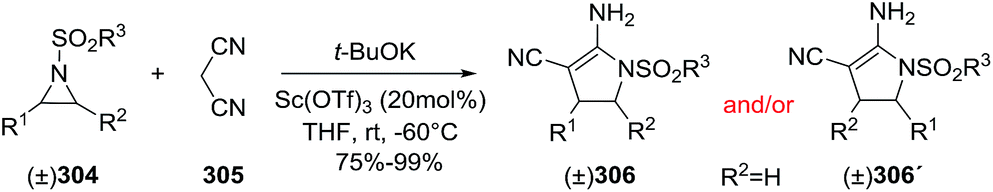
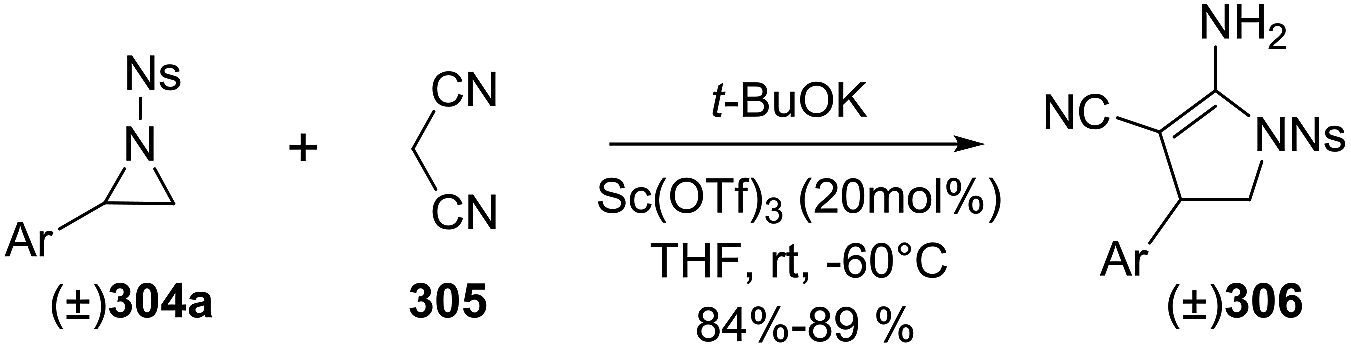
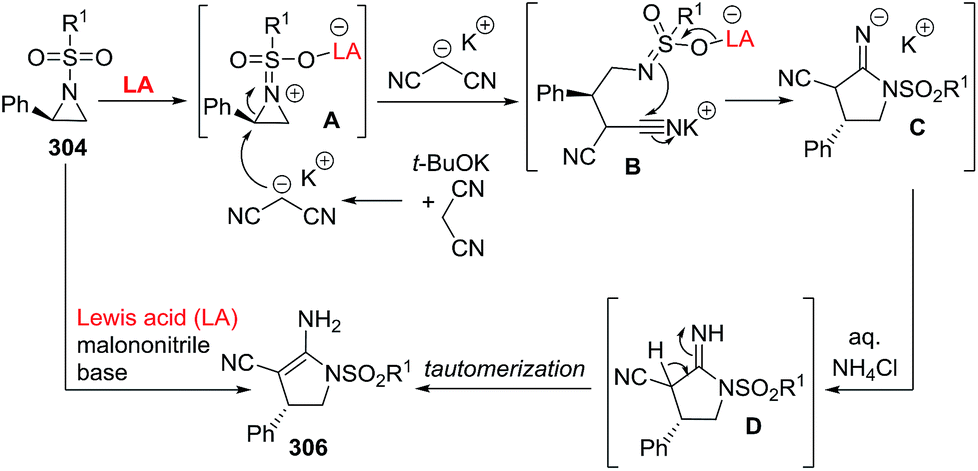
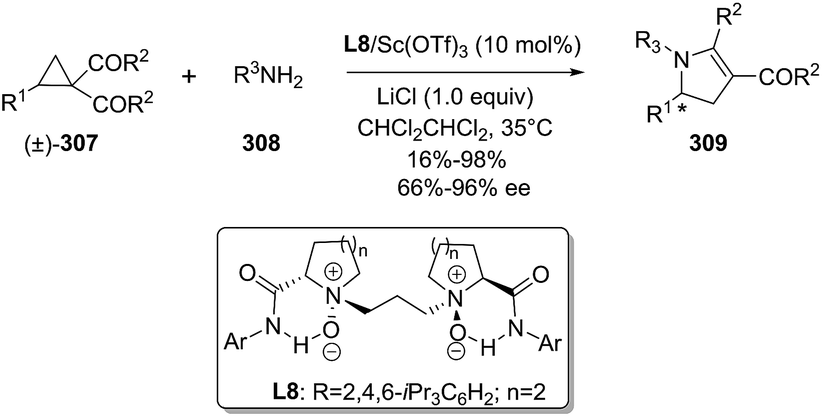
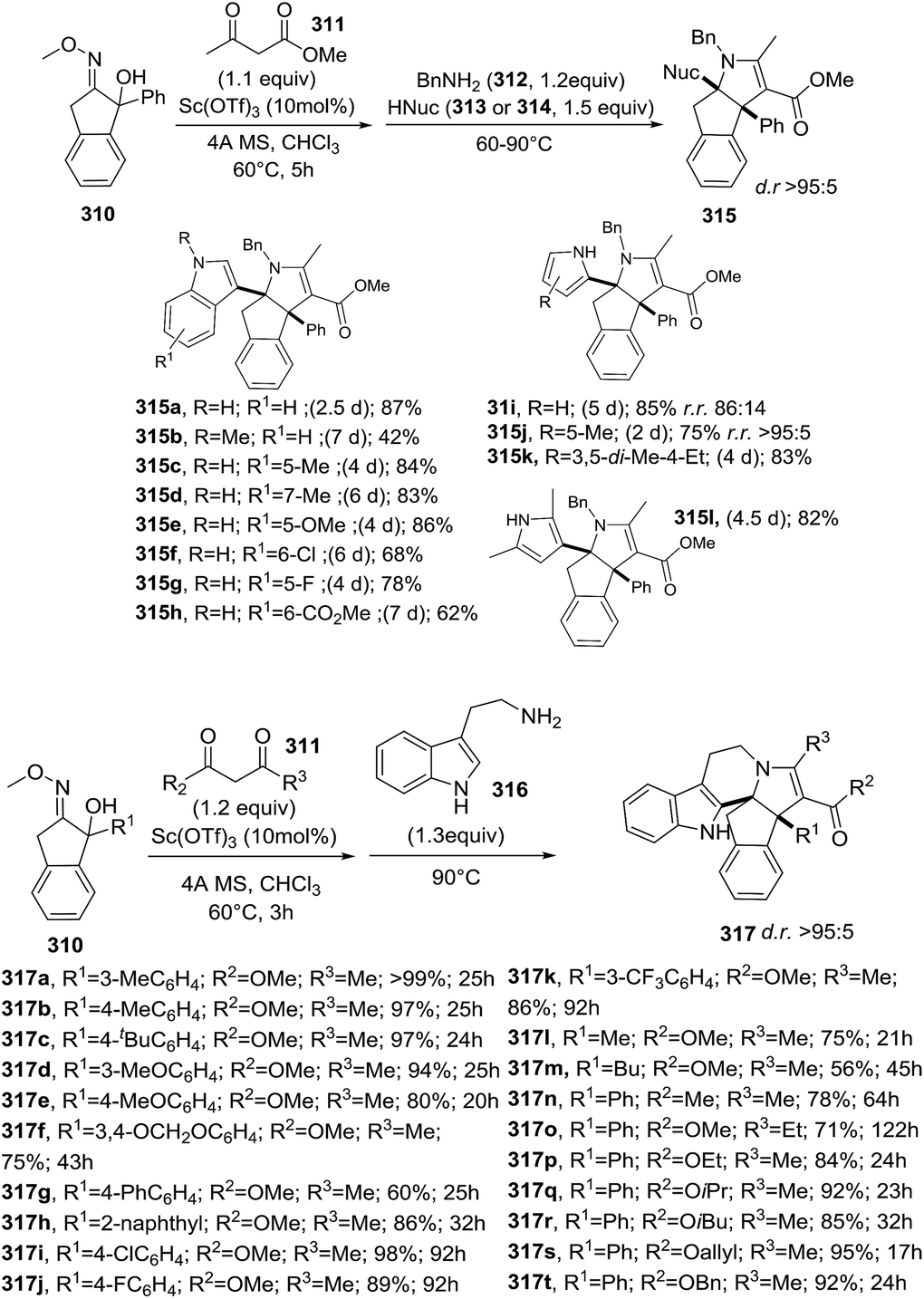
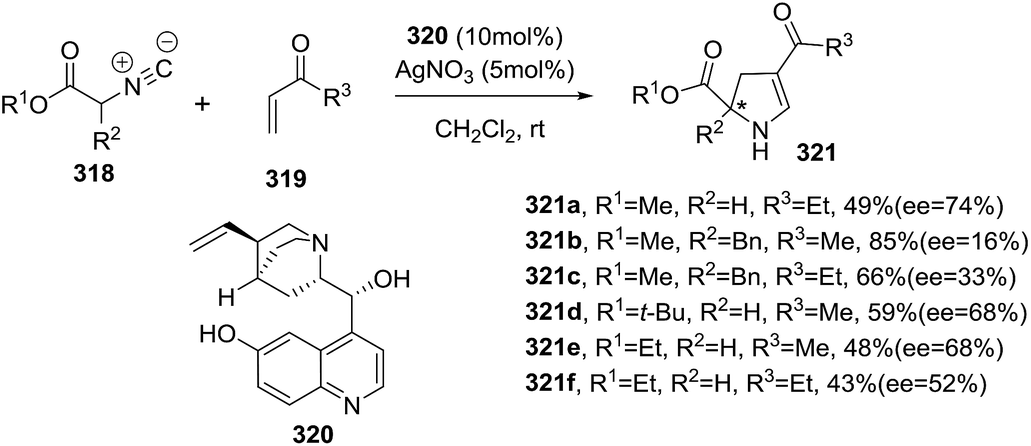
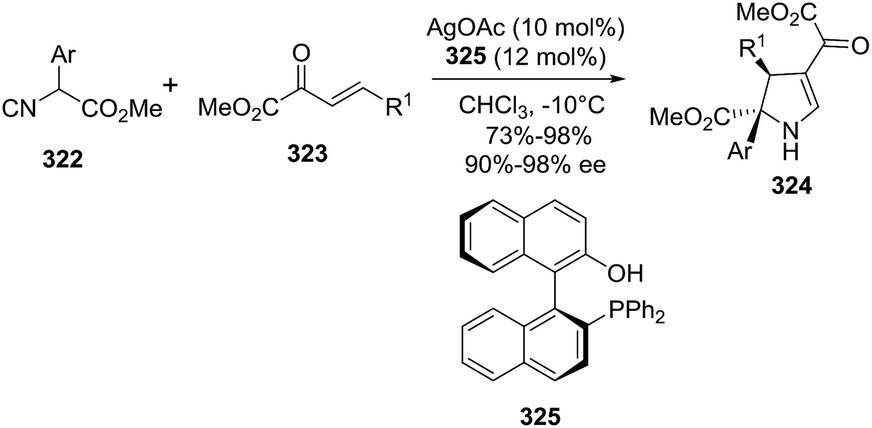
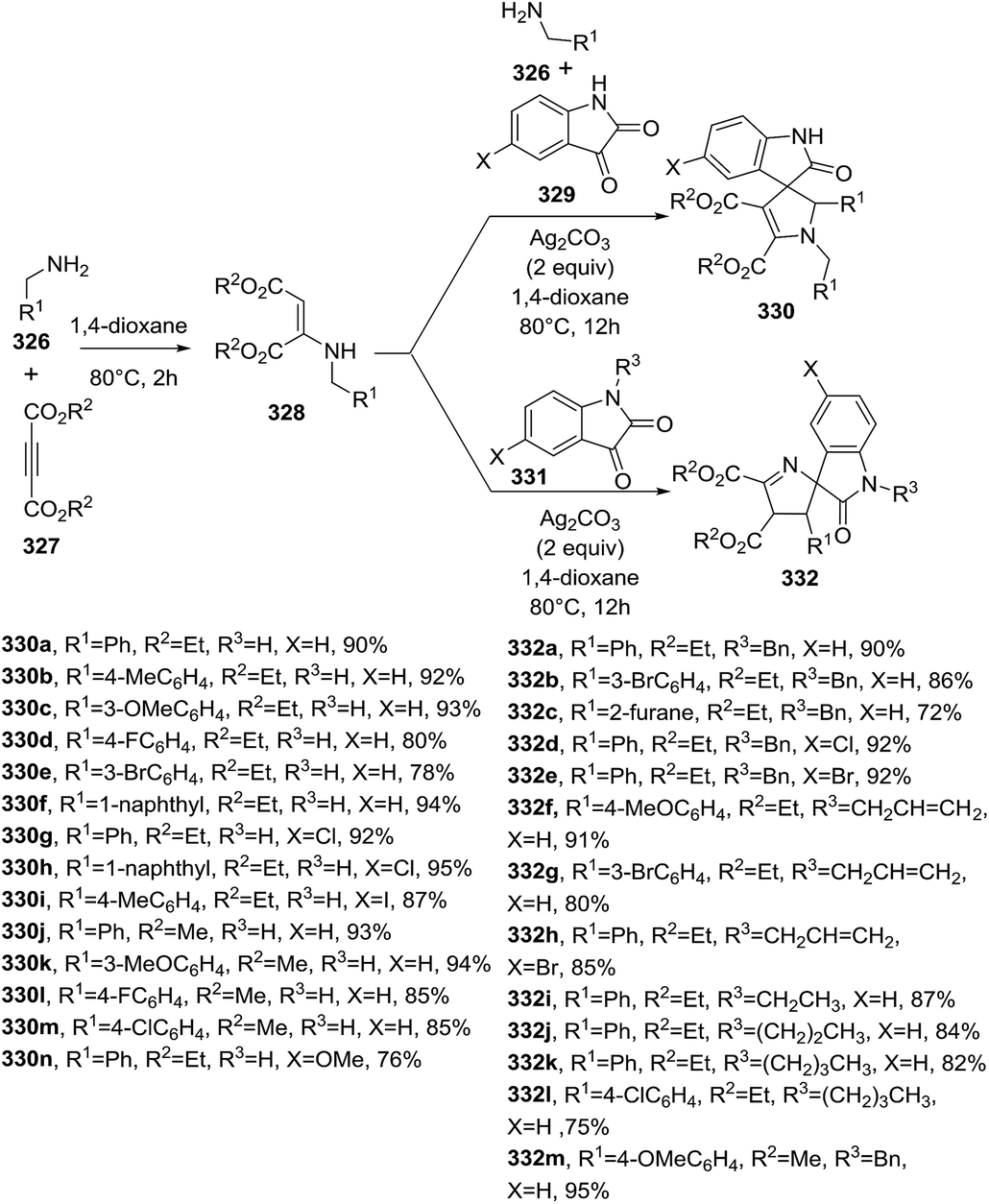
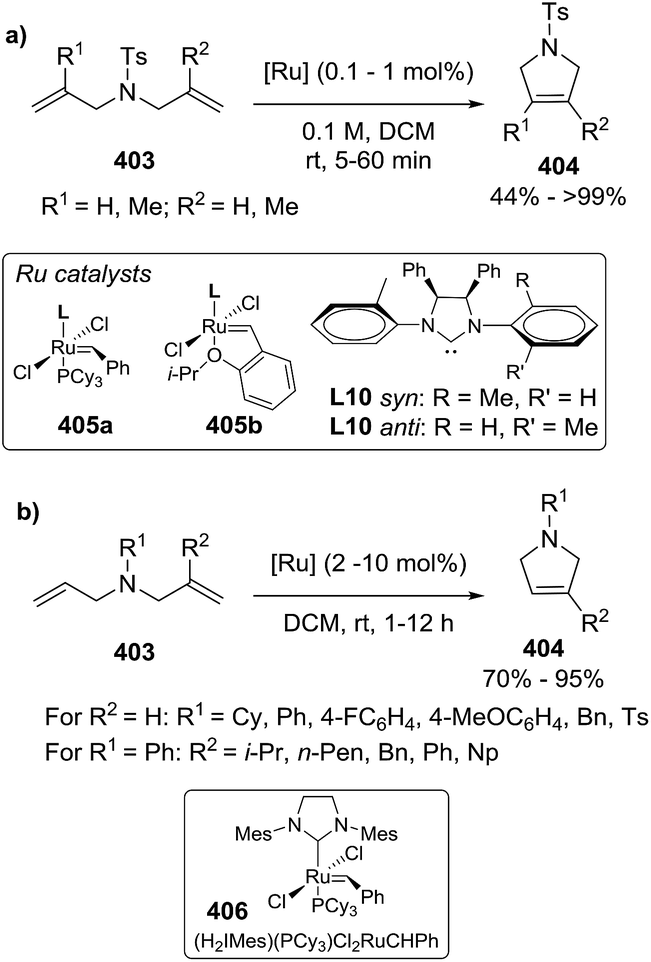
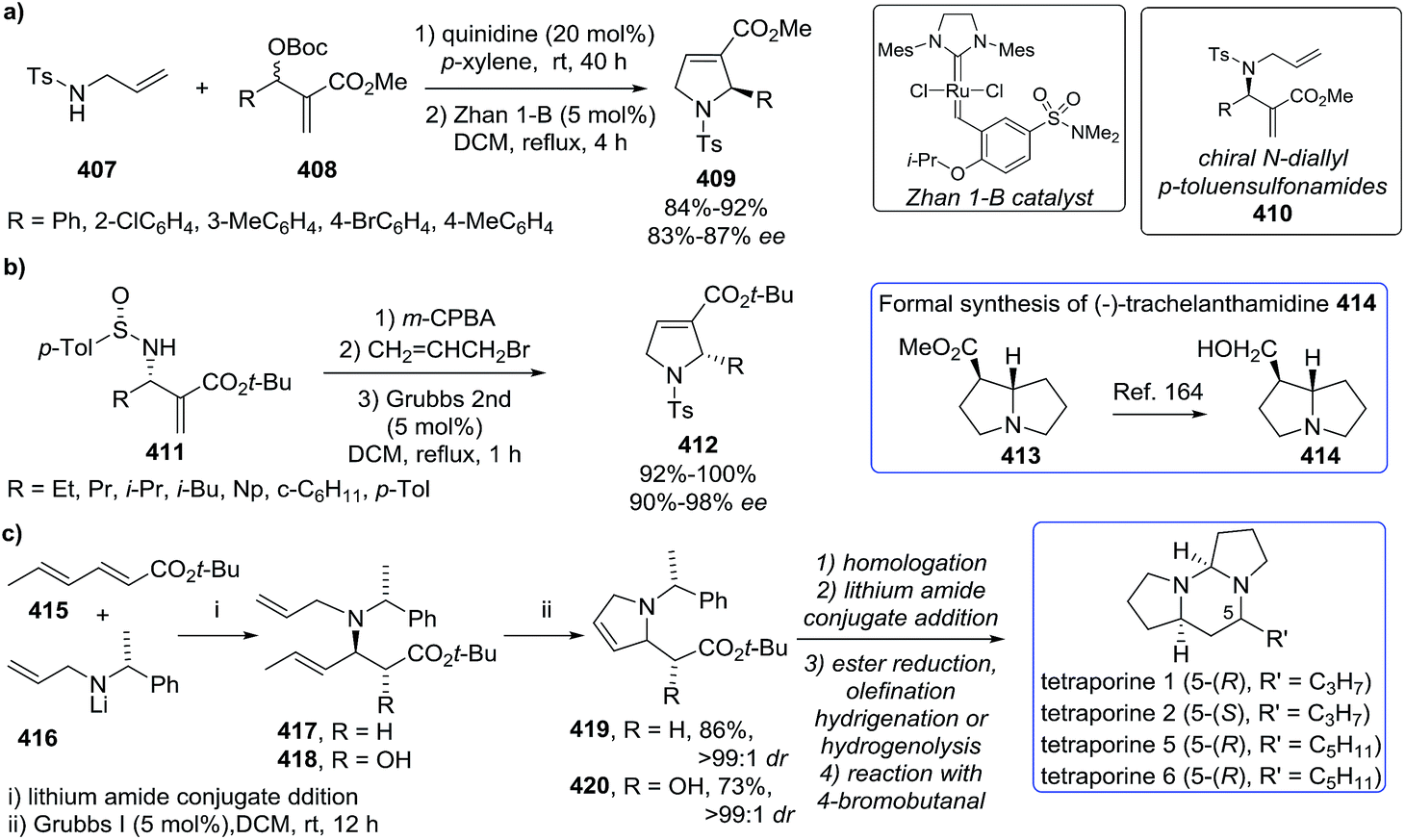
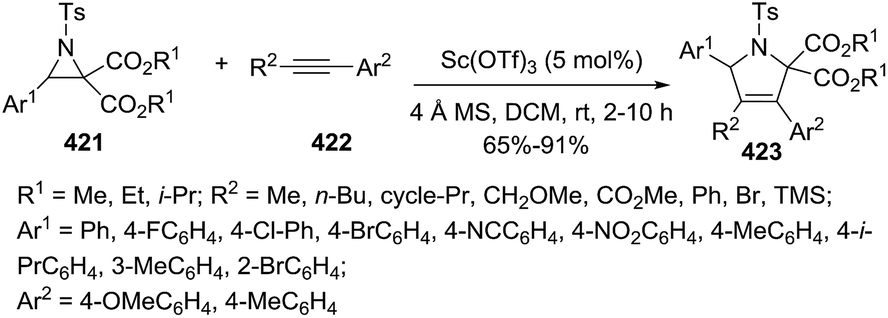
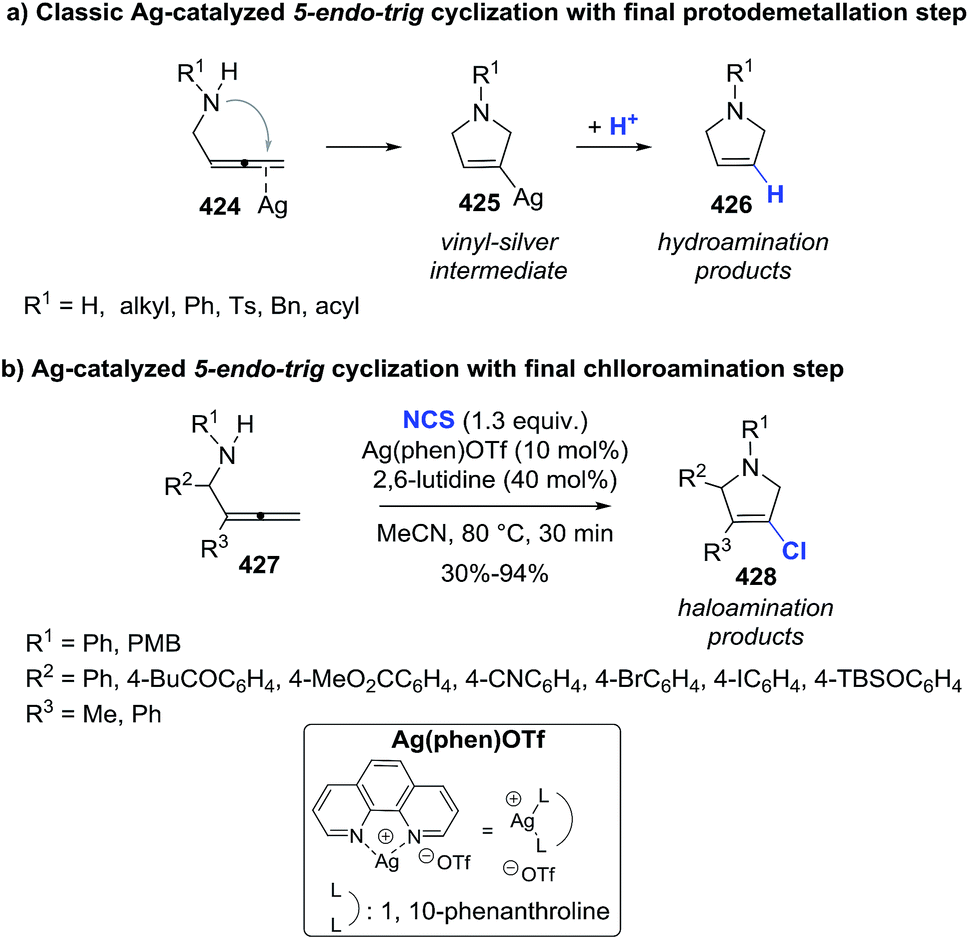
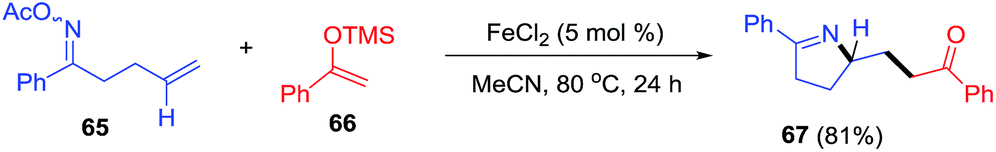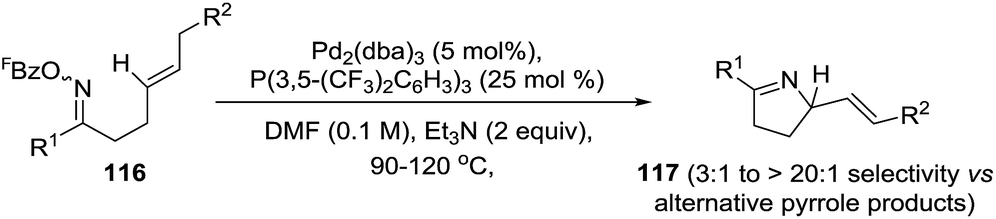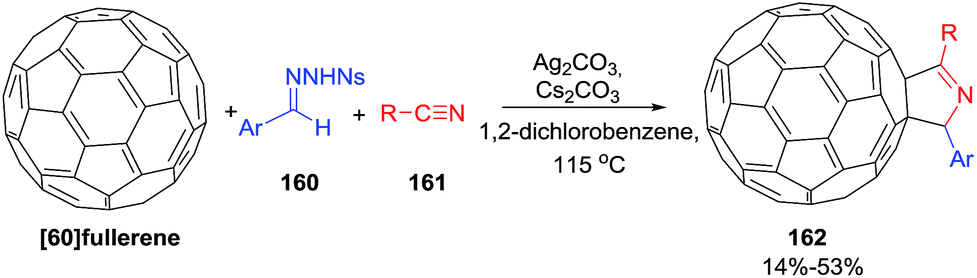DOI:
10.1039/C8RA10247C
(Review Article)
RSC Adv., 2019,
9, 6804-6844
Metal-mediated synthesis of pyrrolines
Received
13th December 2018
, Accepted 18th February 2019
First published on 27th February 2019
Abstract
The five-membered, nitrogen-containing pyrroline ring is a privileged structure. This ring is present in many bioactive compounds from natural sources. Pyrrolines—the dihydro derivatives of pyrroles—have three structural isomer classes, depending on the location of the double bond: 1-pyrrolines (3,4-dihydro-2H-pyrroles), 2-pyrrolines (2,3-dihydro-1H-pyrroles) and 3-pyrrolines (2,5-dihydro-1H-pyrroles). This review aims to describe the latest advances for the synthesis of pyrrolines by transition metal-catalyzed cyclizations. Only reactions in which the pyrroline ring is formed by metal promotion are described. Transformations of the pyrroline ring in other heterocycles, and the structural manipulations of the pyrroline itself are not discussed. The review is organized into three parts, each covering the metal-mediated synthesis of the three pyrroline isomers. Each part is subdivided according to the metal involved, and concludes with a brief description of notable biological activities within the class.
 Noelia S. Medran | Noelia S. Medran completed her PhD in synthetic organic chemistry at the Instituto de Química Rosario in 2017, under the supervision of Prof. Sebastian A. Testero. Since 2017 she is carrying out a postdoctoral research in organoboron chemistry and computational chemistry, particularly applied to Diels–Alder reactions, with Prof. Silvina C. Pellegrinet at the same institute. Since 2015 she is teacher assistant at the department of Medicinal Chemistry of National Argentinean University (UNR). Her research interests center on medicinal chemistry, organometallic chemistry, organoboron chemistry, and computational chemistry. |
 Agustina La-Venia | Agustina La-Venia completed her PhD in synthetic organic chemistry at the Instituto de Química Rosario (IQUIR) in 2011, under the supervision of Prof. Dr Mirta P. Mischne and Prof. Dr Ernesto G. Mata. Between 2011 and 2015, she carried out a postdoctoral research in the combinatorial synthesis of constrained peptidomimetics at the Palacky University (UPOL), Czech Republic under Prof. Dr Viktor Krchňák. Since 2015, she is Junior Researcher of the Argentine National Council of Research at IQUIR. She has been teacher assistant at the department of Organic Chemistry of National Argentinean University (UNR) between 2005 and 2011, and since 2015 she is teaching at the department of Medicinal Chemistry of UNR. Her research interests center on medicinal chemistry, combinatorial chemistry, solid phase synthesis, metal catalysis, and peptidomimetics. |
 Sebastian A. Testero | Sebastian A. Testero completed his PhD in synthetic organic chemistry at the Instituto de Química Rosario in 2005, under the supervision of Prof. R. A. Spanevello. Then he carried out a postdoctoral research in solid-phase synthesis with Prof. E. G. Mata at the same institute. Between 2007 and 2011, he was a postdoctoral research associate at the University of Notre Dame at the Mobashery lab working in medicinal chemistry. Since 2015, he is Adjunct professor of Organic Chemistry and from 2017 an independent researcher of the Argentine National Council of Research. His research interests center on medicinal chemistry, combinatorial chemistry, ozonolysis and organometallic chemistry including metathesis and gold catalysis. |
1. Introduction
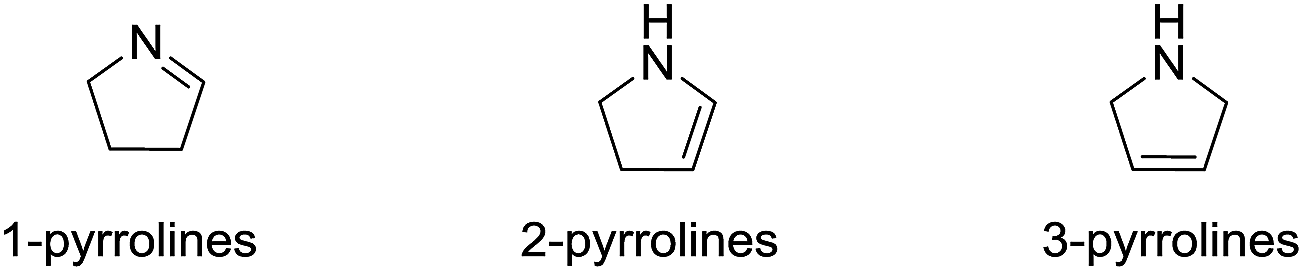
Heterocyclic ring systems are the fundamental building blocks in the vast majority of drugs used to treat animal and human diseases. Among these heterocyclic rings, those containing nitrogen are the most significant. Pyrrolines—the dihydro derivatives of pyrroles—have received considerable attention lately since they exhibit a variety of biological activities. Pyrrolines have three structural isomer classes (Fig. 1), depending on the location of the double bond: 1-pyrrolines (3,4-dihydro-2H-pyrroles), 2-pyrrolines (2,3-dihydro-1H-pyrroles) and 3-pyrrolines (2,5-dihydro-1H-pyrroles).
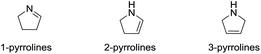 |
| | Fig. 1 Structural isomers of the pyrrolines. | |
Pyrrolines are considered privileged structures as reflected by their presence in many bioactive compounds from natural sources1–9 such as hemes,10 chlorophyll,10 and alkaloids;11,12 as well as in bioactive synthetic molecules.13–19
1-Pyrrolines are cyclic imines whose reactivity allows synthetic manipulation through nucleophilic attack on the prochiral endocyclic imine. Accordingly, stereoselective transformations can occur.20 2-Pyrrolines possess an enamine moiety that allows the further functionalization of the ring system. 2-Pyrrolines are found frequently in the literature under the name “2,3-dihydropyrroles” since the monohydrogenation of pyrroles leads directly to 2,3-dihydropyrroles. In contrast, the cyclic amine and alkene functional groups of the 3-pyrrolines react separately. When this cyclic core is used as precursor, further modifications often involve the double bond, which can be easily transformed e.g. by hydrogenation, (di)halogenation, and dihydroxylation. Thus the 1-, 2- and 3-pyrrolines represent appealing intermediates to obtain pyrroles and pyrrolidines through oxidation21 and reduction,22,23 respectively. Due to the remarkable breadth of their reactivity, pyrrolines are useful intermediates in the preparation of more complex heterocycles.24–37
A variety of well-established methods of pyrrolines synthesis are available.38,39 These methods include intramolecular cyclizations of bifunctional compounds and multi-component cyclizations,39–45 1,3-dipolar cycloadditions,46–51 photo- and thermoinduced reactions,52–54 and ring expansion of aziridines,55,56 among others. However, metal-mediated syntheses have emerged as a valuable complement to these methods20,57,58 due to their high atom economy, their mild reaction conditions, and the high functional group tolerance of the transition metal-catalyzed reactions. This review discusses the latest advances24,38,39,59 (from 2011 to December 2018) for the synthesis of pyrrolines by transition metal-catalyzed cyclizations. Only reactions in which the pyrroline ring is formed by metal promotion are described. Transformations of the pyrroline ring in other heterocycles, and the structural manipulations of the pyrroline itself are not discussed. The review is organized into three parts, each covering the metal-mediated synthesis of the three pyrroline isomers. Each part is subdivided according to the metal involved, and concludes with a brief description of notable biological activities within the class and a synthetic sequence which involve a metal-mediated synthesis of pyrrolines as intermediate towards a more complex heterocycle.
2. Synthesis of pyrrolines
2.1. Synthesis of 1-pyrrolines
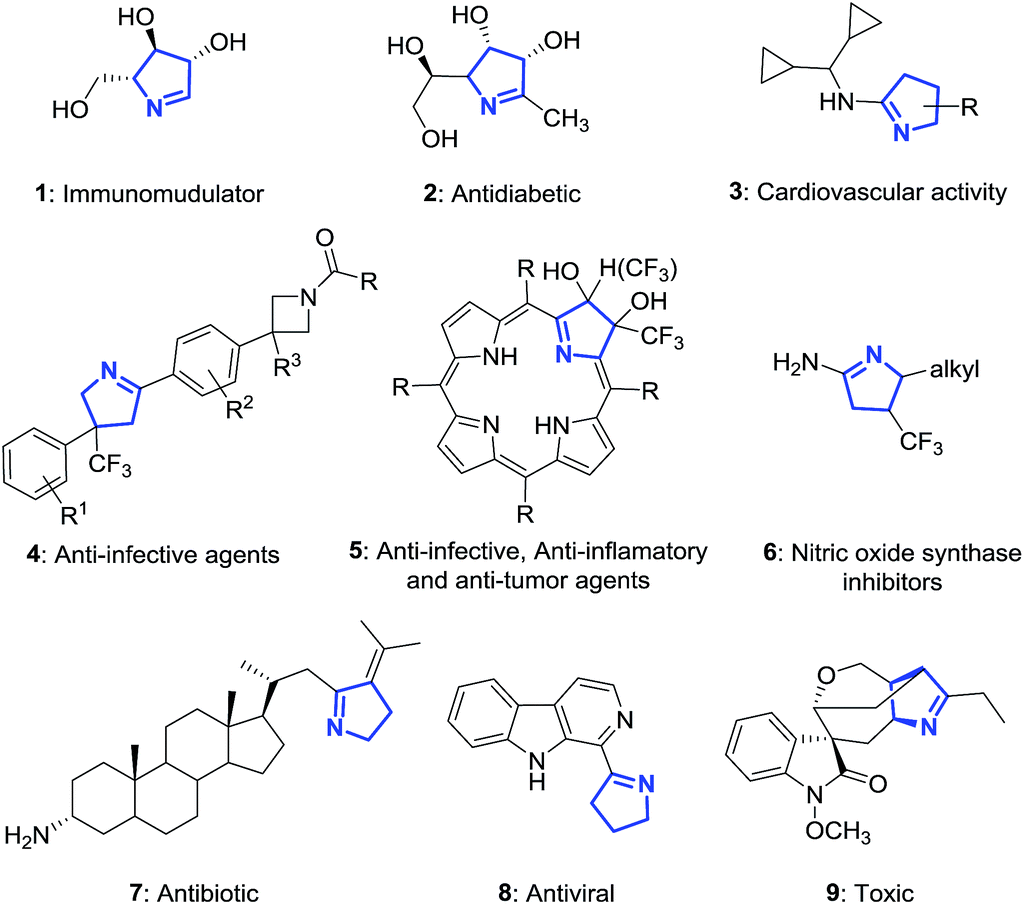
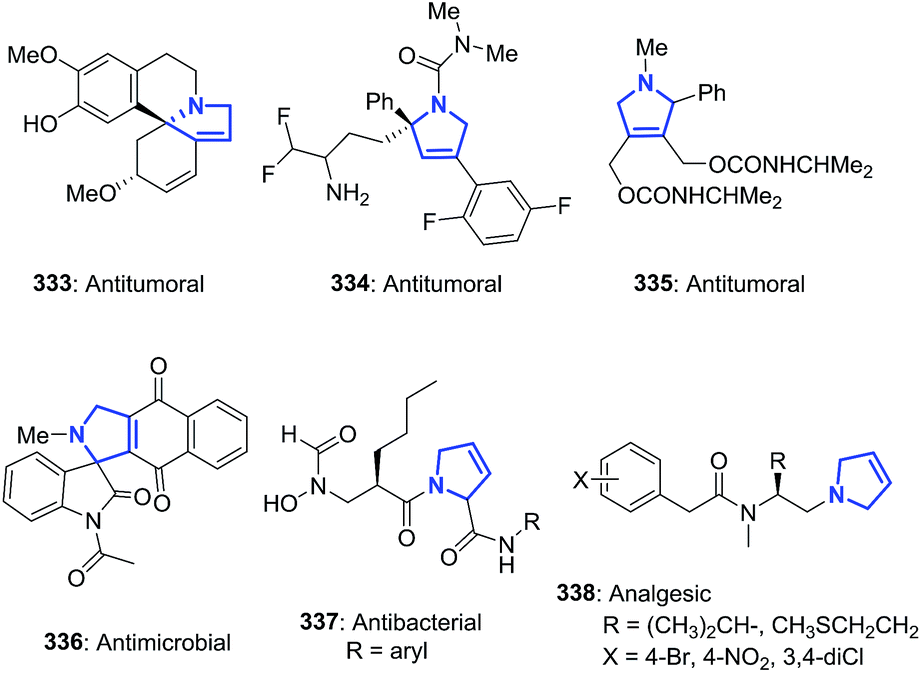
The 1-pyrroline core is exemplified by numerous compounds with biological activity (Fig. 2). Examples include the iminosugar nectrisine (1),60 discovered as an immunomodulator; the iminosaccharide 2 that has glycosidase inhibitory activity;15,61 and the 1-pyrroline 3 that has antihypertensive properties.14 β-Trifluoromethylated 1-pyrrolines (4–6)62 are nitric oxide synthase inhibitors63 and as such possess anti-infective,64–66 anti-tumor,67 and anti-inflammatory activities.65,68 The steroidal alkaloid plakinamine A (7) shows antimicrobial activity against S. aureus and C. albicans,69 whereas eudistomin (8) has antiviral activity.2 The alkaloid gelsenicine (9) is recognized for its high toxicity.70
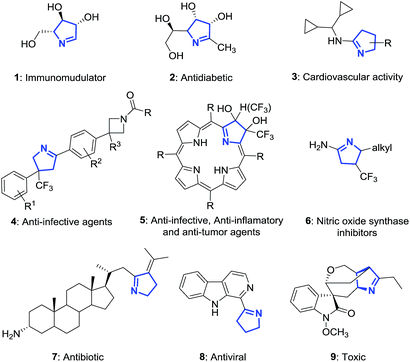 |
| | Fig. 2 Selected examples of biologically active 1-pyrrolines. | |
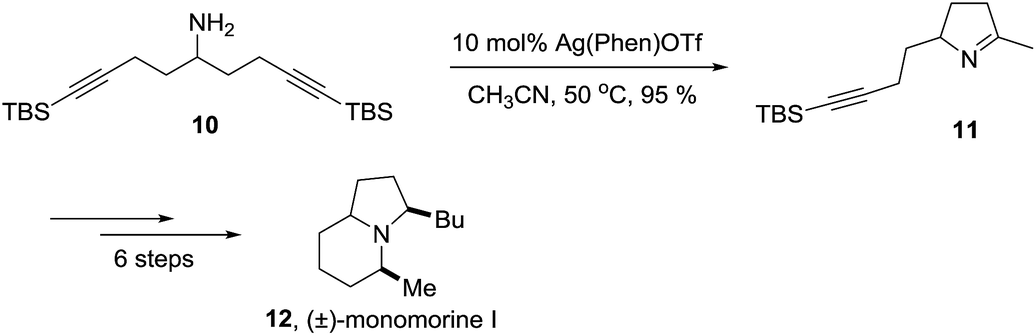
A selected example of 1-pyrroline used as an intermediate towards a more complex heterocycle is described in the Scheme 1. Helquist et al. reported a silver-catalyzed hydroamination of the aminoalkyne 10 that led to 1-pyrroline 11 which was applied to a seven step synthesis of the alkaloid (±)-monomorine I in 26% overall yield.27
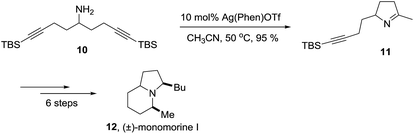 |
| | Scheme 1 Synthesis of the alkaloid (±)-monomorine I through the 1-pyrroline 11. | |
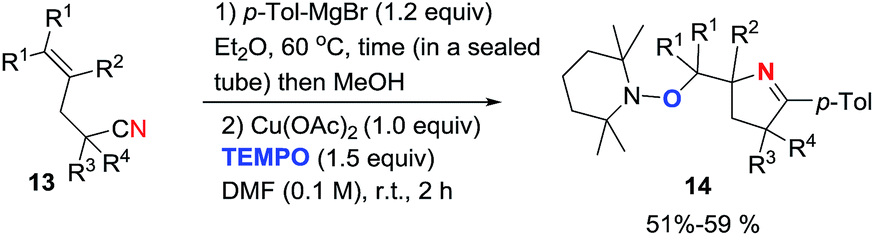
2.1.1. Synthesis of 1-pyrrolines by copper catalysis. Chiba et al.71 developed a method for the synthesis of oxymethyl-substituted pyrrolines employing Cu(OAc)2-mediated intramolecular aminooxygenation of alkenylimines with TEMPO (Scheme 2). The addition of a Grignard reagent (such as p-tolylmagnesium bromide) to a range of alkenyl carbonitriles 13 was performed in a sealed tube at 60 °C. MeOH was used to protonate the products, and DMF was added to reach a concentration of 0.1 M. Immediately, 1 equivalent of Cu(OAc)2 and 1.5 equivalents of TEMPO were added. The aminooxygenation proceeded smoothly at room temperature affording (after 2 h) diverse oxymethyl pyrrolines 14 in moderate yields (typically 50%). Various other Grignard reagents were equally successful.
