this gas is one of the major anthropogenic greenhouse gases, but also because it has been regarded as an abundant, inexpensive, nontoxic, nonflammable, and renewable one-carbon (C1) resource. Along these lines, the synthesis of five-membered cyclic carbonates employing CO2 as a safe alternative to toxic reagents such as phosgene or its derivatives is of great interest because of their wide range of applications in organic synthesis. However, most of CO2 incorporation reactions into carbonates are carried out in toxic and non-recyclable organic solvents. Furthermore, these transformations usually proceed at elevated pressures or supercritical CO2 conditions. Recently, several catalytic systems have been developed that allow the synthesis of functionalized carbonates from the reaction of atmospheric CO2 with corresponding epoxides under solvent-free conditions. This review is an attempt to summarize the most important advances and discoveries in this interesting research arena. The review is divided into three major sections. The first section will discuss ionic liquid catalyzed coupling reactions. The second will cover organocatalyzed reactions. The third focuses exclusively on metal-catalyzed fixations. Notably, the third section has been classified based on the metal element that carries out the catalysis (i.e. copper, palladium, zinc).
 Aazam Monfared | Aazam Monfared was born in Tehran, Iran, in 1965. She received her B.S. degree in Pure Chemistry from University of Shahid Beheshti, Tehran, Iran, and her M.S. degree in Organic chemistry from Shahid Beheshti University, Tehran, Iran, in 1991 under the supervision of Prof. A. Rustaiyan. She received her PhD degree in 1999 under the supervision of Prof. A. Rustaiyan in Shahid Beheshti University, Tehran, Iran. Now, she is working at Payame Noor University of Tehran as Associate Professor. Her research interests include organic synthesis, phytochemistry, drug synthesis, nano chemistry, methodologies and theoretical chemistry. |
 Robab Mohammadi | Robab Mohammadi was born in Tabriz, Iran, in 1979. She received her B.S. degree in Pure Chemistry from University of Tabriz, Iran, and her M.S. degree in applied chemistry from Islamic Azad University, Tabriz Branch, Tabriz, Iran, in 2008 under the supervision of Dr L. Edjlali. She received her PhD degree in 2012 under the supervision of Prof. M. Rabani and Prof. B. Massoumi from Islamic Azad University, Tehran Shomal Branch, Tehran, Iran. Now she is working at Payame Noor University of Tabriz as Associate Professor. Her research interests include nano materials synthesis and new methodologies in material synthesis. |
 Akram Hosseinian | Akram Hosseinian was born in Ahar, Iran, in 1973. She received her B.S. degree in Pure Chemistry from University of Tehran, Iran, and her M.S. degree in inorganic chemistry from Tarbiat Modares University, Tehran, Iran, in 2000 under the supervision of Prof. A. R. Mahjoub. She completed her PhD degree in 2007 under the supervision of Prof. A. R. Mahjoub. Now she is working at University of Tehran as Associate Professor. Her research interests include inorganic and organic synthesis, new methodologies in nano material synthesis. |
 Shahriar Sarhandi | Shahriar Sarhandi was born in Langarud, Iran, in 1975. He received his B.S. degree in applied chemistry from University of Guilan, Rasht, Iran, and his M.S. degree in organic chemistry from University of Guilan, Rasht, Iran, in 2002 under the supervision of Prof. M. Mamaghani and associate Prof. M. Yazdanbakhsh. He received his PhD degree in organic chemistry under the supervision of Prof. E. Vessally and assistant Prof. L. Zare Fekri from Payame Noor University, Zanjan, Iran. Now, He is working in department of chemistry supervision at Guilan combined cycle power plant. His research interests include organic synthesis, drug synthesis, ionic liquid synthesis, green chemistry, methodologies and theoretical chemistry. |
 Parvaneh Delir Kheirollahi Nezhad | Parvaneh Delir Kheirollahi Nezhad was born in Tabriz, Iran, in 1975. She received her B.S degree in applied chemistry from university of Tabriz, Tabriz, Iran, in 1998 and her M.S degree inorganic chemistry from Tarbiat Moallem university of Tehran, Iran, in 2003, under supervision of Dr H. Aghabozorg. She received PhD degree in 2015 under the supervision of Dr K. Nejati and Prof H. Keypour in of Payame Noor University of Mashhad. Her research interests include inorganic chemistry, nanochemistry and theoretical inorganic chemistry. |
1. Introduction
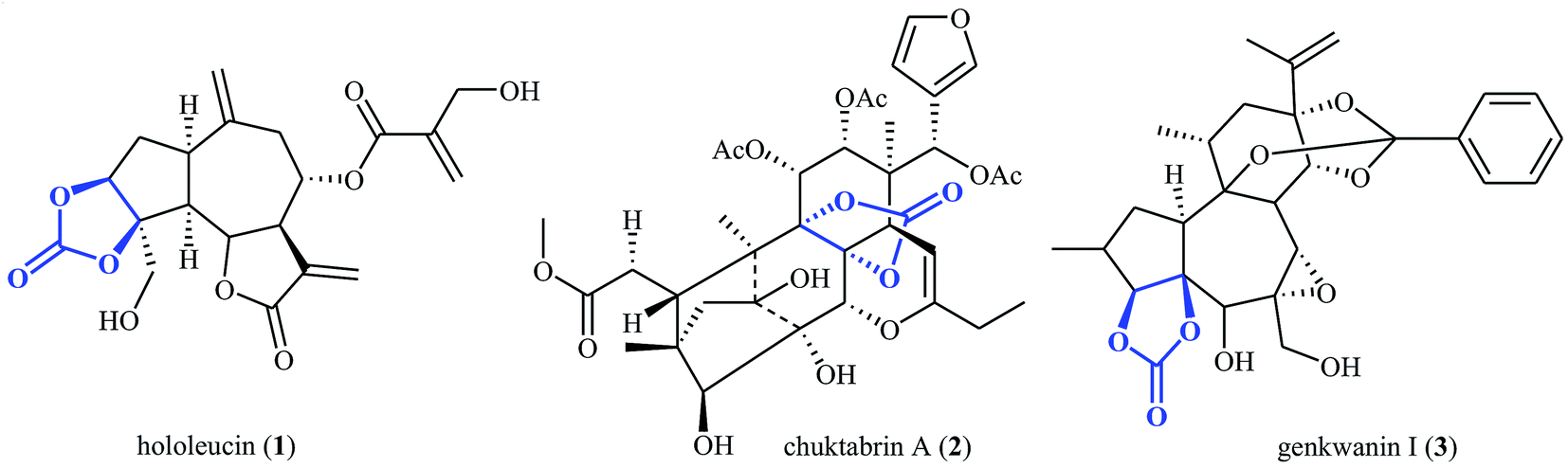
The utilization of carbon dioxide as a one-carbon (C1) feedstock in organic synthesis has attracted ever-increasing attention since it may provide access to a variety of value-added chemicals (e.g. alcohols, carboxylic acids, esters, aldehydes, amides, urethanes, ureas, and carbamates) from a non-toxic, nonflammable, economical, abundant, renewable and environmental friendly resource.1–10 Among various carbon dioxide fixation reactions, cycloaddition of epoxides with CO2 to five-membered cyclic carbonates represents one of the most attractive, straightforward and green protocols.11–15 Needless to say that five-membered cyclic carbonate (1,3-dioxolan-2-one) is one of the most important O-heterocycles having increasing importance in medicinal chemistry16–18 and organic synthesis.19–21 The 1,3-dioxolan-2-one skeleton is also widely found in natural products (Scheme 1).22
 |
| | Scheme 1 Selected examples of natural products containing cyclic carbonate core. | |
For the synthesis of cyclic carbonates from epoxides and CO2, organic solvents were usually necessary.23 The pollution risk of such solvents prompted researchers to look for solvent-free processes.24–28 However, solvent-free fixation of CO2 to carbonate often proceeds at high pressure or supercritical CO2 conditions29,30 and direct use of atmospheric CO2 in this environmentally benign process is of great interest, yet challenging.
In the last few years, numerous catalytic systems have been developed that could effectively catalyze the cycloaddition of epoxides with atmospheric CO2 under solvent-free conditions. Since a number of developments in this interesting and explosively growing research arena have occurred from 2013 to present, a comprehensive review on this hot research topic seems to be timely. In continuation of our recent reviews on the CO2 fixation reactions31 and heterocycles synthesis,32 herein, we will highlight the most important developments on the synthesis of five membered cyclic carbonates through the reaction of corresponding epoxides with atmospheric CO2 under neat conditions (Fig. 1). The review is divided into three major sections. The first section will discuss ionic liquid catalyzed coupling reactions. The second will cover organocatalyzed reactions. The third focuses exclusively on metal-catalyzed fixations. Notably, the third section has been classified based on the metal element that carries out the catalysis (i.e. Cu, Pd, Al, Zn).
 |
| | Fig. 1 Solvent-free fixation of atmospheric CO2 into cyclic carbonates. | |
2. Ionic liquid catalyzed reactions
Ionic liquids are low melting point salts (poorly coordinated cationic and anionic components in the liquid state) that have emerged as an environmentally friendly alternative to the volatile organic solvents. Though primarily used as green reaction media, they are now finding ever-increasing applications as catalysts for a wide variety of chemical reactions.33 Recently, a large number of ionic liquids as solvents/catalysts have been applied in the synthesis of useful organic compounds via CO2 fixation reactions.34
In 2016, Saptal and Bhanage presented an efficient chemical fixation of atmospheric CO2 to epoxides 4 under solvent-free conditions employing bifunctional ionic liquid derived from choline and histidine [Ch][His] as a sustainable catalyst.35 The reactions were carried out in the presence of TBAI (tetra-n-butylammonium iodide) as a co-catalyst at 70 °C and provided the corresponding cyclic carbonates 5 in high yields (Scheme 2). It was observed that the catalyst could be freely recycled and reused for the same reaction at least five times without tangible loss in its catalytic activity and yield. It should be mentioned that other amino-acid-based ionic liquids such as [Ch][Gly], [Ch][Trp], [Ch][Ala], [Ch][Pro], [Ch][Ser], [Ch][Tyr], [Ch][Val], and [Ch][4-OH-Pro] were also found to promote the reaction; however, in lower yields. According to the author proposed mechanism, the presence of OH group of choline and the NH2 group of the amino acids in the ionic liquids has a synergistic effect on the activation of the epoxide and CO2 towards the cycloaddition reactions. The hydroxyl functional group acts as a hydrogen bond donor catalyst and activates the epoxide ring, whereas the presence of the amine functional group of the amino acid activates the CO2 molecules (Scheme 3). It is noted that the above-mentioned ionic liquids were also successfully applied as efficient catalysts for the cyclization of a series of 2-aminobenzonitriles with CO2 (2 MPa) under solvent-free conditions; the expected quinazoline-2,4(1H,3H)-diones were produced in moderate to good yields.
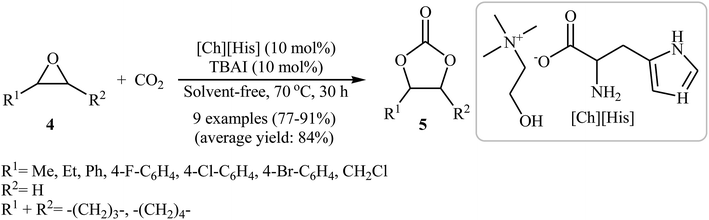 |
| | Scheme 2 [Ch][His]-catalyzed synthesis of carbonates 5 from epoxides 4 under atmospheric pressure of CO2. | |
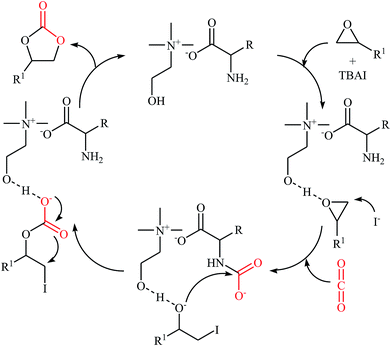 |
| | Scheme 3 Plausible mechanism for the reaction in Scheme 2. | |
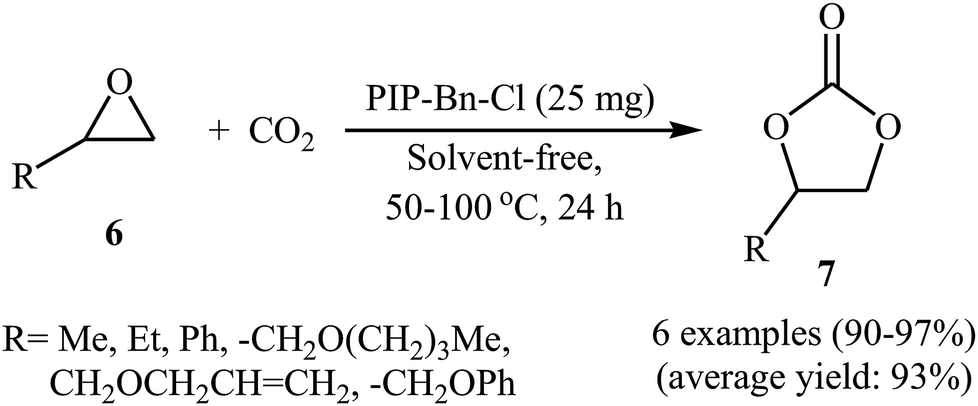
In the same year, Meng and Ma along with their co-workers prepared vinyl-functionalized quaternary phosphonium cation-based porous ionic polymers (PIP-Me-I, PIP-Et-Br, and PIP-Bn-Cl) via a free-radical solvothermal polymerization reaction.36 Due to their high surface areas (402, 625, and 758 m2 g−1, respectively), these polymers exhibited excellent CO2 capacity with a CO2 uptake of 6.1 wt% (1.4 mmol g−1), 6.5 wt% (1.5 mmol g−1), and 6.7 wt% (1.5 mmol g−1) at ambient conditions, respectively. The authors selected PIP-Bn-Cl as a representative catalyst and investigated the catalytic activity of this catalyst for cycloaddition of various epoxides 6 with atmospheric CO2 under solvent-free conditions. The reaction proceeded with no need to any additive and co-catalyst and afforded the desired cyclic carbonates 7 in excellent yields (Scheme 4). Moreover, the catalyst was reusable and could be applied for ten consecutive reaction runs without negligible loss of activity.