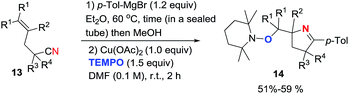 |
| | Scheme 2 Cu(II)-mediated intramolecular aminooxygenation of alkenylimines towards 1-pyrrolines. | |
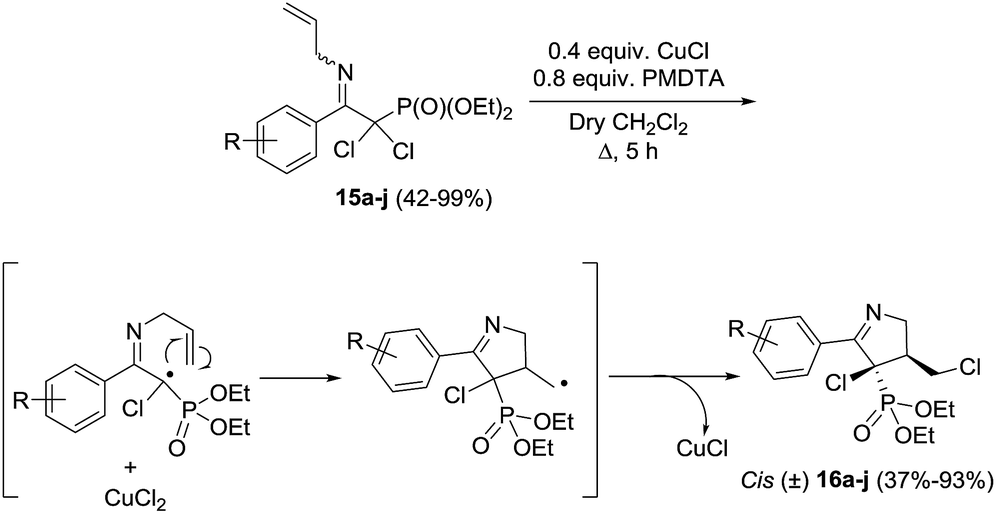
Stevens et al.72 synthesized a library of ten 1-pyrrolines 16a–j from the α,α-dichlorinated imines 15a–j using a heteroatom transfer radical cyclization (HATRC) (Scheme 3). The free-radical ring closure reaction was performed with CuCl in presence of N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDTA) as a ligand. Other ligands such as N,N,N′,N′-tetramethylethylenediamine (TMEDA) proved equally efficient. The addition of the ligands modifies the solubility and the redox potential of the copper catalyst, thus improving its activity. Formation of the five-membered ring proceeds through a radical 5-exo-trig cyclization. Two stereogenic centers are generated during the ring closure. The reaction displays an excellent cis/trans diastereoselectivity (diastereoisomeric ratios are all in the range of 90/10). The authors attribute this diastereoselectivity to the steric hindrance caused by the ethoxy substituents of the phosphonate.
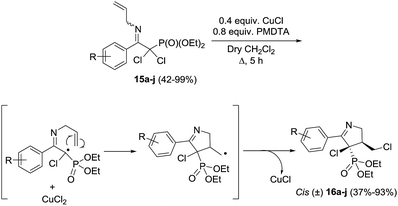 |
| | Scheme 3 Copper-catalyzed heteroatom transfer radical cyclization (HATRC) towards 1-pyrrolines. | |
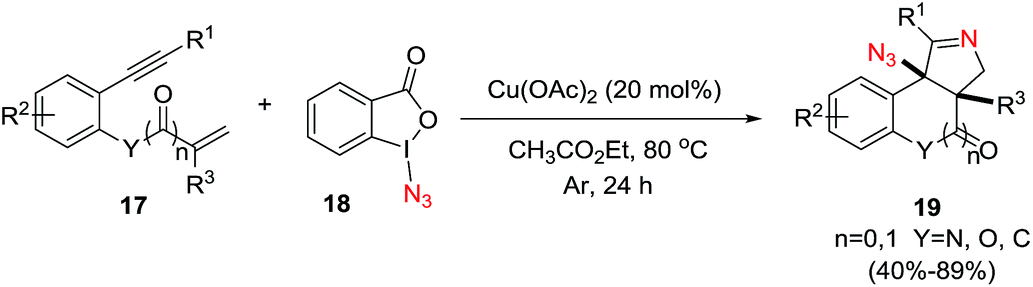
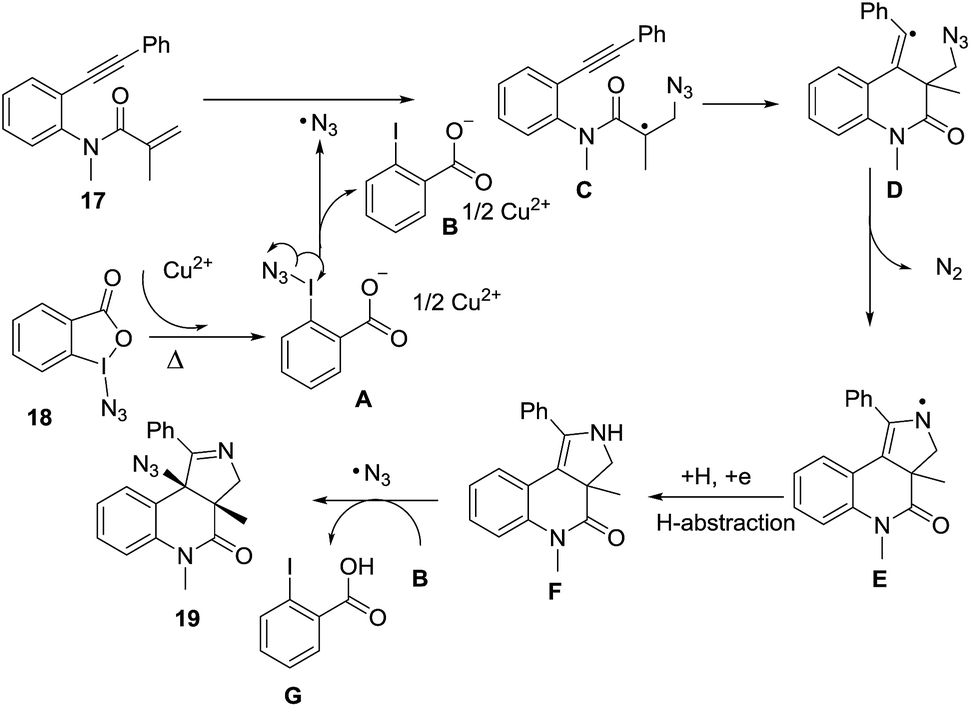
Li et al.73 developed a novel selective copper-catalyzed, azide radical-mediated, [2 + 2 + 1] annulation of benzene-linked 1,n-enynes (n = 6, 7) to give fused pyrrolines 19 (Scheme 4). Azidobenziodoxolone 18 was the source of the azide radical. Other azide reagents such as TMSN3 and NaN3 failed to produce the fused pyrrolines. This one-step synthesis of fused pyrrolines proceeds via the generation of the azide radical from azido-benziodoxolone 18 with the aid of the Cu2+ species as catalyst (Scheme 5). Addition of the azide radical to the alkene moiety of enyne 17 affords an alkyl radical intermediate which undergoes intramolecular addition to the alkyne moiety to give fused pyrroline F as an intermediate. Finally, a second azide radical is incorporated to furnish the final fused pyrroline structures. Internal alkenes with R1 ≠ aliphatic are suitable substrates.
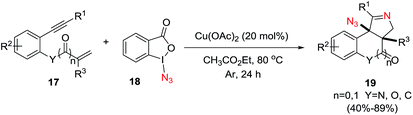 |
| | Scheme 4 [2 + 2 + 1] Annulation/azidation of 1,n-enynes as an entry to fused 1-pyrrolines. | |
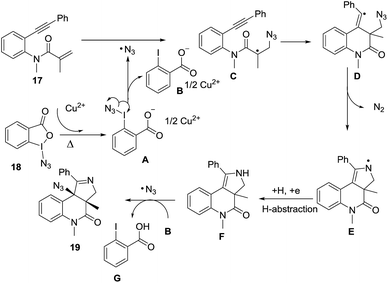 |
| | Scheme 5 Proposed mechanism of [2 + 2 + 1] annulation/azidation of 1,n-enynes. | |
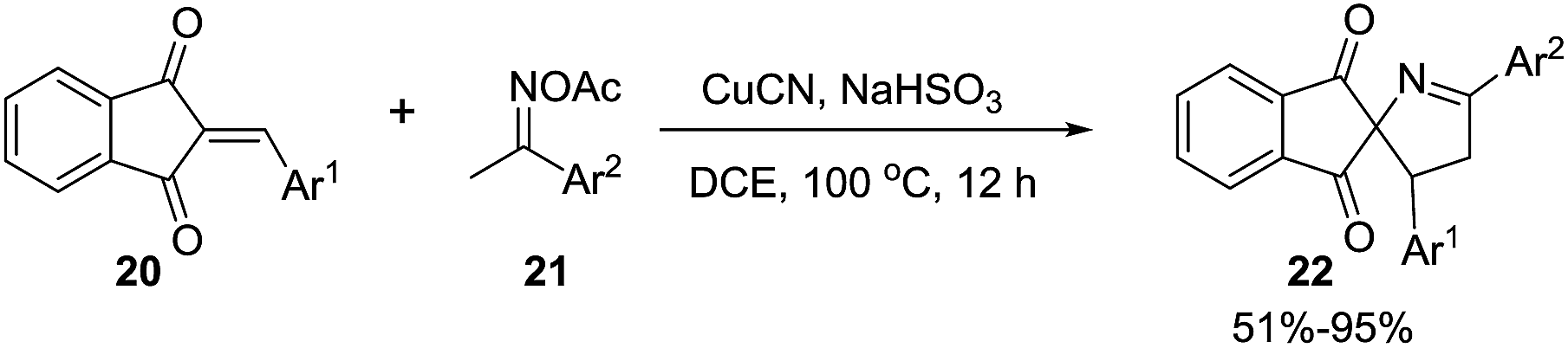
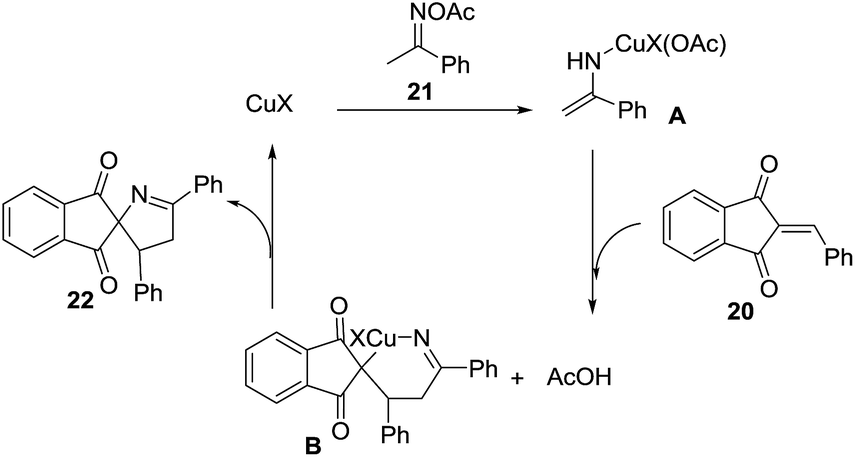
Li et al.74 developed an efficient copper-catalyzed heteroannulation reaction between 2-arylideneindane-1,3-diones (20) and ketoxime acetates (21) for the straightforward synthesis of spiro[indane-1,3-dione-1-pyrrolines] 22 (Scheme 6). The methodology shows broad substrate scope and tolerates a wide range of functionalities in both the 2-arylideneindane-1,3-diones and aromatic ketoxime acetates. Alkyl ketoxime acetates failed to deliver the spiro compounds.
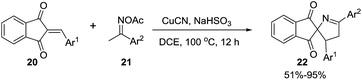 |
| | Scheme 6 Copper-catalyzed heteroannulation reaction between aryl ketone-derived ketoxime acetates and 2-arylideneindane-1,3-dione. | |
The proposed mechanism for this transformation begins with the cleavage of the N–O bond in the ketoxime acetate 21 by cuprous cyanide, through an oxidative addition (Scheme 7). As a result, the copper enamide A intermediate is formed, which reacts with the 2-arylideneindane-1,3-diones 20 to give intermediate B. The product 22 is formed, and the catalyst regenerated, by an intramolecular redox heteroannulation of B.
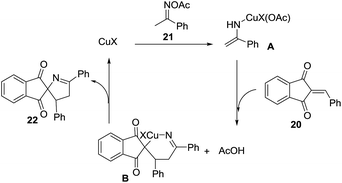 |
| | Scheme 7 Proposed mechanism for the copper-catalyzed heteroannulation reaction between ketoxime acetates and 2-arylideneindane-1,3-dione. | |
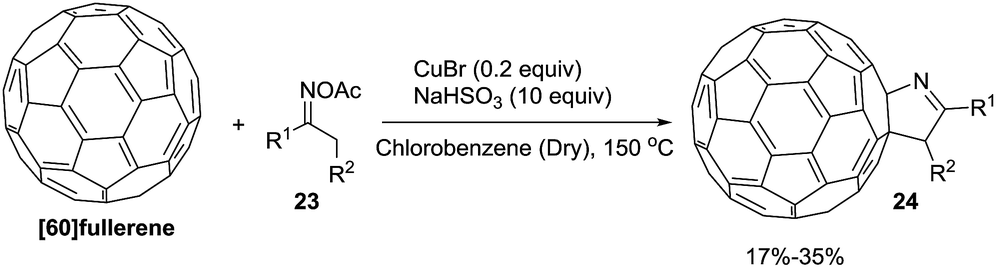
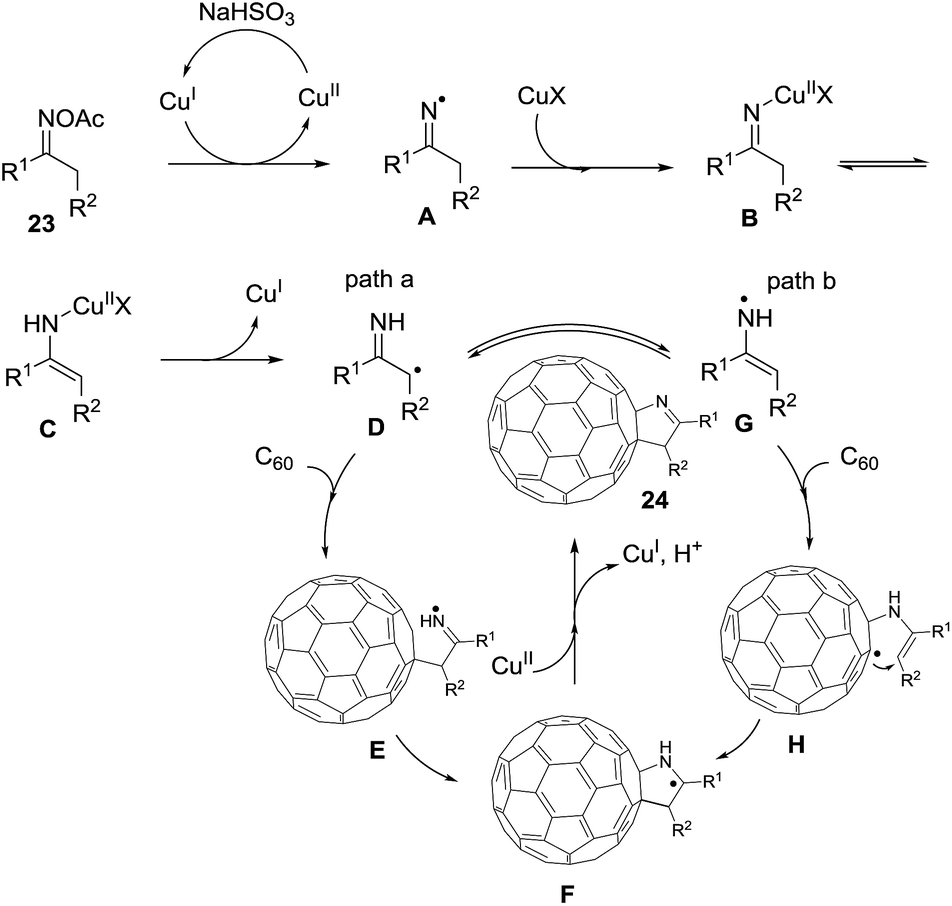
Wang et al.75 constructed novel 1-fulleropyrrolines 24 using a cuprous bromide-catalyzed heteroannulation reaction of [60]fullerene with ketoxime acetates 23 (Scheme 8). The proposed mechanism begins with ketoxime N–O cleavage by Cu(I) to give an imino radical with formation of the C–C and C–N bonds (Scheme 9).
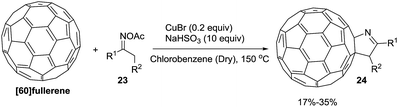 |
| | Scheme 8 Copper(I)-catalyzed heteroannulation of [60]fullerene with ketoxime acetates. | |
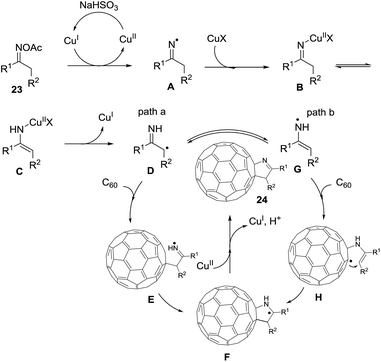 |
| | Scheme 9 Proposed mechanism of the heteroannulation of [60]fullerene with ketoxime acetates. | |
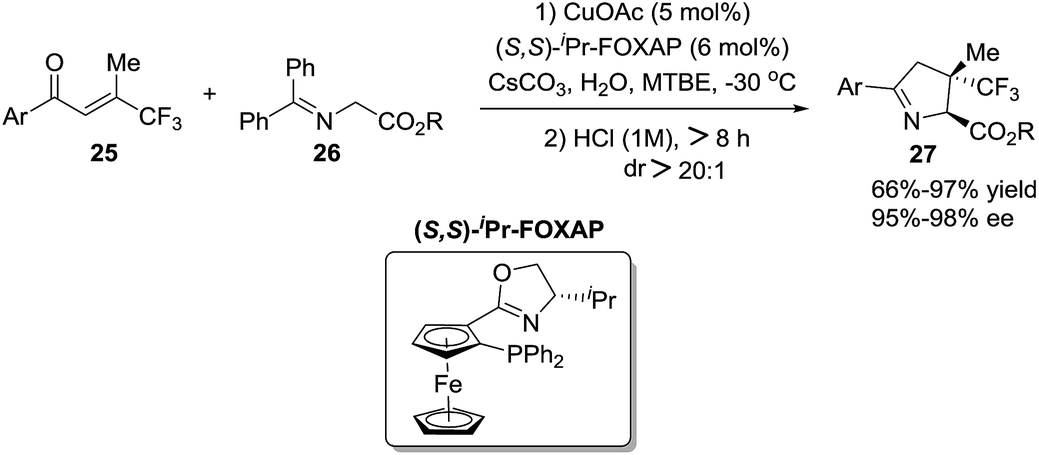
Zhang et al.76 reported a straightforward one-pot synthesis of 1-pyrrolines bearing two contiguous stereocenters exemplified by structure 27, where one is a trifluoromethyl-substituted quaternary carbon (Scheme 10). The copper-catalytic process relies on an asymmetric Michael addition of ketiminoesters 26 to β-trifluoromethyl β,β-disubstituted enones 25 and subsequent hydrolytic cyclization. The optimized asymmetric conditions employed CuOAc as the catalyst and (S,S)-iPr-FOXAP as a chiral ligand in the presence of water (6 equiv.) to achieve this highly chemo-, diastereo-, and enantioselective reaction.
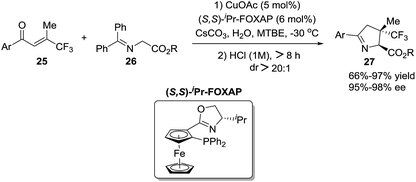 |
| | Scheme 10 Cu(I)-catalyzed Michael addition of ketiminoesters to β-trifluoromethyl β,β-disubstituted enones. | |
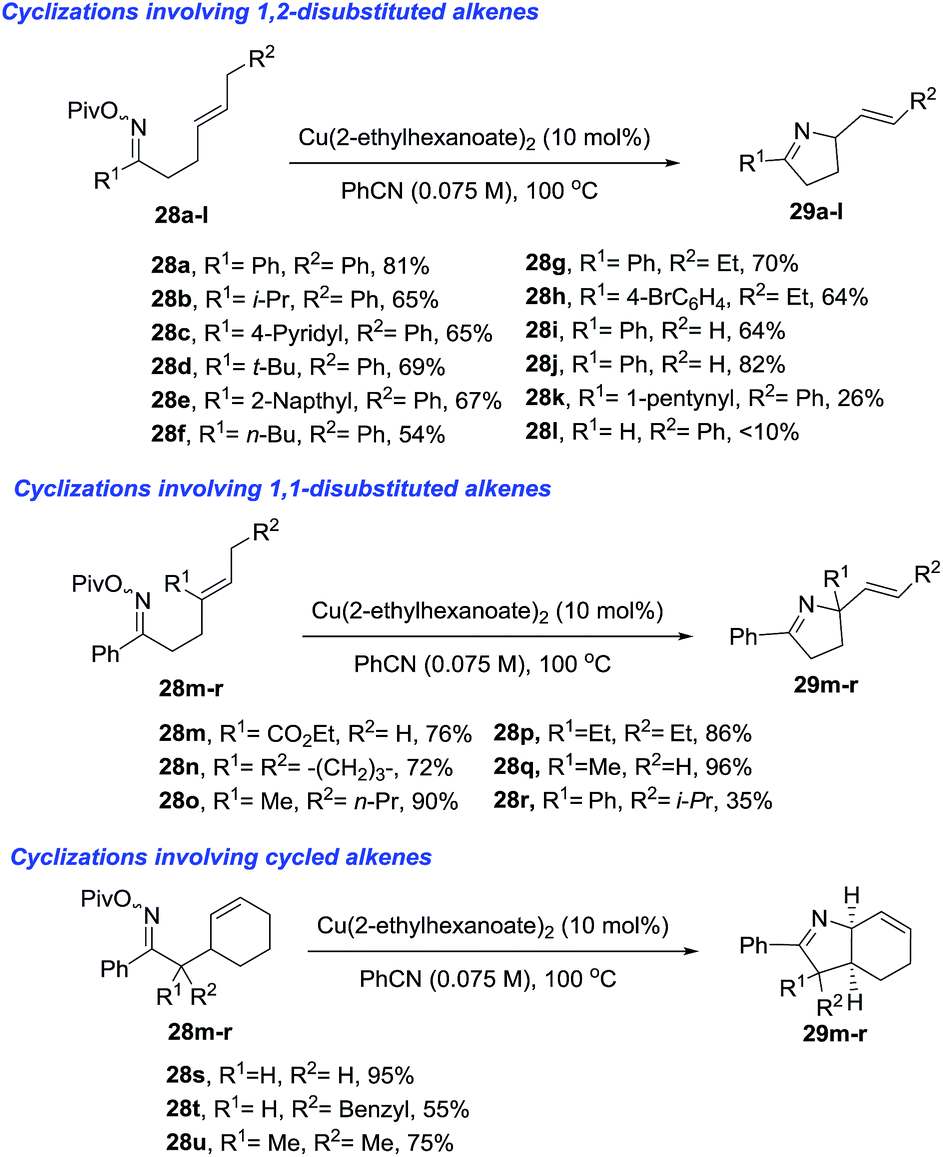
Bower et al.77 developed a copper(I)-catalyzed Heck-like cyclization of oxime esters as an effective alternative to Pd-based protocols (Scheme 11). One advantage of this methodology is that it works with less activated oxime esters such as pivaloyl oxime (28) as the starting material, instead of the more expensive O-pentafluorobenzoyl oximes that are required for the Pd protocol. The range of substrates which delivers 1-pyrrolines (29) includes pivaloyl oxime esters 28a–l that possess pendant 1,2-disubstituted alkenes, the more heavily substituted 1,1-disubstituted alkenes 28m–r, and the cyclohexenes 28s–u. The proposed mechanism involves an intermediate that has iminyl radical character that triggers cyclization to form a C–N bond.
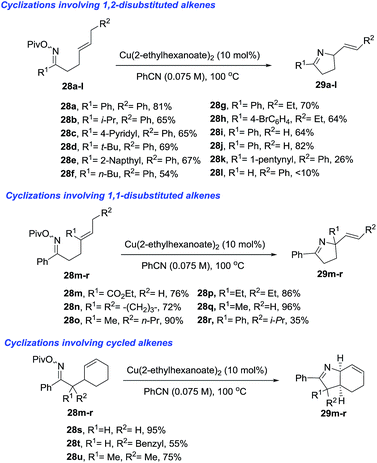 |
| | Scheme 11 Copper(I) catalyzed Heck-like cyclizations of oxime esters towards 1-pyrrolines. | |
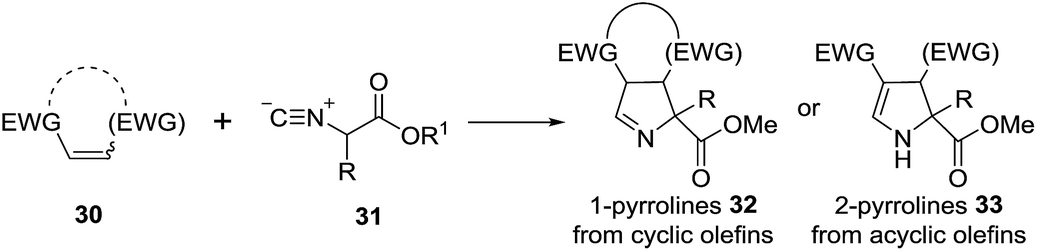
2.1.2. Synthesis of 1-pyrrolines by gold catalysis. A convergent preparation of pyrrolines consists of the formal [3 + 2] cycloaddition of isocyanoacetates 31 with electron-deficient alkenes 30 (Scheme 12). Base, or a metal complex accelerate significantly the reaction.78 With cyclic alkenes the cycloaddition gives the 1-pyrroline (32), whereas with acyclic alkenes the acyclic 2-pyrroline (33) is obtained.
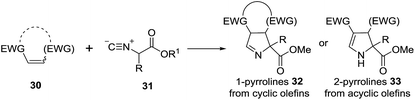 |
| | Scheme 12 [3 + 2] Cycloaddition of isocyanoacetates with electron-deficient alkenes. | |
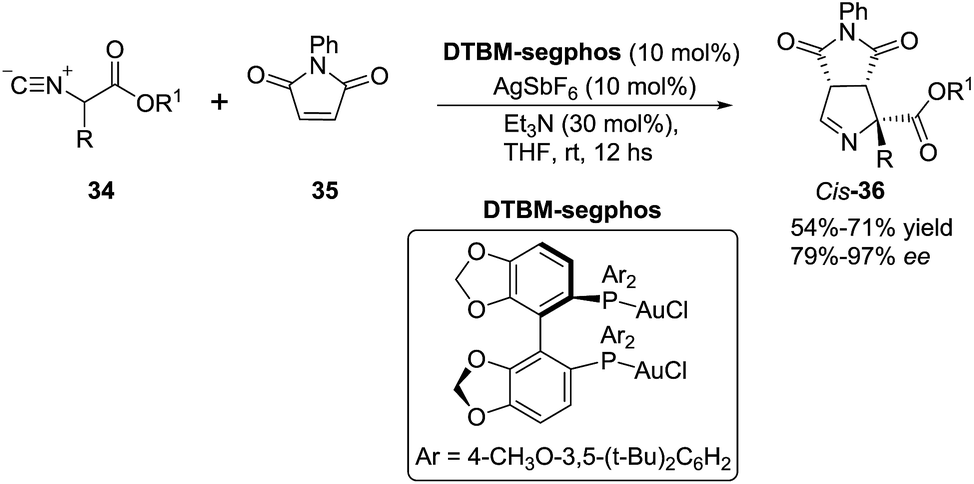
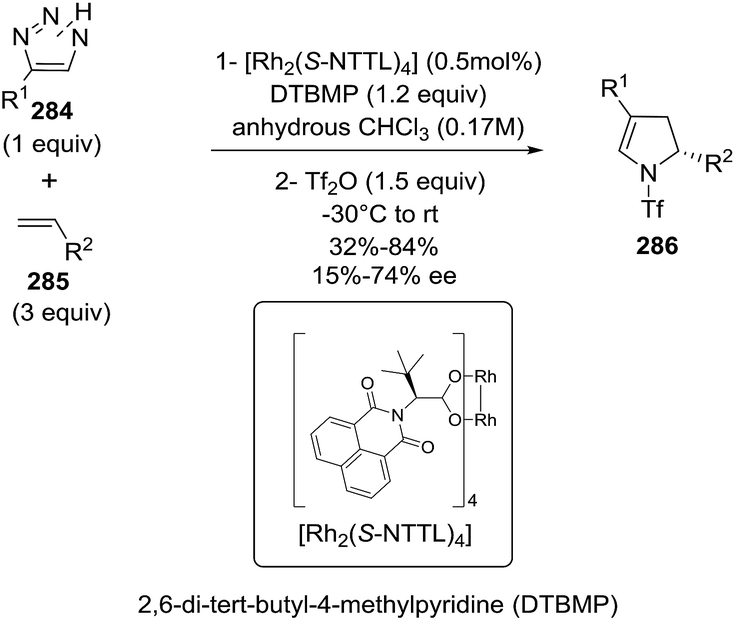
In 2012, Carretero et al. reported79 a highly diastereoselective and enantioselective synthesis of 1-pyrrolines 36 by reaction of isocyanoacetates 34 with phenylmaleimide (35) using an Au(I) catalyst with a chiral DTBM-segphos ligand. The use of substituted isocyanoacetates led to 1-pyrrolines bearing a quaternary stereocenter at C-5 (Scheme 13). The reaction of α-aryl-substituted and α-alkyl-substituted isocyanoacetates gave the 1-pyrroline adduct as a single diastereomer and with high enantiocontrol. This methodology afforded 1-pyrrolines containing a quaternary stereocenter with complete cis diastereoselectivity (both carbonyl substituent oriented in the same direction of the pyrroline ring) and high enantioselectivity (up to 97% ee).
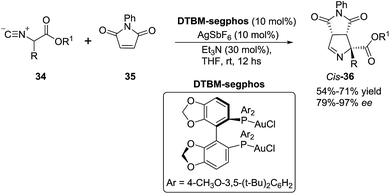 |
| | Scheme 13 Au-catalyzed asymmetric formal [3 + 2] cycloaddition of isocyanoacetates with maleimides. | |
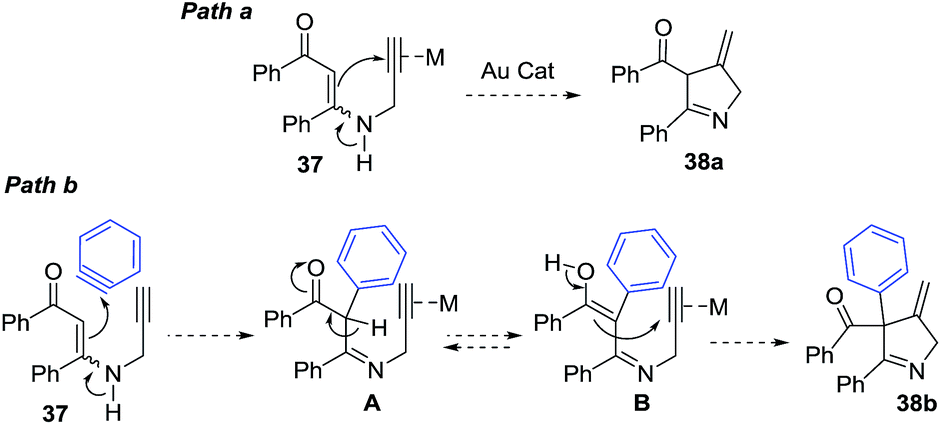
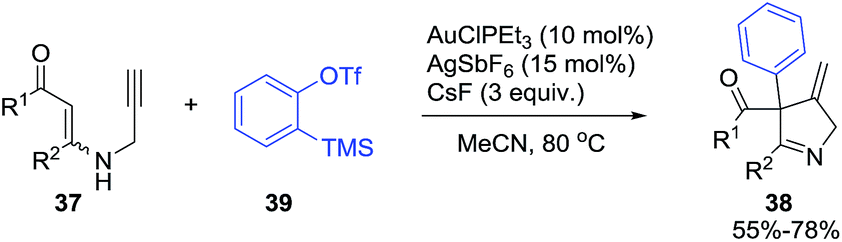
Karunakar et al.80 envisaged a route to 3-methylene-1-pyrrolines from N-propargylic β-enaminones and arynes using gold catalysis. However, only recovered starting material was obtained from the attempted reaction of enaminone 37 and various gold catalysts (Scheme 14, path a). The failure of the reaction was attributed to the poor nucleophilicity of the enaminone or the weak electrophilic character of the propargylic functionality. Then, they had the innovative idea to use an external aryne to enable cyclization (path b). Using benzyne generated in situ and AuCl3/AgSbF6 as catalysts, they observed the formation of the 3-methylene-1-pyrroline 38b (Scheme 14). After a rigorous catalyst screening, they found that the combination of AuCl·PEt3 (10 mol%) and AgSbF6 (15 mol%) in presence of the aryne precursor 39 in CH3CN at 80 °C gave the best yields (Scheme 15). Evaluation of the scope of the reaction using different N-propargylic β-enaminones demonstrated that enaminone with electron-donating groups increased the yield of the cyclisation products.