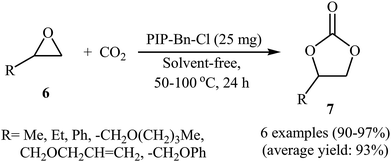 |
| | Scheme 4 (PIP-Bn-Cl)-catalyzed fixation of atmospheric CO2 onto epoxides 6 under solvent-free conditions. | |
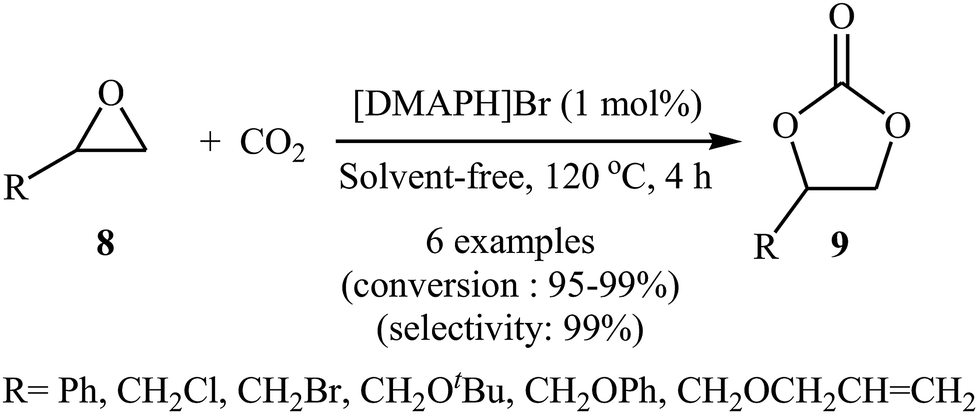
Inspired by these works, the group of Zhang developed of 4-(dimethylamino)pyridine hydrobromide ([DMAPH]Br) as a highly efficient and recyclable ionic liquid catalyst for the synthesis of cyclic carbonates 9 from atmospheric CO2 and terminal epoxides 8 under solvent-free conditions (Scheme 5).37 However, internal epoxides did not work well under this reaction conditions. It is noteworthy that using a mixture of CO2 (15%) and N2 (85%) to mimic flue gas in industry, this ionic liquid catalyst was also able to convert styrene oxide into the corresponding carbonate with excellent conversion (92%) and selectivity (99%).
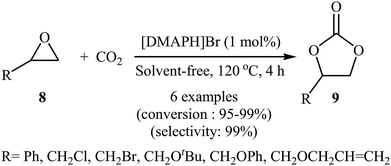 |
| | Scheme 5 Synthesis of carbonates 9 from the [DMAPH]Br-catalyzed reaction of epoxides 8 with CO2. | |
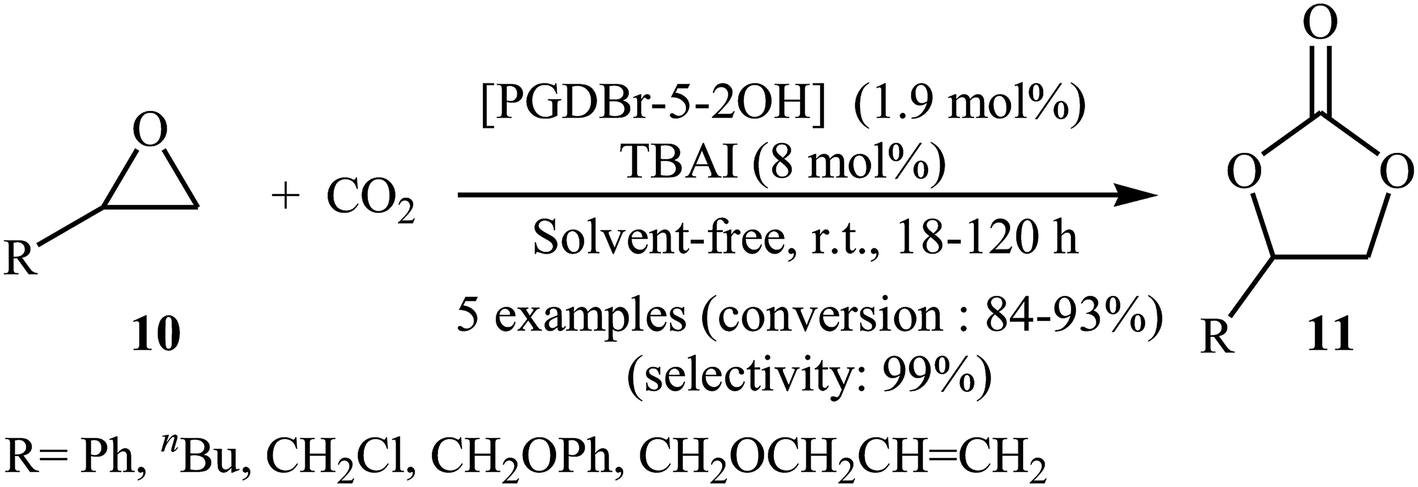
Subsequently, Zhou and Wang along with their co-workers synthesized a novel vicinal dual hydroxyls tethered mesoporous poly-ionic liquid [PGDBr-5-2OH] via free radical polymerization of divinylbenzene and an epoxy-containing imidazolium ionic liquid (glycidyl-3-vinylimidazolium bromide) followed by epoxide ring-opening in hot water.38 The catalyst was successfully used for the cycloaddition of atmospheric CO2 to epoxides 10 in the presence of TBAI as co-catalyst under solvent-free conditions. The authors studied the effects of reaction variables such as reaction time and temperature. It was found that performing the reaction at room temperature for 18–120 h resulted in the best yields (Scheme 6). Just like previous works, this protocol for fixation of CO2 (1 atm) to internal epoxides was considerably less efficient. Based on the DFT calculations, it was found that the vicinal diol moiety in ionic liquid acts as an organocatalytic hydrogen bond donor, whereas, the bromide anion, acts as the nucleophile for the ring-opening of the epoxide.
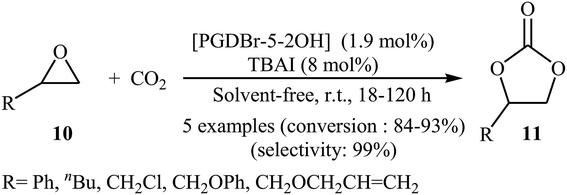 |
| | Scheme 6 Fixation of atmospheric CO2 with epoxides 10 by using [PGDBr-5-2OH] as an organocatalyst. | |
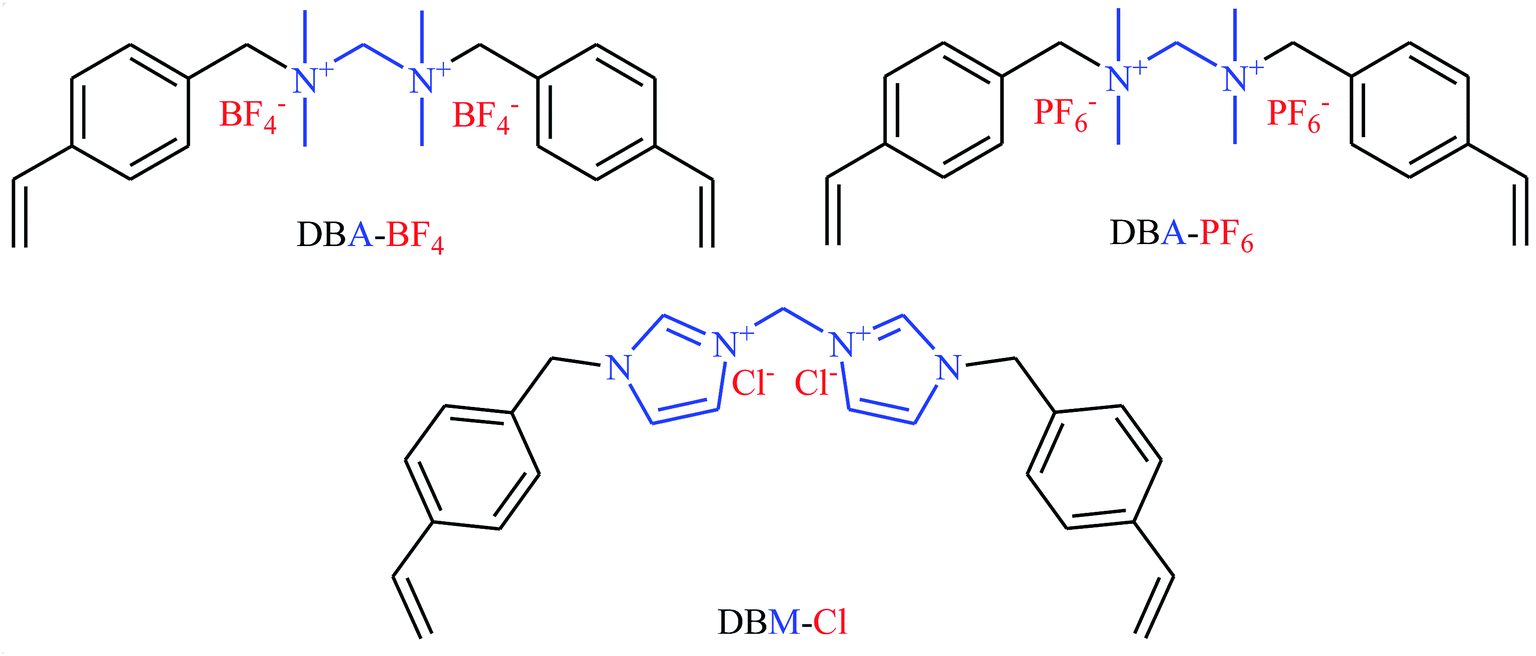
Very recently, Li's research team designed and synthesized a series of novel sponge-like crosslinked quaternary ammonium-based mesoporous poly ionic liquids (PDBA-BF4-SCD, PDBA-PF6-SCD, PDBM-Cl-SCD) through free-radical polymerization of corresponding dicationic monomers with different anions including BF4−, PF6− and Cl− (Scheme 7) and subsequent SCD to form sponge-like polymers.39 The obtained poly ionic liquids were used as catalysts for cycloaddition of atmospheric CO2 to epichlorohydrin under solvent-free conditions. The results proved that PDBA-Cl-SCD exhibited better catalytic performance compared to PDBA-BF4-SCD and PDBA-PF6-SCD. Various terminal epoxides 12 bearing with both alkyl and aryl substituents are well tolerated with this protocol, giving the desired carbonates 13 in almost quantitative yields (Scheme 8). Moreover, the catalyst was reusable and could catalyze at least five reaction cycles similar to the fresh catalyst.
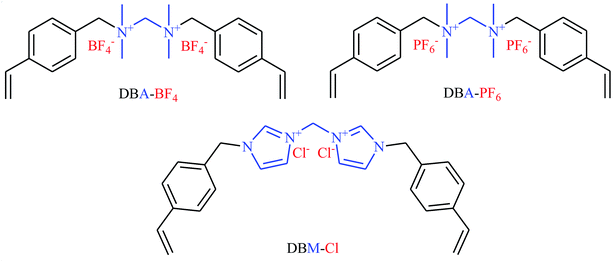 |
| | Scheme 7 Chemical structures of DBA-BF4, DBA-PF6, and DBM-Cl. | |
 |
| | Scheme 8 Synthesis of carbonates 13 from epoxides 12 and atmospheric CO2 under neat conditions developed by Li. | |
3. Organocatalyzed reactions
Over the past few decades, organic catalysts has attracted considerable attention from both academia and industry, and emerged as one of the hot topics in advanced organic chemistry. These catalysts are typically robust, affordable, readily available, non-toxic, and inert towards air and, and thus are easy to handle.40 In 2013, Dufaud and co-workers demonstrated for the first time the usefulness of organocatalysts for the cycloaddition of epoxides with atmospheric CO2 under solvent-free conditions.41 They showed that azaphosphatranes 14 (0.1 mol%) could effectively catalyze the coupling of epoxides and CO2 (1 atm) at 80 °C. Azaphosphatranes can be considered structurally tunable hydrogen-bonding organocatalysts, and as such their catalytic activity can be modulated by changing the substituents attached to the peripheral N-atoms (Scheme 9). The catalysts bearing bulkier substituents (e.g., 4-methoxybenzyl and neo-pentyl) exhibited relatively higher stability than those bearing small substituents (e.g., Me), and the azaphosphatrane bearing 4-methoxybenzyl groups showed the highest catalytic activity which achieved a turnover number of >1000 after 4 days for the conversion of epichlorohydrin.