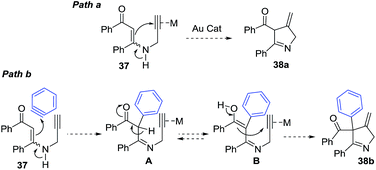 |
| | Scheme 14 Strategies for the conversion of N-propargylic β-enaminone 37 to 1-pyrroline 38b via gold catalysis. | |
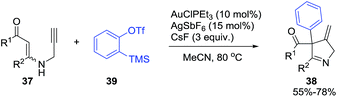 |
| | Scheme 15 Gold-catalyzed cyclization of N-propargylic β-enaminones 37 towards 1-pyrrolines 38b in presence of aryne precursor 39. | |
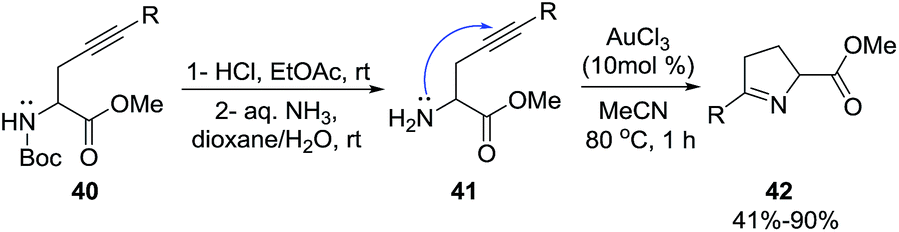
Our group81 developed a versatile strategy to obtain a variety of disubstituted 1-pyrrolines 42 through a gold-catalyzed N-cycloisomerization from alkyne-containing amino acids 40 (Scheme 16). Of the gold catalysts examined, the most effective for this C–N functionalization was AuCl3.
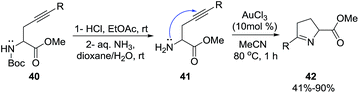 |
| | Scheme 16 Gold-catalyzed C–N functionalization of alkynyl amino acids derivatives. | |
Although this transformation can be performed with other transition metals and with Brønsted acids, AuCl3 is the superior catalyst as it provides a broader scope and better yields. This cycloisomerization of aryl-substituted alkynyl-containing amino acids proceeds exclusively via Au(III)-catalyzed 5-endo-dig N-cyclization. Aryl-substituted alkynyl amino acids bearing electron withdrawing groups provide excellent yields (typically 90%) of the C–N functionalized product whilst derivatives carrying electron-donating groups furnish lower yields (typically 55%). Terminal alkynes fail to afford the pyrroline, they are unreactive under these conditions.
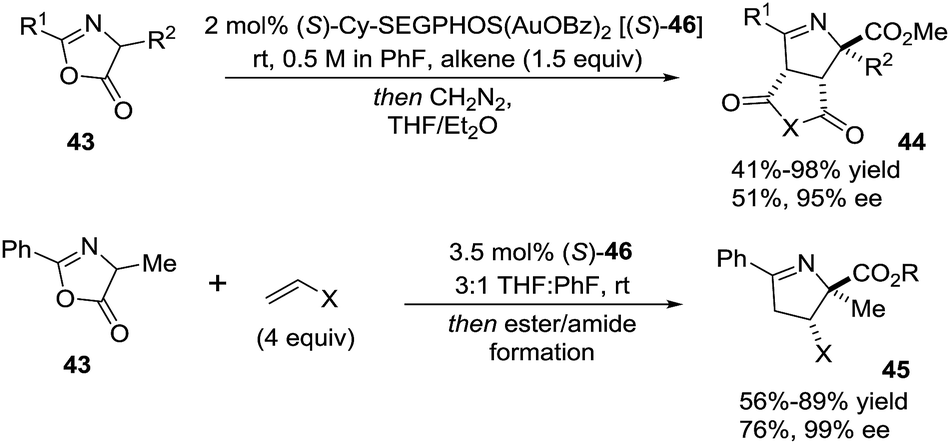
Toste et al.82 evaluated the gold-catalyzed reactions between azlactones 43 and electron-deficient alkenes (such as maleimide and maleic anhydrides, or monosubstituted alkenes) to give stereoselectively the products (44 and 45, respectively) of 1,3-dipolar cycloaddition (Scheme 17). The use of C2-symmetric bis(phosphinegold(I) carboxylate) complexes (S-46) provided good to excellent diastereo- and enantioselectivity to afford 1-pyrrolines 44 and 45. The authors proposed a mechanism in which the gold complexes activate pro-nucleophiles to catalyze the cycloaddition, rather than the more typical mechanism of activation of the carbon–carbon π-bond toward nucleophilic addition.
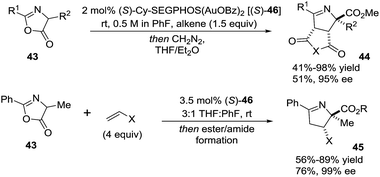 |
| | Scheme 17 Enantioselective gold(I)-catalyzed reactions of azlactones. | |
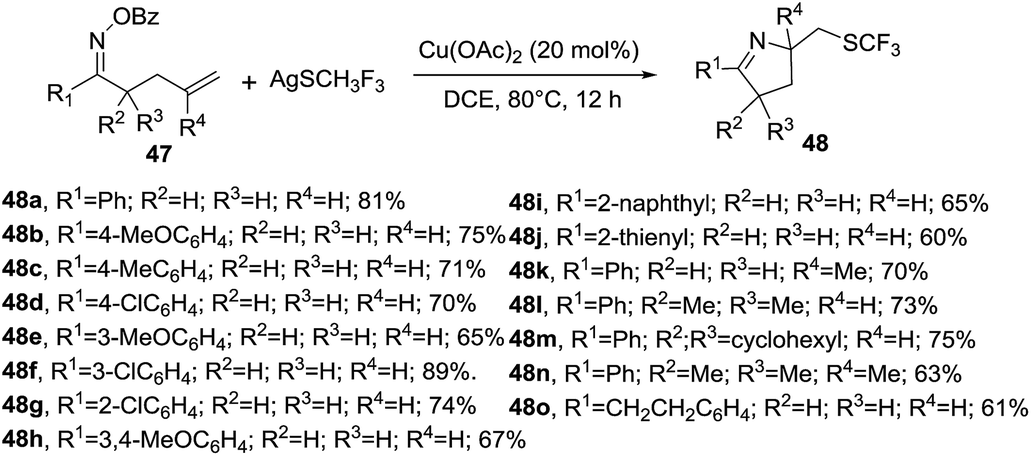
Zhu et al. reported a copper-catalyzed domino cyclization/trifluoromethylthiolation of monosubstituted or 1,1 disubstituted olefins 47 leading to SCF3-substituted 1-pyrrolines 48 in 60–90% yields (Scheme 18).83 AgSCF3 is the reagent of choice as SCF3 source. Benzoyloxy group and Cu(OAc)2 proved to be the best oxime leaving group and catalyst for this transformation. This reaction involves the N–O cleavage of benzoyl oximes and subsequent alkene difunctionalization and tolerates electron-donating and electron-withdrawing groups on the aryl-substituted O-acyl oximes (R1 = Ar, 48a–h) as well as alkyl-substituted O-acyl oxime. R1 = naphthyl and thienyl delivered the SCF3-containing 1-pyrrolines in good yields. The reaction is also compatible with alkyl moieties as the substituents R2 and R3.
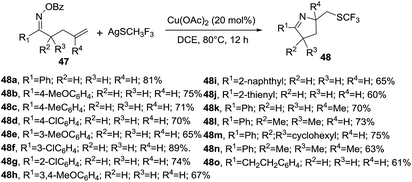 |
| | Scheme 18 Copper-catalyzed synthesis of SCF3-containing 1-pyrrolines 48. | |
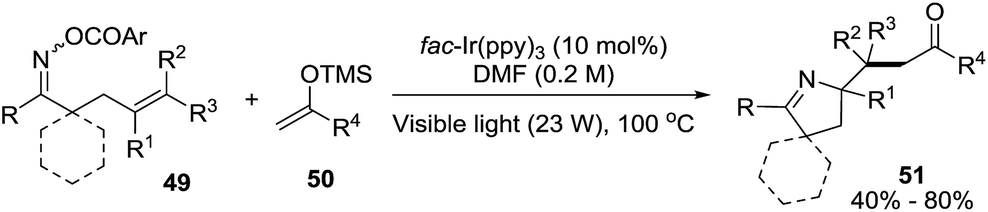
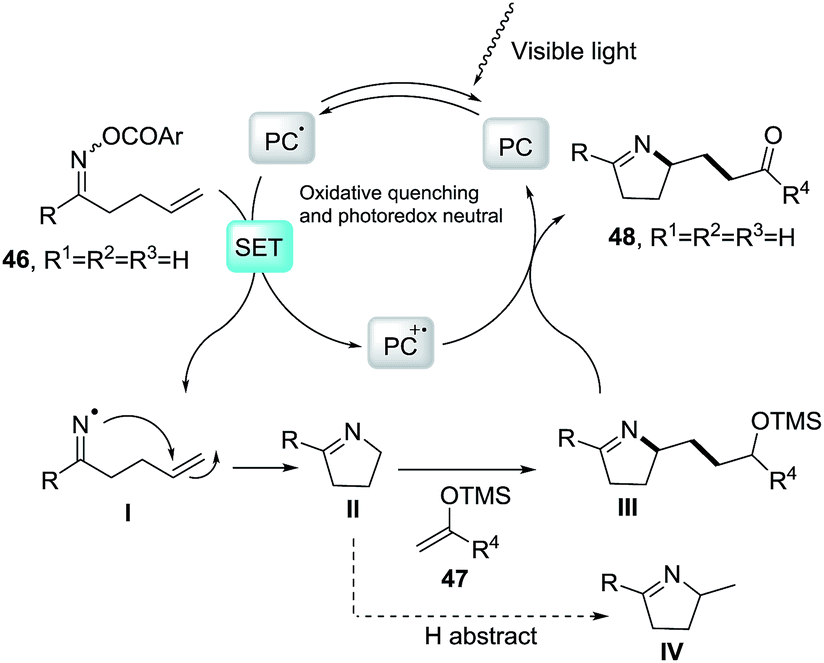
2.1.3. Synthesis of 1-pyrrolines by iridium catalysis. Loh et al.84 developed a strategy based on the iminyl-radical formation for the creation of the C–N bond of the pyrroline (Scheme 19). The iminyl-radical is generated from the O-acyl oxime derivatives 49 in presence of a photocatalyst such as fac-[Ir-(ppy)3] and visible light. The cascade reaction takes place through an intramolecular 5-exo cyclization followed by intermolecular carbon radical trapping with silyl enol ether derivatives 50 (Scheme 20). The procedure allows easy access to densely functionalized pyrroline derivatives 51. The use of silyl enol ethers as coupling partners regenerate the photocatalyst without necessity of an external reductant, while introducing the synthetically useful ketone functionalities. The procedure shows tolerance to a broad range of functionalities and enables diverse substitution patterns with respect to both the starting oxime derivative and the silyl enol ether. Different densely functionalized pyrrolines are obtained in moderate to good yields (typically 40–80%).
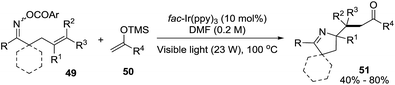 |
| | Scheme 19 Visible-light-promoted carboimination of unactivated alkenes towards 1-pyrrolines. | |
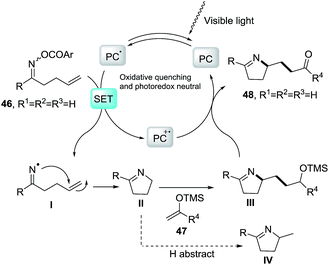 |
| | Scheme 20 Proposed reaction mechanism. | |
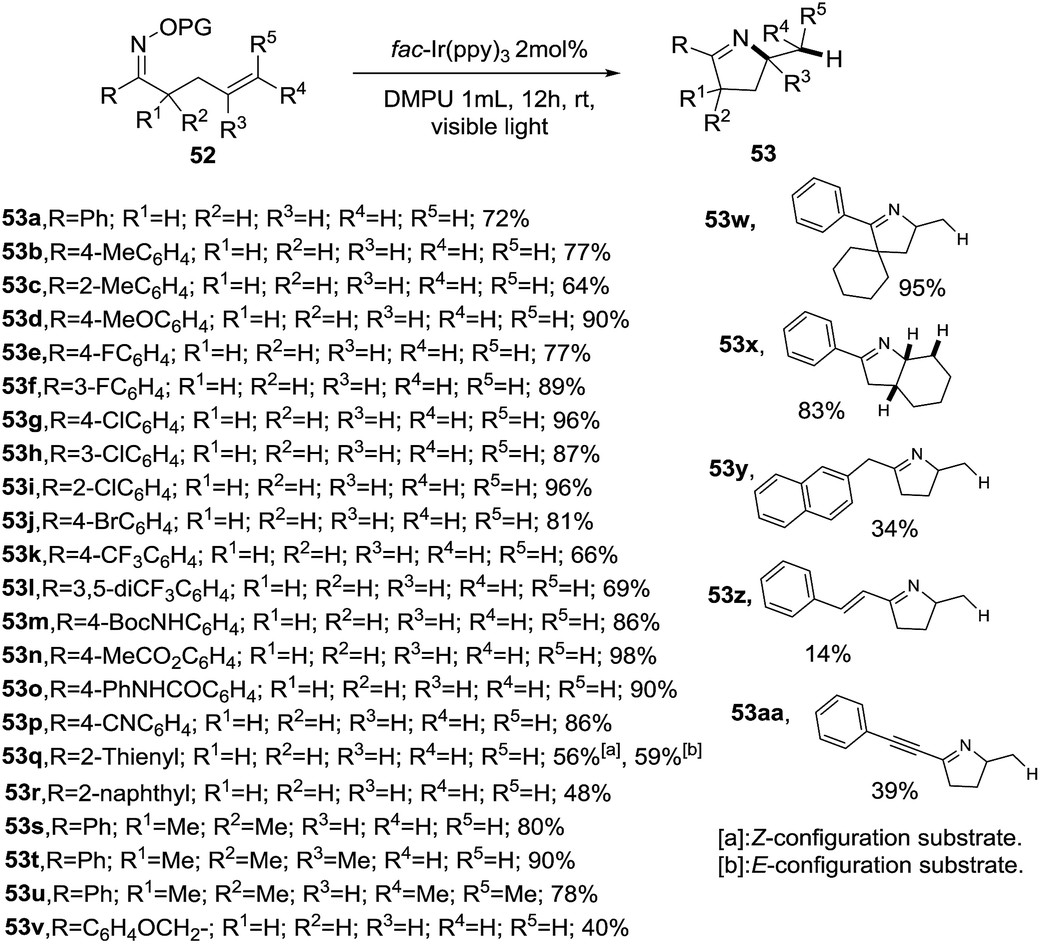
Then, the same group reported a modification of previous protocol adding N,N′-dimethylpropylene urea (DMPU) as solvent, reductant and H donor.85 As result, a visible-light-promoted hydroimination of unactivated alkenes 52 catalyzed by iridium towards the synthesis of 1-pyrrolines 53 was accomplished (Scheme 21). As in the previous report, the procedure shows a broad scope of functionalities and tolerates diverse substitution patterns of the aryl O-acyl oximes to afford 1-pyrrolines XX in good to excellent yields (64–98%). However, a diminished yield was observed when alkyl, naphthyl, alkenyl and alkynyl derived O-acyl oximes (R) were employed as substrates (14–48%).
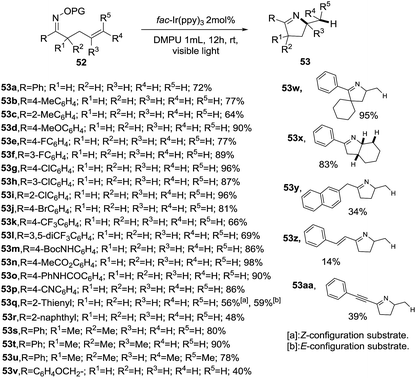 |
| | Scheme 21 Intramolecular cycloaddition of 52 by iridium catalyst. | |
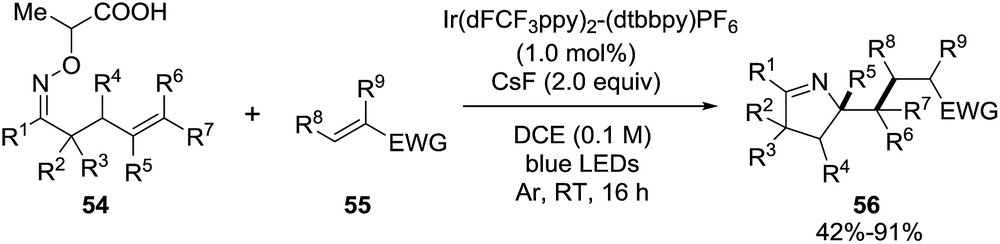
Studer et al. applied visible light to promote the generation of iminyl-radicals by a photoredox decarboxylation of α-imino-oxy propionic acids 54 towards the synthesis of 1-pyrrolines 56 (Scheme 22).86 The reaction mixture between α-imino-oxy propionic acids 54 and olefins with electron withdrawing groups 55 in the presence of 1 mol% of Ir(dFCF3ppy)2-(dtbbpy)PF6 – a photoredox catalyst – and K3PO4 in 1,2-dichloroethane is irradiated with blue LED light to produce 1-pyrrolines 56 in good to excellent yields (typically 42–91%). The reaction takes place without any control of the diasteroselectivity. Different α,β-unsaturated esters with substituents at the α and β position, α,β-unsaturated amides, phosphonate and ketones were efficient Michael acceptors (55). Regarding the iminyl radical precursor 54, phenyl groups with electron-rich or electron-poor substituents, 2-thienyl, 2-naphthyl and alkyl groups at R1 position were well tolerated. α-Iminyl-oxy acids with substituents at R2, R3, R5 and R6 positions were good substrates.
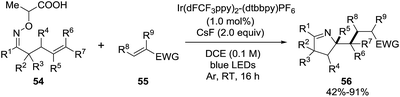 |
| | Scheme 22 Synthesis of 1-pyrrolines 56 using blue LEDs. | |
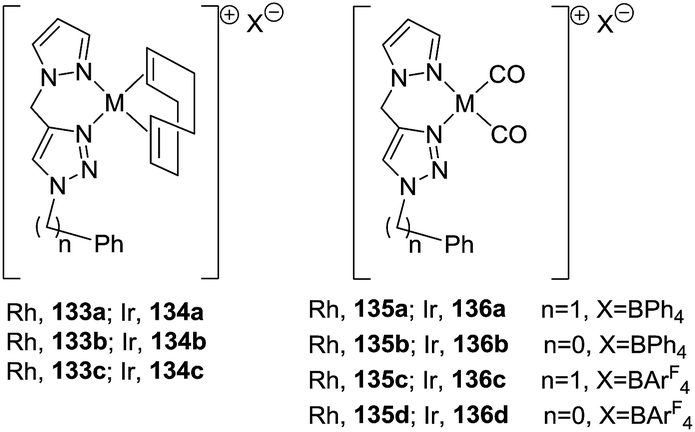
Messerle et al.87 reported the synthesis of 1-pyrrolines via an intramolecular hydroamination of a series of alkynamines catalyzed by both iridium and rhodium complexes with bidentate N–N′ donor ligands (see Fig. 3, Schemes 44 and 45 in Subsection 2.1.7.: Synthesis of 1-pyrrolines by rhodium catalysis).
 |
| | Fig. 3 Pyrazolyl–triazolyl bidentate ligands and their rhodium(I) and iridium(I) complexes. | |
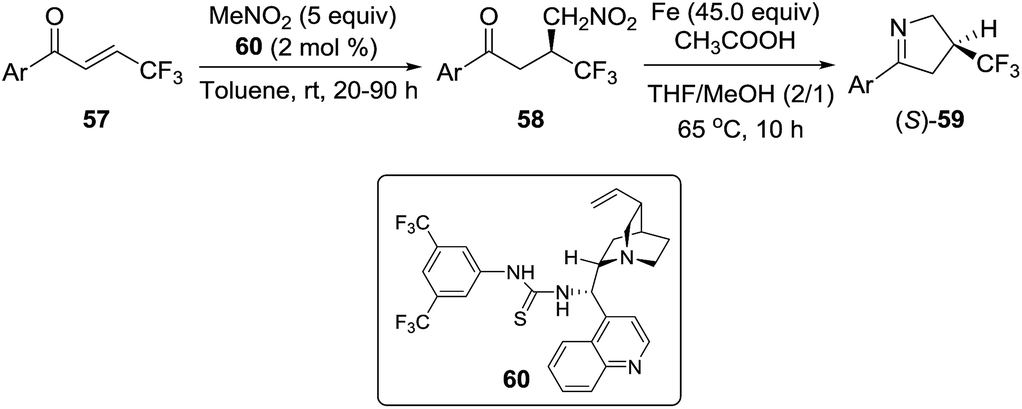
2.1.4. Synthesis of 1-pyrrolines by iron catalysis. In 2012, Shibata et al.88 described the one-pot enantioselective synthesis of β-trifluoromethyl-substituted pyrrolines 59 by an asymmetric conjugate addition of nitromethane to β-trifluoromethylenones 57 catalyzed by a cinchona alkaloid-derived/thiourea (60), followed by an iron-mediated reduction/cyclization/dehydration sequence of the intermediate 58. Excellent yields and high enantioselectivities (up to 98% ee) were obtained (Scheme 23).
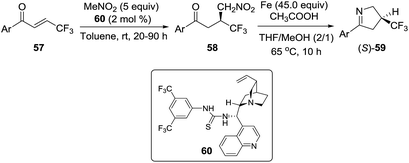 |
| | Scheme 23 One-pot enantioselective synthesis of 3-trifluoromethylated 1-pyrrolines (S)-59. | |
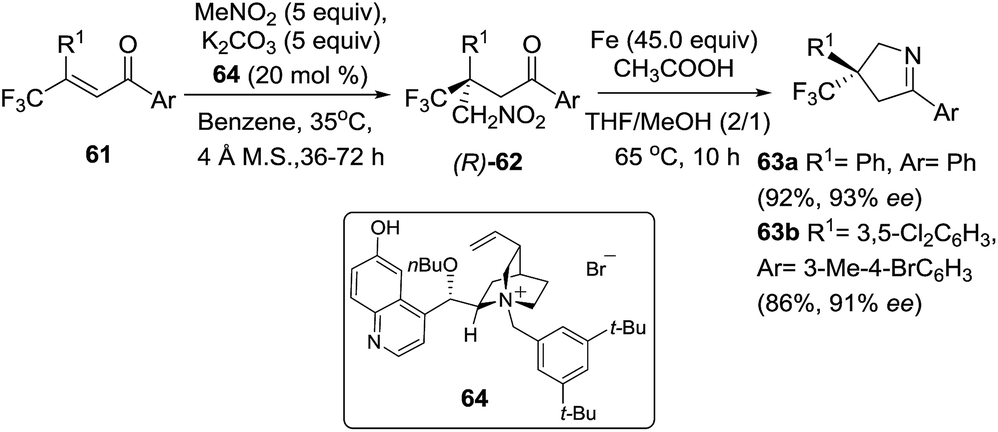
Trifluoromethylated 3,5-diaryl-pyrrolines (63) have attracted much attention because of their promising agrochemical activity as antiparasitics. Since this finding in 2005, more than 7000 variants have been described across numerous patents. In related work Shibata et al.89 disclosed the asymmetric synthesis of 3-trifluoromethyl 3,5-diaryl-pyrrolines 63 using excess Fe(0) for the cyclization step (Scheme 24). The chiral starting materials for this reaction were made by the enantioselective conjugate addition (using a phase-transfer organocatalyst 64) of nitromethane with a variety of β,β-disubstituted enones (61). A family of eighteen 1,4 adducts 62 were obtained in excellent yields and enantioselectivities. Conversion of 62 into the trifluoromethylated 3,5-diaryl-pyrrolines 63 was carried out using excess iron in the presence of acetic acid for the nitro reduction/cyclization/dehydration sequence, and without any loss of the enantiopurity. Transformation to the biologically important trifluoromethylated arylpyrrolines 63 were achieved from the nitromethane adduct 62 with high to excellent yields (typically 90%) in a single step.
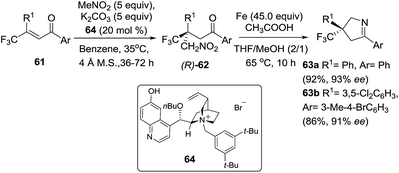 |
| | Scheme 24 Enantioselective conjugate addition of nitromethane to β,β-disubstituted enones 61, followed by an iron-mediated reduction/cyclization/dehydration sequence to 1-pyrrolines 63. | |
Yang et al.90 envisaged oxime esters as electrophilic partners for iron catalysis (Scheme 25). Reductive cleavage of the oxime N–O bond by iron generates useful iminyl radicals.91 Coupling of iminyl radical derivatives of γ,δ-unsaturated oxime (65) with silyl enol ether (66) lead to pyrroline 67 through an intramolecular C–N bond formation.
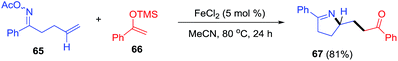 |
| | Scheme 25 Iron-catalyzed coupling of O-acyloximes with silyl enol ethers. | |
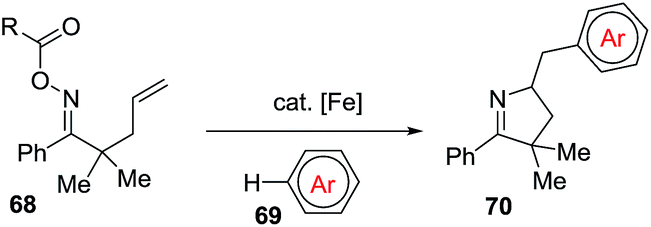
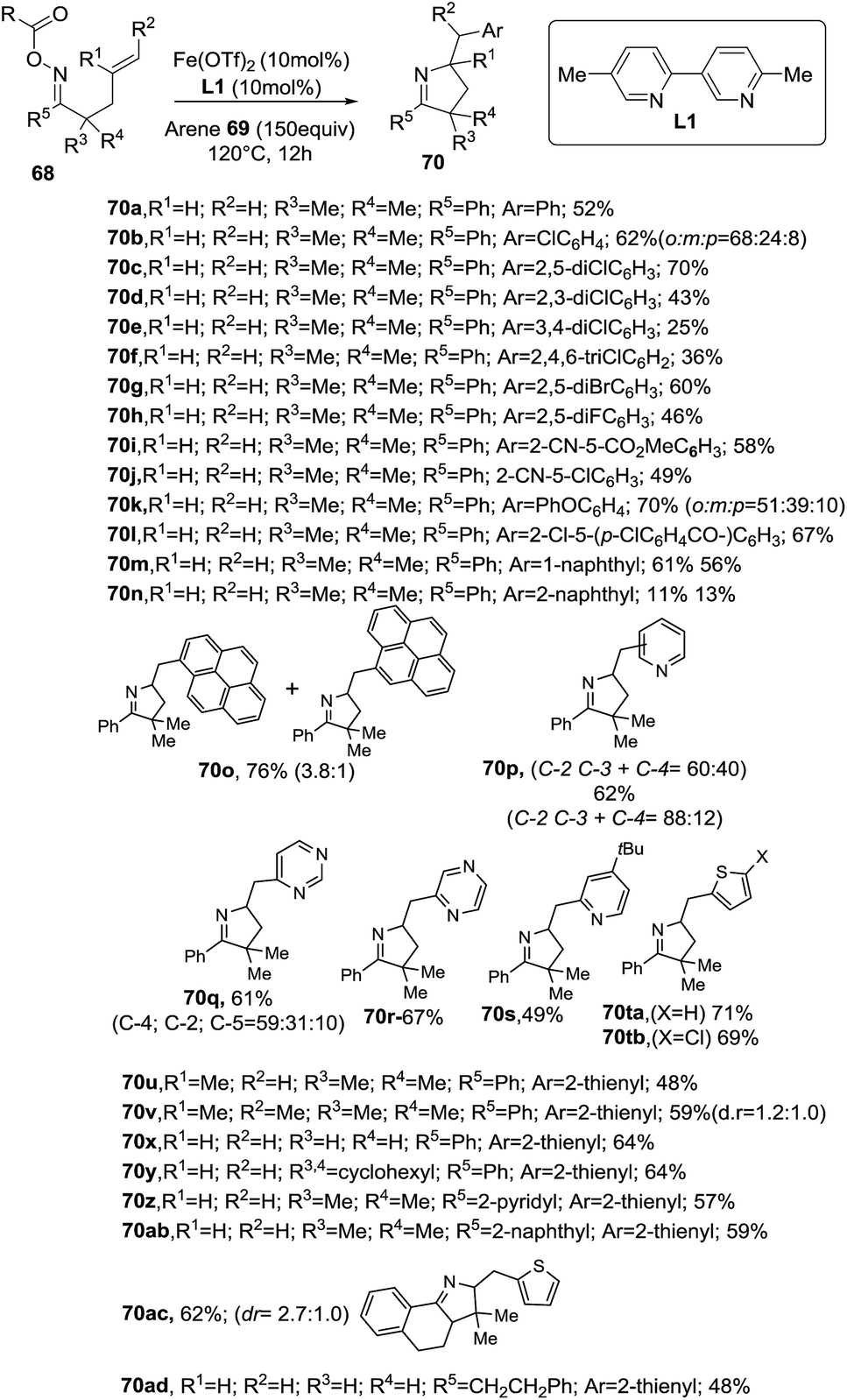
The Oche and the Okamoto group disclosed an iron-catalyzed methodology to achieve 1-pyrrolines 70 that involved the formation of iminyl radicals from alkene-tethered oxime esters 68 and subsequent aminative cyclization and intermolecular homolytic aromatic substitution (Scheme 26).92
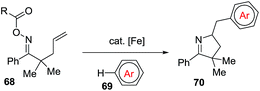 |
| | Scheme 26 Synthesis of 1-pyrrolines 70. | |
The optimized conditions use 10 mol% of Fe(OTf)2 and ligand L1 in presence of a great excess of arenes (150 equiv.). The mixture is heated a 120 °C for 12 h.
The reaction tolerates different arenes 69 with electron-rich and electron-poor substituent, polycyclic aromatic compounds as well as nitrogen- or sulfur-containing heteroarenes. Regarding the scope of alkene-tethered oxime ester 68, R = picolinoyl ester (68a-2-Py) and R = pivaloyl ester (68a-tBu) can be used with the picolinoyl ester giving better yields with substituted arenes. Oxime esters with substituted alkenes (R1 ≠ H and/or R2 ≠ H) provided 1-pyrrolines in good yields. The reaction is applicable to oxime esters 68 with R3 and R4 = H or alkyl groups and R5 = heteroaryl, naphthyl or alkyl groups (Scheme 27). This transformation involving radical intermediates affords 1-pyrrolines 70 with moderate to good yields (36–71%).
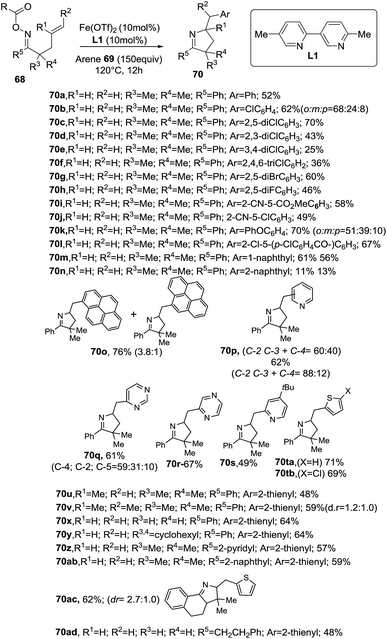 |
| | Scheme 27 Aminative cyclization/intermolecular homolytic aromatic substitution involving iminyl radicals towards 1-pyrrolines 70. | |
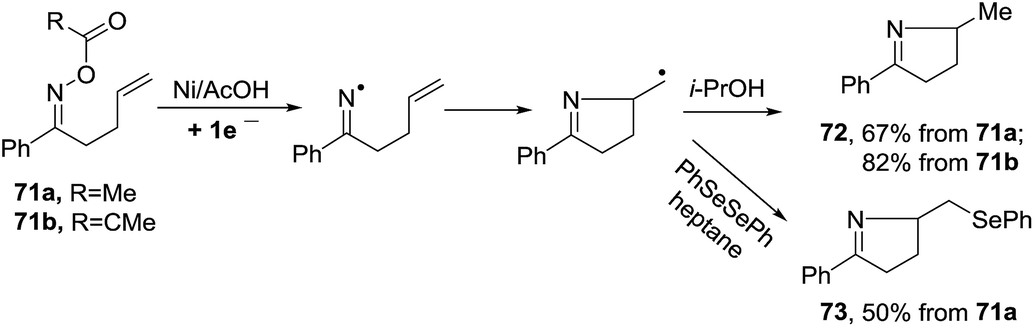
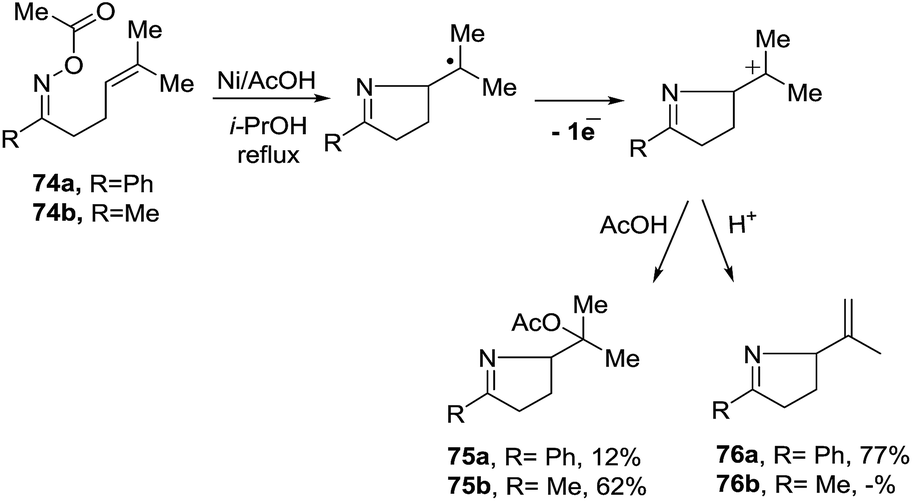
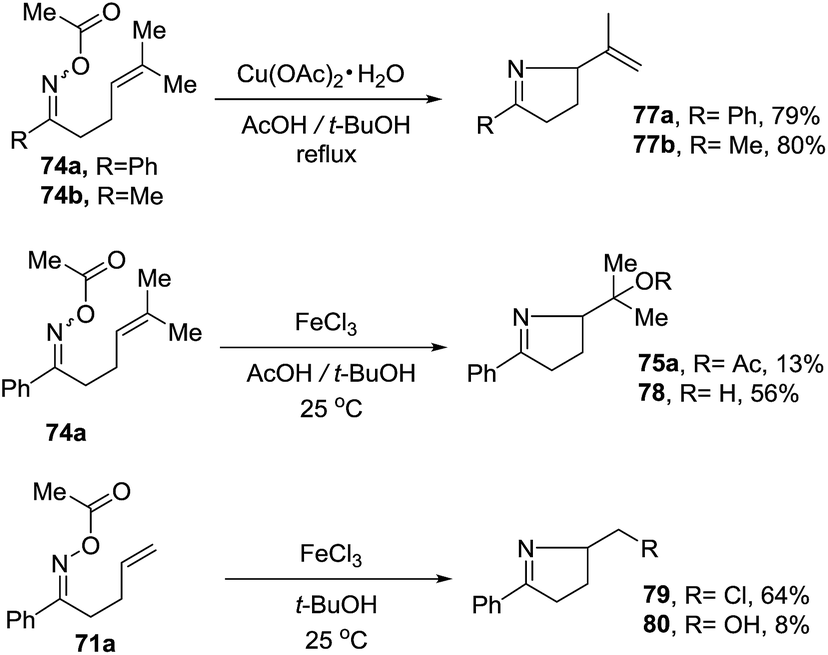
2.1.5. Synthesis of 1-pyrrolines by nickel catalysis. Zard et al.93 conceived of a method for the generation of iminyl radicals based on the reduction of oxime esters (71) with a combination of nickel powder and acetic acid (Scheme 28). The radical center on the nitrogen is intercepted intramolecularly by the well-positioned terminal alkene. Subsequent quenching by isopropanol gives pyrroline 72. The authors proved this radical mechanism with addition of diphenyldiselenide (PhSeSePh), which led to selenide 73. On the other hand, when they replaced the unsubstituted terminal alkene of 71a/b by a trisubstituted alkene of 74a/b, they obtained the isopropenyl alkene 76a and the tertiary acetate 75a in the case of 74a. When starting from 74b, they observed only formation of 75b (Scheme 29). The authors determined that this mechanism is ionic, and not radical, with the following experiment. Reaction of the oxime acetates 74a and 74b containing an isopropylidene group with Cu(OAc)2/AcOH in refluxing t-butanol gave the 1-pyrrolines 77a and 77b with an isopropenyl side-chain with very good yields (Scheme 30). Evaluation of other metallic salts for this reaction showed that from 74a FeCl3/AcOH in t-butanol at room temperature gave the acetate 75a and the alcohol 78. When they treated oxime 71a with FeCl3 in t-butanol in the absence of acetic acid (also at room temperature) they obtained the chloride 79 (64% yield) and a small amount of the alcohol 80 (8% yield) as the products.
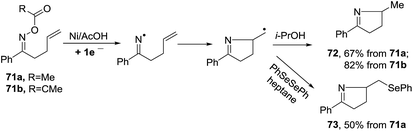 |
| | Scheme 28 Dissolving nickel metal mediated generation of iminyls from oxime esters. | |
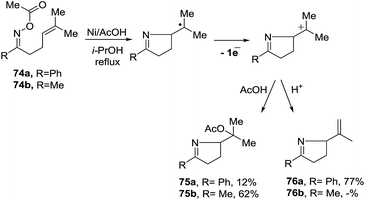 |
| | Scheme 29 Unexpected transformations of an unsaturated oxime acetate. | |
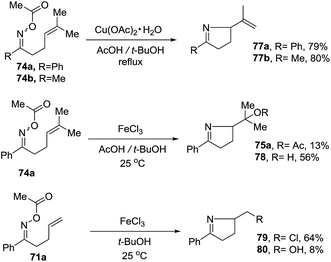 |
| | Scheme 30 Cupric and ferric ion-mediated cyclizations of unsaturated oxime acetates. | |
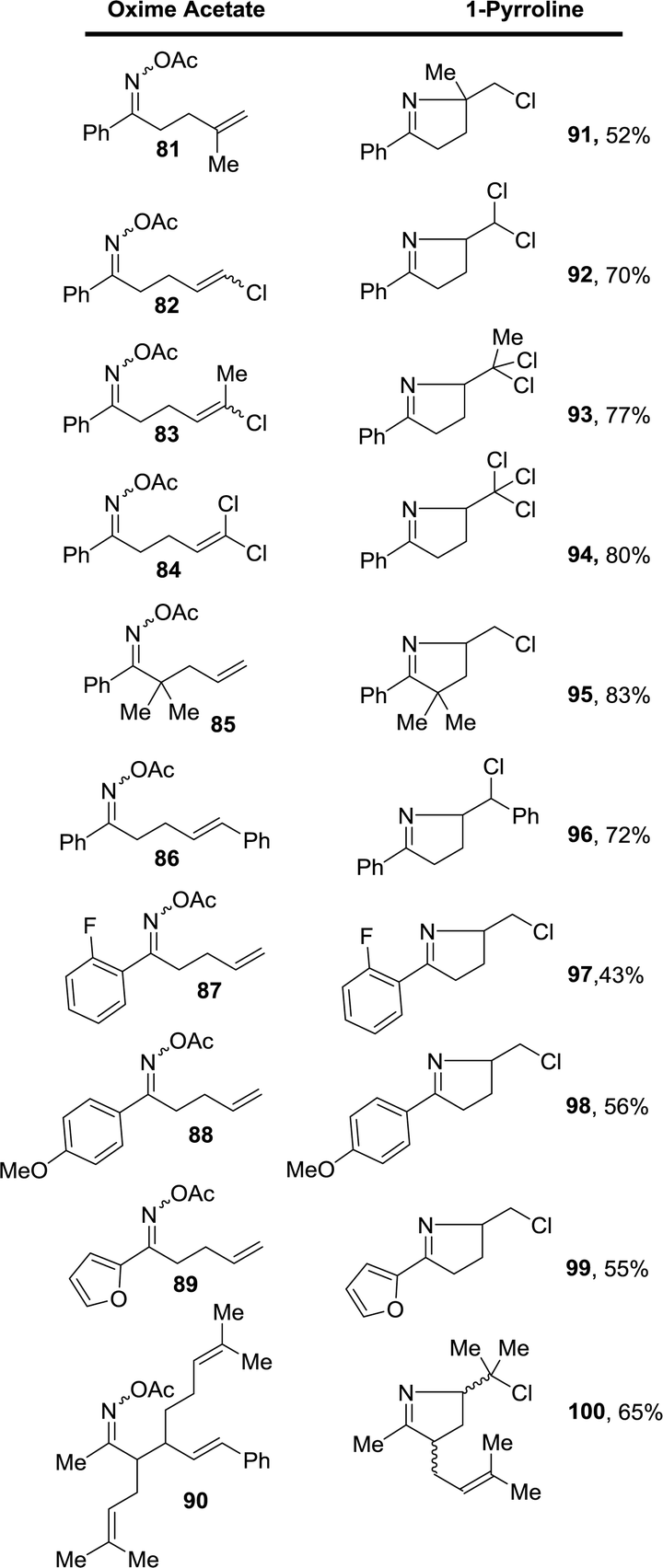
The scope of this FeCl3-promoted ionic transformation in t-butanol was examined with and without the presence of acetic acid (Table 1). While several substitution patterns on the alkene were well tolerated, only the oxime acetate derived from aromatic ketones gave the 1-pyrrolines. The case of oxime acetate 90 derived from a methyl ketone is an exception since other oxime acetate derived from aliphatic ketones produced complex mixtures.
Table 1 Formation of chloroalkyl-1-pyrrolines 90–100 from unsaturated oxime acetates 81–90 mediated by FeCl3
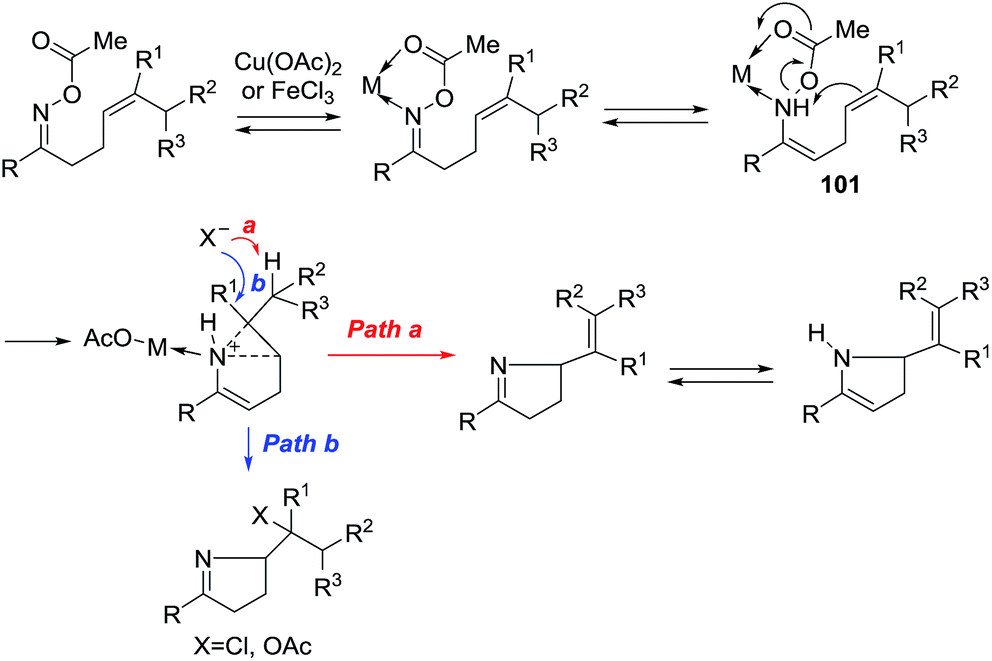
A possible mechanism is given in Scheme 31. The nitrogen from the oxime and the oxygen of ester form an intermediate complex with the metal ion, probably in equilibrium with the enamine that reacts via transition state 101 wherein acetate leaves as the weak N–O bond is broken by nucleophilic attack of the alkene. Electron-rich alkenes are therefore better nucleophiles and improve the reaction. With Cu(II), an alkene is formed by abstraction of a proton (path a). With the Fe(III) of FeCl3, chloride anion competes as a nucleophile to give the chloride as a final product (path b).
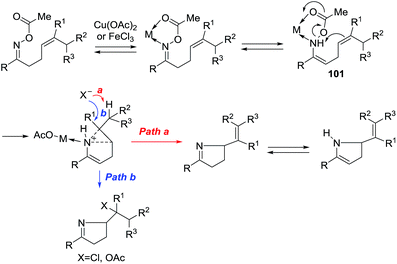 |
| | Scheme 31 Possible mechanism. | |
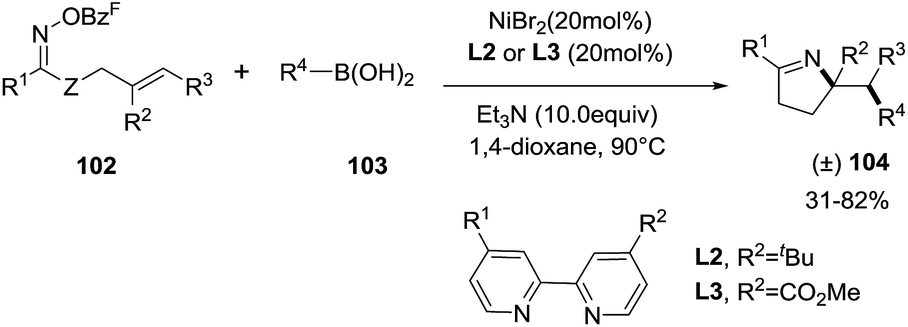
Unlike palladium catalyzed cross-coupling reactions that suffer from β-H-elimination, cross-coupling reactions catalyzed with Ni are not susceptible to β-H-elimination. This feature was exploited by Selander et al. to developed a Nickel-catalyzed 1,2-aminoarylation of oxime ester-tethered alkenes with boronic acids (Scheme 32).94 By reacting γ,δ-unsaturated oxime esters 102 with boronic acids 103 in presence of NiBr2 (20 mol%), Et3N (10 equiv.) and ligands L2 or L3 (20 mol%) in dioxane at 90 °C, 1-pyrrolines 104 were obtained with moderate to good yields (31–82%). The reaction tolerates well several substituted arylboronic acids with electron withdrawing groups or electron donor groups and fused aromatic, heteroaromatic and vinylic boronic acids (R4 = Ar, hetAr, vinyl). Aryl and heteroaryl oxime esters proceed well to 1-pyrrolines (R1 = Ar, HetAr). The reaction is also suitable for non-terminal and cyclic alkenes. This nickel-catalyzed protocol is an excellent alternative to palladium catalyzed cross-coupling reactions (see Subsection 2.1.6.: Synthesis of 1-pyrrolines by palladium catalysis).
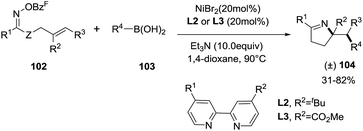 |
| | Scheme 32 Synthesis of 1-pyrrolines 104 by NiBr2. | |
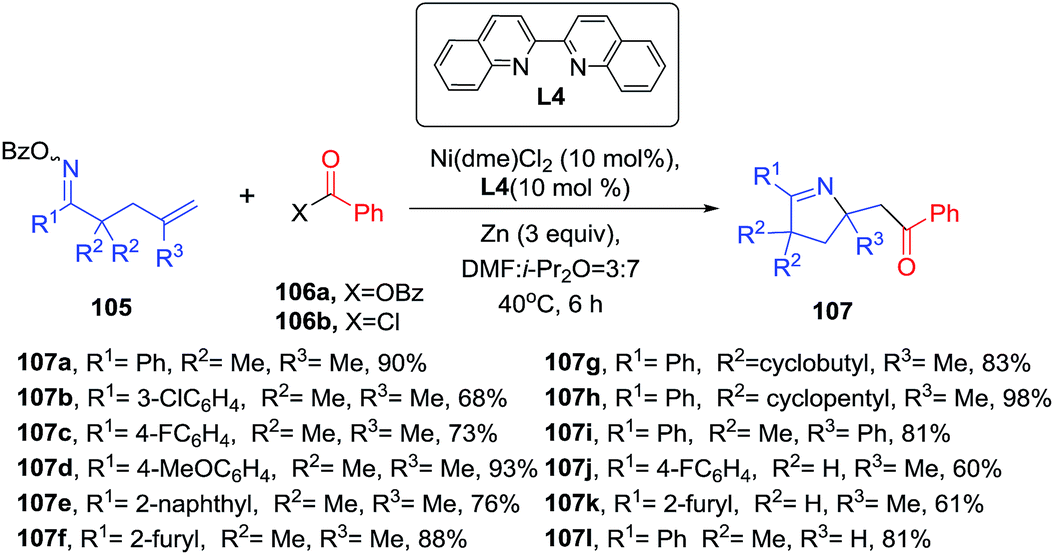
Recently, Wang et al. disclosure a Ni-biquinoline-catalyzed synthesis of multisubstituted 1-pyrrolines 107 through a reductive 1,2-iminoacylation of alkenes (Scheme 33).95 Oxime esters incorporating a pendant terminal olefinic unit 105 were reacted with electrophilic acylating reagents like acid chlorides (106b) or anhydrides (106a) and zinc as a reductive agent to generate 1-pyrrolines 107 in yields ranging from 46 to 98%. Aromatic and heteroaromatic groups in position R1 are well tolerated. Also, oxime esters with R2 = H and monosubstituted alkenes (R3 = H) were suitable substrates. Internal alkenes fail to deliver the product. The iminoacylation reaction can be performed with carboxylic acids in presence of (Boc)2O instead of the acid chlorides or anhydrides. This procedure is an alternative to Bower's Pd catalyzed 1,2-iminoacylation of oxime esters-tethered alkenes with carbon monoxide and organoborons96 (see Subsection 2.1.6.: Synthesis of 1-pyrrolines by palladium catalysis).
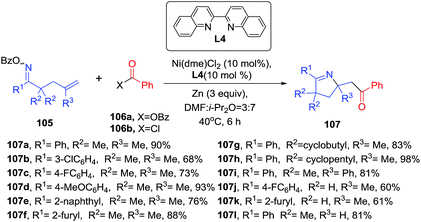 |
| | Scheme 33 Ni-catalyzed 1,2-iminoacylation of alkenes towards 1-pyrrolines. | |
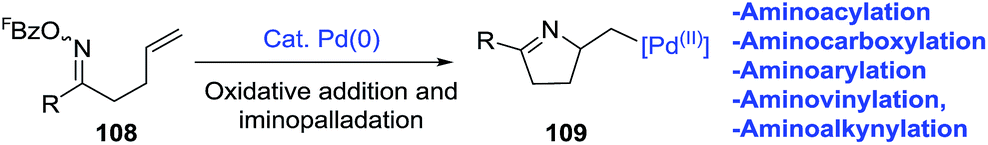
2.1.6. Synthesis of 1-pyrrolines by palladium catalysis. Bower et al.96 developed an extraordinary umpolung approach to obtain a great variety of 1-pyrrolines (Scheme 34). Their methodology is based on the oxidative addition of Pd(0) into the N–O bond of O-pentafluorobenzoyl oxime esters (108) to generate an imino-Pd(II) intermediate 109. This intermediate undergoes an 5-exo cyclization with sterically diverse alkenes to form alkyl-Pd(II) pyrroline intermediates. These intermediates can be decorated by subsequent reaction with organometallic or alcohol nucleophiles (Scheme 35). Moreover, the procedure can be carried out in presence of CO (carbonylative) or absence of CO (non-carbonylative) conditions. Under carbonylative conditions, 1,2-aminoacylation reactions are achieved by using triethylammonium tetraarylborates or pinacol organoboronates as the nucleophiles (products are 110 and 111). With alcohol nucleophiles under carbonylative conditions, a 1,2-aminocarboxylation occurs (product is 112).
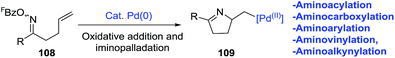 |
| | Scheme 34 Oxidative addition in the oxime ester N–O bond. | |
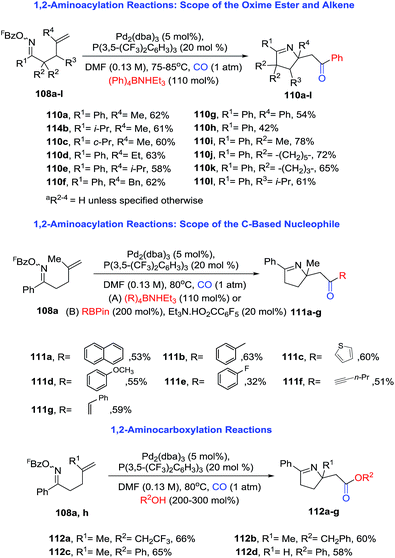 |
| | Scheme 35 Pd-catalyzed 1,2-carboamination of alkenes. | |
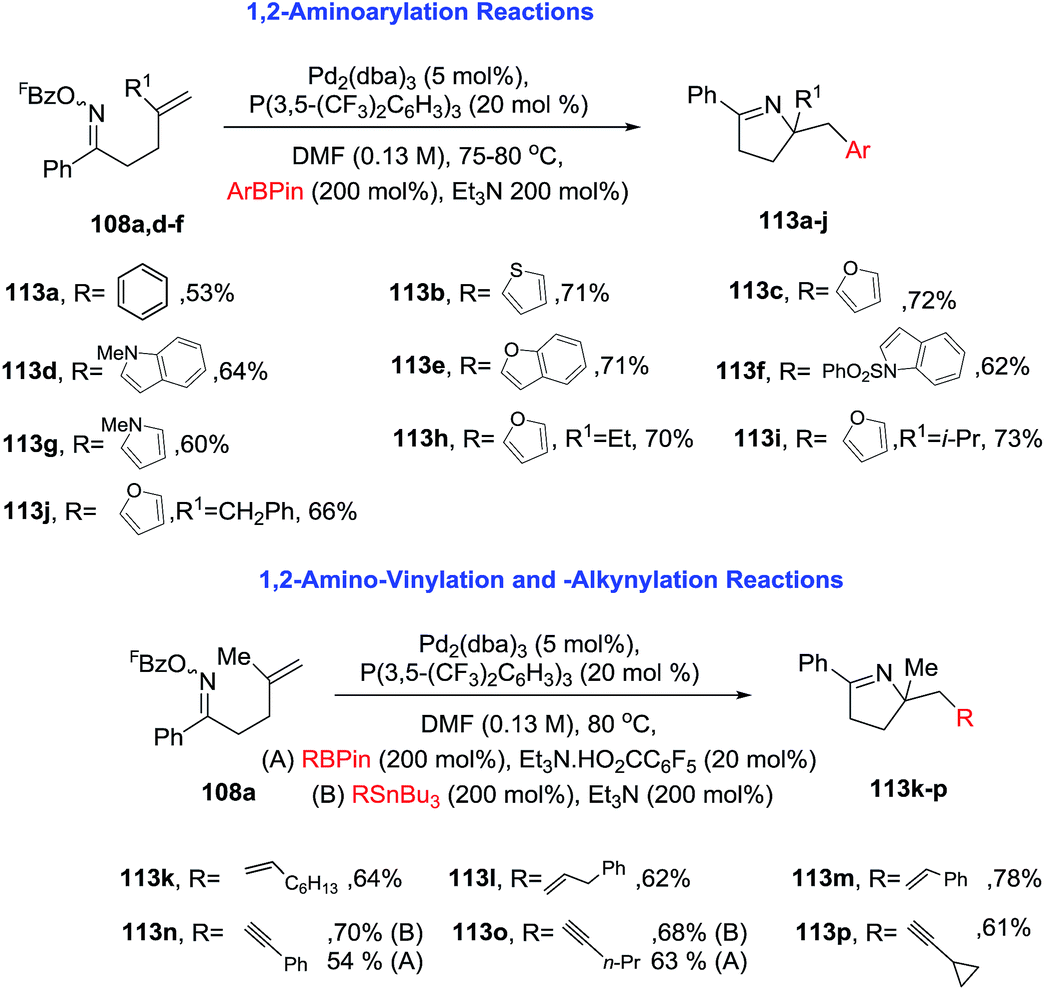
Under noncarbonylative conditions 1,2-aminoarylation is achieved employing a variety of pinacol arylboronates as nucleophiles (Scheme 36, products 113a–j). 1,2-Aminovinylation and 1,2-aminoalkynylation products are obtained using vinyl boronates or alkynyl-stannanes, respectively (products are 113k–p). For alkynylation, less toxic pinacol alkynylboronates can be used instead of alkynyl-stannanes. This approach provides an unified strategy for achieving alkene 1,2-aminoacylation, -carboxylation, -arylation, -vinylation, and -alkynylation.
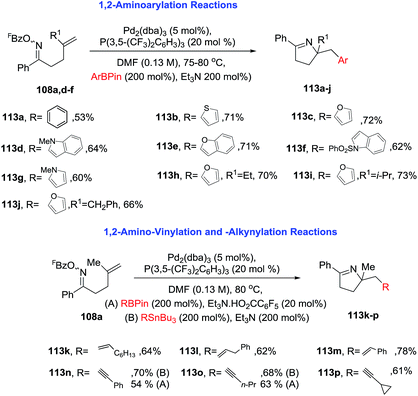 |
| | Scheme 36 Pd-catalyzed 1,2-carboamination of alkenes. | |
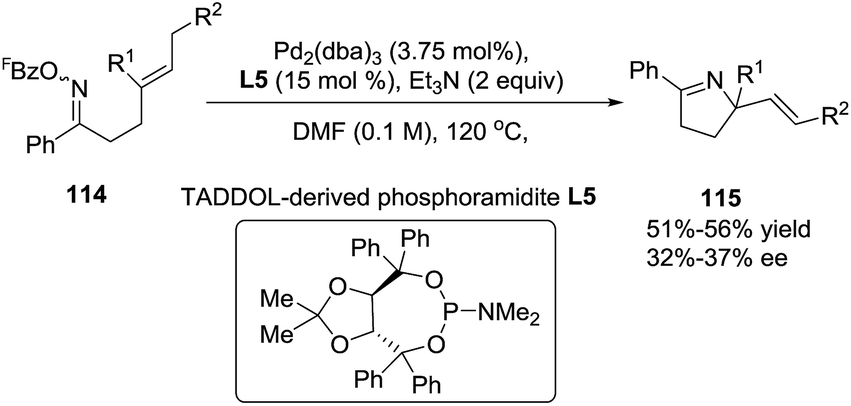
Previously Bower et al. reported97 the Pd-catalyzed cyclization of O-pentafluorobenzoyl oxime esters with 1,1-disubstituted alkenes as a general entry to chiral α,α-disubstituted pyrrolines (115). In this study the key ligand P(3,5-(CF3)2C6H3)3 used in the achiral catalytic systems was replaced by the TADDOL-derived phosphoramidite L5. With 114 as the starting material, modest enantioselectivities (typically 32–37% ee) were achieved (Scheme 37). Nonetheless, these results establish the feasibility of asymmetric Narasaka–Heck cyclizations.
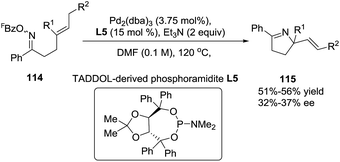 |
| | Scheme 37 Asymmetric Pd-catalyzed cyclization of oxime esters with 1,1-disubstituted alkenes. | |
The same group performed Pd-catalyzed cyclizations of oxime esters with 1,2-dialkylated alkenes (116) as a general entry to chiral dihydropyrroles (117) and avoiding the formation of the undesired pyrrole (Scheme 38).98 In the previous case where 1,1-disubstituted alkenes were used, the nature of the alkene acceptor controlled the direction of β-hydride elimination and the pyrrole formation was bypassed. The selectivity in favor of the pyrroline, and against the pyrrole, derives from the aryl moiety (R2 = aryl). This moiety weakens the benzylic C–H bond and so increases the propensity for β-hydride elimination. Where R2 ≠ aryl, the pyrroline/pyrrole selectivity is lower.
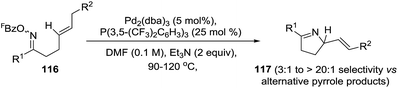 |
| | Scheme 38 Pd-catalyzed cyclization of oxime esters with 1,2-disubstituted alkenes. | |
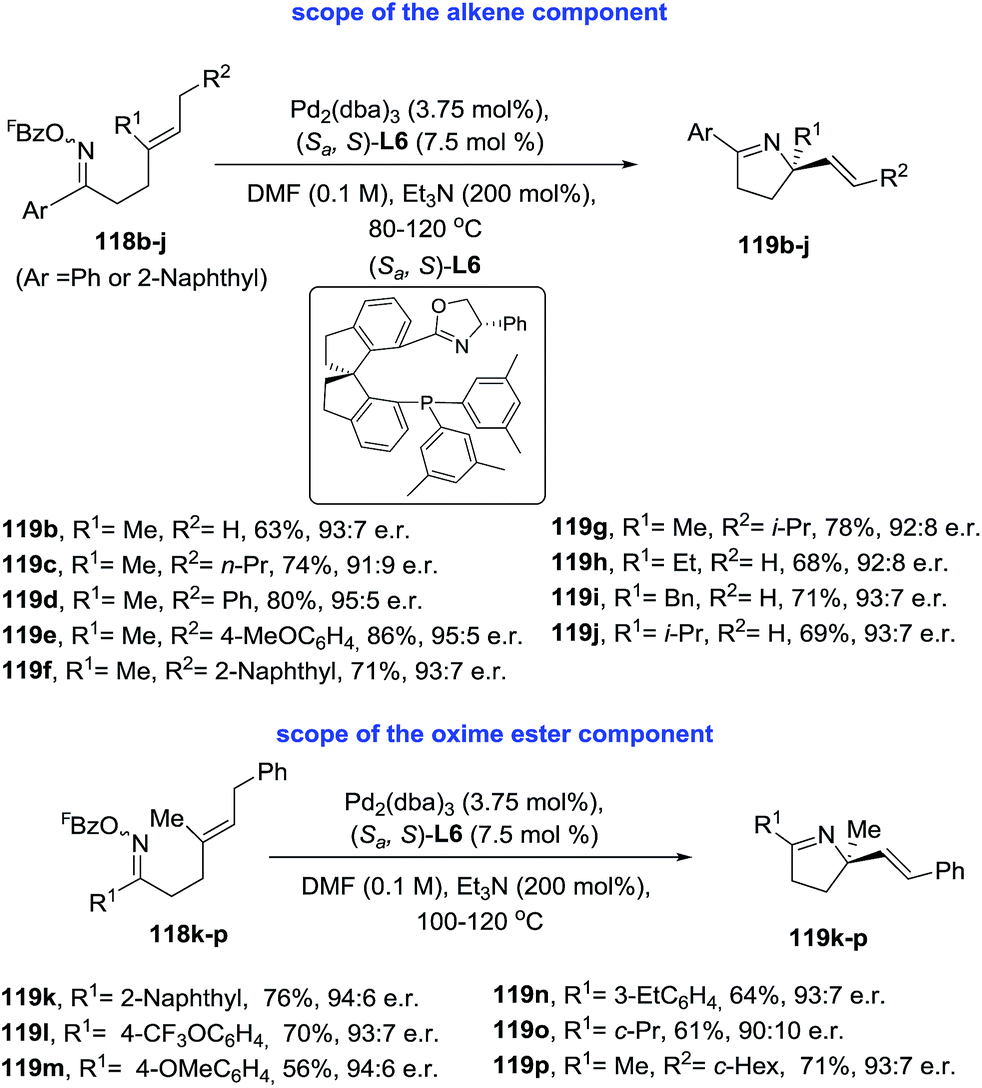
Very recently, Bower and coworkers99 identified a SPINOL-derived P–N ligand system [(Sa,S)-L6] that promoted the first examples of highly enantioselective Narasaka–Heck cyclizations (Scheme 39). This Pd-catalyzed 5-exo cyclization tolerates a range of oxime esters and diverse sterically trisubstituted alkenes (118) and generates otherwise challenging pyrrolidine derivatives containing tetrasubstituted nitrogen-bearing stereocenters (119). Cycled products are obtained in moderate to excellent yields (56–86%) and high enantioselectivity (91![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9 to 95:5 e.r.).
:9 to 95:5 e.r.).
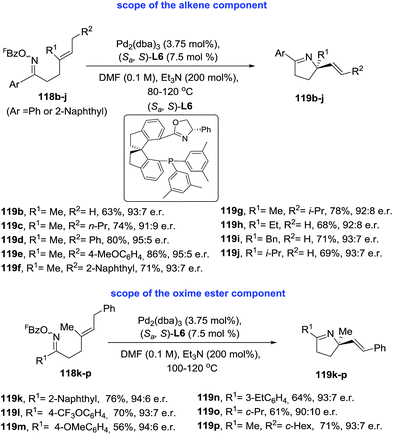 |
| | Scheme 39 Enantioselective Narasaka–Heck cyclizations. | |
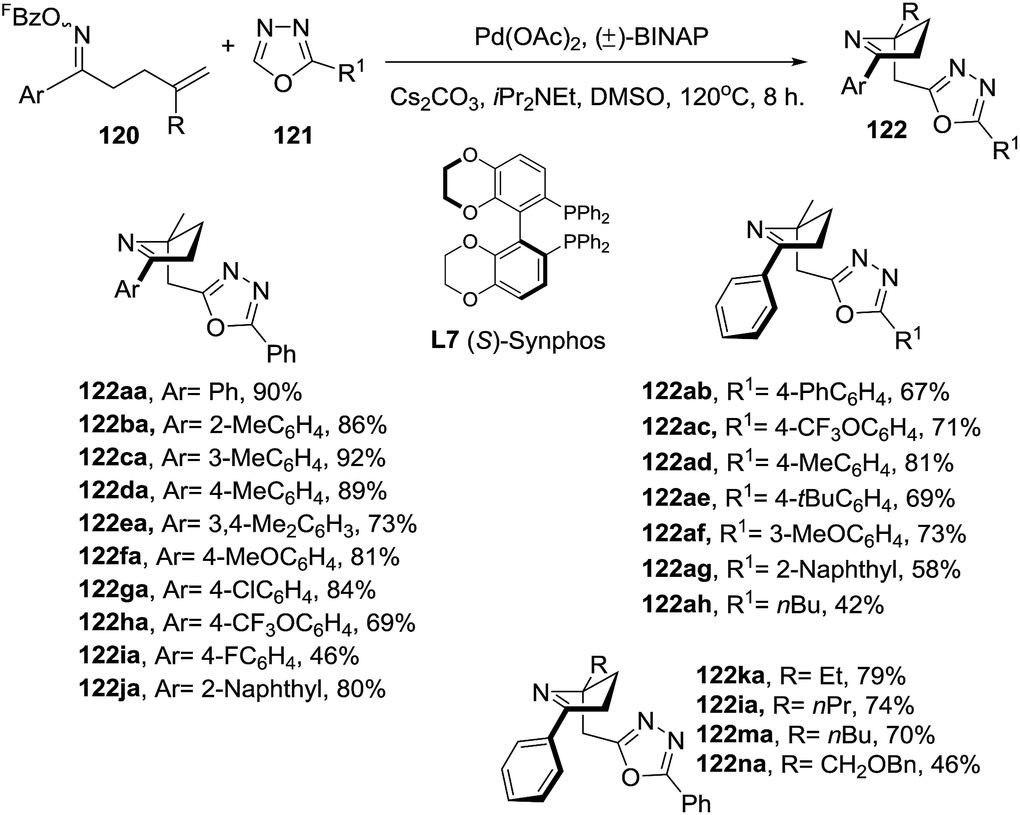
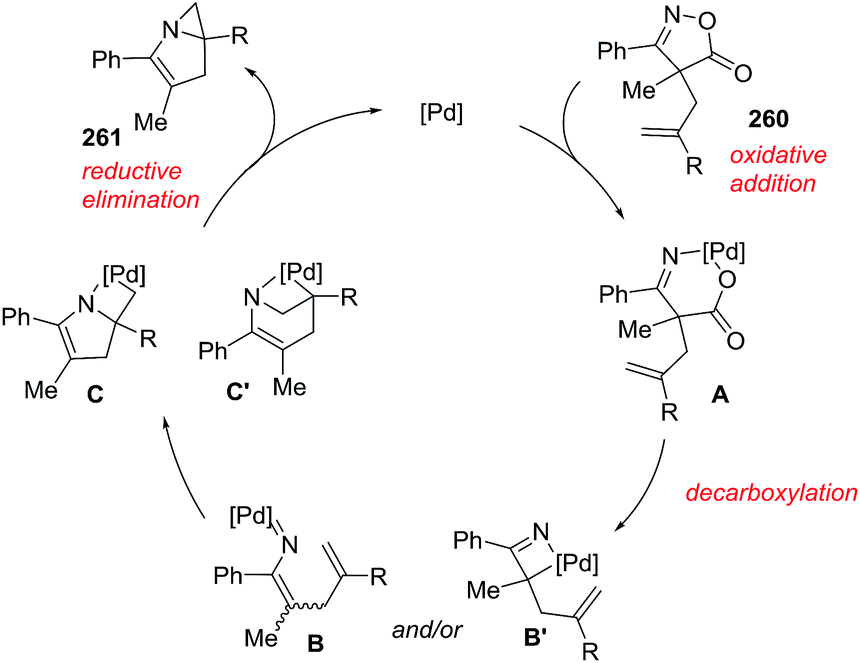
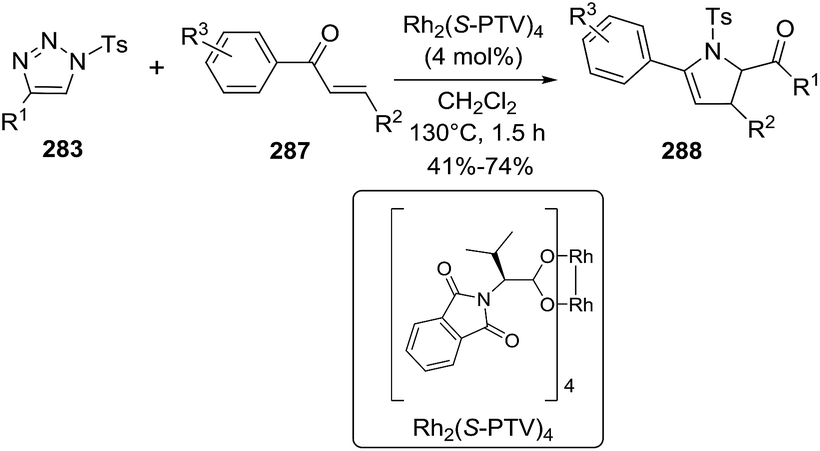
Almost simultaneously with the report of Bower, Bao et al.100 reported a domino processes that involved a palladium-catalyzed Narasaka–Heck reaction followed by direct arene C–H alkylation leading to 2,5,5-trisubstituted dihydropyrroles (122) (Scheme 40). In this process two new bonds were formed: one N(sp2)–C(sp3) bond and one C(sp3)–C(sp2) bond and a quaternary center was generated. The optimized conditions of Pd(OAc)2 (0.1 equiv.), (±)-BINAP (0.2 equiv.), iPr2NEt (4.0 equiv.), Cs2CO3 (3.0 equiv.) in DMSO at 120 °C were applied to substrates with a range of substituted on the aryl (Ar) group of the oxime ester (120), exploring both the electronic nature and position of the substituent. In most of cases, the 1-pyrrolines 122aa–122ja were obtained in very good yields. A variety of substituents on the oxadiazole (121), and different alkyl groups attached to the alkene, were well tolerated. An enantioselective version of this cascade process was developed using (S)-Synphos (L7) as chiral ligand, isolating the 2,5,5-trisubstituted pyrroline 122 in good yields with good to high enantioselectivity.
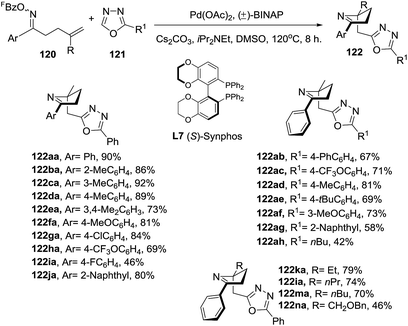 |
| | Scheme 40 Domino Narasaka–Heck reaction/direct arene C–H alkylation. | |
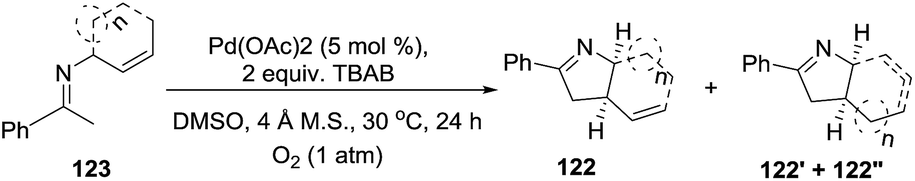
The Glorius group101 developed an entry to 1-pyrrolines (124, 124′, 124′′) starting from the imines 123 derived from acetophenone and cyclic N-allylamines (Scheme 41). The procedure employs a mild oxidative palladium-catalyzed cyclization using O2 as the terminal oxidant. Besides cyclic N-allylamines, N-allylamines containing γ-substituents on the allyl group also deliver the 1-pyrrolines in good yields (61–87%). This atom-economical procedure relies on an intramolecular C–H dehydrogenative 5-exo cyclization.
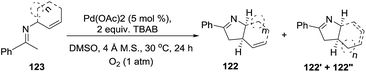 |
| | Scheme 41 Palladium(II)-catalyzed cyclization of imines to 1-pyrrolines. | |
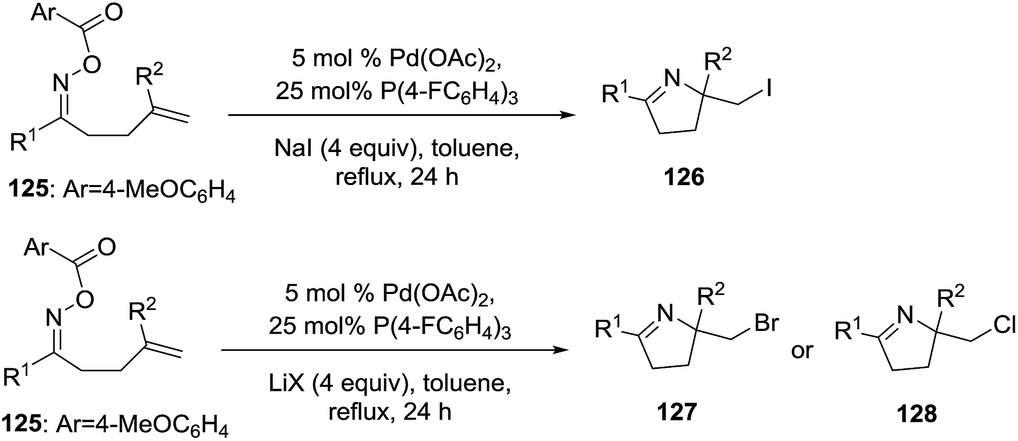
Tong et al.102 developed a methodology to synthesize 2-halomethyl 1-pyrrolines 126 from the oxime ester 125 using a Pd(0)-catalyzed intramolecular iminohalogenations of 1,1-disubstituted alkenes assisted with halide salts (Scheme 42). The use of electron-poor phosphine as ligands proved to be essential to obtain alkyl halogenated dihydropyrroles under reductive elimination. Iminochlorination (128) is less efficient compared to iminobromination (127) and iminoiodination (126). For example, when R1 = Ph and R2 = Me the yields for the iminohalogenation are 75% for iodination, 64% for bromination and 42% for chlorination.