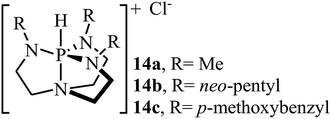 |
| | Scheme 9 Chemical structure of azaphosphatranes 14. | |
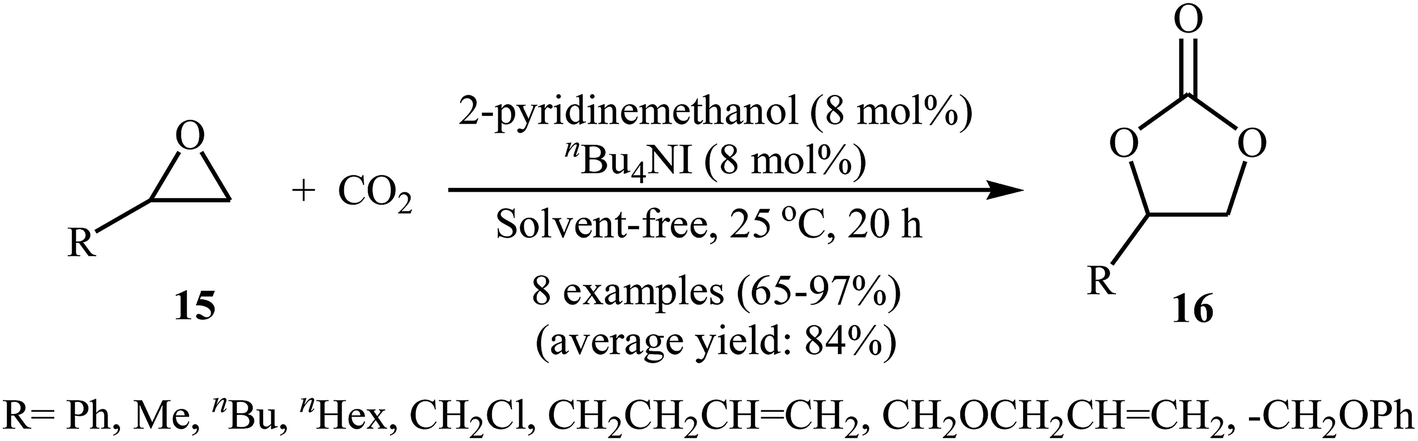
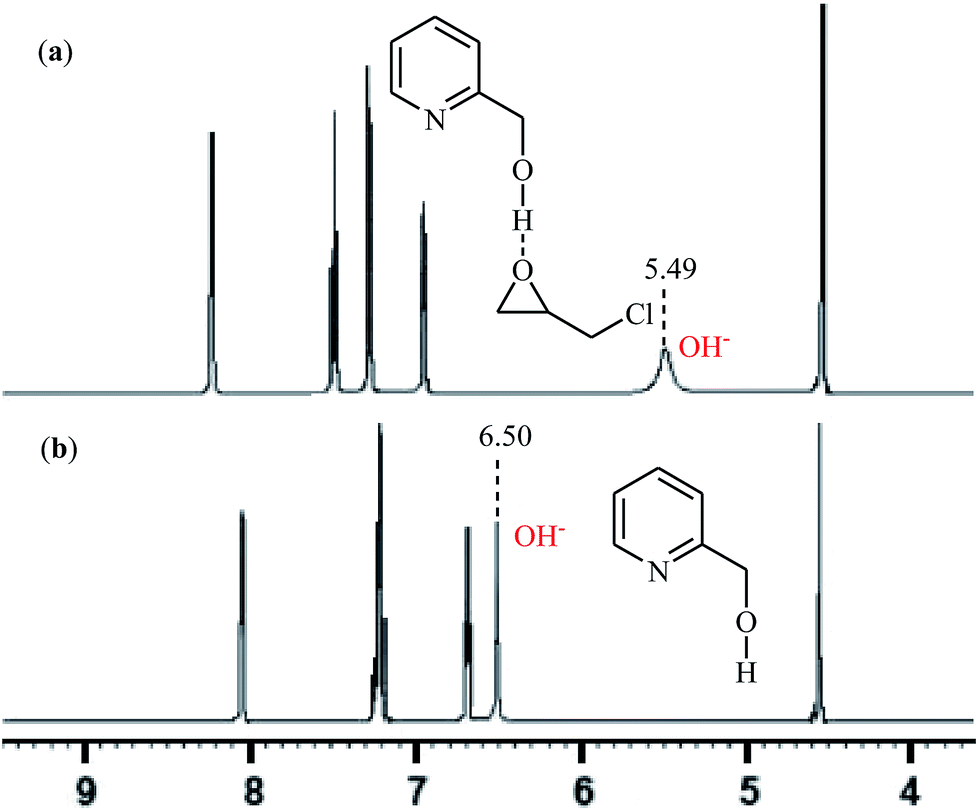
With the objective of designing a greener procedure to five membered cyclic carbonates through cycloaddition of epoxides with CO2, the group of Hirose was able to demonstrate that a range of mono-substituted carbonates 16 could be obtained in high to excellent yields from the reaction of corresponding terminal epoxides 15 with CO2 (1 atm) at room temperature under solvent-free conditions employing 2-pyridinemethanol as a cost-effective organocatalyst and TBAI as a co-catalyst (Scheme 10).42 However, because of the higher steric hindrance of the internal epoxides than terminal epoxides, preparation of the corresponding di-substituted carbonates using this method resulted in poor to moderate yields of desired products. Noteworthy, this inexpensive catalytic system was also successfully applied in the coupling reaction of CO2 and enantiomerically pure epoxides. Moreover, the catalyst was also reusable and could be recovered and reused with negligible loss of the activity. Based on the 1H NMR characterization of the reaction mixture, the authors found that the hydroxyl functional group of 2-pyridinemethanol acts as a hydrogen bond donor catalyst and activates the epoxide ring (Fig. 2). Subsequently, the same authors further improved the efficiency of their protocol by replacing 2-pyridinemethanol with 3-hydroxypyridine.43
 |
| | Scheme 10 2-pyridinemethanol-catalyzed coupling of epoxides 15 with atmospheric CO2. | |
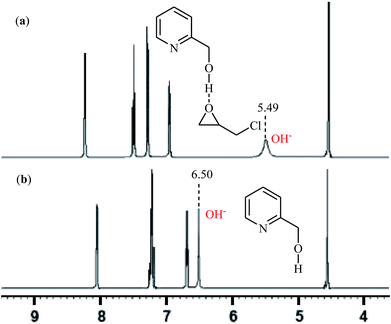 |
| | Fig. 2 1H NMR spectra of: (a) mixture of 2-pyridinemethanol and epichlorohydrin (ratio = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2); (b) 2-pyridinemethanol at 298 K in CDCl3. :2); (b) 2-pyridinemethanol at 298 K in CDCl3. | |
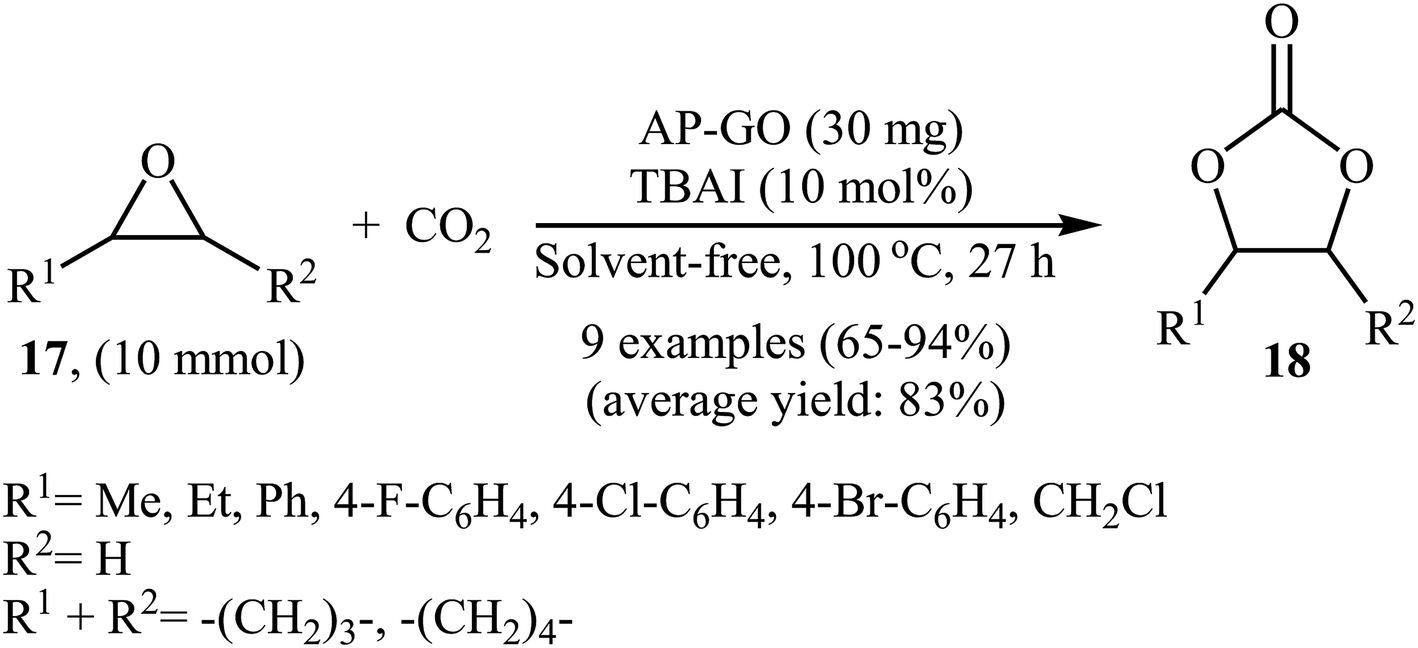
In 2016, Bhanage's research team developed novel amine-functionalized graphene oxide composite by simple heating (80 °C) of graphene oxide with 3-aminopropyltrimethoxysilane (APTMS) in ethanol.44 The authors characterized the synthesized AP-GO by using various analyses such as TEM, SEM, XPS, EDS, XRD, and FTIR. The catalyst has a relatively high surface area of 16.47 ± 0.08 m2 g−1 and a two dimensional structure as shown from the TEM and SEM images. The catalytic utility of the hybrid system was investigated for the coupling reaction of atmospheric CO2 and epoxides 17 under neat conditions at 100 °C. The results established excellent catalytic activity of the AP-GO (Scheme 11) which was reusable and could be recovered and reused for seven runs with negligible loss of performance. The protocol is noteworthy in that both terminal and internal epoxides were well tolerated. Shortly afterwards, the D'Elia laboratory discovered that ascorbic acid could effectively catalyze the conversion of CO2 into cyclic carbonates under ambient and neat conditions with high to excellent yields (64–97% for 10 examples).45
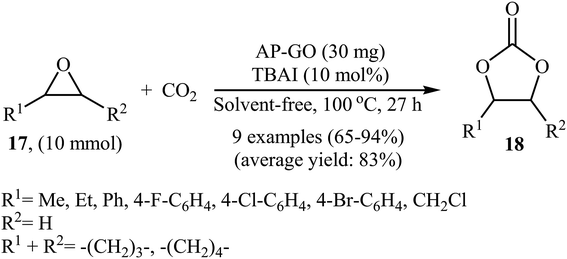 |
| | Scheme 11 Cycloaddition of epoxides 17 with CO2 (1 atm) under neat conditions catalyzed by AP-GO. | |
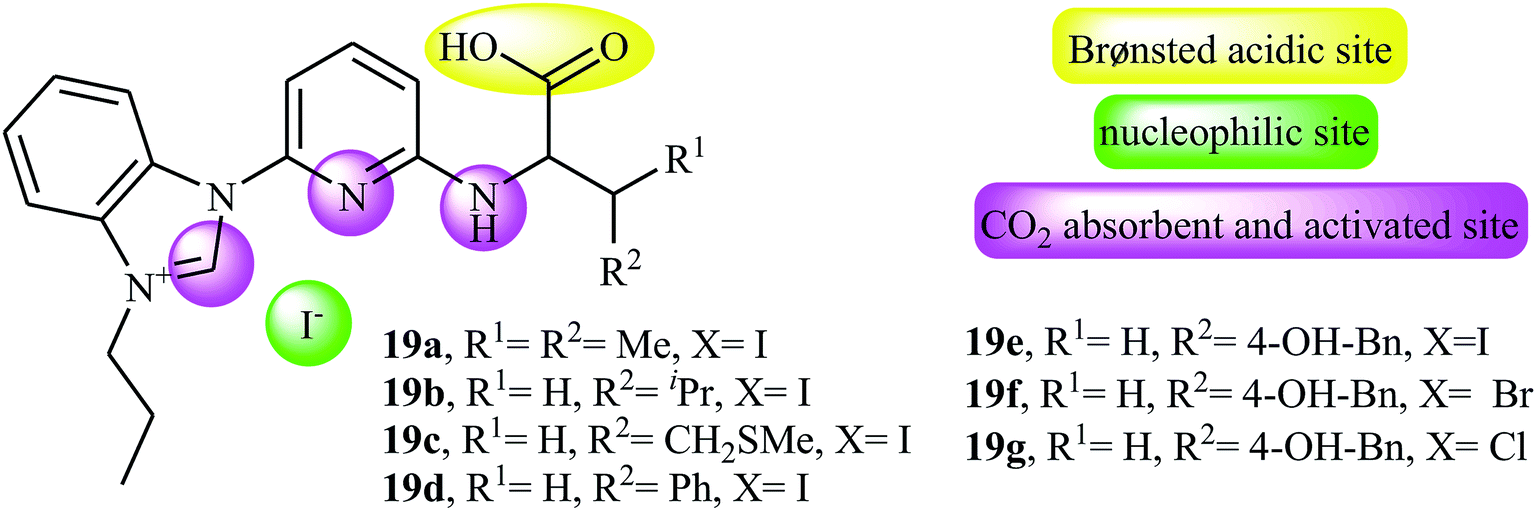
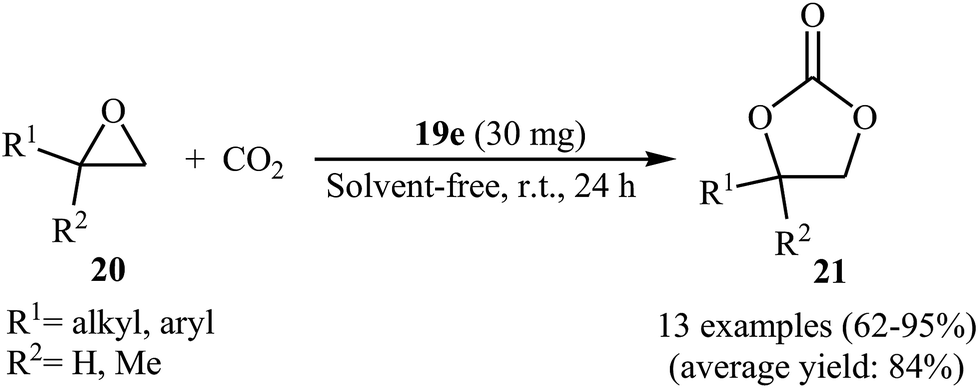
Along this line, very recently, Liu and his team explored the application of seven multi-functional organocatalysts 19a–g in cycloaddition of epoxides with CO2 under neat conditions (Fig. 3).46 The results indicated that the catalyst 19e containing phenolic hydroxyl group was catalytically more active than other catalysts. Thus, in the presence of 4 mol% of organocatalyst 19e under ambient conditions a range of substituted epoxides 20 underwent coupling with CO2 provided the corresponding carbonates 21 in good to excellent yields (Scheme 12). Unfortunately, the reusability of the catalyst did not examined in this study.