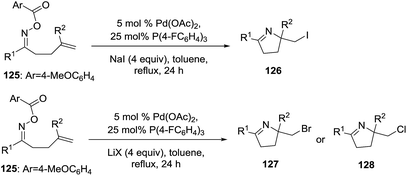 |
| | Scheme 42 Palladium(0)-catalyzed iminohalogenation of alkenes. | |
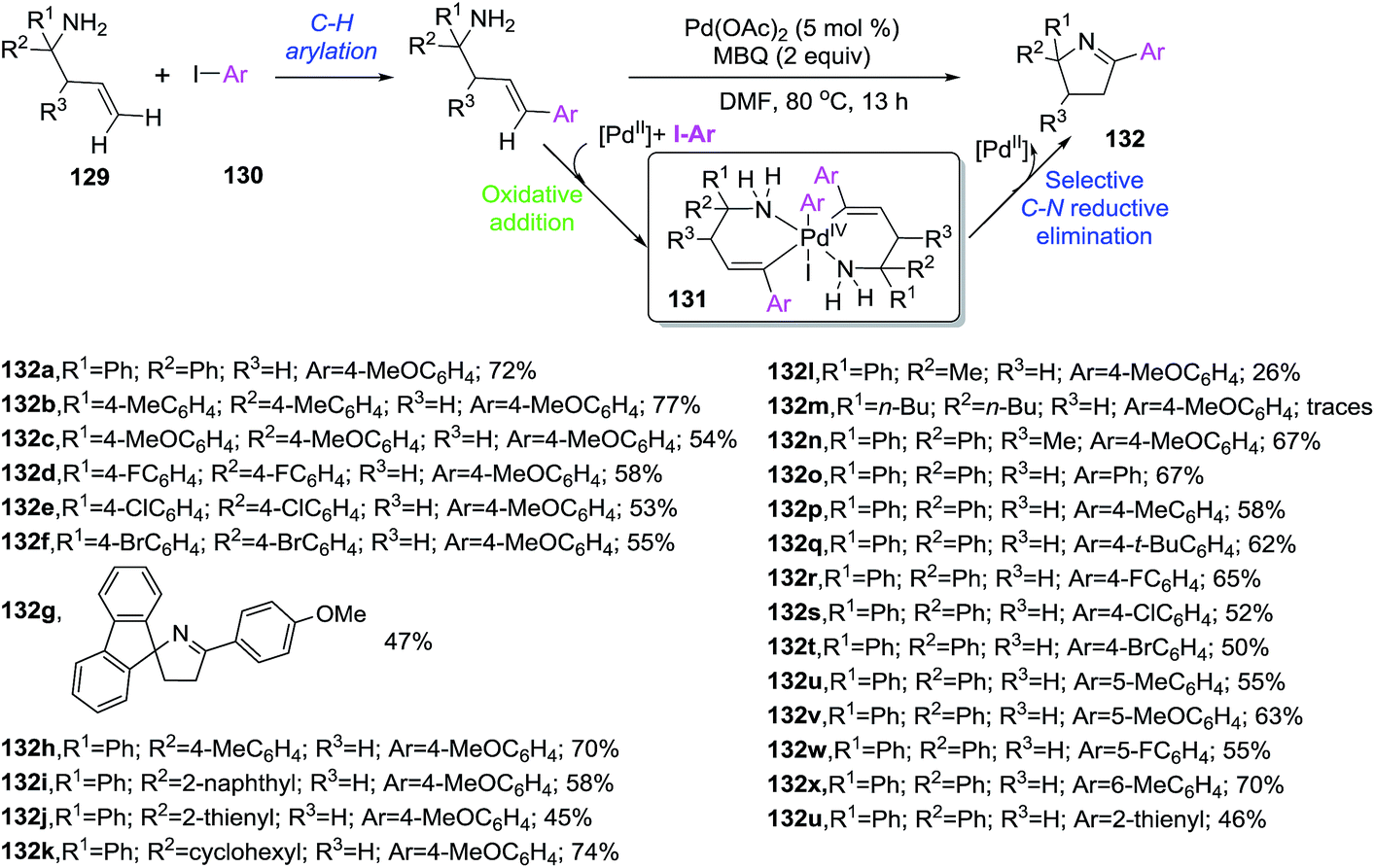
Homoallylic primary amines 129 reacted with aryl iodides 130 in the presence of a Pd catalyst to provide 2-aryl-1-pyrrolines 132 via a cascade C–H arylation/C–H amination sequence (Scheme 43).103 The transformation proceeds thorough a Pd-promoted Heck coupling followed by a C–H amination cocatalyzed by Pd and the aryl iodide, and final tautomarization from 2-pyrrolines to the isolated 1-pyrrolines. Interestingly, this report represents the first example of aryl iodide acting as both coupling reagent and cocatalyst involved in the generation of the active bivalent aryl palladium catalyst 131 capable of promoting the formation of new N–C bond. The optimized protocol involved the use of 2 eq. of aryl iodide 130, Pd(OAc)2 (10 mol%), MBQ (2-methylbenzoquinone, 20 mol%) as oxidant in DMF at 80 °C, leading to a small collection of the final heterocycles 132 with poor to good yields (26–74%). The presence of an oxidant was essential to achieve complete annulation step.
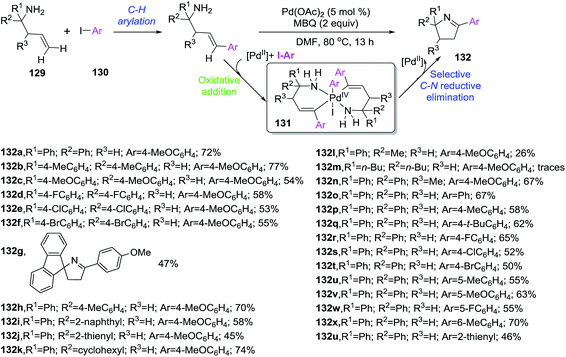 |
| | Scheme 43 Synthesis of 2-aryl-1-pyrrolines 132 via a cascade C–H arylation/C–H amination sequence. | |
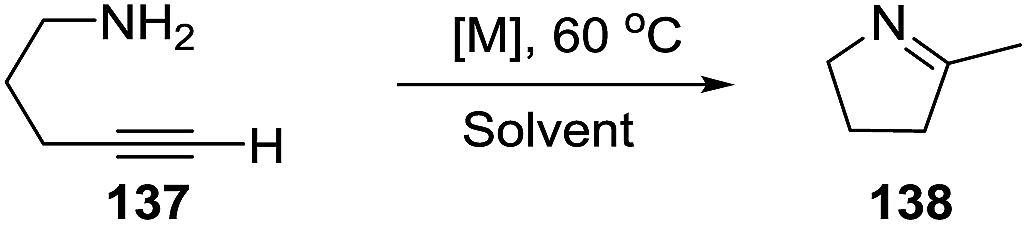
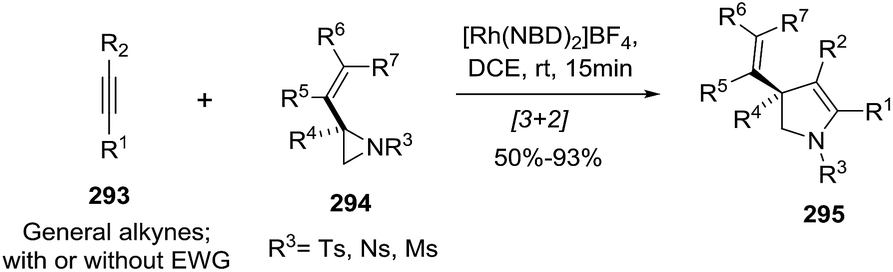
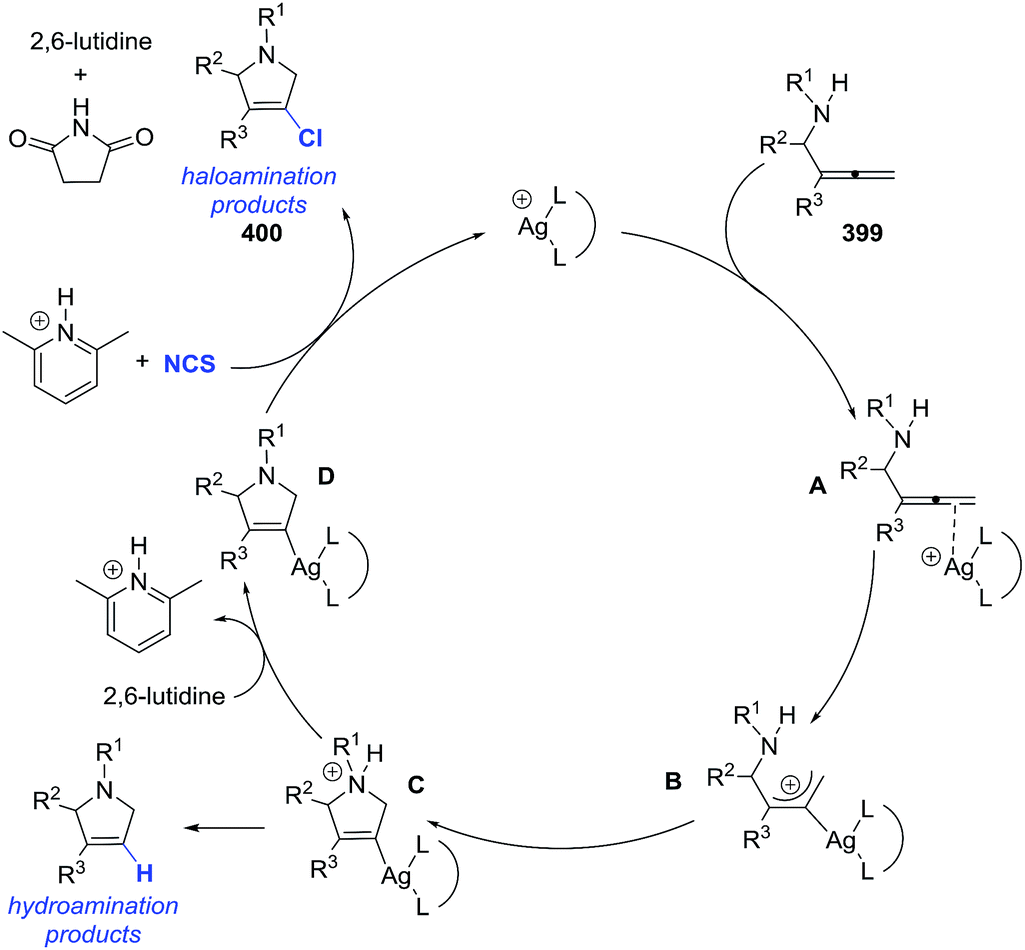
2.1.7. Synthesis of 1-pyrrolines by rhodium catalysis. The hydroamination of alkynes is a highly atom-efficient approach to the synthesis of imines and consist of the N–H addition to a C–C triple bond. If the reaction is intramolecular, the results are cycled imines as 1-pyrrolines. Late transition metal complexes of Ru, Rh, Ir, Pd, Pt, and Cu are effective catalysts for the hydroamination reaction.104,105In 2012, Messerle and coworkers87 reported a series of cationic rhodium (133, 135) and iridium complexes (134, 136) with bidentate N,N′-pyrazolyl–triazolyl ligands (Fig. 3). These complexes were evaluated as catalysts for the intramolecular hydroamination of a series of alkynylamines. In the case of the cyclization of the terminal alkyne as 4-pentyn-1-amine (137) to 2-methyl-1-pyrroline (138) at 60 °C (Scheme 44), the iridium complexes [Ir(N–N)(CO)2]–BArF4 (136c,d) were the most active catalysts giving 5-exo-cyclization exclusively (>98% yield).
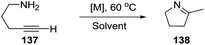 |
| | Scheme 44 Catalyzed cyclization of 4-pentyn-1-amine. | |
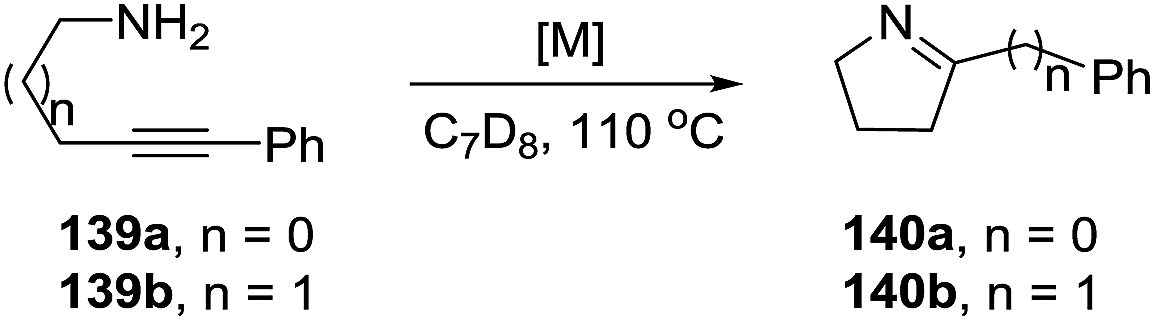
The rhodium complexes generally were less active as catalysts than their iridium analogues. In the case of intermolecular hydroamination of internal alkynes, such as with 4-phenyl-3-butyn-1-amine (139a) and 5-phenyl-4-pentyn-1-amine (139b), the reaction required a higher temperature (110 °C) to reach complete conversion (>98%) towards pyrrolines 140a,b (Scheme 45).
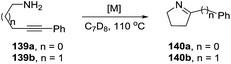 |
| | Scheme 45 Catalyzed cyclization of non-terminal alkynamines. | |
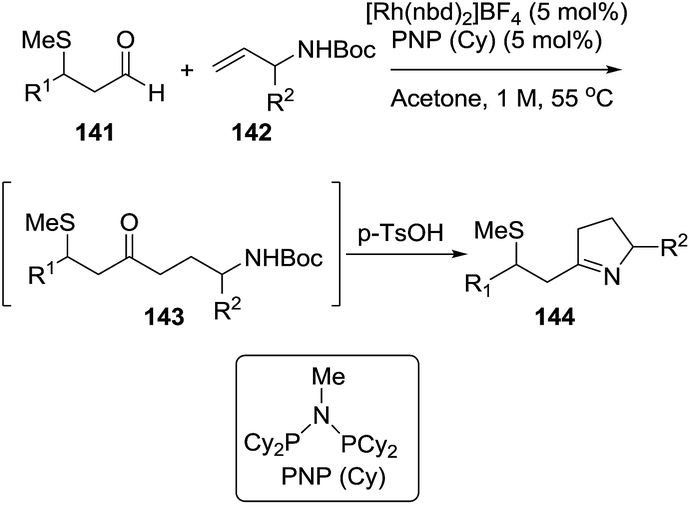
Rhodium(I) catalysts with ligands such as (Cy2P)2NMe and PNP(Cy) are efficient reagents to perform the union of S-chelating aldehydes 141 (aldehydes with a methyl sulfide group in β position) and allylic amines 142 to deliver linear hydroacylation adducts 143. These adducts are treated in situ with p-TsOH to provide the pyrrolines 144 through a dehydrative cyclization (Scheme 46).23 The pyrroline products (144) are formed in good-to-excellent yields (48–90%).
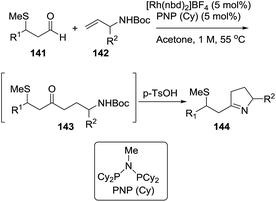 |
| | Scheme 46 Pyrroline synthesis from the combination of S-chelating aldehydes and allylic amines. | |
A variety of alkyl, aryl and heteroaryl substituents (R2 = Ar, HetAr, alkyl) were well tolerated at the allylic position of the alkenes. Also, different aryl or heteroaryl (R1 = Ar, HetAr) S-chelated aldehydes provided the pyrrolines. Aldehydes featuring a chelating group in β-position other than methylthio and disubstituted alkenes were not reactive under these conditions.
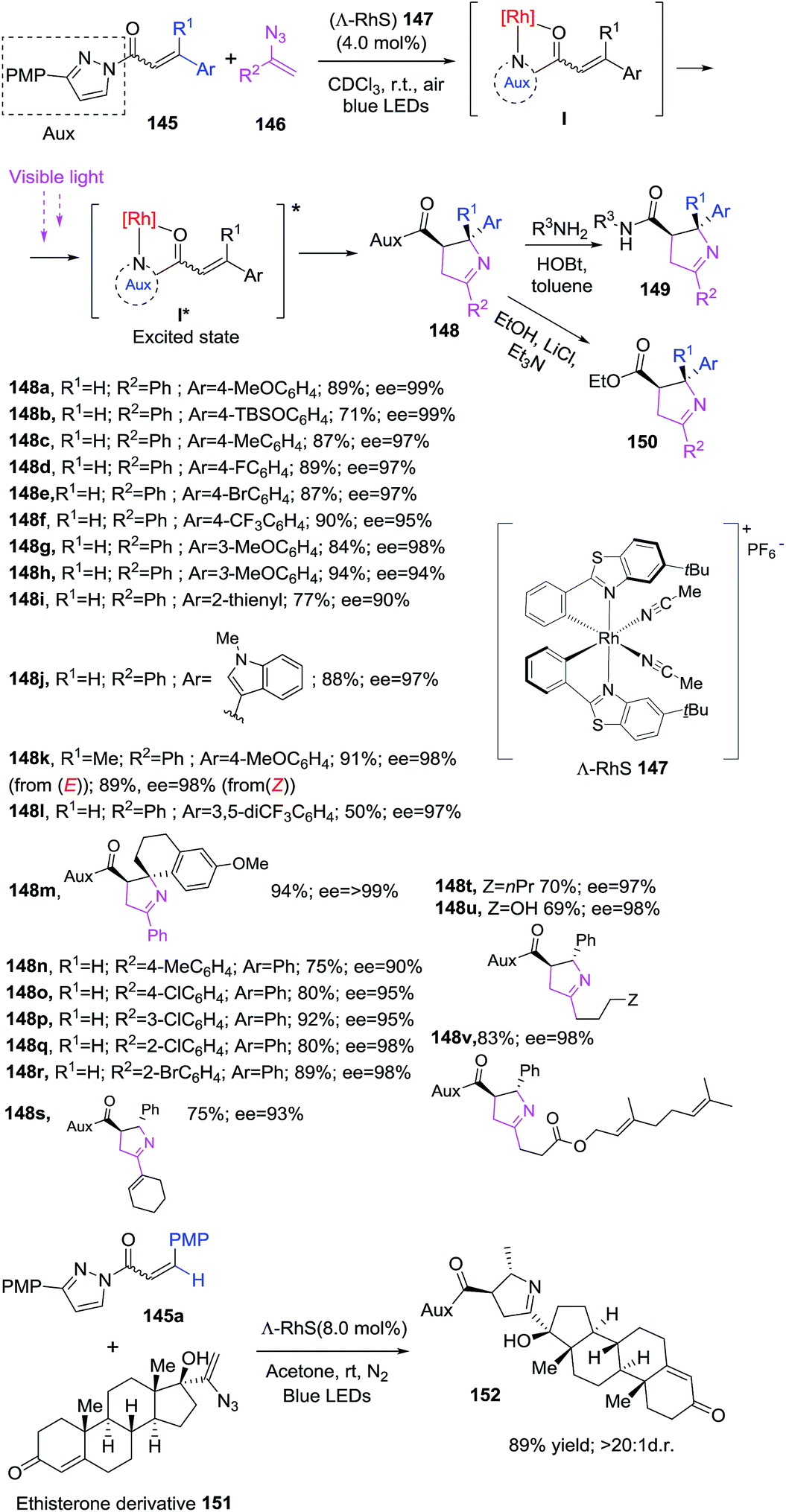
The Meggers group reported for first time a visible-light-induced [2 + 3] photocycloaddition of alkenes 145 with vinyl azides 146 using a chiral rhodium catalyst 147 to obtain enantiomeric pure 1-pyrrolines 148 (Scheme 47).106 The protocol makes use of α,β-unsaturated N-acylpyrazole 145 in combination with vinyl azides 146 in presence of catalyst (Λ-RhS) 147 (4 mol%) using chloroform as solvent under irradiation of blue LEDs. As a result, 1-pyrrolines 148 were obtained in good to excellent yields (69–94%) with complete diastereoselectivity and enantioselectivities of up to >99% ee. The pyrazole auxiliary have a major impact on the reaction result, the best performance is achieved with 3-(p-methoxyphenyl)pyrazole (PMP). The chiral rhodium catalyst 147, developed in Megger's group, coordinates with substrates 145 through the carbonyl oxygen and the pyrazole auxiliary. This complex I generates an excited intermediate I* upon visible-light irradiation which subsequently reacts with a vinyl azides to afford a 1-pyrrolines (Scheme 47). The methodology tolerates various β-aryl substituent on N-acylpyrazole 145 regardless the electronic nature or position as well as heteroaryl moieties and is able to provide 1-pyrrolines with quaternary stereocenter (R ≠ H). α,β-Unsaturated N-acylpyrazoles with E or Z configuration produce the same result. Aryl, vinyl and alkyl azides 146 successfully afforded the desired 1-pyrrolines. The good functional group tolerance was tested with a complex steroid vinyl azide derivative 151 proving the utility of the protocol in late stage synthesis. Finally, the N-acylpyrazole can be easily converted in other functionalities such as amide 149 or ester 150.
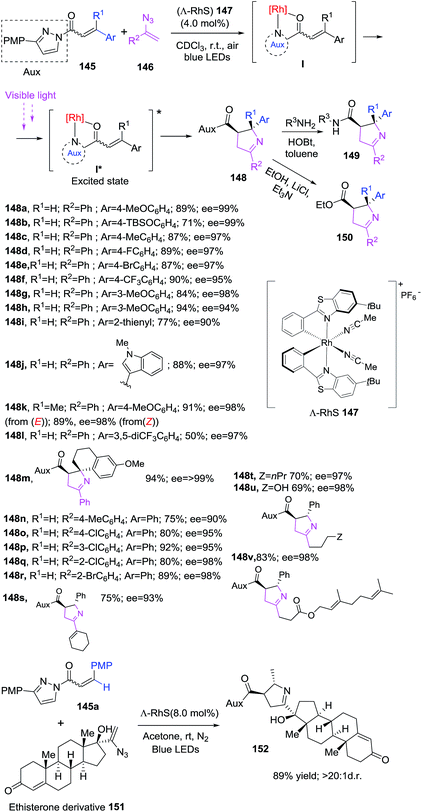 |
| | Scheme 47 Synthesis of 1-pyrrolines using a chiral rhodium catalyst. | |
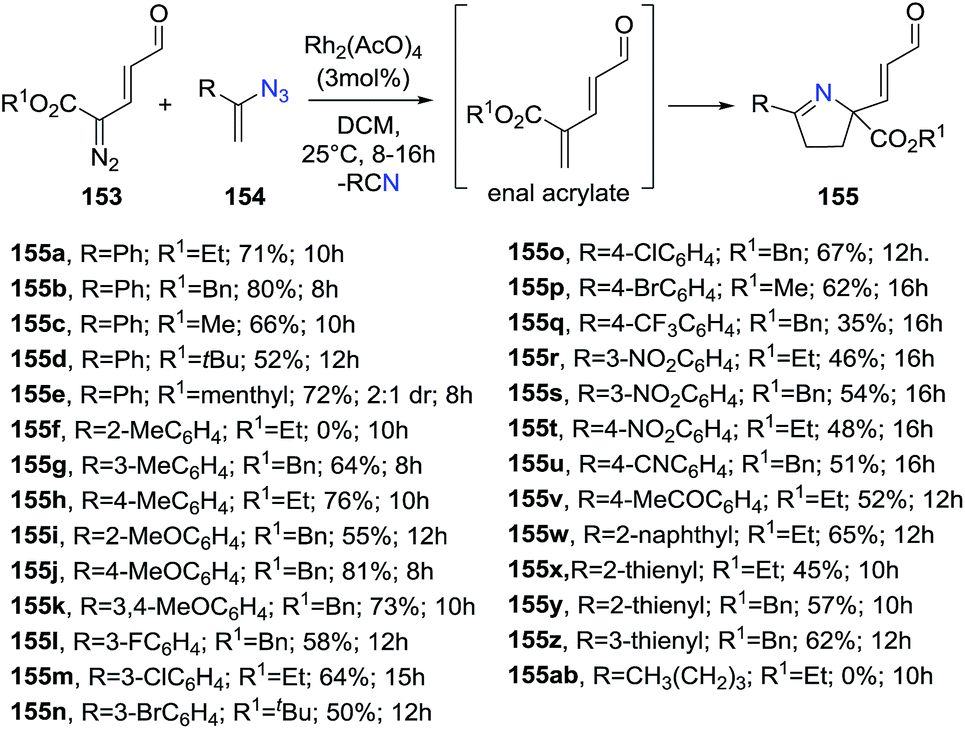
Katukojvala et al. discovered a new rhodium-catalyzed [1 + 1 + 3] annulation reaction of diazoenals 153 and vinyl azides 154 to generate enal-functionalized 1-pyrroline derivatives 155 (Scheme 48).107 These functionalized 1-pyrrolines can be synthetically modified to provide further structural diversity. The reaction occurs through a rhodium-catalyzed olefination of diazoenals with vinyl azides to produce the enal acrylate and subsequent annulation to access to 1-pyrrolines. The optimized conditions involve the use of Rh2(OAc)4 as the catalyst, and of 2.5 equiv. of vinyl azides in dichloromethane at room temperature. Ethyl, benzyl, and methyl ester diazoenals 153 delivered the 1-pyrrolines in very good yields (66–80%). Regarding vinyl azides 154, neutral and electron-rich styryl azides afforded very good yields of functionalized 1-pyrrolines (64–81%) but the used of electron-deficient styryl azides resulted in lower yields (35–54%). Thienyl azides were also compatible with the reactions conditions but aliphatic vinyl azide failed to deliver the 1-pyrrolines 155.
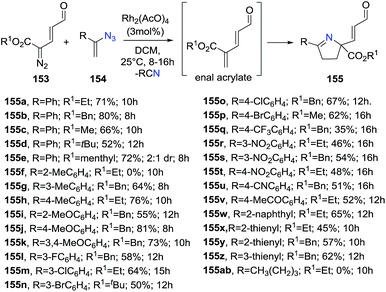 |
| | Scheme 48 Rhodium-catalyzed [1 + 1 + 3] annulation reaction to generate enal-functionalized 1-pyrrolines 155. | |
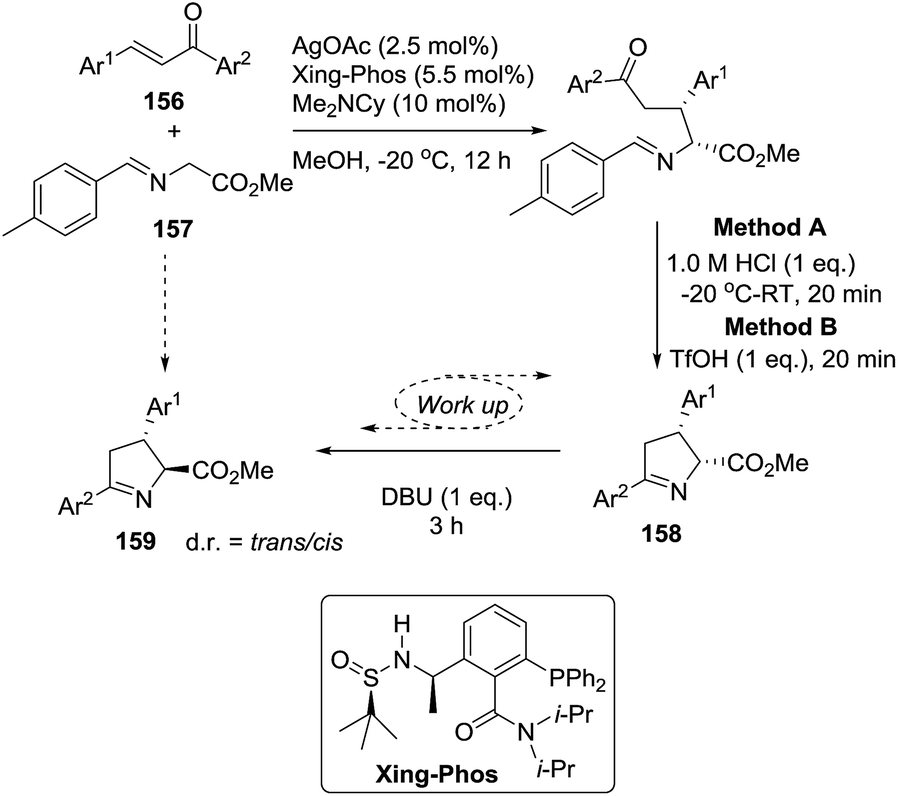
2.1.8. Synthesis of 1-pyrrolines by silver catalysis. Bai et al.22 reported a one-pot asymmetric Michael-type conjugate addition of glycine imino esters (157) to chalcones (156) using a Ag/Xing-Phos-catalyzed [3 + 2] cycloaddition reaction to afford stereoselectively the 1-pyrrolines 158 (Method A, Scheme 49). The combination of Ag/Xing-Phos catalyst with Me2NCy as a base gave the best performance in term of yield (81–98%), diastereoselectivity, and enantioselectivity (up to 98% ee).
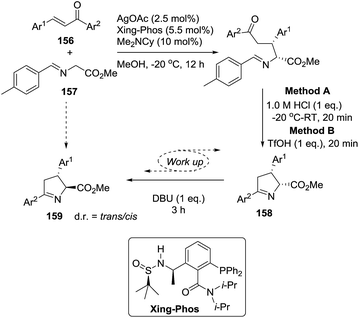 |
| | Scheme 49 Stereodivergent synthesis of cis- and trans-1-pyrrolines (158 and 159). | |
Moreover, trans-pyrrolines could be obtained using the same catalytic system but incorporating an epimerization protocol into the work-up conditions. Method A (HCl) gave the cis-pyrrolines 158 whereas the use of TfOH and DBU (Method B) led to the trans-pyrrolines 159 which were obtained with yields ranging from moderate to excellent (60–95%). The trans isomers 159 were obtained with diastereoselectivities (d.r. = trans/cis ratio) ranging from 83:17 d.r. −92:8 d.r. and enantioselectivities from 81% ee to 98% ee. This stereodivergent synthesis offers the possibility to obtain the cis and trans pyrrolines 158 and 159 as single enantiomers.
Zang and coworkers108 used a novel three-component annulation reaction between [60]fullerene, sulfonylhydrazones 160 and nitriles 161 mediated by Ag(I) as a general entry to disubstituted [60]fullerene-fused pyrrolines 162 (Scheme 50). The reaction exhibits a broad substrate scope and excellent functional-group tolerance. As such it represents a general synthesis of fullerene-bound macromolecules.
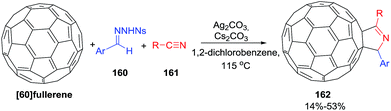 |
| | Scheme 50 Silver(I)-mediated three-component annulation reaction for the synthesis of [60]fullerene-fused 1-pyrrolines. | |
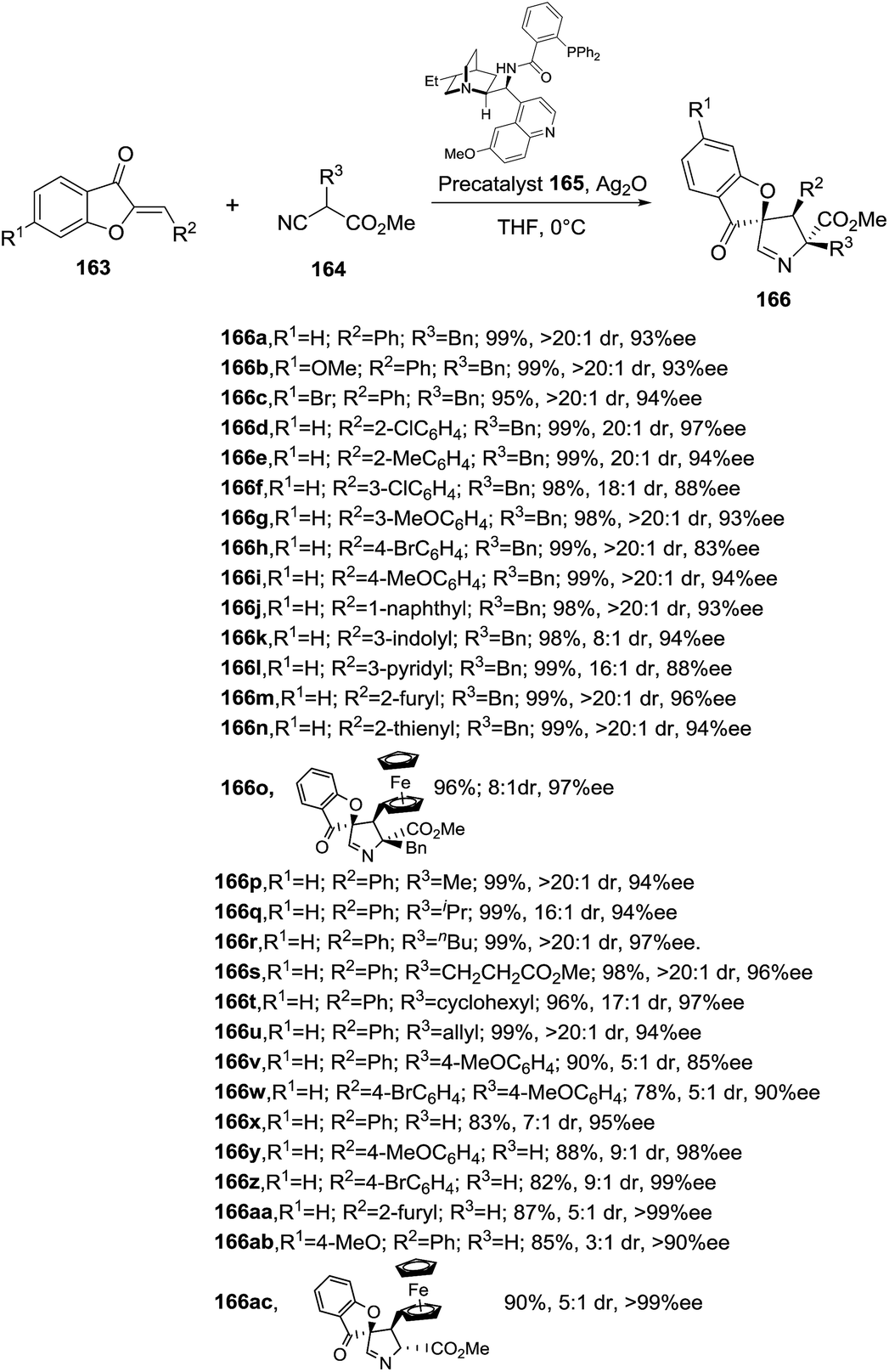
He, Shao and coworkers have recently developed the first enantioselective formal [3 + 2] cycloaddition between isocyanoacetates 164 and aurone anologues 163 catalyzed by chiral silver complexes (Scheme 51).109 Several chiral precatalysts were tested in combination with Ag2O under different reaction conditions to define the optimal procedure which required the use of silver oxide (10 mol%) and precatalyst 165 (20 mol%) in THF at 0 °C. The protocol is robust and simple, tolerating air and moisture. This efficient and atom-economical synthetic methodology provided a set of spiro-1-pyrrolines 166 bearing three stereocenters, with excellent yields (72–99%), and high diastero- and enantioselectivities (up to >20:1 and >99% ee, respectively). Interestingly, this protocol was successfully escalated up to a gram-reaction without a significant loss in yield or stereoselectivity. The mechanism is still under investigation.
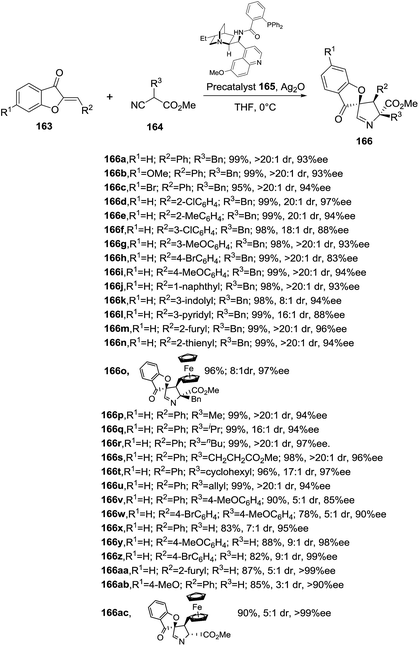 |
| | Scheme 51 Synthesis of spiro-1-pyrrolines 166 catalyzed by chiral silver complexes. | |
Mukhopadhyay et al.110 reported the silver-promoted regioselective synthesis of spiro-1-pyrrolines and spiro-2-pyrrolines (see Subsection 2.2.12.: Synthesis of 2-pyrrolines by silver catalysis, Scheme 113).
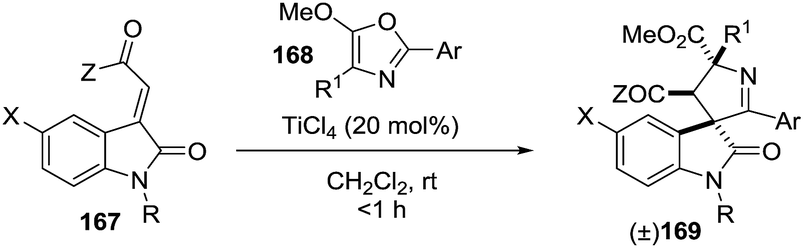
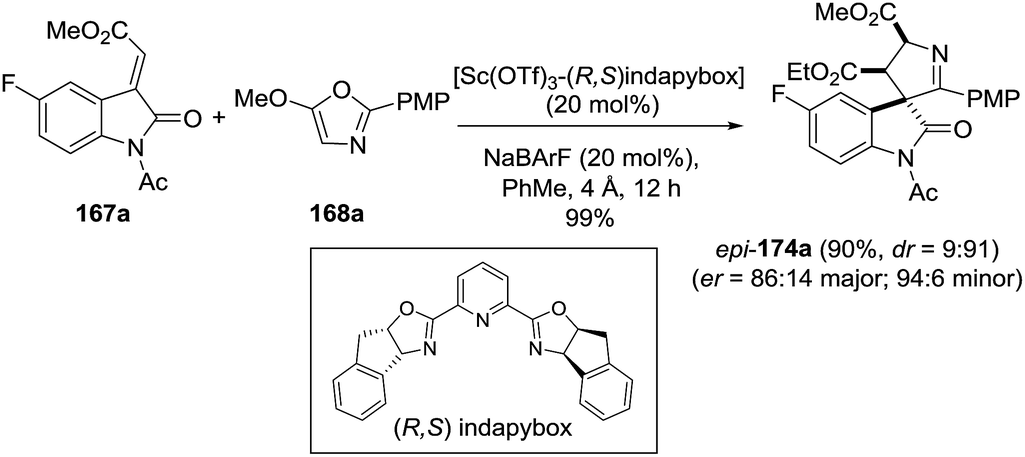
2.1.9. Synthesis of 1-pyrrolines by titanium catalysis. Franz et al.111 devised access to spirocyclic oxindole 1-pyrrolines 169 in excellent yield and diastereoselectivity using a TiCl4-catalyzed formal [3 + 2] cycloaddition between alkylidene oxindoles 167 and 5-alkoxy-2-aryloxazoles 168 (Scheme 52). A chelating group on the nitrogen of the indole is essential for selectivity (R = Ac, Cbz). Substitution at the 4-position of the oxazole afforded a spiro-1-pyrroline (X = F; Z = OEt; R1 = Me; R = Ac; Ar = PMP), containing two stereogenic quaternary centers, with high diastereoselectivity (dr = 94:6)
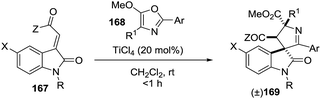 |
| | Scheme 52 Titanium(IV)-catalyzed stereoselective synthesis of spirooxindole-1-pyrrolines. | |
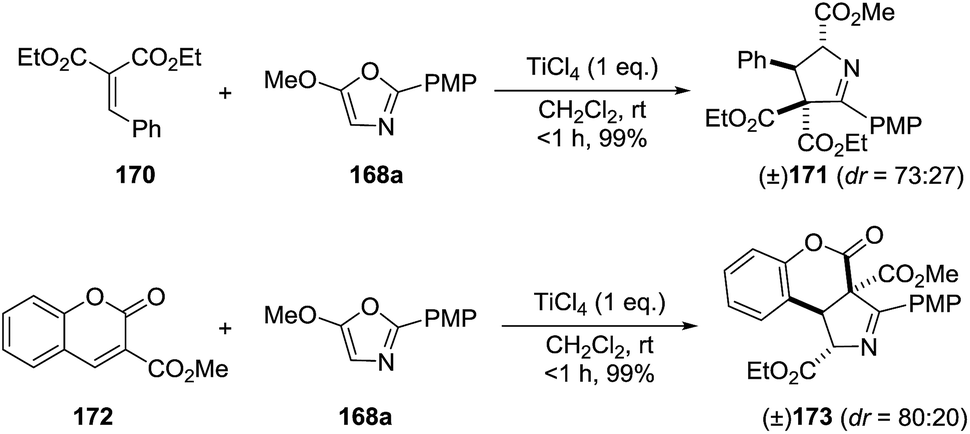
They extended this methodology to the synthesis of the highly functionalized pyrrolines 171 and 173 from ethyl benzylidene malonate (170) and coumarin (172) derivatives, respectively (Scheme 53).