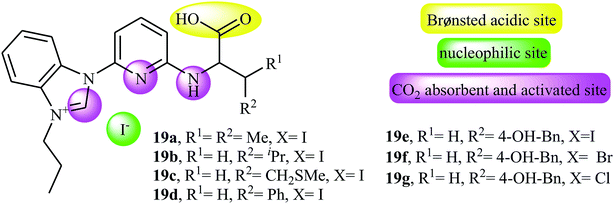 |
| | Fig. 3 Chemical structure of multi-functional organocatalysts 19a–g. | |
 |
| | Scheme 12 Liu's synthesis of cyclic carbonates 21. | |
4. Metal catalyzed reactions
In this section, we describe the current literature on metal and/or metal organic frameworks catalyzed coupling reactions between atmospheric CO2 and epoxides under neat conditions. In the present section, has been classified based on the metal element that carries out the catalysis (e.g. Cu, Ag, Al, Zn, Pd, Co, Cd, Bi, Mo, Sm, Zr).
4.1. Copper
In 2015, Zou et al. designed copper-containing coordination polymer ((CuL)n, L = (Z)-2-(5-chlorin-2-hydroxy benzylideneamino) acetic acid, namely BIT-C) for chemical fixation of atmospheric CO2 to cyclic carbonates (Scheme 13).47 The metal–organic framework (MOF) material exhibited high product selectivity greater than 99% and yield of 99%, in the cycloaddition of terminal epoxides 22 to carbonates 23, in the presence of TBAB co-catalyst under solvent-free conditions. Recycling studies have demonstrated that the catalyst can be easily recovered by simple filtration and reused for five runs with preserving its catalytic activity. Electrospray ionization mass spectrometry (ESI-MS) analyses showed that BIT-C contains a series of fragments ((CuL) to (CuL)6) and the abundances of fragments decrease with increasing degree of polymerization. Therefore, the authors speculated that the mononuclear unit CuL and binuclear unit (CuL)2 are most likely the main active species in catalysis, due to more coordination sites and smaller steric hindrance of Cu atoms compared to multinuclear units ((CuL)3 to (CuL)6). DFT calculations proposed binuclear unit is 62.9 kcal mol−1 more stable than mononuclear unit. This energy difference indicate that (CuL)2 would not be dissociated into CuL by the effects of solvent or co-catalysts and it is worth emphasized that the electrospray ionization in the ESI-MS experiment may provide additional energy to dissociate (CuL)2 into CuL, leading to a signal of CuL. These results clearly proved that the binuclear Cu fragment ((CuL)2) is the most possible active species for the catalysis.
 |
| | Scheme 13 Fixation of atmospheric CO2 with epoxides 22 under solvent-free conditions using BIT-C/TBAB catalytic system. | |
Subsequently, Zou and Zhao along with their co-workers reported an interesting triazole-containing copper-based metal–organic framework, namely Cu4MTTP, in this chemistry (Fig. 4).48 This MOF achieved high yield of cyclic carbonates (88–96%) from small epoxides with the use of TBAB as a co-catalyst under solvent-free, 1 atm and at room temperature. However, due to the confinement of the pore diameter, big-size epoxides were incompatible in this reaction condition.
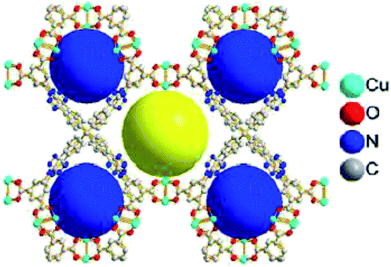 |
| | Fig. 4 Perspective view of 3D porous framework of Cu4MTTP showing the incorporation of unsaturated Cu2 sites and accessible nitrogen-rich triazole groups with two kinds of pores (yellow and blue balls). | |
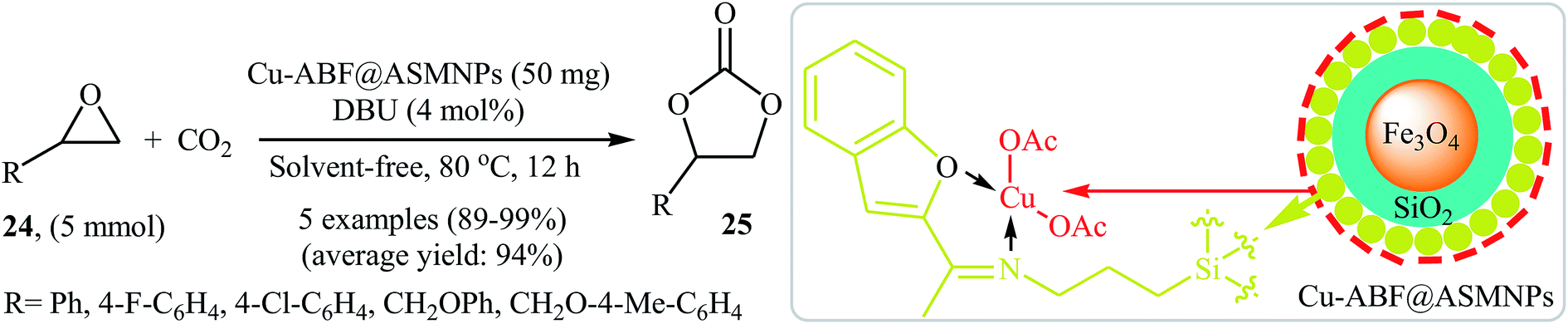
Very recently, the catalytic activity of copper-based amino-functionalised silica-coated magnetic nanoparticles (Cu-ABF@ASMNPs) in the coupling of epoxides with CO2 at ambient temperature and pressure under solvent- and halide-free conditions was studied by Sharma et al.49 The authors carefully studied the effects of reaction variables such as catalyst loading, base and temperature. It was found that using 50 mg of the catalyst (for 5 mmol of epoxide) at 80 °C resulted in the best yields. Among the various bases like Et3N, PPh3, DBU, DMAP, TBD; DBU was the most efficient for this reaction. Various terminal epoxides 24 reacted well under the reaction conditions to produce the corresponding carbonates 25 in excellent to almost quantitative yields (Scheme 14). Additionally, the catalyst could be readily separated from the reaction mixture by using an external magnet and reused five times without a loss in its activity.
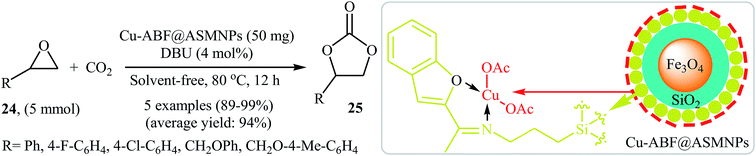 |
| | Scheme 14 (Cu-ABF@ASMNPs)-catalyzed synthesis of carbonates 25 from epoxides 24 and CO2 developed by Sharma et al. | |
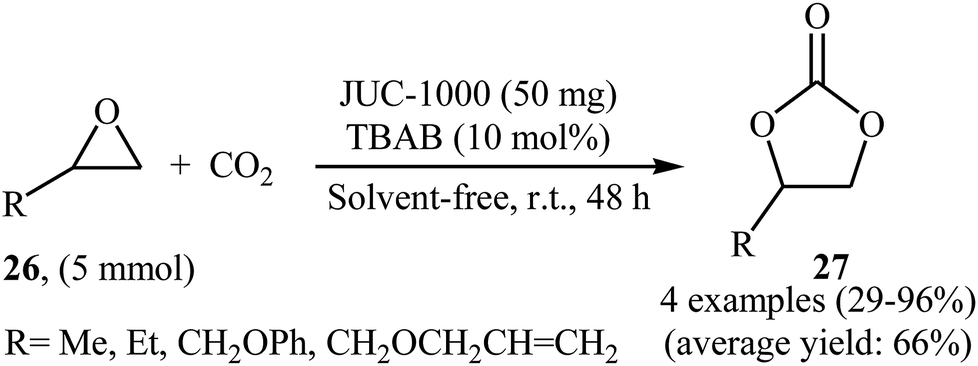
In an innovative design, the group of Ma synthesized an organic ligand 2,4-bis(3,5-dicarboxyphenylamino)-6-ol triazine (H4BDPO) that behave as a buffer, in which the phenol group is a weak acid with the amino and triazine groups serving as weak bases. This buffer molecule was then reacted with Cu(NO3)2·2.5H2O in the mixture of DMF/H2O at 85 °C for 3 days.50 The obtained microporous MOF, [Cu24(BDPO)12(H2O)12]·30DMF·14H2O (namely JUC-1000) was employed as an efficient catalyst for promoting cycloaddition of terminal epoxides 26 with atmospheric CO2 in the presence of TBAB under solvent-free conditions. The procedure furnished the desired carbonates 27 with use of very low amount of the catalyst (0.25 mol% per exposed copper site) at ambient temperature (Scheme 15). The comparison of the synthesized catalyst with the previously reported ones (MOF-505 and HKUST-1) demonstrated the superior catalytic activity of the novel catalyst.
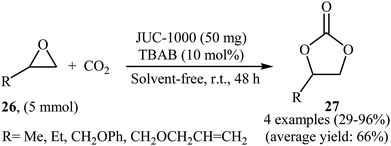 |
| | Scheme 15 (JUC-1000)-catalyzed coupling of atmospheric CO2 with epoxides 26 under neat conditions. | |
4.2. Silver
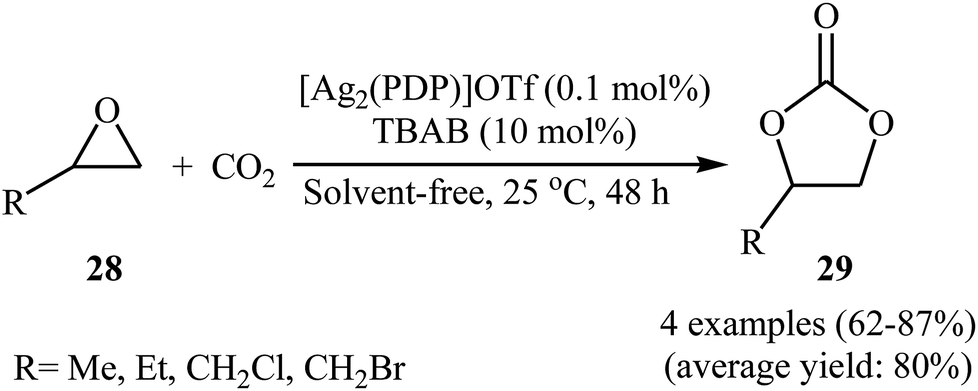
Silver-based metal–organic frameworks have recently attracted considerable attention as efficient catalysts for the synthesis of cyclic carbonates via cycloaddition of corresponding propargyl alcohols with atmospheric CO2 under neat conditions.51 However, the catalytic performance of these MOF in the coupling reaction of epoxides with CO2 under above-mentioned conditions has been scarcely studied; in fact, only one example of such a reaction was reported thus far. In this report Chen, Gan, and Yi showed that treatment of alkyl substituted terminal epoxides 28 with CO2 (1 atm) in the presence of dinuclear silver complex [Ag2(PDP)]OTf at ambient temperature under solvent-free condition, resulted in functionalized carbonates 29 in good to high yields (Scheme 16).52 However, epoxides bearing an aryl group were not well compatible in this reaction. The reusability test established that the complex is decomposed due to the excess Br− in co-catalyst TBAB.
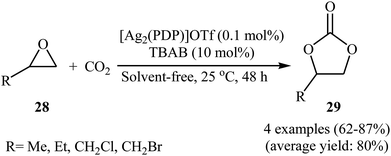 |
| | Scheme 16 Ag-catalyzed coupling of epoxides 28 with CO2 (1 atm) under solvent-free conditions. | |
4.3. Aluminum
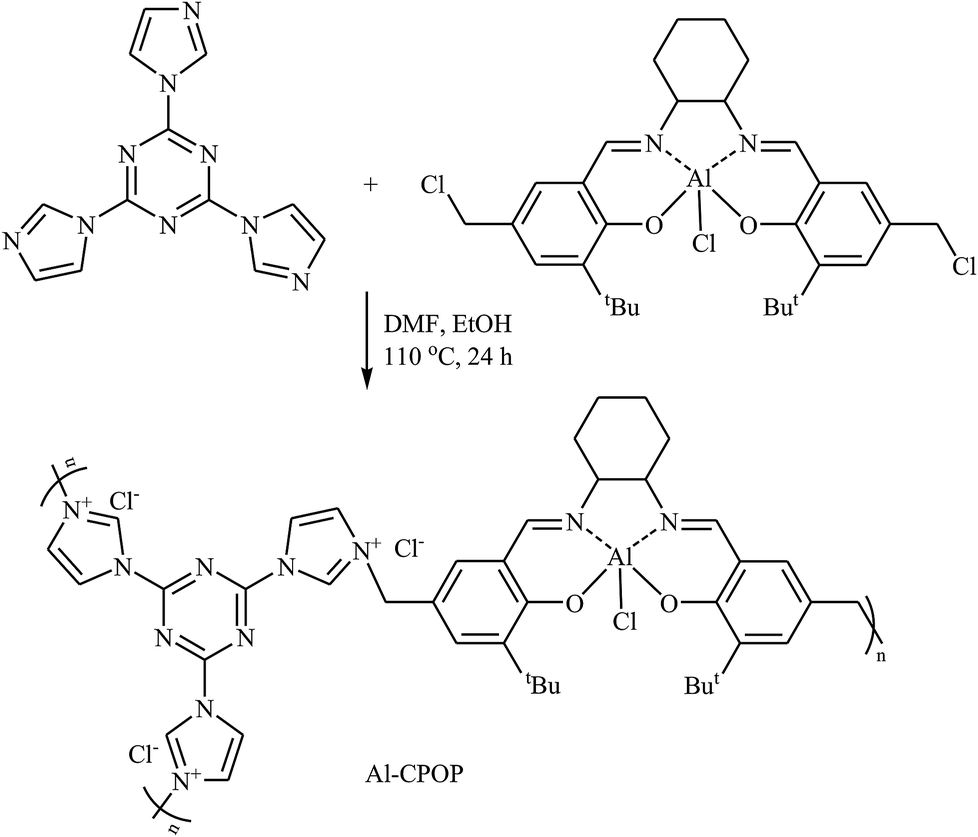
In 2016, Cao's research team developed a novel cationic porous organic polymer containing Salen-Al and imidazolium functionalities (Al-CPOP), through one-pot reaction of chloromethylation Salen-Al monomer and 2,4,6-tris(imidazol-1-yl)-1,3,5-s-triazine (TIST) in a mixture of DMF and EtOH (Scheme 17). The obtained polymer had a BET surface area of 136 m2 g−1 and was successfully used as a bifunctional catalyst for cycloaddition of terminal epoxides with CO2 at ambient pressure under solvent-free conditions.53
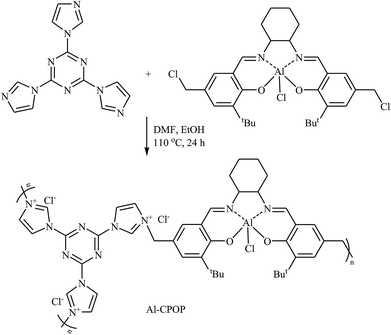 |
| | Scheme 17 Synthesis route for the preparation of Al-CPOP. | |
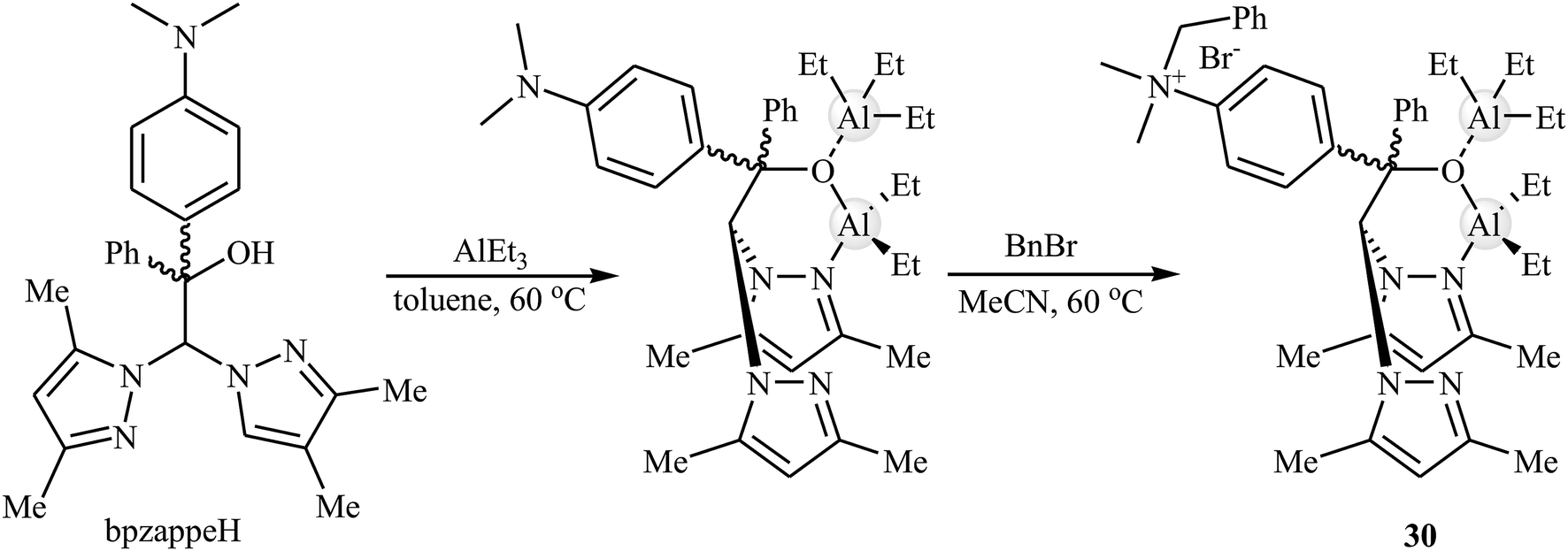
Later, Martínez et al. designed and synthesized a novel aluminum-based heteroscorpionate complex 30 through the reaction of heteroscorpionate precursor 2,2-bis(3,5-dimethylpyrazol-1-yl)-1-[4-(dimethylamino)phenyl]-1-phenylethanol (bpzappeH) with AlEt3 in dry toluene followed by treatment with benzyl bromide in MeCN at 60 °C (Scheme 18).54 Low catalytic loading (0.5–1 mol%) of the complex was used for promoting coupling reaction of atmospheric CO2 and a range of terminal epoxides in the presence of Bu4NBr (0.5 mol%) as a co-catalyst at room temperature. The desired products were slowly obtained in good to excellent yields (62–95% for 12 examples) under solvent-free conditions. The catalyst also showed excellent catalytic activity for the synthesis of disubstituted cyclic carbonates from challenging internal epoxides. However, these reactions were required to higher CO2 pressure (20 bar).
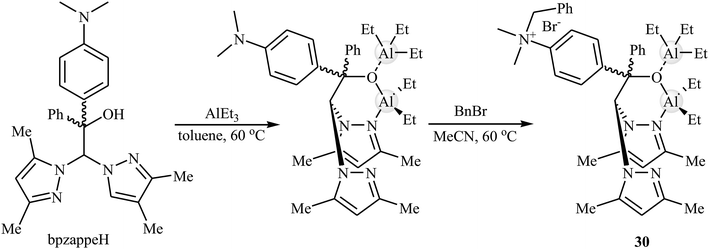 |
| | Scheme 18 Synthesis route for the preparation of aluminum-based heteroscorpionate complex 30. | |
In the closely related investigation, the same research team showed that performing the process in the presence of 5 mol% of aluminum-based complex 31 as a catalyst and without any co-catalyst at 35 °C afforded the expected carbonates 33 in high to excellent yields (Scheme 19).55
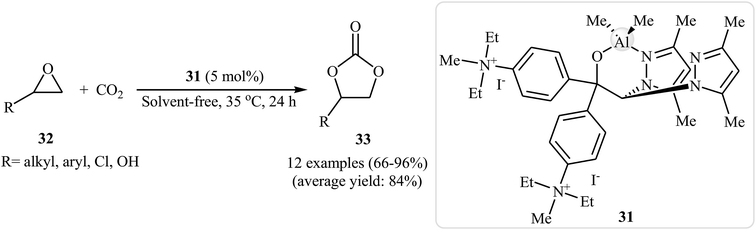 |
| | Scheme 19 Cycloaddition of epoxides 32 with atmospheric CO2 under solvent-free conditions catalyzed by aluminum-based complex 31. | |
4.4. Zinc
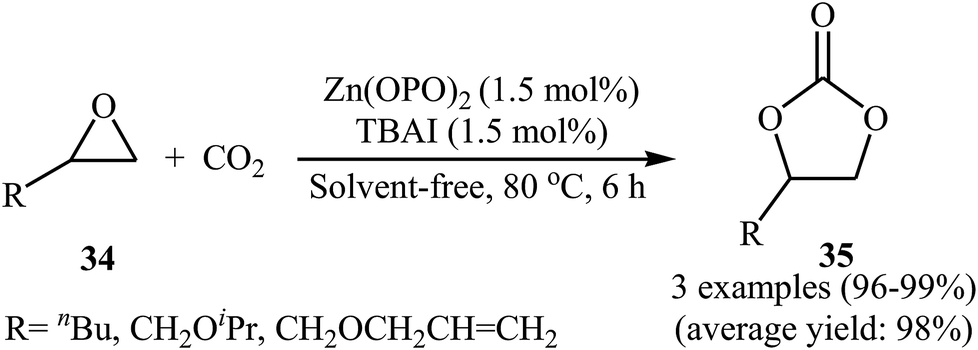
One of the early attempts to chemical fixation of CO2 at atmospheric pressure with epoxides under neat conditions utilizing Zn-based-catalysts was reported in 2016 by Ma et al.56 In this report, a recyclable tetraoxo-coordinated zinc catalyst (Zn(OPO)2, HOPO = 1-hydroxy-2-pyridone) was employed to the cycloaddition of a small series of terminal epoxides 34 with CO2 in the presence of TBAI as a co-catalyst. Under the optimized protocol [Zn(OPO)2 (1.5 mol%), TBAI (1.5 mol%), 80 °C, 6 h] the desired products 35 were obtained in almost quantitative yields (Scheme 20). However, in the cases of sterically hindered and internal epoxides, higher pressure of CO2 (20–50 atm) were needed for the outcome of the reaction. The authors found that other Zn-catalysts also promoted the reaction (e.g., ZnSO4, ZnI2, Salen-Zn); albeit in lower yields. Notably, the presence of co-catalyst was crucial for the success of this reaction. No product was detected in the absence of TBAI. To study the reusability of the Zn(OPO)2/TBAI system, the reaction of 1,2-epoxyhexane was considered as a model of these cycloadditions. After completion of the reaction, the catalytic system was recovered from the reaction mixture by centrifugation and directly used for the next run. It was found that this system could be reused up to five consecutive times with a slight decrease in its activity.
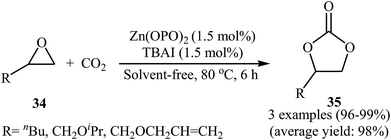 |
| | Scheme 20 Ma's synthesis of cyclic carbonates 35. | |
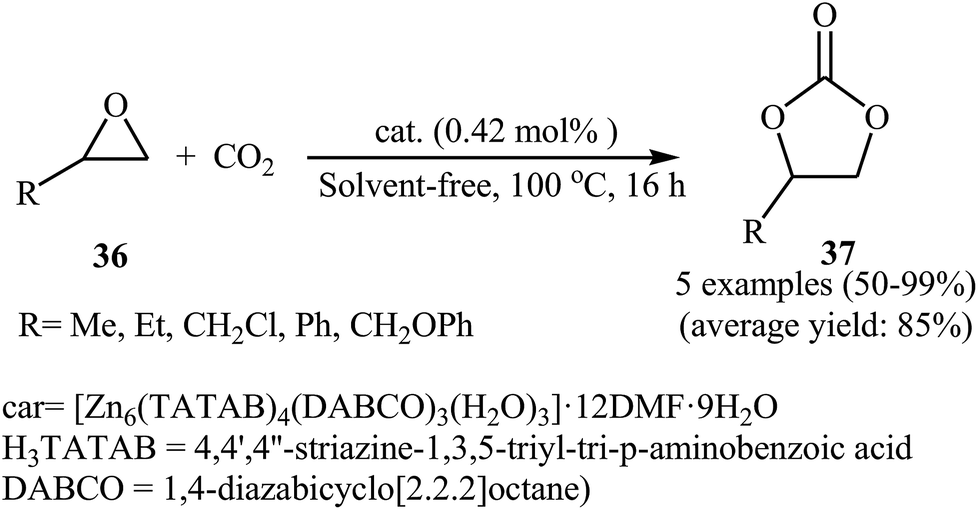
Subsequently, Du and co-workers developed a novel 3D → 3D interpenetrated Zn-polyhedral MOF [Zn6(TATAB)4(DABCO)3(H2O)3]·12DMF·9H2O with an internal diameter of about 3.2 nm which contains 48 Zn2+ Lewis acid catalytic sites and 48 –NH– Lewis base sites and used in the same cycloaddition reaction.57 This reusable dual-walled cage MOF exhibited high catalytic performance for a coupling of a range of terminal epoxides 36 with atmospheric CO2 in the absence of any co-catalyst or solvent (Scheme 21). However, internal epoxides did not work well under this reaction conditions.
 |
| | Scheme 21 Coupling of terminal epoxides 36 with CO2 using [Zn6(TATAB)4(DABCO)3(H2O)3]·12DMF·9H2O as catalyst. | |
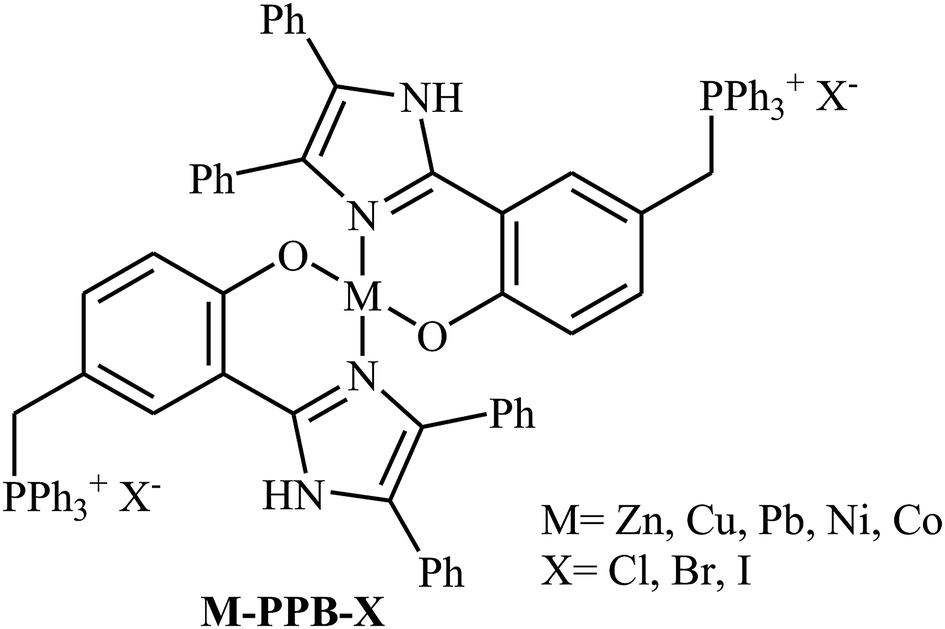
Along this line, in 2018, Guo's group designed and synthesized a series of metal complexes containing a metal ion (Zn, Cu, Pb, Ni, Co) and two quaternary phosphonium salt units anchored on the ligands (M-PPB-X) as shown in Scheme 22.58 The authors compared the catalytic activity of these complexes in the reaction between propylene oxide and CO2 at ambient pressure under solvent-free conditions. The results showed that the catalytic activity is highly dependent on the type of metal center. The decreasing order of the catalytic activity for these single-component bifunctional metal complexes was found to be Zn–PPBCl > Co–PPBCl ≈ Ni–PPBCl > Pb– PPBCl > Cu–PPBCl. The comparative studies also proved that the leaving ability of bifunctional halogen anion group (Cl, Br, I) did not played an important role in the catalytic performance of these bifunctional complexes. Thus, Zn–PPBCl was selected as the best catalyst for this transformation. Under optimized conditions (0.33 mol% of Zn–PPBCl, neat, 1 atm, 110 °C, 13–60 h), the reaction tolerated various terminal epoxides and gave the expected carbonates in fair to excellent yields. However, simple 2-methyloxirane, sterically hindered isobutylene oxide and internal cyclohexene oxide failed to participate in the reaction.