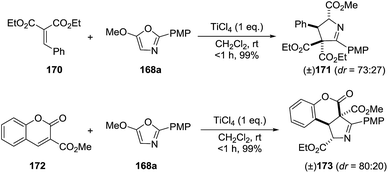 |
| | Scheme 53 Cyclization with malonate alkylidene and coumarin electrophiles. | |
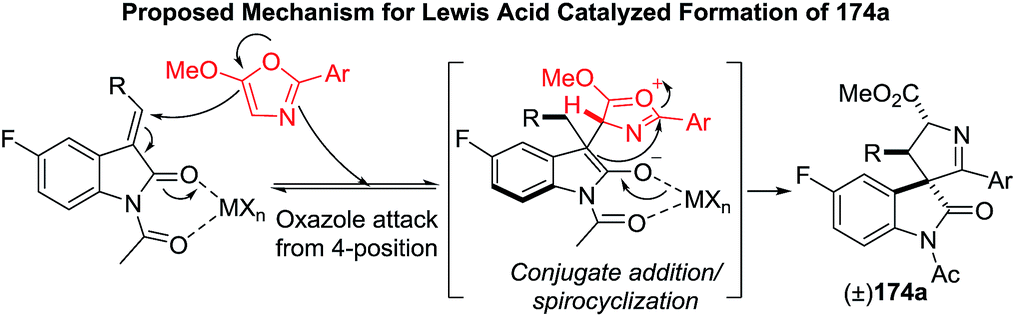
The use of a chiral scandium(III)–indapybox/BArF complex provided the enantioenriched spirooxindole-1-pyrroline 174a (86:14 er), and a ligand-induced reversal of diastereoselectivity was observed (Scheme 54).
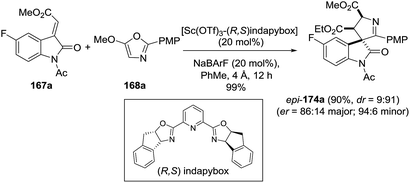 |
| | Scheme 54 Synthesis of enantioenriched pyrroline epi-174a. | |
The proposed mechanism is depicted in the Scheme 55.
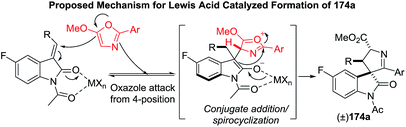 |
| | Scheme 55 Proposed mechanism for Lewis acid catalyzed spirooxindole-1-pyrrolines. | |
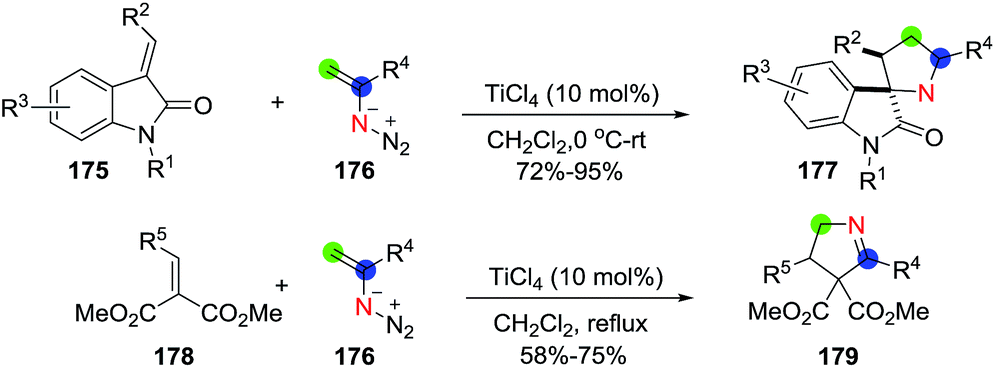
Chiba et al.112 carried out a TiCl4-catalyzed addition of vinyl azides (176) onto α,β-unsaturated carbonyl compounds to construct the 1-pyrroline (Scheme 56). Employing 3-alkylidene-2-oxoindoline (175) as the α,β unsaturated carbonyl and TiCl4 (10 mol%), the 1-pyrroline-containing spiro structures 177 were obtained in excellent yields (72–95%). When a 2-alkylidenemalonate (178) was used, a series of 1-pyrrolines with various aryl, alkenyl and alkyl motifs (179) were obtained in satisfactory yields (58–75%). The proposed mechanism involves titanium-catalyzed conjugate addition of vinyl azides to the α,β unsaturated carbonyl and subsequent denitrogenative ring-expansion of the transient azidocyclobutane intermediates formed to obtain the 1-pyrroline ring.
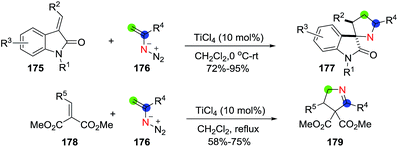 |
| | Scheme 56 Titanium-catalyzed construction of 1-pyrrolines using vinyl azides. | |
2.2. Synthesis of 2-pyrrolines
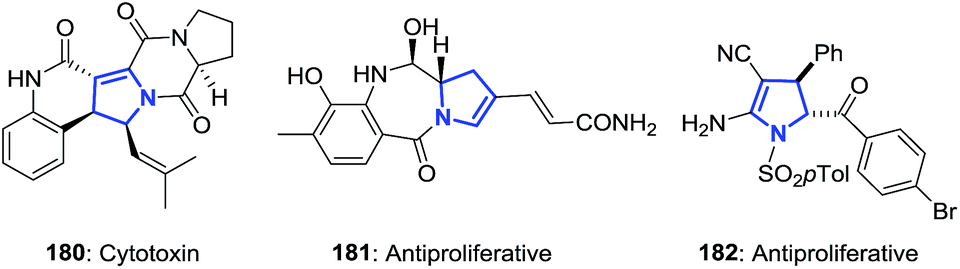
The 2-pyrrolines ring is found in many biologically-active compounds (Fig. 4). Notable examples include the cytotoxin spirotryprostatin B (180, showing complete inhibition of the cell cycle progression of tsFT210 cells in the G2/M phase and growth inhibition toward leukemia K562 and HL-60 cells);7 the antibiotic and cytotoxin anthramycin (181, produced by Streptomyces refuineus);8 and the synthetic, highly substituted racemic 2-pyrroline 182 demonstrated to have antiproliferative activity.16
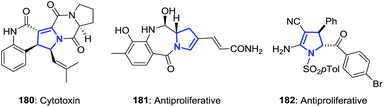 |
| | Fig. 4 Selected examples of biologically active 2-pyrrolines. | |
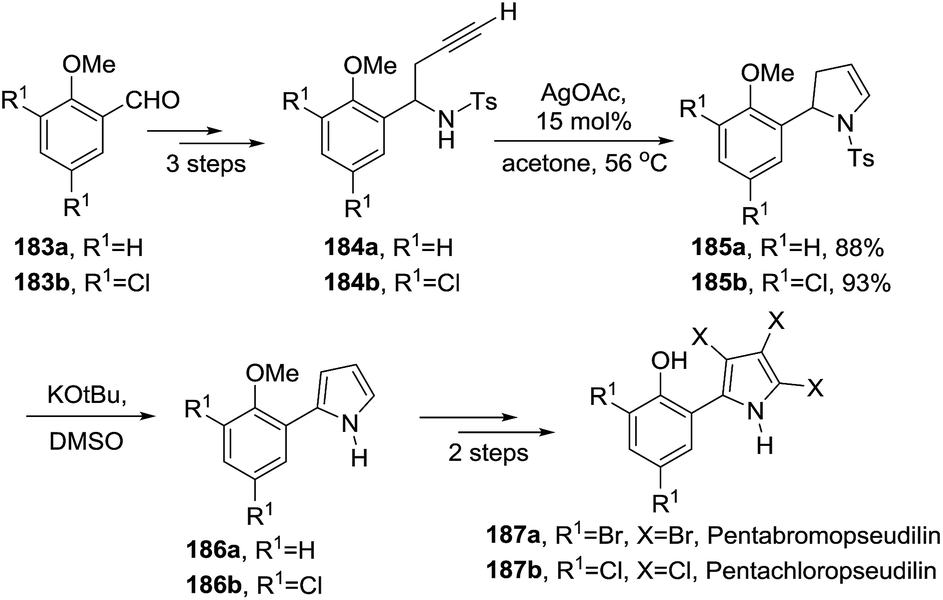
An example of 2-pyrroline used as an intermediate towards a more complex heterocycle was described by Knolker et al.30 The antibiotics pentabromopseudilin 187a and pentachloropseudilin 187b were synthesized in seven steps from the benzaldehydes 183a and 183b respectively (Scheme 57). Both syntheses made use of a silver(I)-catalyzed cyclization of N-tosylhomopropargylamines 184a and 184b that led to 2-pyrrolines 185a and 185b. These pyrrolines were oxidized to the pyrroles 186a and 186b which were finally transformed in pentabromo- (187a) and pentachloropseudilin (187b) in 23% and 19% overall yield, respectively.
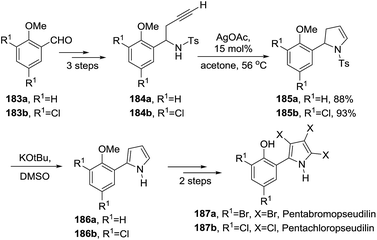 |
| | Scheme 57 Synthesis of pentabromopseudilin 187a and pentachloropseudilin 187b through the 2-pyrrolines 186a and 186b, respectively. | |
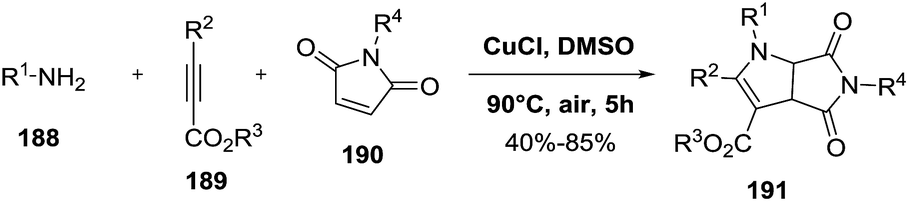
2.2.1. Synthesis of 2-pyrrolines by copper catalysis. Zhu et al. described the copper-catalyzed oxidative cyclization of maleimides 190 with different amines 188 and alkynyl esters 189 to give a variety of 2-pyrrolines 191 (Scheme 58).113 CuCl in dimethylsulfoxide (DMSO) at 90 °C in presence of air were the most effective conditions. The presence of oxygen in the reaction medium was necessary to effect the transformation. The reaction has a good tolerance towards different functional groups, allowing both N-protected and free NH maleimide, amines containing electron-donating and electron-withdrawing substituents, and alkyl- as well as aryl-substituted alkynyl esters. In all cases, the desired product was obtained in good to moderate yield (40–85%). A radical mechanism was proposed.
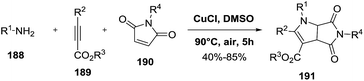 |
| | Scheme 58 Cu-catalyzed synthesis of fully substituted 2-pyrrolines. | |
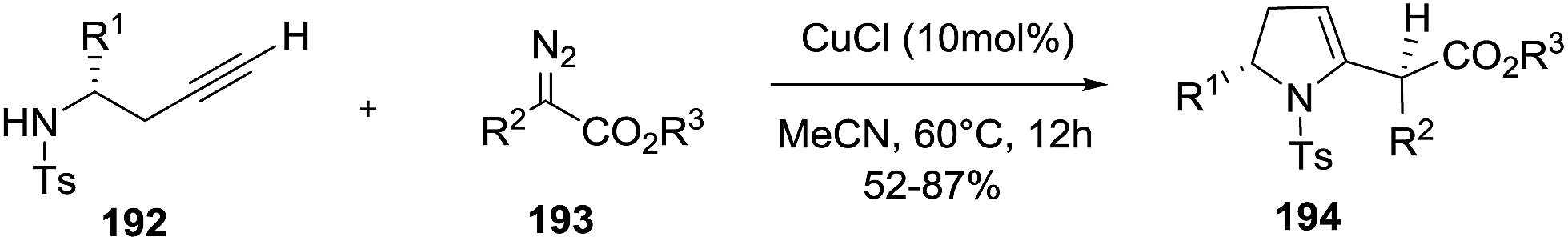
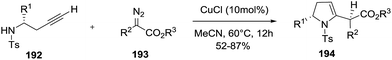
Sun et al. reported the copper-catalyzed 5-exo-dig hydroamination of homopropargyl sulfonamides 192 with diazo compounds 193 to give 2-pyrrolines 194 (Scheme 59).114 The possible first step involves formation of allenoate intermediates by metal-catalyzed cross-coupling of an alkyne with diazo compounds which then undergo a 5-exo-dig intramolecular hydroamination.
 |
| | Scheme 59 Copper-catalyzed tandem annulation towards 2-pyrrolines. | |
When chiral homopropargyl sulfonamides with a variety of substituent adjacent to the N-atom are subjected to the optimal reaction conditions (10 mol% CuCl in acetonitrile), 2,5 disubstituted 2-pyrrolines are obtained with good yields (52–87%) and good diasteroselectivity (dr typically 4:1). A library of twenty-four 2,5-disubstituted 2-pyrrolines 194 was synthesized.
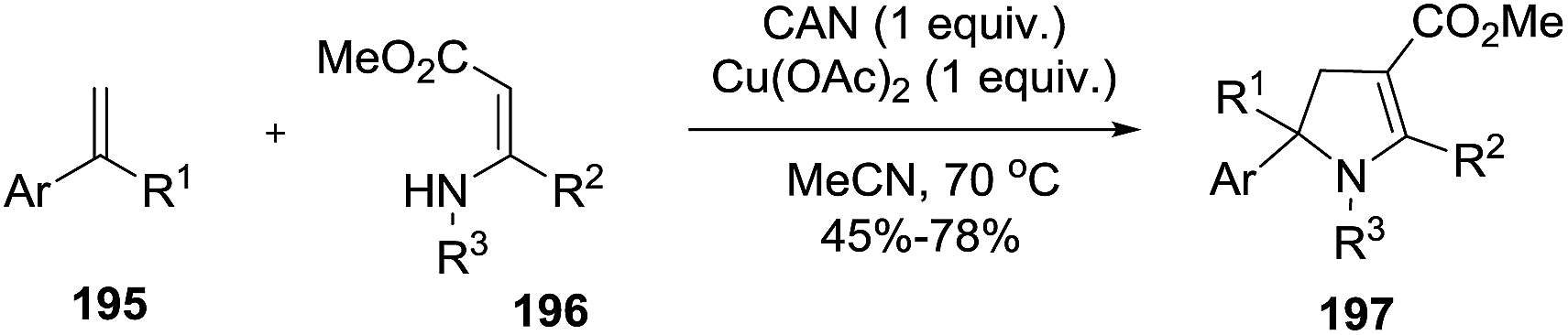
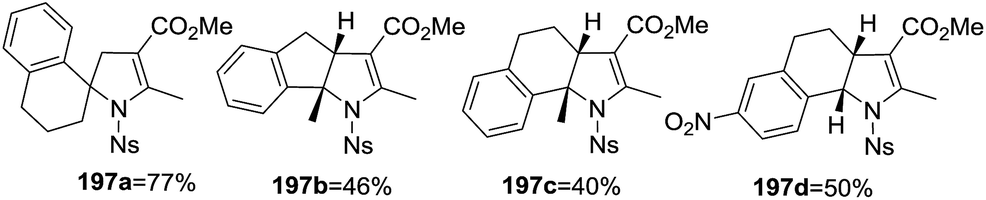
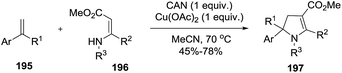
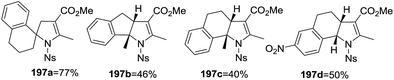
An alternative synthesis of multi-substituted 2-pyrrolines 197 was described by Yoshida et al. in 2016 (Scheme 60).115 They reported the oxidative radical cyclization of N-sulfonyl β-enamino esters 196 with different aryl-substituted alkenes 195. When cyclic alkenes are used, spirocyclic and tricyclic dihydropyrroles like 197a, 197b, 197c, and 197d were obtained (Scheme 61). Under optimal conditions with ceric ammonium nitrate and Cu(OAc)2 as oxidants, the desired products were obtained in moderate to high yields (45–78%).
 |
| | Scheme 60 Oxidative radical cyclization of N-sulfonyl β-enamino esters with alkenes towards 2-pyrrolines. | |
 |
| | Scheme 61 Spirocyclic and tricyclic dihydropyrrole obtained by the reaction with cyclic alkenes. | |
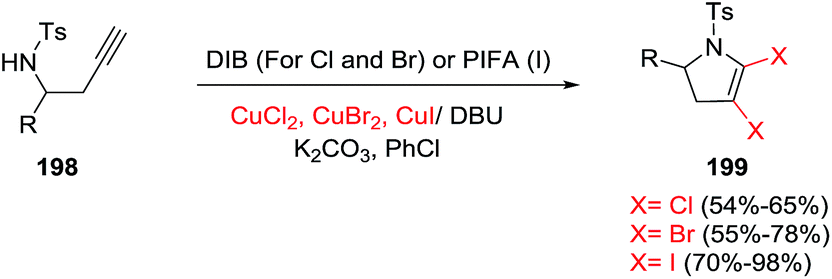
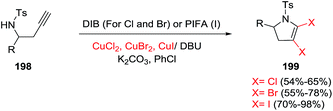
In 2017, the Xiang research group reported the synthesis of 4,5-dihalo-substituted 2,3-dihydropyrroles 199 from a variety of homopropargyl sulfonamides 198 (Scheme 62).116 This transformation uses two equivalents of copper halides and one equivalent of a hypervalent iodine compound, in the presence of an organic base (such as DBU) and an inorganic base (such as K2CO3) in chlorobenzene as the solvent. The combination of CuI and PIFA gives the 2,3-dihydro-4,5-diiodopyrroline products. The dibromopyrrolines (dichloropyrrolines) are obtained using CuBr2 (CuCl2) and oxidant DIB [PhI(OAc)2]. A breadth of different aromatic, heteroaromatic, and alkyl-substituted alkynyl amines under the former conditions give the diiodopyrrolines in very good to excellent yields, defining a broad scope to the reaction. In general, the diiodopyrrolines were obtained in higher yields than the dibromopyrroline, with the dichloropyrroline obtained in the lowest yield.
 |
| | Scheme 62 Transformation of homopropargyl sulfonamides into dihalo-2-pyrrolines. | |
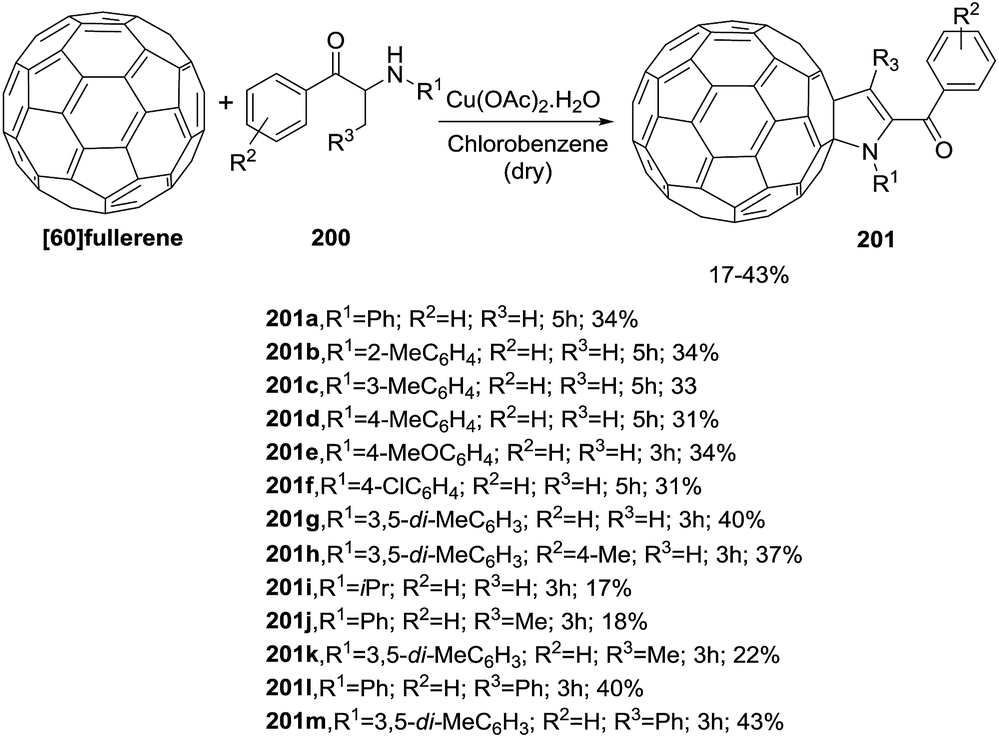
Wang et al. reported a copper-catalyzed synthesis of 2-fulleropyrrolines 201 bearing a trisubstituted or a tetrasubstituted C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond promoted by copper from α-amino ketones 200 and [60]fullerene (Scheme 63).117 This is the first synthesis of a 2-fulleropyrrolines with R3 = H. The yields are in the range of 17% to 43%.
C bond promoted by copper from α-amino ketones 200 and [60]fullerene (Scheme 63).117 This is the first synthesis of a 2-fulleropyrrolines with R3 = H. The yields are in the range of 17% to 43%.
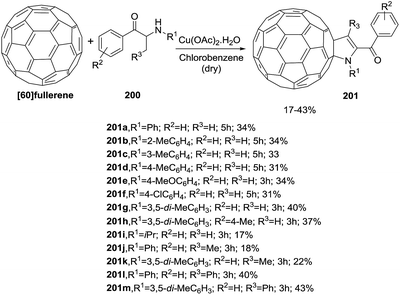 |
| | Scheme 63 Synthesis of 2-fulleropyrrolines 201. | |
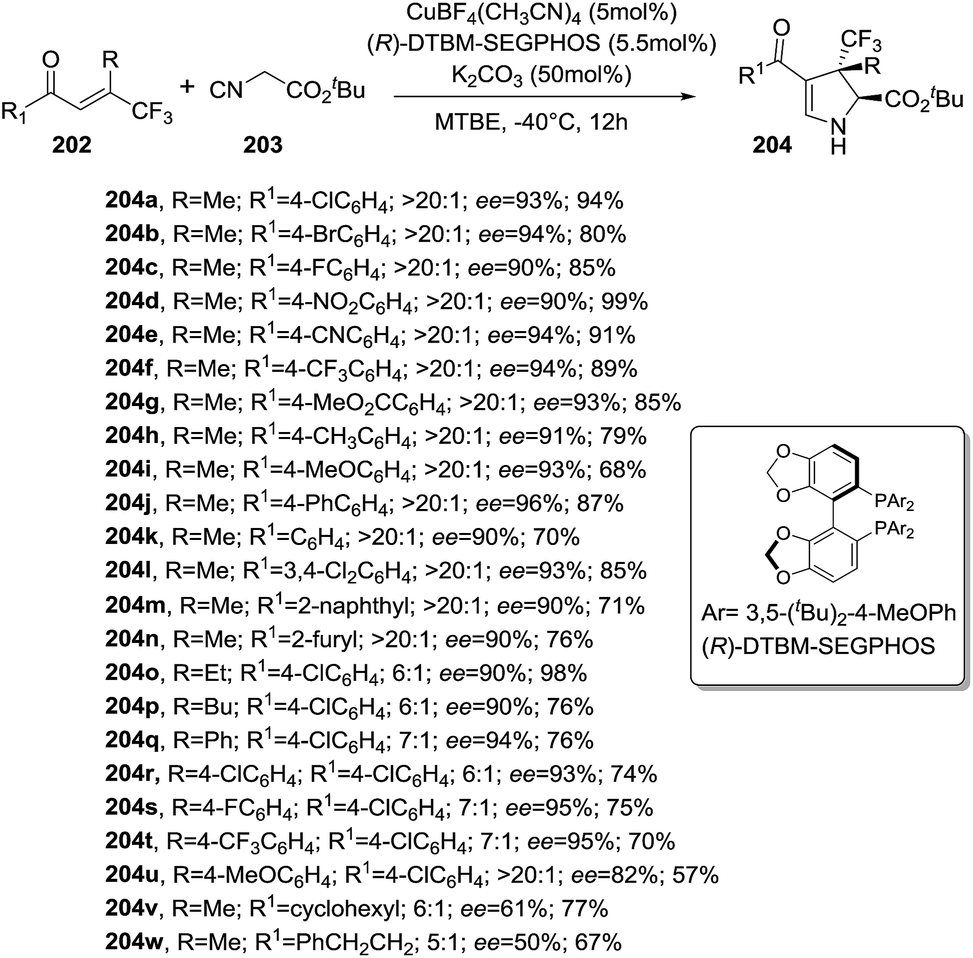
Recently, Zhang et al. disclosed an enantioselective [3 + 2] cycloaddition synthesis of 2-pyrrolines 204 bearing a trifluoromethylated all-carbon quaternary stereocenter from α-tert-butyl isocyanoacetates 203 and β-trifluoromethyl β,β-disubstituted enones 202 in the presence of a copper catalysis (Scheme 64).118 The combination of Cu(CH3CN)4BF4 and the chiral ligand (R)-DTBM-SEGPHOS provided high diastereoselectivities (>20:1) and enantioselectivities (50–96% ee) along with very good yields (57–99%). The protocol tolerates a variety of enones 202 bearing electron-rich or -poor substituted aryl groups (R1) but good diasteroselectivities are only obtained when R = Me. The presence of the β-trifluoromethyl group in the enone 202 is necessary for attaining high reactivity and stereocontrol.
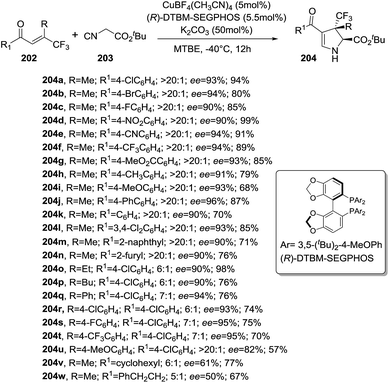 |
| | Scheme 64 Synthesis of 2-pyrrolines 204 by enantioselective [3 + 2] cycloaddition. | |
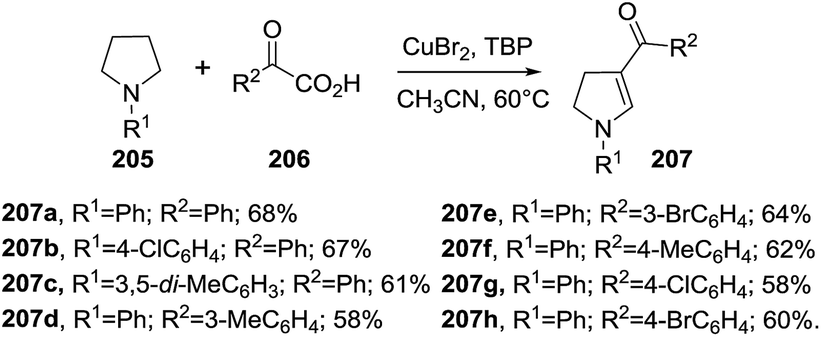
Fan et al. reported a copper-catalyzed cascade reaction synthesis of 3-acyl-2-pyrrolines 207 from pyrrolidines 205 and 2-oxo-2-arylacetic acids 206 (Scheme 65).119 The reaction is believed to occur through a C(sp3)–H bond dehydrogenation of the pyrrolidine to give an enamine intermediate which is subsequently cross-coupled with the acyl species coming from the decarboxylation of 2-oxo-2-arylacetic acid 206. It is postulated that the copper catalyst plays an essential role in the dehydrogenation, decarboxylation and cross coupling steps. N-Aryl substituted pyrrolidines 205 and various 2-oxo-2-arylacetic acids 206 deliver the 3-acyl-2-pyrrolines 207 in good yields (58–68%). N-Alkyl substituted pyrrolidines give pyrroles instead of 3-acyl-2-pyrrolines.
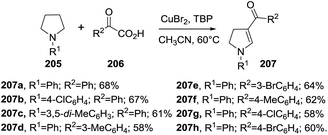 |
| | Scheme 65 Copper-catalyzed synthesis of 3-acyl-2-pyrrolines 207. | |
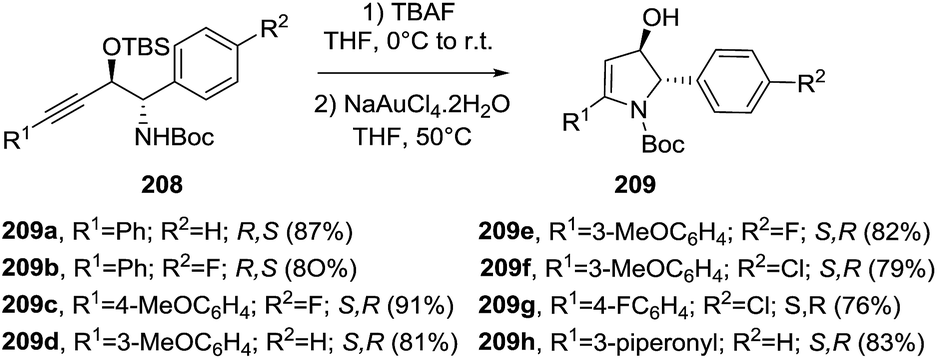
2.2.2. Synthesis of 2-pyrrolines by gold catalysis. The Rutjes group developed an enantioselective gold-catalyzed 5-endo-dig cyclization into the 2,3-dihydro-3-hydroxy-substituted 2-pyrrolines 209 using the alkynyl-substituted amino alcohols 208 as the starting material (Scheme 66).120 This starting material originated through enzyme-catalyzed hydrocyanation of the α,β-acetylenic aldehydes. Deprotection of the hydroxyl group with TBAF followed by reaction with NaAuCl4·2H2O (10 mol%) in THF at 50 °C gave the 4-hydroxy-2-pyrrolines 209 in good yields over the two steps.
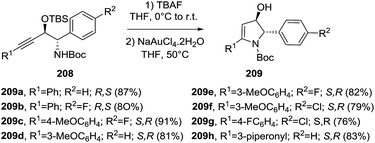 |
| | Scheme 66 Gold-catalyzed cyclization of substituted alkyne-containing amino alcohols towards 2-pyrrolines. | |
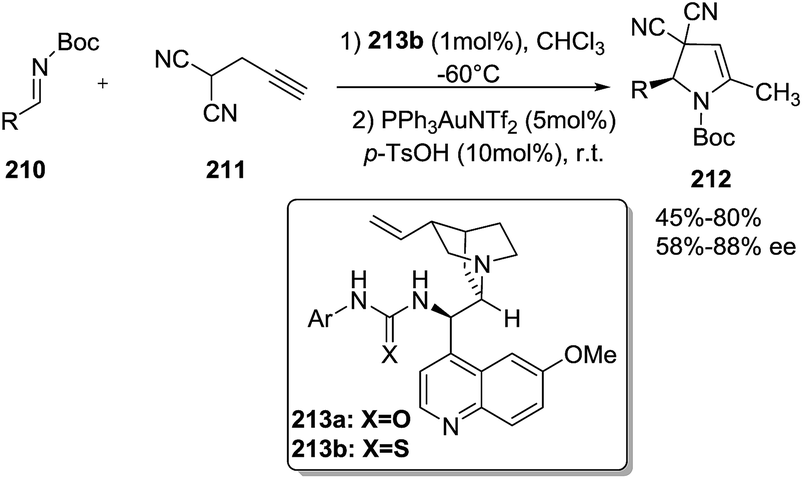
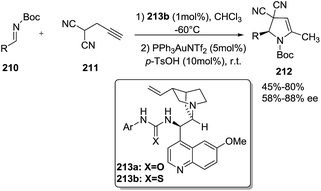
Jørgensen reported121 an enantioselective synthesis of 2-pyrrolines 212 from propargyl malononitrile (211) and N-Boc-protected imines 210 through an organocatalytic Manich-type reaction followed by gold-catalyzed alkyne hydroamination and subsequent isomerization (Scheme 67). This one-pot sequential protocol that combine transition-metal catalysis with organocatalysis furnished the 2-pyrrolines 212 in good yields (up to 80%), high selectivities endo/exo (10 > 1), and enantioselectivities up to 88%. The protocol works only for terminal alkynes. Once the organocatalyst 213b forms the Mannich product, the latter is protonated by the excess of p-TsOH, and in this way deactivation of the gold catalyst is prevented. The two catalytic systems are compatible with one-pot operation.
 |
| | Scheme 67 Asymmetric one-pot sequential organo- and gold catalysis for the enantioselective synthesis of 2-pyrrolines 212. | |
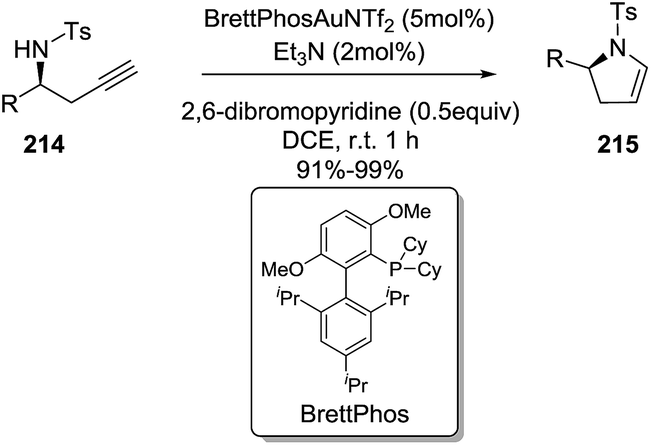
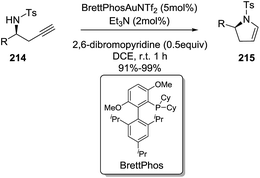
Ye et al. reported an enantioselective gold-catalyzed synthesis of 2-pyrrolines 215 from chiral homopropargyl sulfonamides 214 using a combination of bases as catalytic additives.122 The intramolecular hydroamination occurs via a 5-endo-dig cyclization, through an anti-Markovnikov addition. The optimal reaction conditions use BrettPhos·AuNTf2 (5 mol%), a combination of Et3N (2 mol%) and 2,6-dibromopyridine (0.5 equivalent) as the basic additives, in 1,2-dichloroethane at room temperature (Scheme 68). The 2,3-dihydropyrroles 215 are isolated in excellent yields and without epimerization of the stereocenter. This practical procedure offers a route to both 2-pyrrolines enantiomers, simply by selecting the starting chiral homopropargyl sulphonamides (R or S) 214. The reaction showed wide generality. Diversely substituted homopropargyl sulfonamides produced the 2-pyrrolines in excellent yields (91–99%).
 |
| | Scheme 68 Enantioselective synthesis of 2-pyrrolines via gold-catalyzed cycloisomerization of chiral homopropargyl sulfonamides. | |
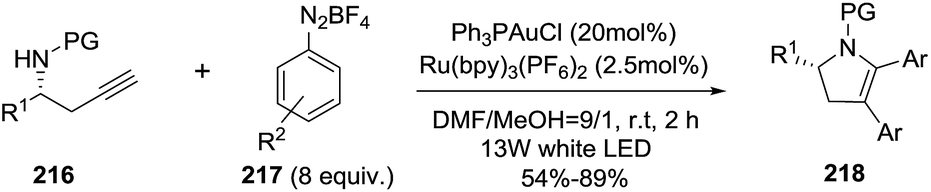
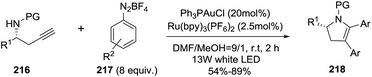
Later, the same group reported an enantioenriched access to multisubstituted 2-pyrrolines 218 by a combination of gold catalysis and visible-light photoredox catalysis.123 Using different chiral homopropargyl sulfonamides 216 and a variety of aryldiazonium salts 217 in the presence of Ph3P·AuCl and a visible light photocatalysts such Ru(bpy)3(PF6)2, and irradiating with 13 W white LEDs, a bis-arylative 5-endo-dig cyclization led to enantioenriched 2,3-dihydropyrroles with high enantiomeric excesses (96–99% ee) and moderate yields (54–89%) (Scheme 69). The proposed mechanism is an Au(I)/Au(III) redox cycle accomplished by visible-light photoredox catalysis, without using a strong oxidant, and accomplished in a mild and selective manner.
 |
| | Scheme 69 Dual photoredox/gold catalysis towards enantioenriched 2-pyrrolines. | |
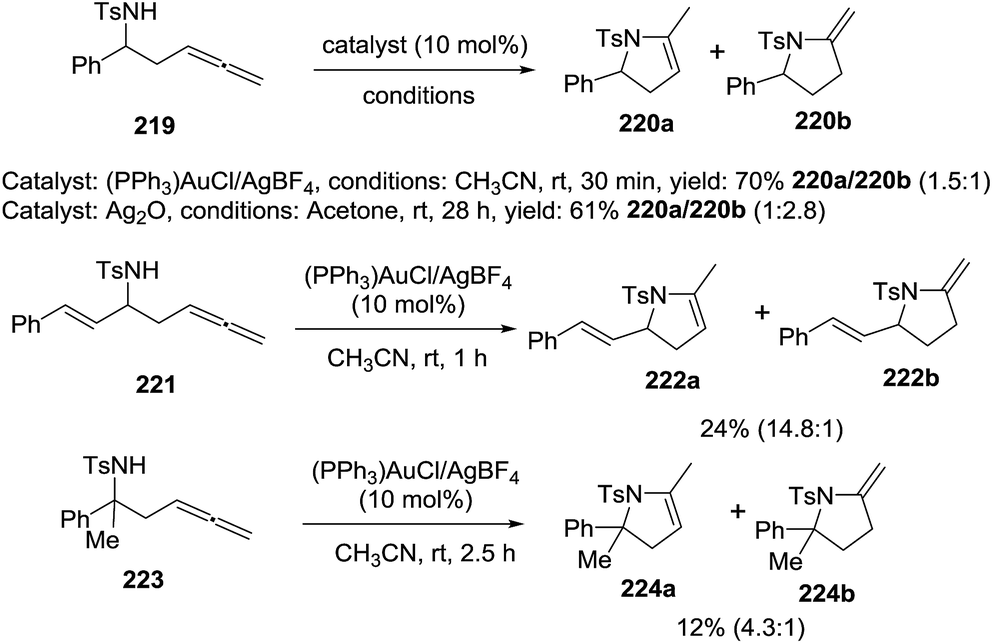
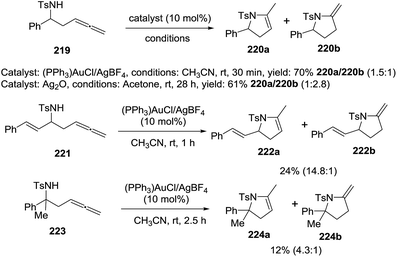
Pyne et al. reported the cyclization of β-amino allenes 219 by gold and silver catalysis to afford the pirrolines 220a via a 5-endo-dig cyclization to 220b and subsequent isomerization to the more stable isomer 220a (Scheme 70).124 Under gold catalysis, the optimal conditions consist of using 10 mol% of (PPh3)AuCl/AgBF4 in acetonitrile at room temperature. β-amino allenes 219 provided the pyrroline 220a and the pyrrolidine 220b with 70% isolated yield in a ratio (220a/220b = 1.5:1). Treating the same allene with Ag2O in acetone a room temperature generated a mixture 220a/220b = 1:1.28 in 61% overall yield. The β-amino allene 221, substituted with the conjugated cinnamyl group, was subjected to the optimized gold catalyzed conditions obtaining a mixture between the pyrroline 222a and pirrolidine 222b in low yield but favoring the formation of 222a. Under optimized conditions, the β,β-disubstituted allene 223 produced the pyrroline 224a and the isomer 224b in low yield (12%) in a ratio 224a/224b = 4.3:1.
 |
| | Scheme 70 Gold- and silver-catalysed cyclisation reactions of β-amino allenes. | |
Hou et al.125 have studied the gold and palladium catalyzed intramolecular hydroamination of N-(3-butynyl) sulfonamides leading to 2,3-dihydropyrroles (see Scheme 83, Subsection 2.2.7.: Synthesis of 2-pyrrolines by palladium catalysis).