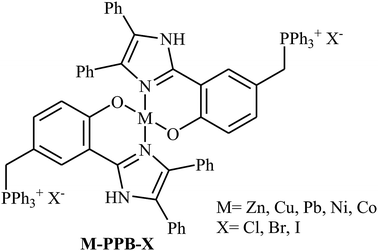 |
| | Scheme 22 Chemical structure of M-PPB-X. | |
Following up to the Zn-catalyzed reaction of atmospheric CO2 with epoxides under neat conditions, various Zn-based metal–organic frameworks have been developed in the current year (Table 1). These include: [Zn2(TCA)(BIB)2.5]·(NO3),59 [(PS-anthra)Zn(Cl)(H2O)],60 [Zn4Tb3L4],61 [Zn(BDC)(L)],62 and a zeolite-like zinc-tetrazole framework with 24-nuclear zinc cages.63 Interestingly, all of these catalysts were reusable and could be recovered easily and reused for several reaction runs without appreciable loss their catalytic activity.
Table 1 Cycloaddition of epoxides with atmospheric CO2 under solvent-free conditions using Zn-based metal–organic frameworks as catalysts
| Entry |
Catalyst |
Conditions |
NEa |
Yield (%) |
| Average |
Range |
| Number of examples. H3TCA = tricarboxytriphenyl amine, BIB = 1,3-bis(imidazol-1-ylmethyl)benzene. PS = polystyrene, anthra = anthranilic acid. PS = polystyrene, anthra: anthranilic acid. L = 4-pyridyl carboxaldehyde isonicotinoyl hydrazone; H2BDC = benzene-1,4-dicarboxylic acid. |
| 1 |
[Zn2(TCA)(BIB)2.5]·(NO3)b |
TBAB (1 mol%), 80 °C, 4 h |
5 |
81.5 |
51–99 |
| 3 |
[(PS-anthra)Zn(Cl)(H2O)]c |
TBAB (5 mol%), r.t., 6 h |
6 |
94 |
89–97 |
| 4 |
[Zn4Tb3L4]d |
TBAB (5 mol%), r.t., 48 h |
7 |
74.5 |
21–96 |
| 5 |
[Zn(BDC)(L)]e |
TBAB (1.8 mol%), r.t., 8 h |
6 |
70 |
14–99 |
4.5. Palladium
In 2017, Khatun and co-workers reported an efficient coupling of various internal and terminal epoxides 38 with CO2 using mesoporous TiO2 supported palladium nanoparticles (Pd@MTiO2) as catalyst under atmospheric pressure.64 These reactions were carried out using TBAB as a co-catalyst under neat conditions and generally provided corresponding functionalized carbonates 39 in excellent to almost quantitative yields (Scheme 23). This supported catalyst was easily prepared by simple heating (170 °C) of mesoporous TiO2 with PdCl2 in ethylene glycol and has a high surface area of over 172 m2 g−1 and a spherical structure as shown from the SEM image (Fig. 5). The recycling test established that the catalyst could be recovered and reused for five reaction runs. Notably, the atomic absorption spectroscopy analysis indicated the leaching of the active catalytic species was negligible.
 |
| | Scheme 23 Pd-catalyzed fixation of atmospheric CO2 with epoxides 38 in the absence of any solvent. | |
 |
| | Fig. 5 The FE-SEM images of Pd@MTiO2 material at high (a) and low (b) resolutions. | |
4.6. Cobalt
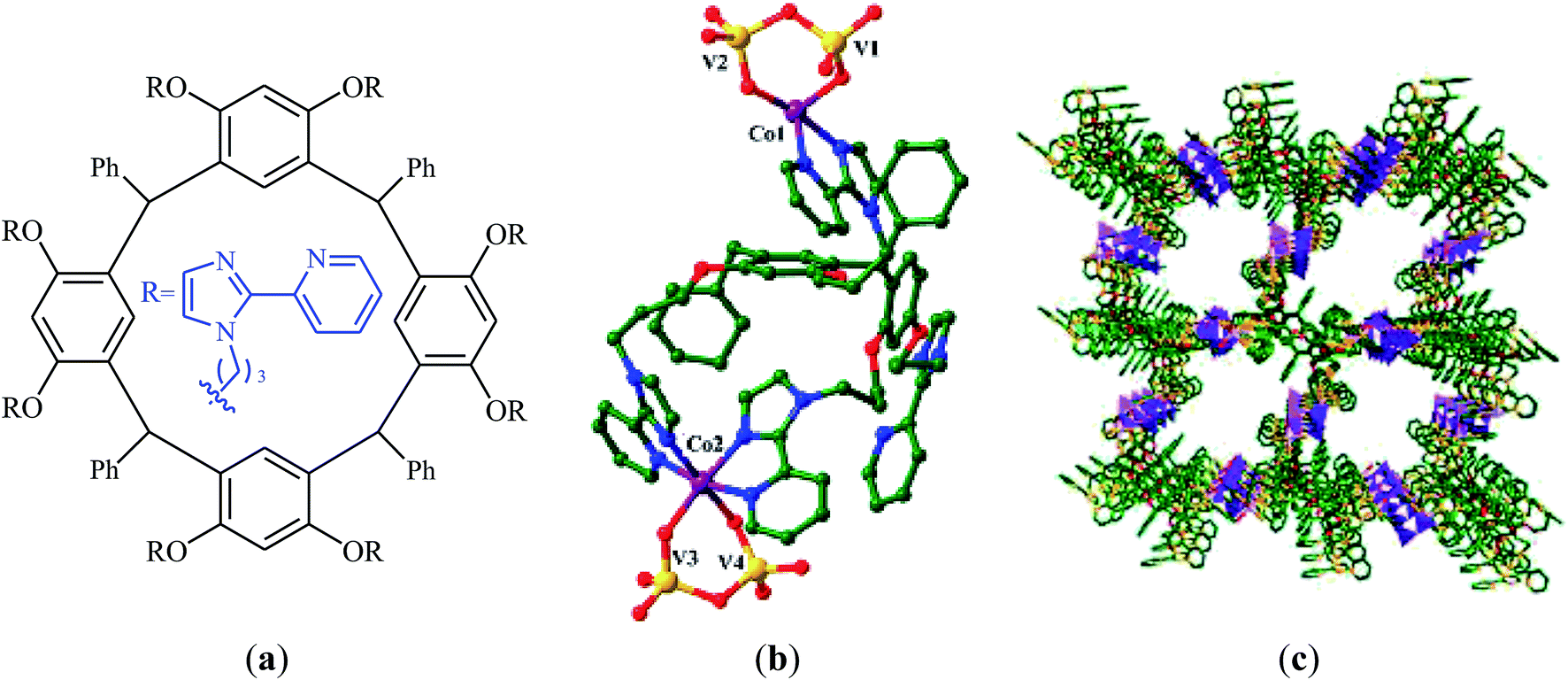
A novel functionalized wheel-like resorcin[4]arene ligand (Fig. 6a) was designed and synthesized through the reaction of resorcinol with 1-bromo-3-chloropropane in the presence of MeONa followed by functionalization with benzaldehyde and finally treatment with 12 equiv. of 2-(2-pyridyl)imidazole.65 The novel ligand was successfully utilized for the construction of a promising polyoxovanadate–resorcin[4]arene-based metal–organic framework, [Co2L0.5V4O12]·3DMF·5H2O (Fig. 6b and c) by incorporation of Co and V through the reaction with a mixture of NH4VO3 and Co(NO3)2·6H2O in a binary solvent DMF/H2O with ratio 1:1. Various mono-substituted cyclic carbonates were successfully synthesized (72–99% for 8 examples) through the reaction of corresponding terminal epoxides with atmospheric CO2 employing this heterogeneous catalyst under solvent-free conditions. The catalyst could be easily recycled and reused five times without significant loss of activity and selectivity.
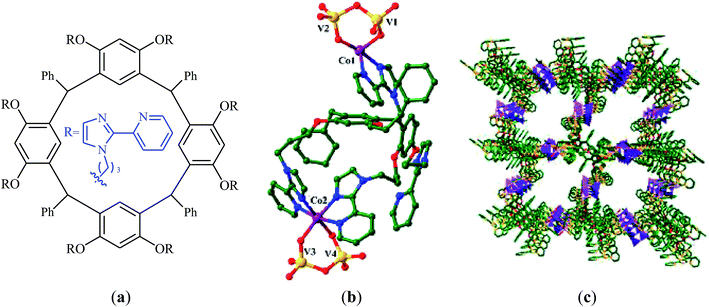 |
| | Fig. 6 (a) Chemical structure of ligand; (b) Coordination environments of the CoII and VV cations in [Co2L0.5V4O12]·3DMF·5H2O; (c) View of the porous packing structure of [Co2L0.5V4O12]·3DMF·5H2O. | |
4.7. Cadmium
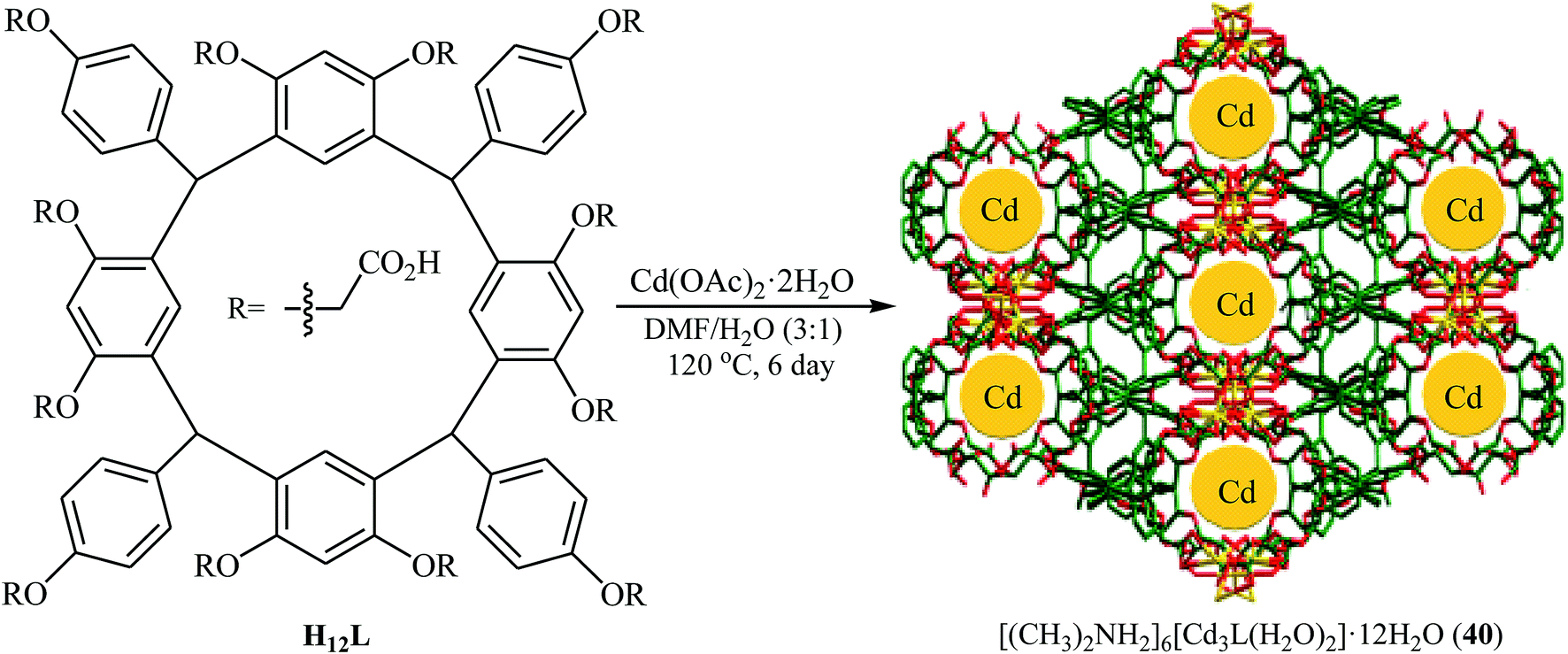
Recently, Yang and Ma along with their co-workers developed a novel microporous anionic metal–organic framework, [(CH3)2NH2]6[Cd3L(H2O)2]·12H2O (40) through solvothermal assembly of a new resorcin[4]arene-functionalized dodecacarboxylic acid (H12L) and Cd(II) cations (Fig. 7).66 The microporous framework 40 was found to be an efficient catalyst in the synthesis of cyclic carbonates through the cycloaddition of atmospheric CO2 to corresponding epoxides in the presence of TBAB as co-catalyst under solvent-free conditions. The catalyst also showed excellent reusability in this system with no remarkable decrease in performance after five consecutive runs. Moreover, fluorescent 40 could be used a luminescent sensor to probe Fe3+ and Cr2O72− in aqueous solutions with high selectivity and sensitivity.