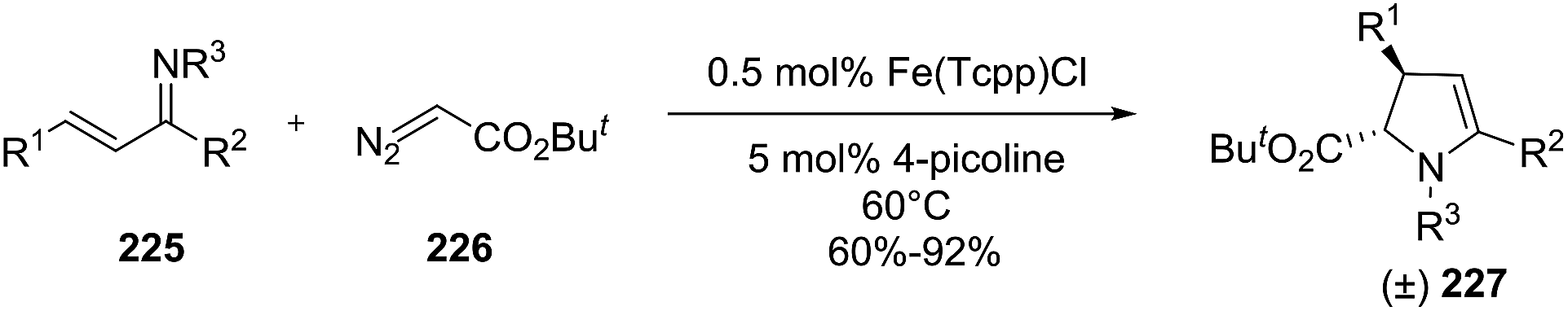
2.2.3. Synthesis of 2-pyrrolines by iron catalysis. Tang et al. reported the diastereoselective synthesis of trans-2,3-disubstituted 2-pyrrolines 227 from α,β-unsaturated imines 225 and alkyl diazoacetate 226 in presence of catalytic tetra(p-chlorophenyl)porphyrin iron chloride (Fe(Tcpp)Cl) and 4-picoline (Scheme 71).126 The formal [4 + 1] annulations occur via an iron carbenoid intermediate, which is readily available from alkyl diazo acetate. The yields are ranging from 60% to 92% with diastereoselectivities higher than 50:1.
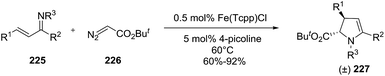 |
| | Scheme 71 Diastereoselective synthesis of 2-pyrrolines via a catalytic formal [4 + 1] annulations. | |
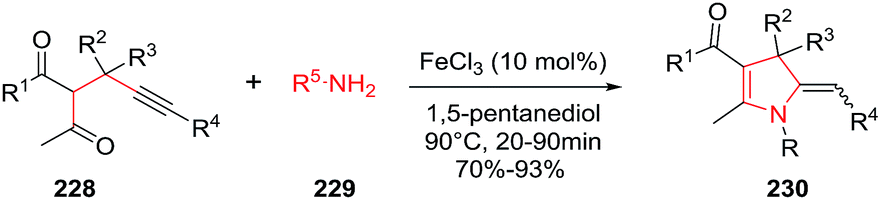
The development of iron-catalyzed organic reactions is attractive since iron is an abundant, cheap, environmentally friendly, and efficient catalyst. In 2014, Bi et al. described the FeCl3 catalyzed stereoselective synthesis of 5-(aryl)alkylidene-4,5-dihydropyrroles 230 through a [4C + 1N] cyclization of 4-alkynyl ketones 228 with primary amines 229 (Scheme 72).127 The optimized conditions used FeCl3 (10 mol%) in 1,5-pentanediol at 90 °C. The protocol tolerates a wide range of 4-alkynyl ketones to give the multisubstituted 2-pyrroline 230 in very good to excellent yields (70–93%) and high stereoselectivities (Z/E ratio typically 1:0.05). The Z-isomer of the exocyclic alkene dominates the product mixture. The procedure is also useful to obtain 3-spirodihydropyrroles 230a and 230b from the cyclization of 4-alkynyl ketones containing a cyclic quaternary carbon center, again using primary amines to initiate the cyclization (Scheme 73).
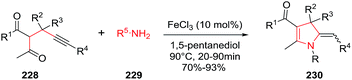 |
| | Scheme 72 Iron-catalyzed [4C + 1N] cyclization of 4-acetylenic ketones with primary amines. | |
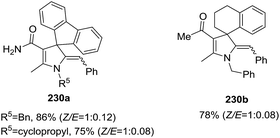 |
| | Scheme 73 Synthesis of 3-spiro 2-pyrrolines by Fe-catalyzed [4C + 1N] cyclization. | |
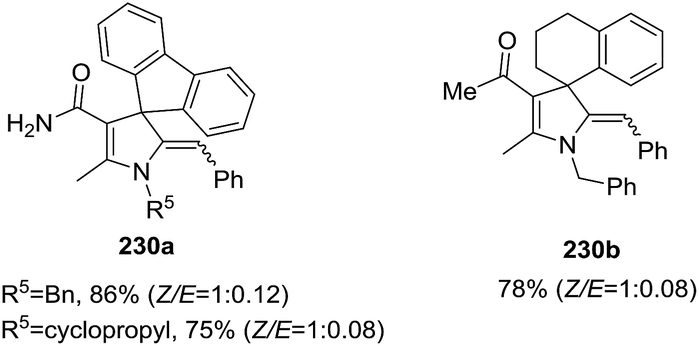
Recently, the Rueping group reported an intramolecular hydroamination of α-allenic amines 231 to 2-pyrrolines 233 catalyzed by an air- and moisture-stable iron cyclopentadienone complex (Scheme 74).128 The protocol consists in adding 5 mol% of iron complex 232 and 6 mol% of trimethylamine N-oxide to Cbz-allenic amines 231 in THF at 70 °C for 24 h. The yields obtained varied from 57% to 86% of the 2-pyrrolines 233.
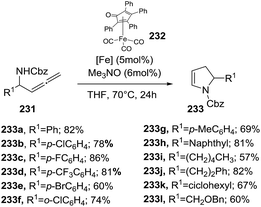 |
| | Scheme 74 Iron-catalyzed hydroamination of α-allenic amines 231 towards 2-pyrrolines 233. | |
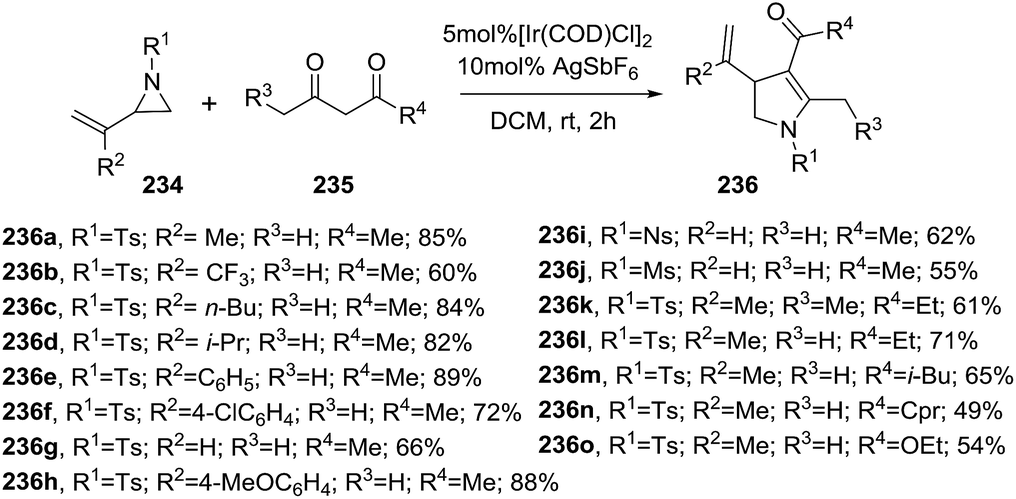
2.2.4. Synthesis of 2-pyrrolines by iridium catalysis. Zhang et al. described an iridium-catalyzed synthesis of 2-pyrrolines 236 from vinylaziridines 234 and α-unsubstituted 1,3-dicarbonyls 235 through a domino-ring-opening cyclization in good yields (49–89%)(Scheme 75).129 The reaction conditions involve the use of 5 mol% of [Ir(COD)Cl]2, 10 mol% of AgSbF6 as additive in dichloroethane at room temperature for 2 h. Alkyl or aryl substituents on the vinylaziridine (R2) are well tolerated. Regarding the N-protecting group (R1), tosyl, nosyl and mesyl provide the 2-pyrrolines with similar yields. Symmetrical and unsymmetrical 1,3 diketones produced the corresponding 2-pyrrolines with excellent regioselectivity. β-ketoesters are suitable substrates.
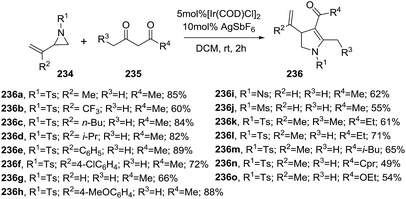 |
| | Scheme 75 Reaction between vinylaziridines 234 and 1,3-dicarbonyls 235 towards 2-pyrrolines 235. | |
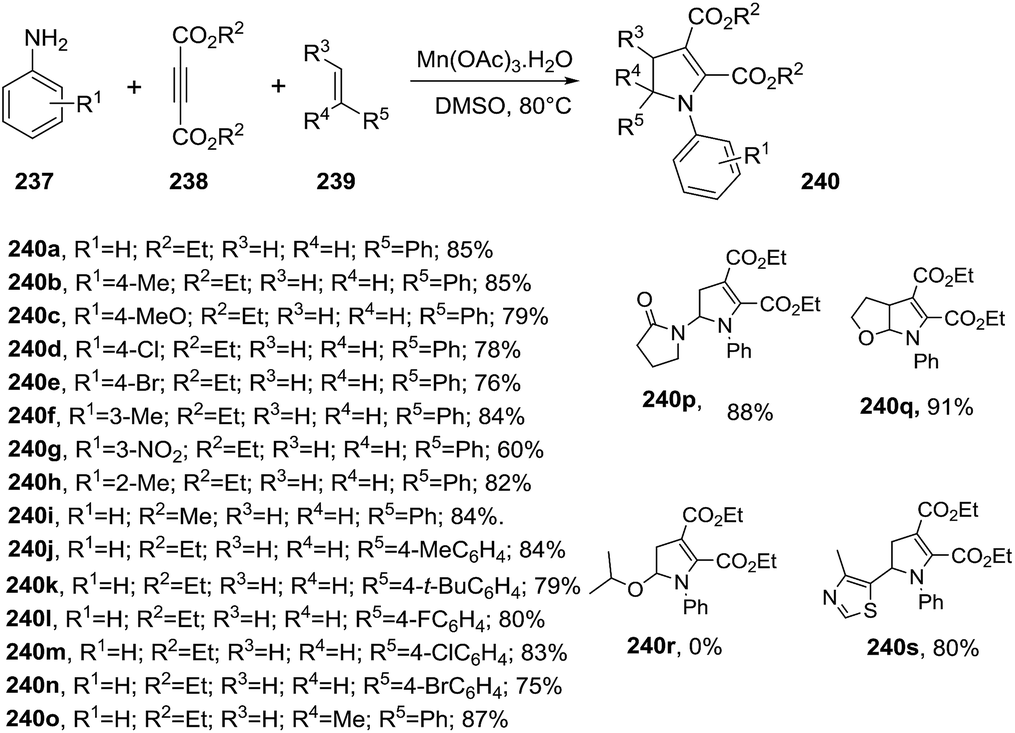
2.2.5. Synthesis of 2-pyrrolines promoted by manganese. Zheng et al. disclosed the first oxidative cyclization promoted by Mn(OAc)3 to access to polysubstituted 2-pyrrolines 240 from anilines 237, alkynes esters 238 and alkenes 239 (Scheme 76).130 This free radical multi-component reaction consists in adding 2 equiv. of Mn(OAc)3 to the three substrates in DMSO at 80 °C. The reaction accepts a broad scope of anilines furnishing 2-pyrrolines in good to excellent yields irrespective of the electronic nature or position of substituent. Regarding the alkenes 239, the methodology displays a broad scope for terminal olefins with yields ranging from 60% to 91%. Alkyl amines and 1,2 disubstituted alkenes fails to delivers the 2-pyrrolines.
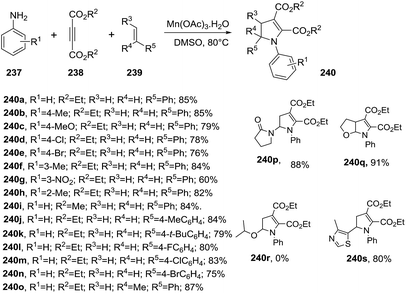 |
| | Scheme 76 Synthesis of polysubstituted 2-pyrrolines 240. | |
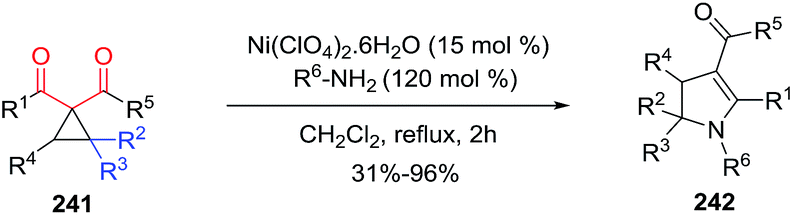
2.2.6. Synthesis of 2-pyrrolines by nickel catalysis. France et al.131 developed a general synthetic approach to 4-carboxy- and 4-keto-2,3-dihydropyrroles 242 using Ni(ClO4)2·6H2O as a Lewis acid catalyst via nucleophilic primary amine ring-opening cyclizations of donor–acceptor (D–A) cyclopropanes 241 (Scheme 77). The optimized conditions were 15 mol% of Ni(ClO4)2·6H2O with 1.2 equiv. of the primary amine in refluxing CH2Cl2 or 1,2-dichloroethane. In this way, several primary amine nucleophiles and different substituted D–A cyclopropanes 241 provided highly substituted 2,3-dihydropyrroles 242 with an electron-withdrawing group in position four in good to excellent yields (31–96%). The particularity of 2,3 dihydropyrroles bearing an electron-withdrawing group at position four is the extended conjugation with the enamine that makes possible vinylogous reactivity.
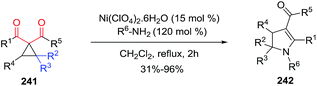 |
| | Scheme 77 Amine ring-opening cyclizations of cyclopropanes to form 2-pyrolines. Donor moiety in blue, acceptor moiety in red. | |
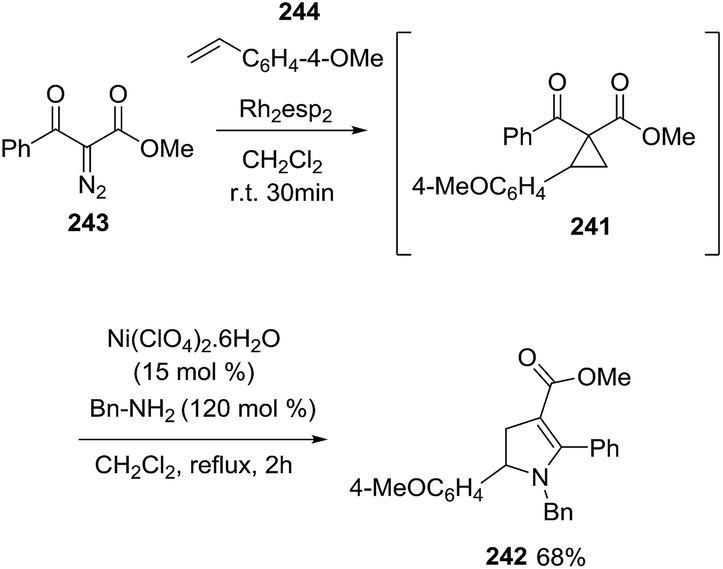
Taking advantage of the fact that the synthesis of the D–A cyclopropanes 241 from alkenes 244 and α-diazo carbonyls 243 and the subsequent pyrroline formation are both carried out in CH2Cl2, the authors performed the tandem one-pot cyclopropanation/amine ring-opening cyclization to obtain the desired vinylogous 2,3-dihydropyrroles 242 in very good yield for the two steps (Scheme 78).
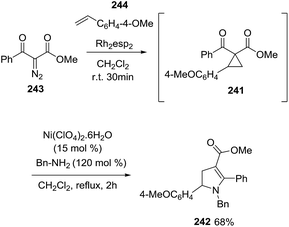 |
| | Scheme 78 One-pot tandem cyclopropanation/amine ring-opening cyclization. | |
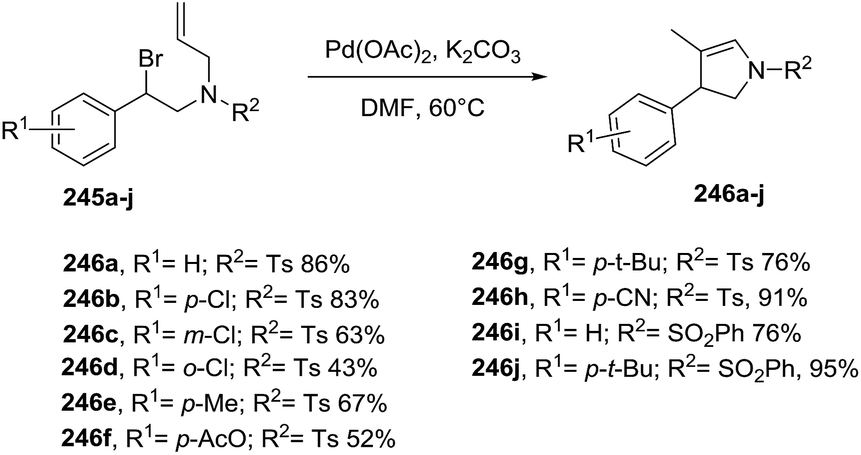
2.2.7. Synthesis of 2-pyrrolines by palladium catalysis. A broad variety of approaches towards the synthesis of 2-pyrrolines using palladium catalysts has been developed. Several of these involve the cyclization of alkenyl-substituted amines. In particular, bromoalkenyl amines provide the 2-pyrroline via Pd-catalysis of the intramolecular Heck reaction. For instance, Pan et al.132 described a ligand-free palladium catalyst for the cyclization of secondary benzylic bromides bearing β-hydrogens 245 (Scheme 79). The optimal reaction conditions were Pd(OAc)2 (1 mol%) and K2CO3 in DMF at 60 °C for 24 h. The desired 2-pyrrolines 246 were obtained with moderate to excellent yields and with high regioselectivities. Suitable substrates included different aromatic substituents, as well as various N-sulfonyl amines.
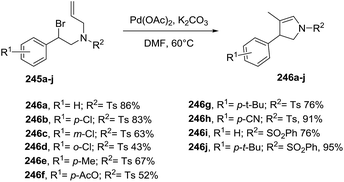 |
| | Scheme 79 Palladium-catalyzed intramolecular Heck reaction towards 2-pyrrolines. | |
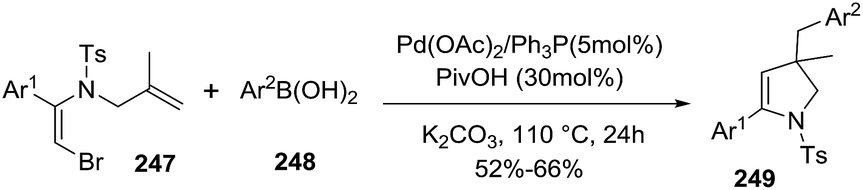
A variety of 2-pyrrolines 249 derivatives were synthesized from the reaction of vinyl bromide 247 with arylboronic acids 248 by a palladium-catalyzed tandem intramolecular Heck/intermolecular Suzuki cross-coupling reaction (Scheme 80).133 The optimized conditions were 5 mol% of Pd(OAc)2, 5 mol% of PPh3, 2 equiv. K2CO3 and 30 mol% of pivalic acid in N,N-dimethylacetamide at 110 °C for 24 hours. The 2-pyrrolines 249 were obtained in moderate to good yields (typically 52–66%).
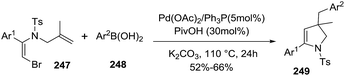 |
| | Scheme 80 Palladium-catalyzed tandem synthesis of 2-pyrrolines. | |
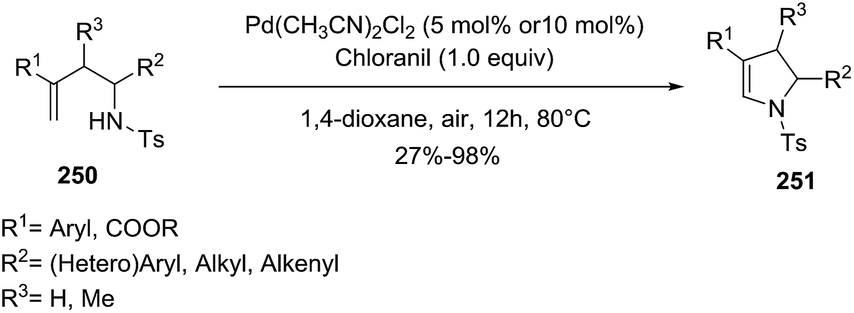
Loh et al.134 described the use of N-homoallyl-N-tosyl amines 250 to generate multisubstituted dihydropyrroles 251 via direct amination of the alkene (Scheme 81). The optimal reaction conditions of this oxidative amination used Pd(CH3CN)2Cl2 (10 mol%) and chloranil (1 equiv.) as the oxidant in 1,4-dioxane, at 80 °C for 24 h under air atmosphere. Different derivatizations at several positions (R1–R3) achieved the pyrroline with moderate to high yield (27–98%), proving the versatility of the reaction. Notably, the 5-endo-trig cyclization proceeded efficiently when the alkenes were substituted with electron-withdrawing groups at R1 position. On the contrary, when R1 is a long alkyl chain the annulations did not take place.
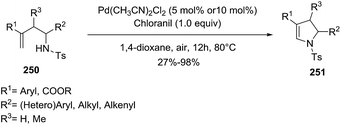 |
| | Scheme 81 Pd-promoted intramolecular oxidative amination towards 2-pyrrolines. | |
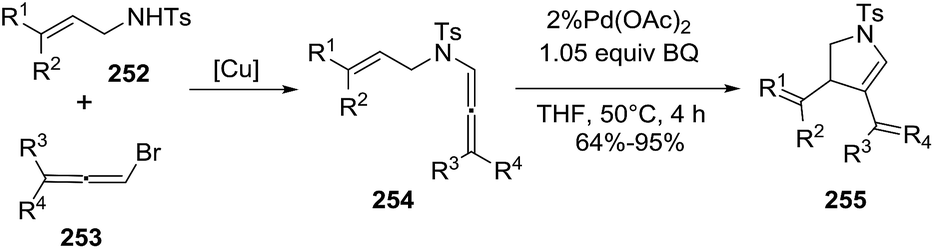
Bäckvall reported the carbocyclization of aza-enallenes 254 in the presence of catalytic Pd(OAc)2 to give the 2-pyrrolines 255 (Scheme 82).135 Several substitution patterns on both the alkene 252 and allene 253 were well tolerated, providing moderate to high yields of the final pyrrolines (64–95%) (Scheme 82).
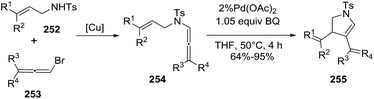 |
| | Scheme 82 Carbocyclization of aza-enallenes 254 promoted by [Pd]. | |
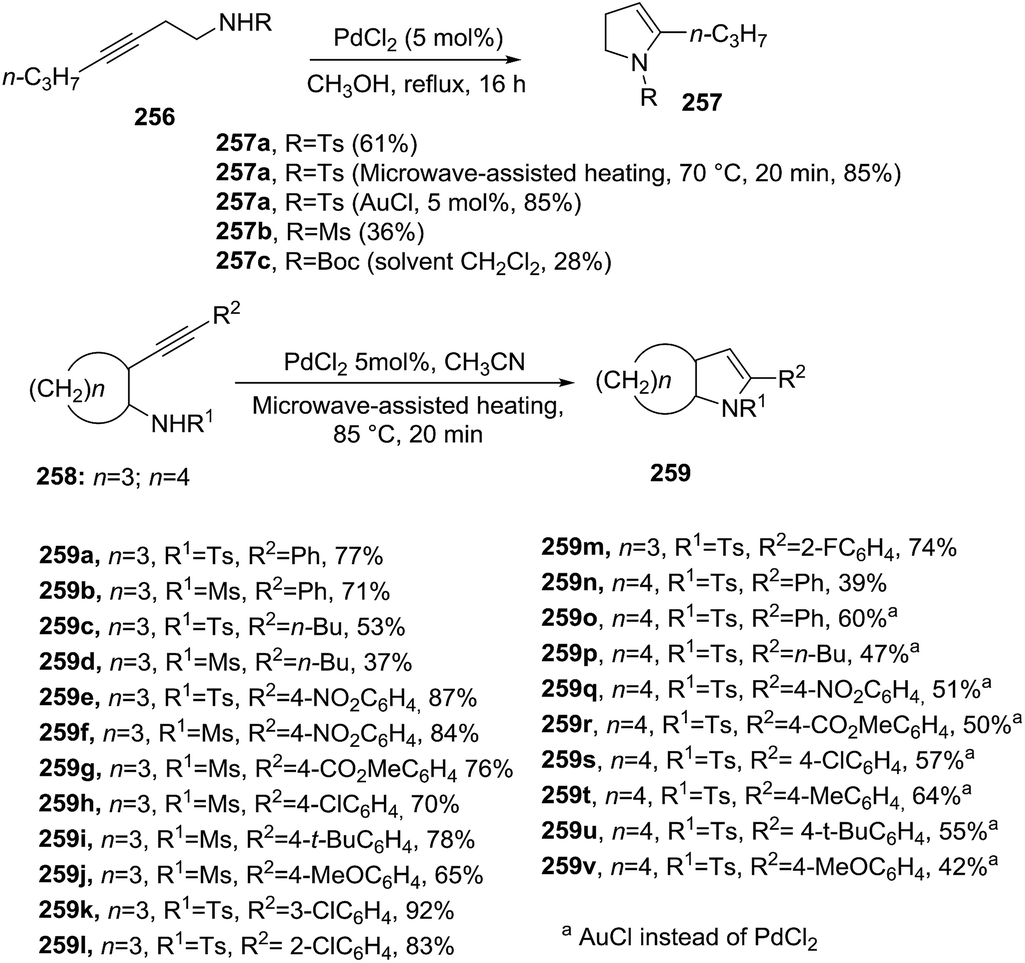
Hou et al.125 studied the Pd-catalyzed intramolecular hydroamination of N-(3-butynyl) sulfonamides leading to 2,3-dihydropyrroles (Scheme 83). Both N-substituted 3-heptynamines 256 and the cyclopentanamine (and cyclohexanamine) analogues 258 underwent 5-endo-dig cyclization providing 5-propyl-2,3-dihydro-1H-pyrroles 257a–c and 259a–v, respectively, with good yields (up to 92%). The optimal reaction conditions for 256 used PdCl2 (5 mol%) in anhydrous methanol at reflux for 16 hours. In contrast, the amines 258 required microwave heating in acetonitrile for 20 minutes to achieve the bicyclic-2-pyrrolines 259 with higher yields. Optimum results were obtained with aryl-alkynes substituted with electron-withdrawing groups on the aromatic ring (R2 = 4-NO2–Ph, 3-Cl–Ph). Interestingly, the annulation of 256 into 257 and 258 into 259 proceeded efficiently in presence of AuCl catalyst.
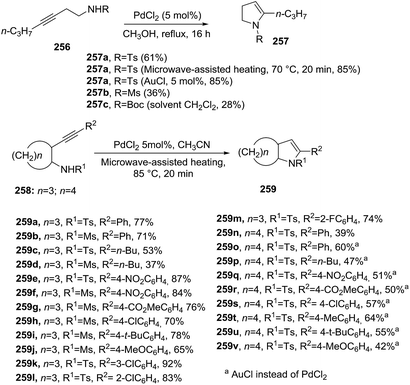 |
| | Scheme 83 Pd-catalyzed intramolecular hydroamination of N-(3-butynyl) sulfonamides towards mono and bicyclic 2-pyrrolines. | |
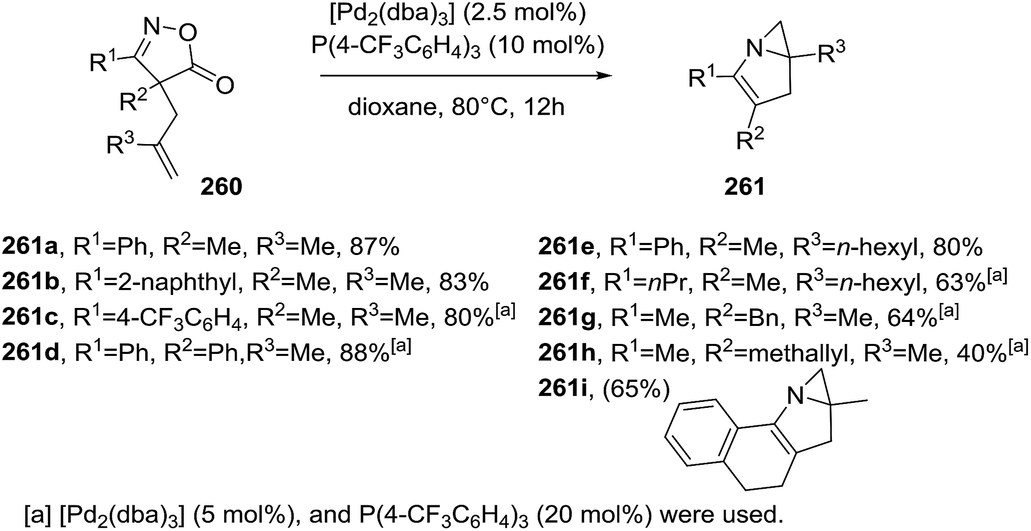
Okamoto et al. developed the Pd-catalyzed intramolecular aziridination of 4H-isoxazol-5-ones 260 leading to N-fused bicyclic aziridines 261a–i (Scheme 84) as a different approach.136 The optimal reaction conditions used Pd2(dba)3 (2.5 mol%) and (4-CF3C6H4)3P (10 mol%) in dioxane at 80 °C for 12 hours, affording the fused bicyclic structures in good yields (63–88%). Aromatic substituents at R1 position proved to be very reactive substrates. Furthermore, the tetracyclic product 261i was obtained in 65% yield. The proposed mechanism starts with an oxidative addition of isoxazolone 260 leading to a six-membered palladacycle A, which readily undergoes decarboxylation towards vinylnitrene/palladium complex B and/or four-membered azapalladacyclobutene intermediate B′ (Scheme 85). Subsequent cycloaddition of these alkenes provides two possible azapalladacycles C and C′, which can undergo the reductive elimination to produce bicyclic aziridine 261.
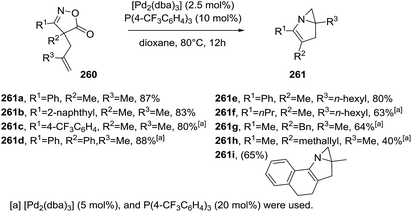 |
| | Scheme 84 Pd-catalyzed intramolecular aziridination of 4H-isoxazol-5-ones 1 leading to N-fused bicyclic aziridines 261. | |
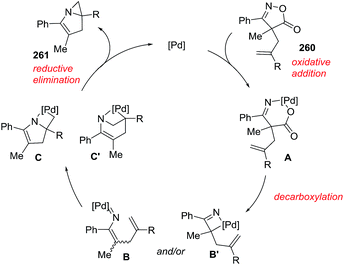 |
| | Scheme 85 Proposed mechanism for the intramolecular aziridation transformation. | |
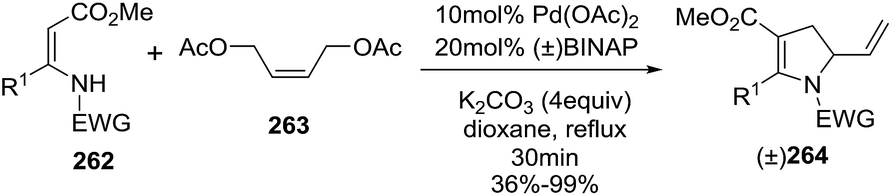
Palladium-mediated intermolecular strategies affording 2-pyrrolines were developed as well. As an example, Yoshida et al. prepared the 2-vinyl-2,3-dihydropyrroles 264 from reaction of the β-enaminocarbonyls 262 with 1,4-diacetoxy-2-butene 263, using Pd(OAc)2 as catalyst and BINAP as an additive (Scheme 86).137 The reaction tolerates a variety of sulfonyl groups as the EWG moiety on nitrogen, and as well different alkyl and aryl substituents on the β-enamino ester. In addition, both E or Z 2-butene-1,4-diol bisacetates can be used to obtain 2-vinyl-2,3-dihydropyrroles 264.
 |
| | Scheme 86 Synthesis of functionalized 2-pyrrolines by oxidative radical cyclization of N-sulfonyl β-enamino esters with alkenes. | |
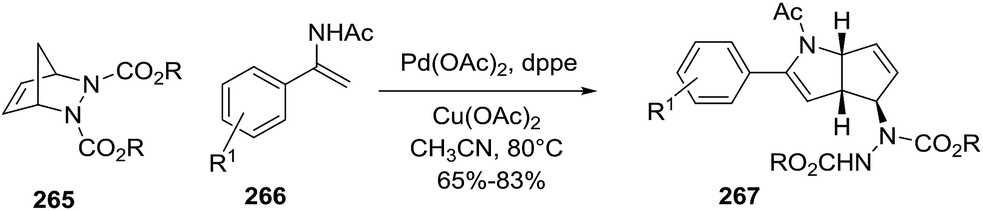
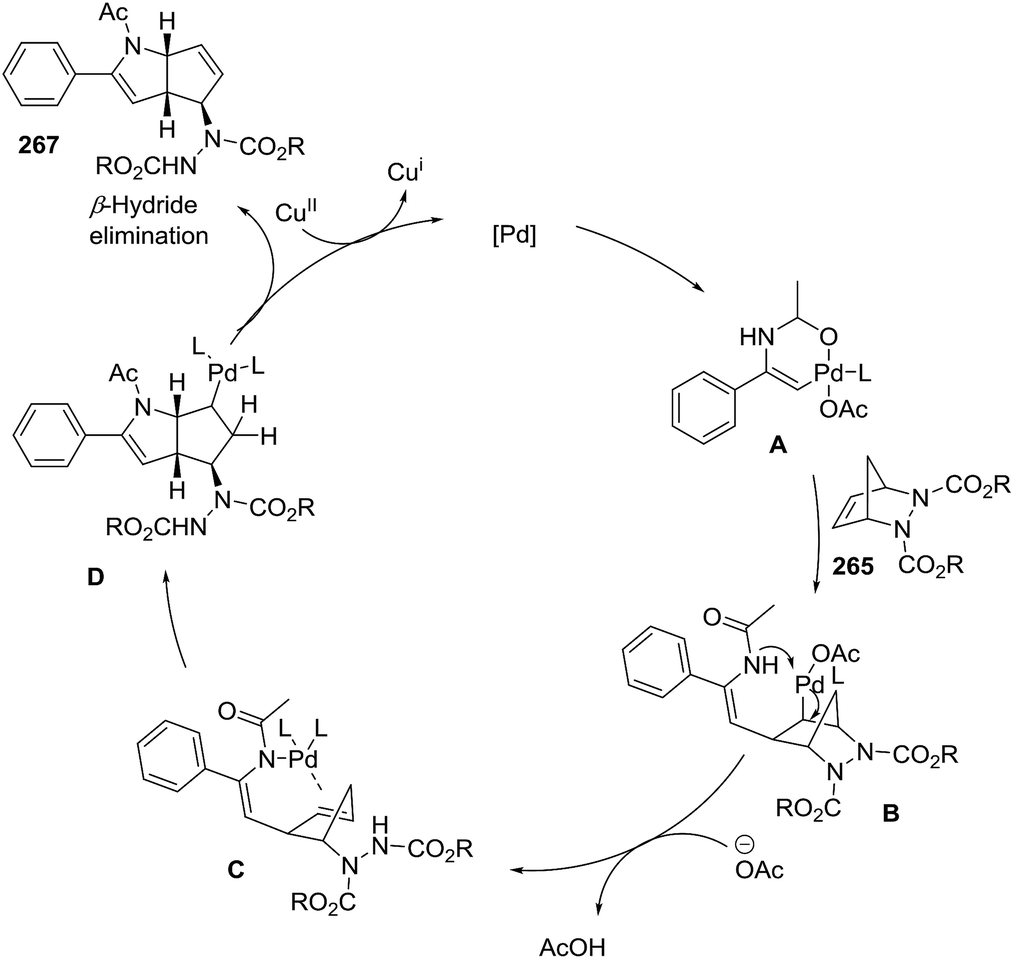
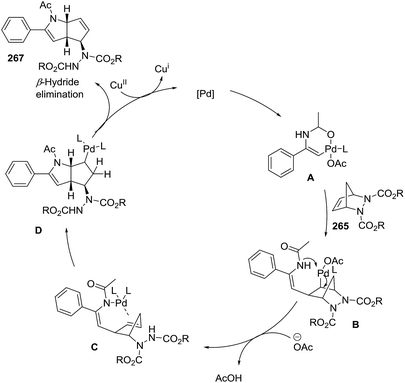
A second example of Pd-catalyzed C–H activation/oxidative is the coupling of enamides 266 with diazabicyclic olefins 265, leading to the cyclopentane-fused N-protected 2-pyrrolines 267 (Scheme 87).138 The optimal reaction conditions (using 265 with enamide 266) were Pd(OAc)2 (10 mol%), Cu(OAc)2 (2.0 equiv.), and dppe (10 mol%) in acetonitrile at 80 °C for 12 hours. In general, aryl enamides with electron-withdrawing substituents on the aromatic ring afforded the highest yields (65–83%). A plausible mechanism would include two stages (Scheme 88). The first is C–H bond activation of the enamide 266 by Pd(OAc)2 to form cyclic intermediate A. Coordination of the alkene 265 to A followed by carbopalladation would produce intermediate B. Aminopalladation and subsequent ring opening of A could generate intermediate C, which would furnish the bicyclic product 267 via a final β-hydride elimination. Reoxidation of palladium by the Cu(II) completes the catalytic cycle.