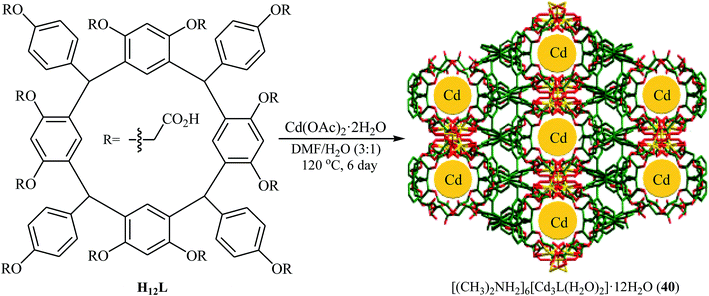 |
| | Fig. 7 Schematic diagram showing the formation of [(CH3)2NH2]6[Cd3L(H2O)2]·12H2O (40). | |
4.8. Bismuth
In 2017, Peng and co-workers introduced a series of Bi(III) porphyrin complex catalysts 41 for solvent-free fixation of atmospheric CO2 and epoxides.67 The complexes were prepared by simple metalation of the corresponding porphyrin ligands with BiX3 (X = Cl, Br, I) in THF. The catalytic activities of the obtained complexes were tested for the coupling of 2-methyloxirane with CO2 under solvent-free conditions in the presence of TBAI as co-catalyst. The results established that the catalytic performances were strongly influenced by the substitute of porphyrin framework and axial group X−. It was found that complex 41a was the most active catalyst for this transformation. Under the optimized conditions [41a (0.2 mol%), TBAI (2.4 mol%), CO2 (1 atm), neat, 90 °C, 6–24 h] a variety of terminal epoxides 42 react to give moderate to almost quantitative yields of the desired products 43 (Scheme 24). However, internal epoxides showed very low reactivity under this reaction conditions. The results demonstrated that substrates with aliphatic groups gave higher yields than those with aromatic groups.
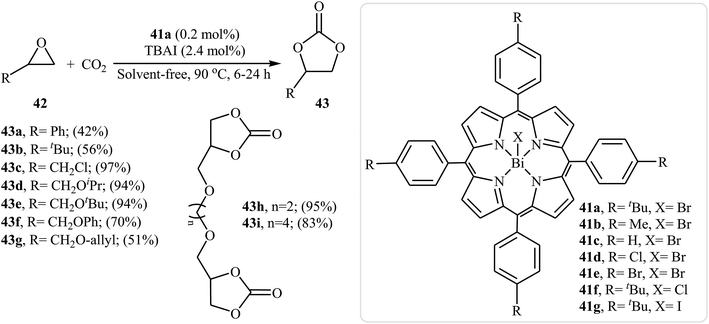 |
| | Scheme 24 Peng's synthesis of carbonates 43. | |
4.9. Molybdenum
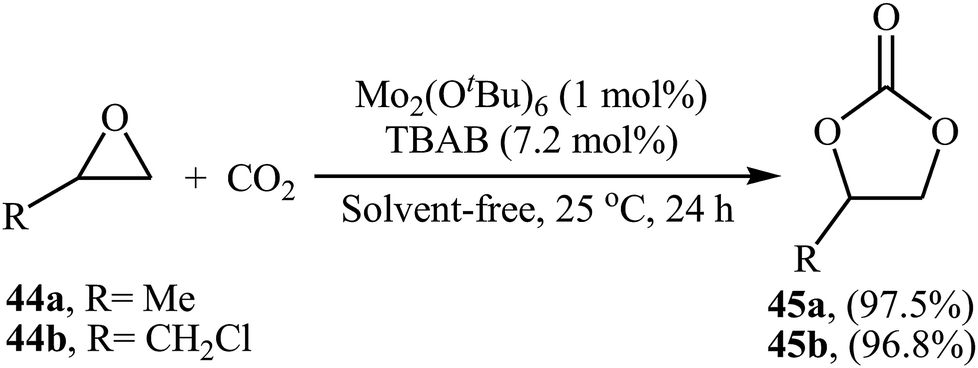
Very recently, the group of Ma-Cheng demonstrated for the first time the usefulness of molybdenum alkoxides as highly active catalysts for the chemical fixation of epoxides with atmospheric CO2 under neat conditions.68 Among four investigated triply-bonded dimolybdenum compounds [i.e., Mo2(OSiMe3)6, Mo2(OiPr)6, Mo2(OtBu)6, and Mo2(ONe)6], Mo2(OtBu)6 was found to be the best catalyst. The authors studied the effects of reaction variables such as catalyst loading, co-catalyst and temperature. It was found that using 1 mol% of the catalyst and 7.2 mol% of TBAB as a co-catalyst at room temperature resulted in the best yields (Scheme 25). However, in the case of epoxides bearing an aryl group, higher loading of co-catalyst and DCM solvent was needed for the success of the reaction. It should be mentioned that this catalytic system was also successfully applied in the fixation of terminal alkynes and aromatic heteroarenes with atmospheric CO2 to the synthesis of corresponding carboxylic acids.
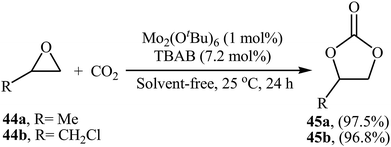 |
| | Scheme 25 Mo2(OtBu)6-catalyzed synthesis of carbonates 45 through coupling of epoxides 44 with CO2 (atm) in the absence of any solvent. | |
4.10. Samarium
Recently, Zhao et al. reported ionic rare-earth metal (lanthanum, ytterbium, yttrium and samarium) complexes 46–49 bearing an imidazolium cation for the cycloaddition of CO2 to epoxides under neat conditions.69 Very low catalytic loadings (0.2 mol%) of the latter complex (49, Scheme 26) could promote the cycloaddition of atmospheric CO2 to various monosubstituted epoxides in the absence of any co-catalyst in 60–97% yields. When 49 (0.2 mol%) was applied for the cycloaddition of bulky/internal epoxides at 90 °C (20 bar CO2 pressure) the authors observed moderate to excellent yields (40–95%) of expected carbonates in 12 h. More importantly, the catalyst can be easily separated from the reaction mixture by simple distillation, and then be reused five times without significant losses in the catalytic activity.
 |
| | Scheme 26 Chemical structures of complexes 46–49. | |
4.11. Zirconium
Recently, the group of Cao has reported a novel bifunctional imidazolium functionalized Zr-based metal–organic-framework, (I−)Meim-UiO-66, by post-synthetic ionization (PSI) of a new microporous imidazole-containing Zr-MOF Im-UiO-66.70 Transmission electron microscopy (TEM) image of the synthesized MOF showed that the average size of the Im-UiO-66 particles was about 20–30 nm. Owing to the Brønsted-acid (Zr–OH/OH2) sites and nucleophilic iodide ions in (I−)Meim-UiO-66, this framework was shown to be an efficient and recyclable heterogeneous catalyst for the cycloaddition of terminal epoxides 50 with atmospheric CO2 without using any co-catalyst and solvent (Scheme 27).
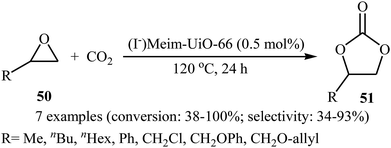 |
| | Scheme 27 Cao's synthesis of carbonates 51. | |
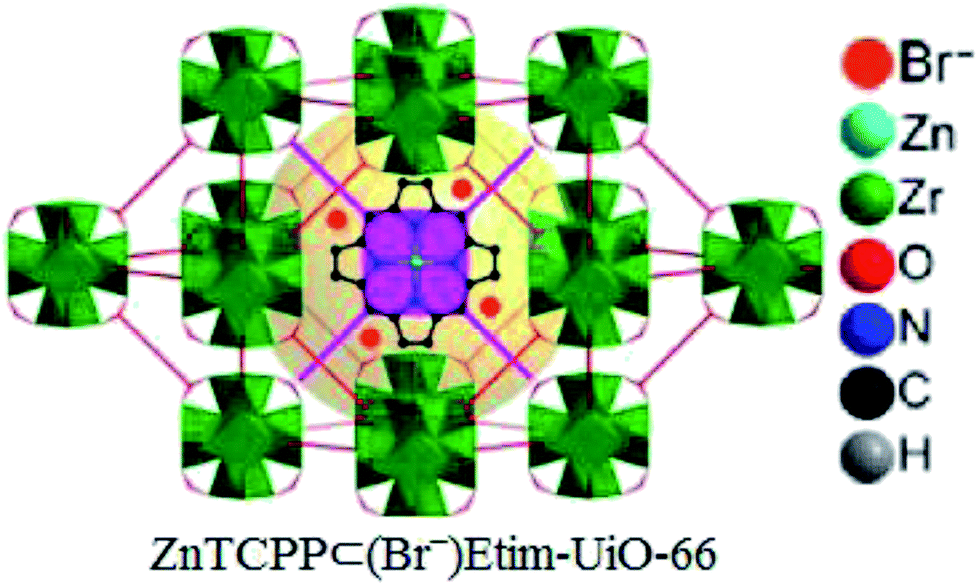
Later, zinc porphyrin/imidazolium integrated multivariate zirconium metal–organic framework (ZnTCPP⊂(Br−)Etim-UiO-66) has been successfully synthesized and applied as a reusable and efficient heterogeneous catalyst for this chemistry by the same research team (Fig. 8).71
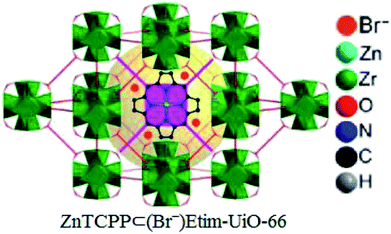 |
| | Fig. 8 Perspective view of structure of ZnTCPP⊂(Br−)Etim-UiO-66. | |
5. Conclusion
The 1,3-dioxolan-2-one (five-membered cyclic carbonate) compound is not only prevalent in a large variety of important classes of natural products and biologically active molecules, but also used as a versatile intermediate in organic synthesis. Among the various methods for the synthesis of titled compounds, cycloaddition of epoxides with CO2 represents one of the most attractive, straightforward and green protocols. From green and sustainable chemistry perspectives, solvent-free reactions are indisputably more desirable than transformations in any kind of solvents. The most important advantages of solvent-free reactions over classical methods include: cost savings, reduced energy consumption, and reactor size. In this review, we have summarized the most important advances and discoveries in the cycloaddition of epoxides with atmospheric CO2 under solvent-free conditions. To the best of our knowledge, this mini-review is the first of its kind to collect literature in the arena of solvent-free CO2 fixation reactions. As illustrated, most of the conversion of CO2 covered in this review have been carried out at ambient temperature and provided the expected carbonates in high yields. Moreover, most of the catalysts presented here could be recovered and reused for several reaction runs without tangible loss in their catalytic activity and selectivity, providing more sustainable processes for the chemical fixation of CO2. We conclude this review article by hoping that it will stimulate interest and encourage further research in this area.
Conflicts of interest
There are no conflicts to declare.
References
- B. Yu and L. N. He, ChemSusChem, 2015, 108, 52–62 CrossRef PubMed.
- J. Rintjema and A. W. Kleij, Synthesis, 2016, 48, 3863–3878 CrossRef CAS.
- G. Fiorani, W. Guo and A. W. Kleij, Green Chem., 2015, 17, 1375–1389 RSC.
- X.-F. Wu and F. Zheng, Top. Curr. Chem., 2017, 375, 4 CrossRef PubMed.
- Q.-W. Song, Z.-H. Zhou and L.-N. He, Green Chem., 2017, 19, 3707–3728 RSC.
- X.-D. Lang, X. He, Z.-M. Li and L.-N. He, Curr. Opin. Green. Sus., 2017, 7, 31–38 CrossRef.
- Q.-W. Song, Z.-H. Zhou and L.-N. He, Green Chem., 2017, 19, 3707–3728 RSC.
- M. Bonchio, A. Cherubini-Celli, J. Mateos, L. Dell'Amico and X. Companyó, ChemSusChem, 2018, 11, 3056–3070 CrossRef PubMed.
- R. Martin, A. Tortajada, F. Juliá-Hernández, M. Borjesson and T. Moragas, Angew. Chem., Int. Ed., 2018, 57, 15948–15982 CrossRef PubMed.
- R. Norhasyima and T. Mahlia, J. CO2 Util., 2018, 26, 323–335 CrossRef CAS.
- D. J. Darensbourg and M. W. Holtcamp, Coord. Chem. Rev., 1996, 153, 155–174 CrossRef CAS.
- X.-D. Lang, X.-F. Liu and L.-N. He, Curr. Org. Chem., 2015, 19, 681–694 CrossRef CAS.
- X. D. Lang and L. N. He, Chem. Rec., 2016, 16, 1337–1352 CrossRef CAS PubMed.
- D.-H. Lan, N. Fan, Y. Wang, X. Gao, P. Zhang, L. Chen, C.-T. Au and S.-F. Yin, Chin. J. Catal., 2016, 37, 826–845 CrossRef CAS.
- H. Buettner, L. Longwitz, J. Steinbauer, C. Wulf and T. Werner, Top. Curr. Chem., 2017, 375, 50 CrossRef PubMed.
- O. D. Hensens, A. J. Kempf, R. E. Schwartz, R. S. Sykes, C. F. Wichmann, K. E. Wilson, S. B. Zimmerman and D. L. Zink, Antifungal tri-yne carbonates, US Pat. 4806565, 1989 Search PubMed.
- T. Asaka, M. Kashimura, Y. Misawa, S. Morimoto and K. Hatayama, 5-O-desosaminylerythronolide a derivatives, US Pat. 5602239, 1997 Search PubMed.
- R. C. C. Huang, A. Gittis, E. Moudrianakis, J. A. Dohm, J. R. Hwu and M.-H. Hsu, Heterocyclic and carbonate derivatives of NDGA and their use as new anti-HIV and anti-cancer agents, US Pat. 7741357B1, 2010 Search PubMed.
- A.-A. G. Shaikh and S. Sivaram, Chem. Rev., 1996, 96, 951–976 CrossRef CAS PubMed.
- H. Tomita, F. Sanda and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 3678–3685 CrossRef CAS.
- H. Liu, Z. Huang, Z. Han, K. Ding, H. Liu, C. Xia and J. Chen, Green Chem., 2015, 17, 4281–4290 RSC.
- H. Zhang, H.-B. Liu and J.-M. Yue, Chem. Rev., 2014, 114, 883–898 CrossRef CAS PubMed.
- C. Martin, G. Fiorani and A. W. Kleij, ACS Catal., 2015, 5, 1353–1370 CrossRef CAS.
- C. X. Miao, J. Q. Wang, Y. Wu, Y. Du and L. N. He, ChemSusChem, 2008, 1, 236–241 CrossRef CAS PubMed.
- J. Sun, L. Han, W. Cheng, J. Wang, X. Zhang and S. Zhang, ChemSusChem, 2011, 4, 502–507 CrossRef CAS PubMed.
- M. Liu, L. Liang, X. Li, X. Gao and J. Sun, Green Chem., 2016, 18, 2851–2863 RSC.
- R. R. Kuruppathparambil, T. Jose, R. Babu, G.-Y. Hwang, A. C. Kathalikkattil, D.-W. Kim and D.-W. Park, Appl. Catal., B, 2016, 182, 562–569 CrossRef CAS.
- Y. Jiang, C. Zhang, Y. Li, P. Jiang, J. Jiang and Y. Leng, New J. Chem., 2018, 42, 16829–16835 RSC.
- Y. Du, F. Cai, D.-L. Kong and L.-N. He, Green Chem., 2005, 7, 518–523 RSC.
- J.-Q. Wang, X.-D. Yue, F. Cai and L.-N. He, Catal. Commun., 2007, 8, 167–172 CrossRef CAS.
-
(a) S. Arshadi, E. Vessally, M. Sobati, A. Hosseinian and A. Bekhradnia, J. CO2 Util., 2017, 19, 120–129 CrossRef CAS;
(b) S. Arshadi, E. Vessally, A. Hosseinian, S. Soleimani-amiri and L. Edjlali, J. CO2 Util., 2017, 21, 108–118 CrossRef CAS;
(c) E. Vessally, M. Babazadeh, A. Hosseinian, S. Arshadi and L. Edjlali, J. CO2 Util., 2017, 21, 491–502 CrossRef CAS;
(d) E. Vessally, K. Didehban, M. Babazadeh, A. Hosseinian and L. Edjlali, J. CO2 Util., 2017, 21, 480–490 CrossRef CAS;
(e) E. Vessally, S. Soleimani-Amiri, A. Hosseinian, L. Edjlali and M. Babazadeh, J. CO2 Util., 2017, 21, 342–352 CrossRef CAS;
(f) K. Didehban, E. Vessally, M. Salary, L. Edjlali and M. Babazadeh, J. CO2 Util., 2018, 23, 42–50 CrossRef CAS;
(g) E. Vessally, R. Mohammadi, A. Hosseinian, L. Edjlali and M. Babazadeh, J. CO2 Util., 2018, 24, 361–368 CrossRef CAS;
(h) S. Farshbaf, L. Z. Fekri, M. Nikpassand, R. Mohammadi and E. Vessally, J. CO2 Util., 2018, 25, 194–204 CrossRef CAS;
(i) A. Hosseinian, S. Ahmadi, R. Mohammadi, A. Monfared and Z. Rahmani, J. CO2 Util., 2018, 27, 381–389 CrossRef CAS;
(j) A. Hosseinian, S. Farshbaf, R. Mohammadi, A. Monfared and E. Vessally, RSC Adv., 2018, 8, 17976–17988 RSC;
(k) E. Vessally, A. Hosseinian, L. Edjlali, M. Babazadeh and K. Didehban, Mini-Rev. Org. Chem., 2018, 15, 315–323 CrossRef CAS;
(l) E. Vessally, A. Hosseinian, M. Babazadeh, L. Edjlali and R. Hosseinzadeh-Khanmiri, Curr. Org. Chem., 2018, 22, 315–322 CrossRef CAS;
(m) M. Daghagheleh, M. Vali, Z. Rahmani, S. Sarhandi and E. Vessally, Chem. Rev. Lett., 2018, 1, 23–30 Search PubMed.
-
(a) E. Vessally, L. Edjlali, A. Hosseinian, A. Bekhradnia and M. D. Esrafili, RSC Adv., 2016, 6, 49730–49746 RSC;
(b) E. Vessally, A. Hosseinian, L. Edjlali, A. Bekhradnia and M. D. Esrafili, RSC Adv., 2016, 6, 71662–71675 RSC;
(c) E. Vessally, A. Hosseinian, L. Edjlali, A. Bekhradnia and M. D. Esrafili, RSC Adv., 2016, 6, 99781–99793 RSC;
(d) E. Vessally, S. Soleimani-Amiri, A. Hosseinian, L. Edjlali and A. Bekhradnia, RSC Adv., 2017, 7, 7079–7091 RSC;
(e) S. Arshadi, E. Vessally, L. Edjlali, E. Ghorbani-Kalhor and R. Hosseinzadeh-Khanmiri, RSC Adv., 2017, 7, 13198–13211 RSC;
(f) S. Shahidi, P. Farajzadeh, P. Ojaghloo, A. Karbakhshzadeh and A. Hosseinian, Chem. Rev. Lett., 2018, 1, 37–44 Search PubMed;
(g) S. Arshadi, E. Vessally, L. Edjlali, R. Hosseinzadeh-Khanmiri and E. Ghorbani-Kalhor, Beilstein J. Org. Chem., 2017, 13, 625–637 CrossRef CAS;
(h) S. Mohammadi, M. Musavi, F. Abdollahzadeh, S. Babadoust and A. Hosseinian, Chem. Rev. Lett., 2018, 1, 49–55 Search PubMed;
(i) S. Soleimani-Amiri, E. Vessally, M. Babazadeh, A. Hosseinian and L. Edjlali, RSC Adv., 2017, 7, 28407–28418 RSC;
(j) E. Vessally, A. Hosseinian, L. Edjlali, E. Ghorbani-Kalhor and R. Hosseinzadeh-Khanmiri, J. Iran. Chem. Soc., 2017, 14, 2339–2353 CrossRef CAS;
(k) M. Babazadeh, S. Soleimani-Amiri, E. Vessally, A. Hosseinian and L. Edjlali, RSC Adv., 2017, 7, 43716–43736 RSC;
(l) E. Vessally, A. Hosseinian, L. Edjlali, M. Babazadeh and R. Hosseinzadeh-Khanmiri, Iran. J. Chem. Chem. Eng., 2017, 36, 1–13 CAS;
(m) E. Vessally, M. Babazadeh, K. Didehban, A. Hosseinian and L. Edjlali, Curr. Org. Chem., 2017, 21, 2561–2572 CrossRef CAS;
(n) E. Vessally, M. Babazadeh, A. Hosseinian, L. Edjlali and L. Sreerama, Curr. Org. Chem., 2018, 22, 199–205 CrossRef CAS;
(o) S. Farshbaf, L. Sreerama, T. Khodayari and E. Vessally, Chem. Rev. Lett., 2018, 1, 56–6532 Search PubMed;
(p) S. Sarhandi, M. Daghagheleh, M. Vali, R. Moghadami and E. Vessally, Chem. Rev. Lett., 2018, 1, 9–15 Search PubMed.
- R. L. Vekariya, J. Mol. Liq., 2017, 227, 44–60 CrossRef CAS.
- Z.-Z. Yang, Y.-N. Zhao and L.-N. He, RSC Adv., 2011, 1, 545–567 RSC.
- V. B. Saptal and B. M. Bhanage, ChemSusChem, 2017, 10, 1145–1151 CrossRef CAS PubMed.
- Q. Sun, Y. Jin, B. Aguila, X. Meng, S. Ma and F. S. Xiao, ChemSusChem, 2017, 10, 1160–1165 CrossRef CAS PubMed.
- Z. Zhang, F. Fan, H. Xing, Q. Yang, Z. Bao and Q. Ren, ACS Sustainable Chem. Eng., 2017, 5, 2841–2846 CrossRef CAS.
- Z. Guo, Q. Jiang, Y. Shi, J. Li, X. Yang, W. Hou, Y. Zhou and J. Wang, ACS Catal., 2017, 7, 6770–6780 CrossRef CAS.
- Y. Xie, Q. Sun, Y. Fu, L. Song, J. Liang, X. Xu, H. Wang, J. Li, S. Tu and X. Lu, J. Mater. Chem. A, 2017, 5, 25594–25600 RSC.
-
(a) D. W. MacMillan, Nature, 2008, 455, 304–308 CrossRef CAS PubMed;
(b) J. Alemán and S. Cabrera, Chem. Soc. Rev., 2013, 42, 774–793 RSC.
- B. Chatelet, L. Joucla, J.-P. Dutasta, A. Martinez, K. C. Szeto and V. R. Dufaud, J. Am. Chem. Soc., 2013, 135, 5348–5351 CrossRef CAS PubMed.
- L. Wang, G. Zhang, K. Kodama and T. Hirose, Green Chem., 2016, 18, 1229–1233 RSC.
- X. Wang, L. Wang, Y. Zhao, K. Kodama and T. Hirose, Tetrahedron, 2017, 73, 1190–1195 CrossRef CAS.
- V. B. Saptal, T. Sasaki, K. Harada, D. Nishio-Hamane and B. M. Bhanage, ChemSusChem, 2016, 9, 644–650 CrossRef CAS PubMed.
- S. Arayachukiat, C. Kongtes, A. Barthel, S. V. Vummaleti, A. Poater, S. Wannakao, L. Cavallo and V. D'Elia, ACS Sustainable Chem. Eng., 2017, 5, 6392–6397 CrossRef CAS.
- N. Liu, Y.-F. Xie, C. Wang, S.-J. Li, D.-H. Wei, M. Li and B. Dai, ACS Catal., 2018, 8, 9945–9957 CrossRef CAS.
- B. Zou, L. Hao, L.-Y. Fan, Z.-M. Gao, S.-L. Chen, H. Li and C.-W. Hu, J. Catal., 2015, 329, 119–129 CrossRef CAS.
- P.-Z. Li, X.-J. Wang, J. Liu, J. S. Lim, R. Zou and Y. Zhao, J. Am. Chem. Soc., 2016, 138, 2142–2145 CrossRef CAS PubMed.
- R. K. Sharma, R. Gaur, M. Yadav, A. Goswami, R. Zbořil and M. B. Gawande, Sci. Rep., 2018, 8, 1901 CrossRef PubMed.
- H. He, Q. Sun, W. Gao, J. A. Perman, F. Sun, G. Zhu, B. Aguila, K. Forrest, B. Space and S. Ma, Angew. Chem., Int. Ed., 2018, 57, 4657–4662 CrossRef CAS PubMed.
- Q.-W. Song, B. Yu, X.-D. Li, R. Ma, Z.-F. Diao, R.-G. Li, W. Li and L.-N. He, Green Chem., 2014, 16, 1633–1638 RSC.
- J.-J. Chen, Z.-L. Gan and X.-Y. Yi, Catal. Lett., 2018, 148, 852–856 CrossRef CAS.
- T.-T. Liu, J. Liang, Y.-B. Huang and R. Cao, Chem. Commun., 2016, 52, 13288–13291 RSC.
- J. Martínez, J. A. Castro-Osma, C. Alonso-Moreno, A. Rodríguez-Diéguez, M. North, A. Otero and A. Lara-Sánchez, ChemSusChem, 2017, 10, 1175–1185 CrossRef PubMed.
- F. de la Cruz-Martínez, J. Martínez, M. A. Gaona, J. Fernández-Baeza, L. F. Sánchez-Barba, A. M. Rodríguez, J. A. Castro-Osma, A. Otero and A. Lara-Sánchez, ACS Sustainable Chem. Eng., 2018, 6, 5322–5332 CrossRef.
- R. Ma, L.-N. He and Y.-B. Zhou, Green Chem., 2016, 18, 226–231 RSC.
- Y.-H. Han, Z.-Y. Zhou, C.-B. Tian and S.-W. Du, Green Chem., 2016, 18, 4086–4091 RSC.
- J. Peng, H.-J. Yang, S. Wang, B. Ban, Z. Wei, B. Lei and C.-Y. Guo, J. CO2 Util., 2018, 24, 1–9 CrossRef CAS.
- C. Yao, S. Zhou, X. Kang, Y. Zhao, R. Yan, Y. Zhang and L. Wen, Inorg. Chem., 2018, 57, 11157–11164 CrossRef CAS PubMed.
- S. Ghosh, P. Mondal, D. Das, K. Tuhina and S. M. Islam, J. Organomet. Chem., 2018, 866, 1–12 CrossRef CAS.
- L. Wang, C. Xu, Q. Han, X. Tang, P. Zhou, R. Zhang, G. Gao, B. Xu, W. Qin and W. Liu, Chem. Commun., 2018, 54, 2212–2215 RSC.
- P. Patel, B. Parmar, R. I. Kureshy, N. U. Khan and E. Suresh, ChemCatChem, 2018, 10, 2401–2408 CrossRef CAS.
- C.-S. Cao, Y. Shi, H. Xu and B. Zhao, Dalton Trans., 2018, 47, 4545–4553 RSC.
- R. Khatun, P. Bhanja, P. Mondal, A. Bhaumik, D. Das and S. M. Islam, New J. Chem., 2017, 41, 12937–12946 RSC.
- B.-B. Lu, J. Yang, Y.-Y. Liu and J.-F. Ma, Inorg. Chem., 2017, 56, 11710–11720 CrossRef CAS PubMed.
- B.-B. Lu, W. Jiang, J. Yang, Y.-Y. Liu and J.-F. Ma, ACS Appl. Mater. Interfaces, 2017, 9, 39441–39449 CrossRef CAS PubMed.
- J. Peng, Y. Geng, H.-J. Yang, W. He, Z. Wei, J. Yang and C.-Y. Guo, Mol. Catal., 2017, 432, 37–46 CrossRef CAS.
- J.-H. Chen, C.-H. Deng, S. Fang, J.-G. Ma and P. Cheng, Green Chem., 2018, 20, 989–996 RSC.
- Z. Zhao, J. Qin, C. Zhang, Y. Wang, D. Yuan and Y. Yao, Inorg. Chem., 2017, 56, 4568–4575 CrossRef PubMed.
- J. Liang, R.-P. Chen, X.-Y. Wang, T.-T. Liu, X.-S. Wang, Y.-B. Huang and R. Cao, Chem. Sci., 2017, 8, 1570–1575 RSC.
- J. Liang, Y.-Q. Xie, Q. Wu, X.-Y. Wang, T.-T. Liu, H.-F. Li, Y.-B. Huang and R. Cao, Inorg. Chem., 2018, 57, 2584–2593 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Shahriar Sarhandi
a and
Parvaneh Delir Kheirollahi Nezhad
*a
b,
Shahriar Sarhandi
a and
Parvaneh Delir Kheirollahi Nezhad
*a