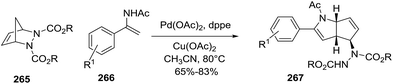 |
| | Scheme 87 Pd-catalyzed CH activation/oxidative coupling towards cyclopentene fused 2-pyrrolines. | |
 |
| | Scheme 88 Proposed mechanism for the Pd-catalyzed CH activation/oxidative coupling. | |
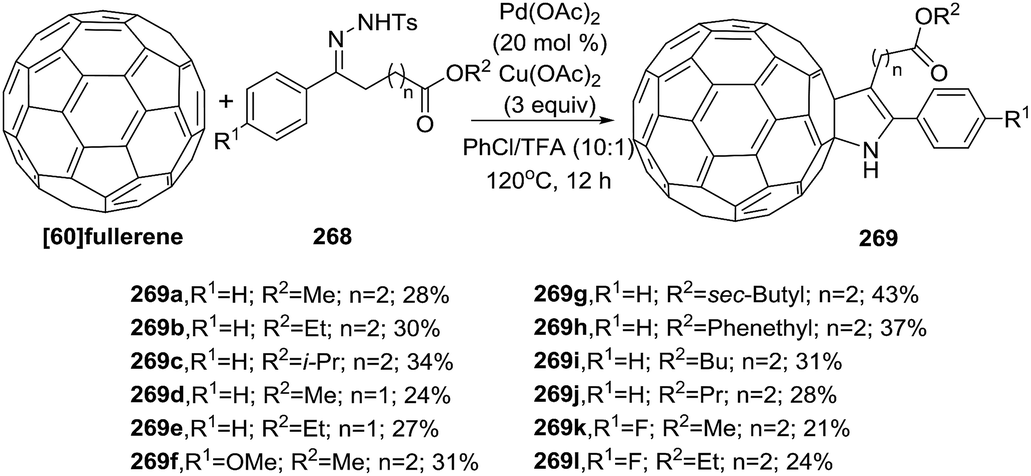
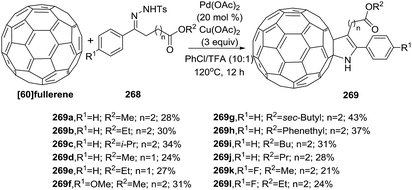
Peng et al. reported an efficient Pd-catalyzed synthesis of the scarce N-unsubstituted 2-fulleropyrrolines 269 employing [60]fullerene and benzoyl hydrazone esters 268 (Scheme 89).139 The reaction involves the use of 20 mol% of Pd(OAc)2 and 3 equivalent of Cu(OAc)2 as oxidant in a solvent mixture of chlorobenzene/TFA (10:1) at 120 °C. The presence of TFA generates a highly electrophilic cationic Pd(II) species and provides better results. The reaction of benzoyl hydrazones esters 268 with electron-donating or electron-withdrawing substituent on the phenyl ring and a variety of esters proceeded efficiently in acceptable yields (21–43%).
 |
| | Scheme 89 Pd-catalyzed syntheses of the fulleropyrrolines 269. | |
2.2.8. Synthesis of 2-pyrrolines by platinum catalysis. A new synthesis of 2-pyrrolines was recently developed by Shi et al. via platinum-catalyzed cyclization of a variety of (hetero)aryl-allenes 270, through the migration of the (hetero)arylmethylene group (Scheme 90).140 After screening several catalysts across a number of conditions, the use of PtCl2 (5 mol%) with an electron-deficient phosphine ligand such as P(C6F5)3 in anisole at 70 °C were identified as the optimal conditions. This Pt(II)-catalyzed migration protocol tolerates a wide range of (hetero)aryl-allenes 270. The desired 2-pyrrolines 271 are obtained in yields up to 97%.
 |
| | Scheme 90 Platinum-catalyzed intramolecular annulation of (hetero)aryl-allenes. | |
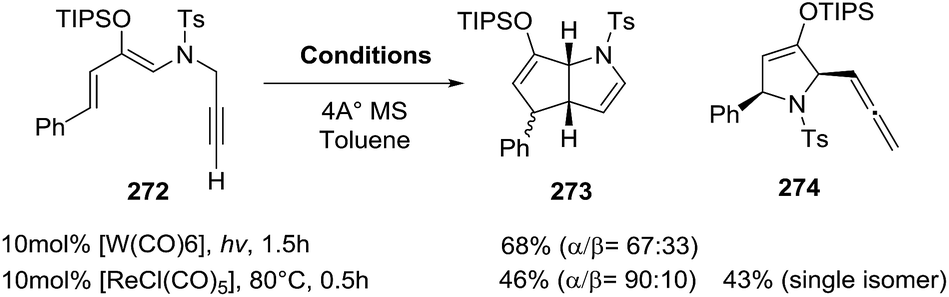
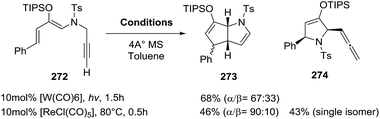
2.2.9. Synthesis of 2-pyrrolines by rhenium catalysis. Kusama et al.141 reported the [ReCl(CO)5]-catalyzed (10 mol%) synthesis of the 2-azabicyclo[3.3.0]octane 273 and the allenyl-substituted dihydropyrrole 274, from the sulfonamide 272 (Scheme 91). This transformation can also be carried out by tungsten(0) catalysis. By evaluating different sulfonyl groups and alternative rhenium catalysts, the selective preparation of either 273 or 274 was possible. The optimal conditions for 273 used an N-nosyl amine and [ReCl(CO)4·PPh3] as a neutral catalyst. On the other hand, dihydropyrrole 274 was obtained as the major product with the p-methoxybenzenesulfonyl (Mbs) sulfonamide and the cationic rhenium catalyst prepared in situ from [ReCl(CO)5] (1 mol%) and AgSbF6 (10 mol%). These reactions demonstrate the high synthetic utility of geminal carbo-functionalization of alkynes and rhenium and tungsten complexes in pyrroline synthesis.
 |
| | Scheme 91 Tungsten(0)- and rhenium(I)-catalyzed tandem cyclization of acetylenic dienol silyl ethers. | |
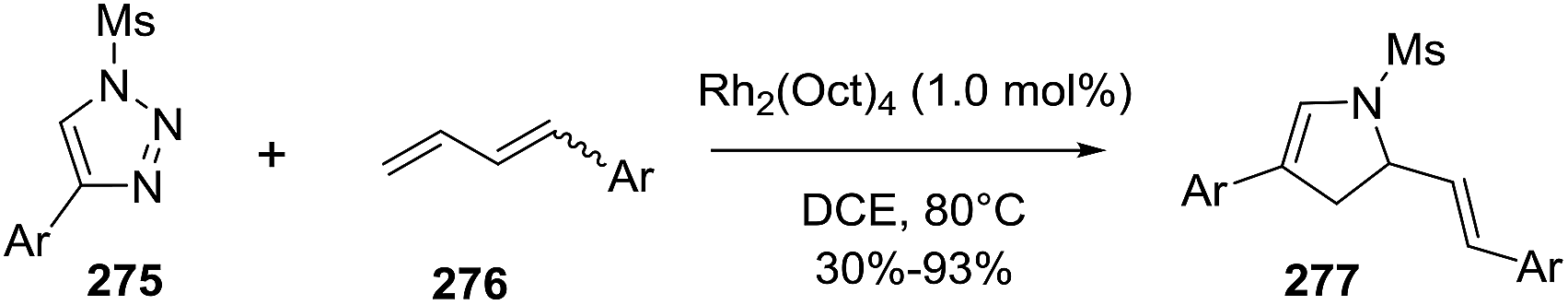
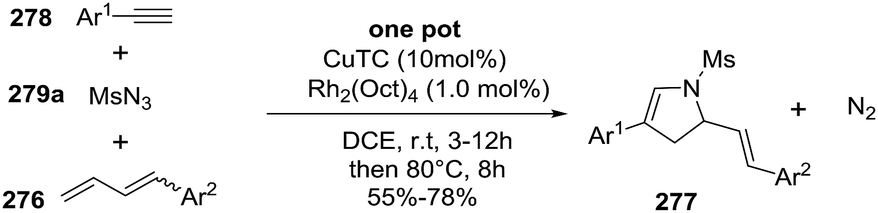
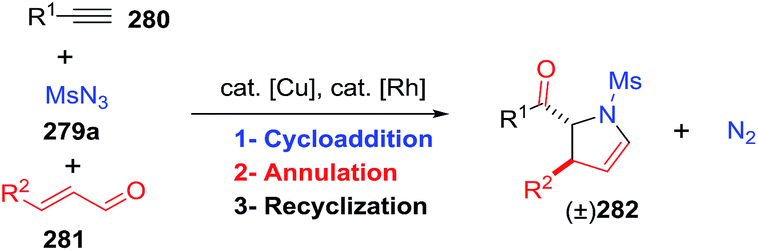
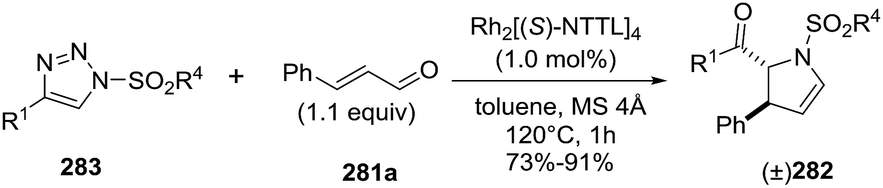
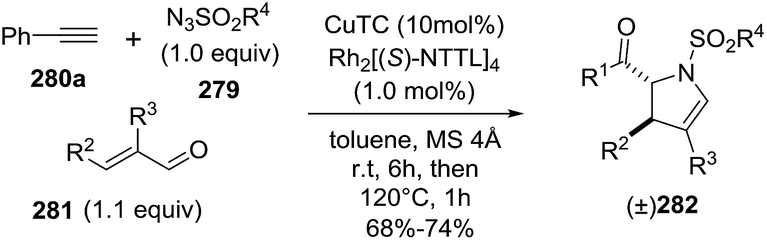
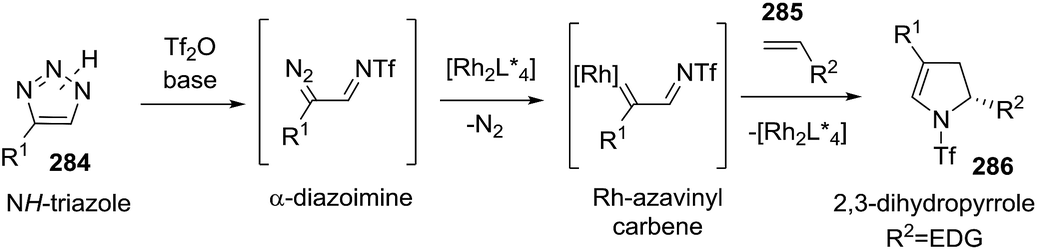
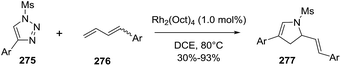
2.2.10. Synthesis of 2-pyrrolines by rhodium catalysis. Kim et al. reported the straightforward synthesis of 2-pyrrolines 277 from sulfonyl triazoles 275 and 1,3-dienes 276, catalyzed by Rh2(Oct)4, under liberation of N2 as the single byproduct (Scheme 92). This product results from sequential [3 + 2]–[2 + 1] cycloadditions giving a net [4 + 3] aza-annulation.142 The sulfonyl group structure affected appreciably the yield of the reaction. The N1-methanesulfonyl triazole was best, while a tosyl group was not tolerated. In contrast, different substituents at C-4 of the phenyl ring (Ar1 of Scheme 88) of the triazole did not affect the yield. Both the Z- or E-1,3 diene gave similar yields of 277. Finally, the authors applied this procedure starting from terminal alkynes 278, mesyl azide, and 1,3-dienes 276 (Scheme 93). This one-pot, three-component synthesis of dihydropyrroles is a synthetically attractive annulation. In the first step, the mesyl triazole 275 is generated through a copper-catalyzed click reaction between the terminal alkyne 278 and the mesyl azide 279. Rhodium-catalyzed annulation then leads to the 2-pyrrolines 277. This result demonstrates that the copper does not interfere with this final annulation.
 |
| | Scheme 92 Aza-[4 + 3] annulation through sequential [3 + 2]–[2 + 1] cycloadditions leading to 2-pyrrolines. | |
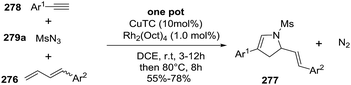 |
| | Scheme 93 One-pot synthesis of 2-pyrrolines starting from arylacetylenes, mesyl azide, and buta-1,3-dienylbenzenes. | |
A rhodium(II)-catalyzed cycloaddition of 1-sulfonyl 1,2,3-triazoles 283 with α,β-unsaturated aldehydes 281 was disclosed in 2013 by the Murakami group.143 The combination of terminal alkynes 280, N-sulfonyl azides 279, and α,β-unsaturated aldehydes 281 as starting materials resulted in the diasteroselective formation of the racemic trans-2,3-disubstituted 2-pyrroline 282 (Scheme 94).
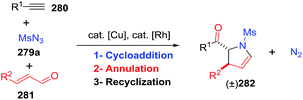 |
| | Scheme 94 Diastereoselective synthesis of trans-2-pyrrolines starting from terminal alkynes, N-sulfonyl azides, and α,β-enals. | |
N-Sulfonyl-1,2,3-triazoles 283 are useful intermediates in the synthesis of heterocycles. In the presence of rhodium catalysts these triazoles generate α-imino rhodium carbene (azavinylcarbenes) which reacts with the α,β-unsaturated aldehydes 281 to give the 2-pyrrolines 282 (Scheme 95). With the chiral and bulky [Rh2(S-NTTL)4] as the catalyst, the exclusive formation of the racemic trans-2,3 disubstituted 2-pyrroline is observed. With less bulky Rh catalysts, an undesired 4,5-dihydro-1,4-oxazepine subproduct is observed. The production of the racemic dihydropyrroles even though the use of a chiral catalyst is explained by a non-stereospecific ionic mechanism. A screening of α,β-unsaturated aldehydes 281 revealed that a variety of (E) and (Z) β-monosubstituted enals and acyclic α,β-disubstituted enals were converted effectively into the 2-pyrrolines. Different groups at the 4-position of triazoles 283 and on the sulfonyl group all participated in the annulation reaction to give good yields (typically 80%).
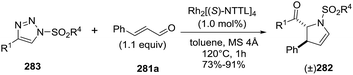 |
| | Scheme 95 Rh(II)-catalyzed denitrogenative annulation of triazoles to synthesize racemic trans-2,3-disubstituted 2-pyrrolines. | |
The reaction was made more practical in the form of a one-pot synthesis starting from terminal alkynes 280a, sulfonyl azides 279, and α,β-unsaturated aldehydes 281, using a mixture of CuTC (10 mol%) and Rh2[(S)-NTTL]4 (1.0 mol%) as co-catalysts (Scheme 96).
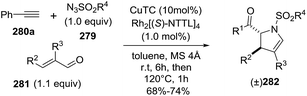 |
| | Scheme 96 One-pot synthesis of 2-pyrrolines 282 starting from phenylacetylene. | |
Using a similar strategy the Fokin group synthesized 2-pyrrolines 286 from in situ generated N-triflyl triazoles 284 and alkenes 285 (Scheme 97).144 Azavinylcarbenes can be obtained directly from N-sulfonyl triazoles or N-triflyltriazoles in presence of a rhodium catalyst. These azavinyl carbenes react with 4-methoxystyrene to produce enantioenriched 2-pyrrolines 286 when a chiral rhodium catalyst is used. The highest enantioselectivity (72% ee) was achieved with [Rh2(S-NTTL)4] (Scheme 98). The authors suggest that the moderate enantioselectivity arises from a rapid bond rotation that erodes the enantioselectivity during the mechanism. The protocol affords 2-pyrrolines when electron-rich olefins such as 4-methoxystyrene or 2-methoxystyrene are used as partners.
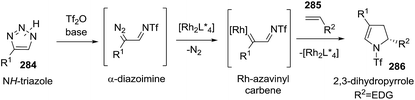 |
| | Scheme 97 Reactions of N-triflyl-Rh-azavinyl carbenes with olefins. | |
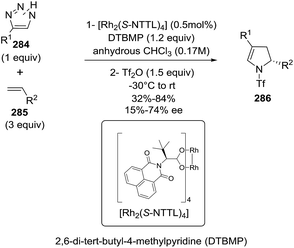 |
| | Scheme 98 Catalytic asymmetric transannulation of NH-1,2,3-triazoles with alkenes. | |
An efficient Rh-catalyzed annulation between α,β-unsaturated ketones 287 and N-sulfonyl-1,2,3-triazoles 283 have been developed leading to multisubstituted 2-pyrrolines 288 (Scheme 99).145 In this methodology, the generated α-rhodium imino carbene species served as the electrophiles against the α,β-unsaturated ketones, which produced a nucleophilic attack through their oxygen atom. Interestingly, small structural differences in the ligands of the explored rhodium catalysts led to drastic changes in the outcome of the reaction. The optimized conditions for aryl enones (287) involved the use of Rh2(S-PTV)4 (4 mol%), using 2 equivalents of the triazole substrate in dry DCM, at 130 °C during 1.5 hours, under nitrogen atmosphere.
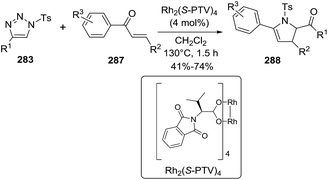 |
| | Scheme 99 Carbenoid strategy to generate multisubstituted 2-pyrrolines. | |
Tang et al. reported a rhodium(II)-catalyzed formal aza-[3 + 2] cycloadditions of 1-sulfonyl 1,2,3-triazoles 283 with (E)-1-aryl-1,3-butadienes 289 leading to 2-pyrrolines 290 (Scheme 100).146 The key intermediaries, the α-imino rhodium carbenes (azavinyl carbenes), were readily prepared from N-sulfonyl-1,2,3-triazoles using [Rh2(oct)4] as the rhodium catalyst (in 1,2-DCE at 140 °C for 12 h). The racemic 2-pyrrolines 290 are isolated as the sole products in very good yields (typically 60–95%). When (Z)-1-aryl-1,3-butadiene instead of the E analog are used, a [4 + 3] cycloaddition to give 2,5-dihydroazepine products was predominant. With 1,1-diphenyl-, 1-phenyl-2-methyl-, and 1-TBSO-substituted 1,3-dienes as the diene, 2-pyrrolines are the main product (typical yields of 23–90%).
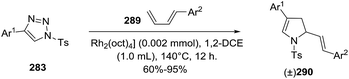 |
| | Scheme 100 Rhodium(II)-catalyzed formal aza-[3 + 2] cycloadditions of 1-sulfonyl 1,2,3-triazoles with (E)-1-aryl-1,3-butadienes leading to 2-pyrrolines. | |
Zhang reported the one-pot rhodium-catalyzed intramolecular hydroaminomethylation of substituted cinnamylamines 291 to generate the 4-aryl-2,3-dihydropyrroles 292 with moderate to excellent yields (typically 52–99%, Scheme 101).147 The reaction is performed with H2/CO in a 1/1 ratio at 20 bar pressure. Triphenylphosphine was the best phosphorous ligand for this reaction. Variations in the amine substituents with different R2 alkyl group and electron-withdrawing substituents at the R1 phenyl ring of the cinnamyl group all gave very good yields.
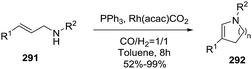 |
| | Scheme 101 Rh-catalyzed intramolecular hydroamino-methylation reaction leading to 2-pyrrolines. | |
Zhang and co-workers148 described a rhodium-catalyzed intermolecular [3 + 2] cycloaddition of chiral vinylaziridines 294 and alkynes 293 to give optically active 2-pyrrolines 295 (Scheme 102). The optimized conditions used 5 mol% of [Rh(NBD)2]BF4 in 1,2-dichloroethane (room temperature, 15 min). The procedure gives good to excellent yields of product for both internal and terminal alkynes 293 and for several substituted vinylaziridines 294. In addition, a complete transfer of chirality from the vinylaziridine to the 2-pyrroline was observed with 90–99% ee. The use of aliphatic alkynes gave moderate to excellent NMR yields, but the cyclic enamines were unstable to purification since they are hydrolytically labile. With non-symmetric internal alkynes, the product of the reaction (in good yields) is a single regioisomer (R1 = TMS, R2 = Me, 64%, 96% ee; R1 = 4-MeOC6H4, R2 = CH2OMe, 69%, 96% ee).
 |
| | Scheme 102 Rhodium-catalyzed intermolecular [3 + 2] cycloaddition of chiral vinylaziridines and alkynes towards optically active 2-pyrrolines. | |
A diastereoselective methodology for the synthesis of tetrahydro-furanodihydropyrroles 298 and tetrahydropyranodihydropyrroles 300 containing N,O-acetal moieties was proposed by Lee et al. These bicyclic heterocyclic compounds were prepared by a rhodium-catalyzed denitrogenative transannulation of N-sulfonyl-1,2,3-triazoles 296 with oxacycloalkenes 297 and 299 (Scheme 103).149 Optimal conditions for this transformation use Rh2(OAc)4 (1 mol%) as the catalyst, N-sulfonyl-1,2,3-triazoles, and oxacycloalkenes in dichloroethane solution (0.05 M) at 80 °C. Under this condition, different N-sulfonyl-1,2,3-triazoles 296 provided the transannulation product without significant variation of yields. The generality of the reaction was proven with the employment of different substituted dihydrofurans 297. Variously substituted racemic 1,2,3-triazoles 296 were obtained as the single diastereomer in moderate to good yield. This protocol was evaluated using 2,3-dihydropyran 299. The desired transannulated product was obtained in good yields (50–60%) (Scheme 103). Finally, the versatility of this rhodium-catalyzed [3 + 2] cycloaddition was demonstrated by the generation of the 1,2,3-triazoles in situ from terminal alkynes, tosyl azides in presence of 2,3-dihydrofuran and with a CuTC and Rh2(OAc)4 as co-catalysts. This three-component one-pot reaction gave the tetrahydrofuranodihydropyrroles in acceptable yields (typically 50–52%) and again proves compatibility between the copper and rhodium-carbenoid catalysts (Scheme 104).
 |
| | Scheme 103 Rhodium-catalyzed denitrogenative transannulation of N-sulfonyl-1,2,3-triazoles with oxacycloalkenes. | |
 |
| | Scheme 104 Rh-catalyzed sequential transannulation. | |
Alcaide and Almendros group reported a rhodium-catalyzed synthesis of 2-pyrrolines 303 from 1-benzenesulfonyl-4-aryl-1,2,3-triazoles 302 with allenols 301 (Scheme 105).150 The protocol makes use of 1 mol% of Rh2(oct)4 in toluene at reflux to obtain a separable mixture of 2-pyrrolines 303 (only the mayor diastereoisomer is shown) with yields in the range of 31–73% (R2 ≠ H). When R1 is aromatic, the diastereoselectivities were modest (d.r. = 55:45 to d.r. = 85:15) but when R1 is aliphatic only the trans diastereomer is observed. However, the yields obtained with R1 = aliphatic are modest (typically 40%). The presence of the hydroxyl group in the allene is necessary for the success of the reaction. The mechanism involves the formation of azavinylcarbenes which suffer a nucleophilic addition of the allenol and subsequent azacyclization.
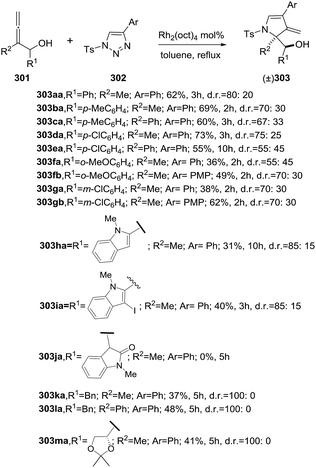 |
| | Scheme 105 Rhodium-catalyzed synthesis of 2-pyrrolines 303. | |
2.2.11. Synthesis of 2-pyrrolines by scandium catalysis. Ghorai et al.151 disclosed a practical and enantioselective synthesis of the highly substituted 4,5-dihydropyrroles 306 and/or 306′ via a Domino Ring-Opening Cyclization (DROC) of N-activated aziridines 304 with malononitrile 305 using Sc(OTf)3-catalysis (Scheme 106). The optimal reaction conditions used t-BuOK as a base. Initial screening with monosubstituted N-sulfonyl aziridines 304 gave satisfactory yields of the racemic 4,5-dihydropyrroles (typically 75–99%).
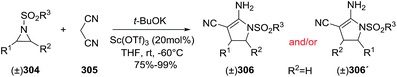 |
| | Scheme 106 Synthesis of 2-pyrrolines via an Sc(III)-catalyzed Domino Ring-Opening Cyclization (DROC) of N-activated aziridines with malononitrile. | |
The aziridine having the more easily removable N-nosyl group gave excellent yields (84–89%) (Scheme 107).
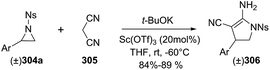 |
| | Scheme 107 Domino Ring-Opening Cyclization (DROC) with N-nosyl aziridines. | |
Enantiomerically pure 4,5-dihydropyrroles 306 were obtained as a single regioisomer from enantiomerically pure alkyl monosubstituted and 2,3-disubstituted aziridines 304. The proposed mechanism involves an SN2 nucleophilic attack of the malononitrile anion on the Lewis activated aziridine to generate intermediate B, which undergoes intramolecular cyclization with subsequent protonation and tautomerization to deliver the product (Scheme 108).
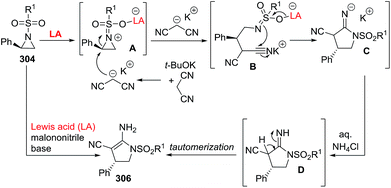 |
| | Scheme 108 Proposed mechanism for the Domino Ring-Opening Cyclization (DROC). | |
In 2015, Xia et al. reported the asymmetric scandium-catalyzed synthesis of chiral 2,4,5-trisubstituted 2-pyrrolines 309 through the ring-opening cyclization reaction of cyclopropyl ketones 307 with primary amines 308.152 The use of Sc(OTf)3 with the chiral N,N′-dioxide ligand L8 gave the best results (Scheme 109). The optimal reaction conditions used the Sc(III)/L8 complex (10 mol%) as catalyst and LiCl as additive (in CHCl2CHCl2 at 35 °C for 96 h). The methodology tolerates a broad range of cyclopropyl ketones 307 and primary amines 308 with exception of aliphatic amines. In all cases (R3 = Ar), the multisubstituted 2-pyrrolines 309 were obtained in good to excellent enantioselectivities (66–96% ee) and poor to excellent yields (16–98% yield). The origin of the enantioselectivity is caused by a kinetic resolution process.
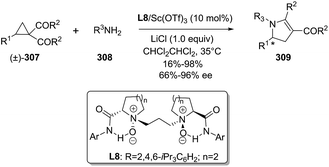 |
| | Scheme 109 Asymmetric scandium-catalyzed synthesis of chiral 2,4,5-trisubstituted 2-pyrrolines 309. | |
Schneider et al. reported an Sc(OTf)3-catalyzed multicomponent reaction towards multicyclic 2-pyrrolines 315 from 2-hydroxy oxime ethers 310, 1,3-dicarbonyls 311, primary amines 312 and indoles 313 or pyrroles 314 (Scheme 110).153 This (2 + 2 + 1)-cycloannulation/aza-Friedel–Crafts alkylation sequence provide complex 2-pyrrolines with excellent yields (42–99%) and diasteroselectivities (d.r. >95:5). The procedure tolerates either electron-rich or electron-deficient indoles. However, the use of less nucleophilic N-methyl indole resulted in lower yields. Other than that, alkyl substituted pyrroles were sufficiently reactive to form the multicyclic 2-pyrrolines. An intramolecular version of this aza-Friedel–Crafts alkylation was achieved with the amine component tethered to the reactive π-nucleophile. This is exemplified with the use of 2-(heteroaryl)ethylamine as tryptamine 316. Regarding the scope of 2-hydroxy oxime ethers 310, the protocol tolerates electron-rich or electron-deficient aryl groups, naphthyl and alkyl groups (R1).
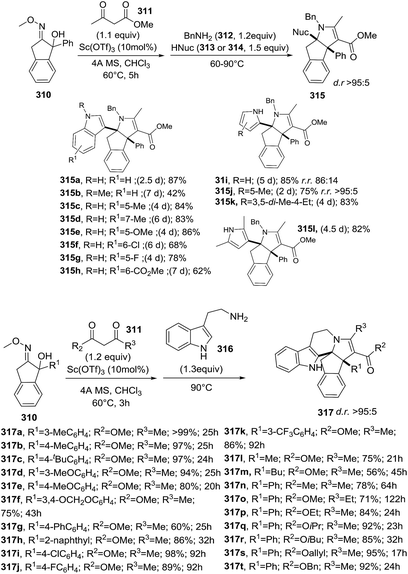 |
| | Scheme 110 Sc(OTf)3-catalyzed multicomponent reaction towards multicyclic 2-pyrrolines 317. | |
2.2.12. Synthesis of 2-pyrrolines by silver. Amat et al.154 disclosed the first example of an asymmetric cooperative multicatalytic cascade between isocyanoacetates 318 and α,β-unsaturated ketones 319 for the rapid construction of enantioenriched 2,3-dihydropyrroles 321, employing the combination of a chiral cinchona alkaloid-derived organocatalyst and silver nitrate as the additive. The scope of the methodology was evaluated with respect to different substituted isocyanoacetates 318 and α,β-unsaturated ketones 319 which in combination with cupreine 320 as the chiral base, led to products in moderate enantiomeric excess (Scheme 111).
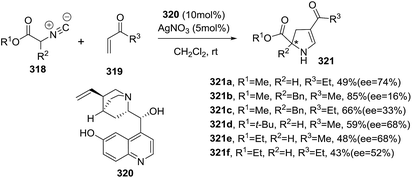 |
| | Scheme 111 Cooperative catalytic synthesis of enantioenriched pyrrolines 321. | |
Simultaneously to the previous report, Gong et al.155 published an analogous transformation towards optically active 2-pyrrolines 324 but using α,β-unsaturated esters 323 instead of α,β-unsaturated ketones as the starting materials (Scheme 112). This study represented the first example of a highly enantioselective [3 + 2] cycloaddition of isocyanoesters 322 to 2-oxobutenoate esters 323 catalyzed by the silver complex formed from silver acetate and (S)-(2′-hydroxy-1,1′-binaphthyl-2-yl)diphenylphosphine 325. Different substituents present in both reactants were evaluated. In most of the cases 2,3-dihydropyrroles 324 were obtained with excellent yields (73–98%) and high enantioselectivity (90–98% ee).
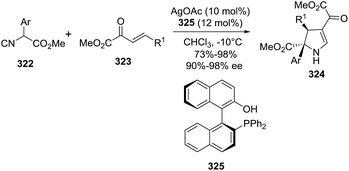 |
| | Scheme 112 Asymmetric formal [3 + 2] cycloaddition reaction towards chiral 2-pyrrolines. | |
Mukhopadhyay et al.110 reported a multicomponent reaction promoted by Ag(I) through activation of the Cα(sp3)–H bond of the corresponding benzylamine 326 in presence of but-2-ynedioate 327 and isatin 329 (X = H). A regioselective [1,5]-rearrangement gave the spiro-2,3-dihydropyrroles 330 while an alternative [1,3]-rearrangement produced the spiro-1-dihydropyrroles 332. The regioselective generation of each heterocyclic core (330 versus 332) was achieved under the same reaction conditions involving Ag(I) and depending on the nature of the group bond to the nitrogen of isatin (Scheme 113). When isatin or N-methylisatin is used, a [1,5] rearrangement occurs to afford 2-pyrrolines 330. If the isatin is N-substituted with benzyl, allyl, ethyl, propyl, or an n-butyl group, a [1,3]-rearrangement occurs to give 1-pyrrolines 332. NMR experiments are consistent with enamine 328 as the common intermediary to both products. The Ag(I) source of choice was Ag2CO3, one of the most available and least expensive sources of silver. In addition, the Ag2CO3 was recycled successfully. The scope of the reaction was evaluated using various derivatives of each starting material (R1, R2, R3). A steric effect of the N-substituents is crucial for the fate of the reaction, for instance when R3 = allyl or n-butyl produce lower yields compared with R3 = benzyl. When R3 = ethyl or propyl the reaction gives good yields (87% and 84% respectively).
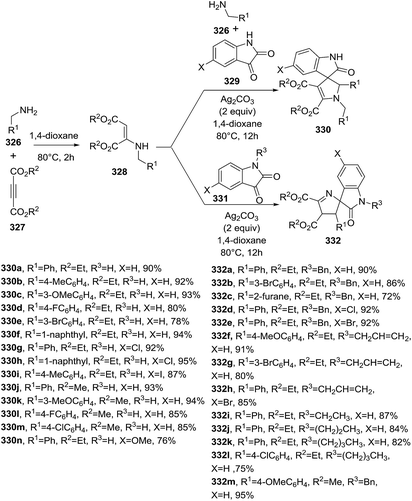 |
| | Scheme 113 Regioselective synthesis of spiro-dihydropyrroles 330 and 332. | |
2.3. Synthesis of 3-pyrrolines
Illustrative examples of the importance of the 3-pyrroline ring in natural products and bioactive compounds are displayed in Fig. 5. The 3-pyrroline scaffold is part of the polycyclic core of erysovine (333), a natural erythrinan alkaloid which presents chemotherapeutic properties.9 The substituted 2,5-dihydropyrrole is the central ring of the kinesin spindle protein inhibitor 334 (ref. 19) and of compound 335.156 Both of these compounds exhibit antitumoral activity. Spirooxindol derivative 336 (ref. 157) has potent anti-microbial activity. The 2,5-dihydropyrrole formyl hydroxyamino derivatives 337 are peptide deformylase inhibitors achieving better in vitro antibacterial activities than existing drugs.158 A set of synthetic N-3-pyrrolines amino arylacetamide derivatives 338 exhibited potent and high selectivity as kappa-agonists with potentially safer analgesic activity.17
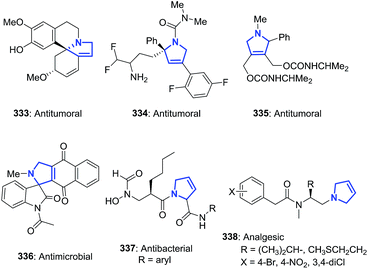 |
| | Fig. 5 Selected examples of biologically active 3-pyrrolines. | |
Pyne et al. demonstrated the versatility of using 3-pyrrolines as intermediaries in their synthesis of complex alkaloids. The chiral 3-pyrroline 340 was used as an common intermediate towards the synthesis of the glycosidase inhibitors uniflorine A (341), casuarine (342), australine (343), and 3-epi-australine (344).32 The chiral pyrroline 340 is available in gram scale starting from L-xylose and using a ring-closing metathesis of 339 as a key step. The pyrrolizidine alkaloids 341–344 were obtained in satisfactory overall yields in several steps from 340 (Scheme 114).
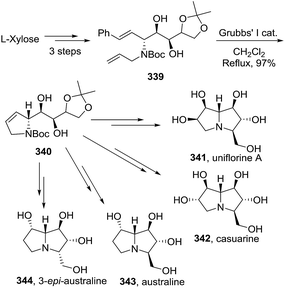 |
| | Scheme 114 Synthesis of pyrrolizidine alkaloids through the 3-pyrroline 340. | |
2.3.1. Synthesis of 3-pyrrolines by copper catalysis. Between 2010 and 2011, several groups used known (but adapted) synthetic approaches to generate 3-pyrrolines under green, copper-catalyzed conditions. The Njardarson group159 developed the Cu(hfacac)2 (hfacac: hexafluoroacetylacetonate) catalyzed ring expansion of chiral vinyl aziridines 345 to produce a set of mono and fused 3-pyrroline products 346 (Scheme 115a). This transformation exhibited high stereocontrol, with each starting material produced a single stereoisomer. Using the same catalyst, Zhou and coworkers160 generated the 3-pyrrolines 349 but via the different mechanism of overall intermolecular [4 + 1] cycloaddition of 1,3-dienes 347 and the nitrene precursors 348 (Scheme 115b). The more likely mechanism, however, involved [2 + 1] cycloaddition of a vinylaziridine intermediate (350), followed by ring expansion. This mechanism justifies the stereochemistry of the final pyrroline as determined exclusively by the geometry of the conjugated dienes (as inferred from the proposed metal-chelated vinylaziridines 350A, 350B.). In particular, the 1,4-disubstitued dienes were the best substrates to reach high diastereomeric ratio (cis-product with R2 and R3 substituents pointing towards the same side of the heterocycle compared to the trans-product with those substituents pointing to opposite faces of the ring). In the same year, a report described the synthesis of 3-pyrrolines 353 via tandem allenylation/cyclization reactions catalyzed by the NHC (N-heterocyclic carbene)-ligated copper complex [Cu(IPr)Cl] (Scheme 115c).161 The proposed mechanism for this sequence of reactions invokes an amine allenyl intermediate that cyclizes in presence of the copper catalyst. Rao et al.162 developed a synthetic route directed to trisubstituted 3-pyrrolines 355 via an intramolecular hydroamination of homoallylic amino alcohols 354 mediated by Cu(OTf)2 (Scheme 115d). In this case, the use of enantioenriched aminoalcohols as starting materials produced chiral heterocycles with excellent dr [trans (R2 and methyl group on the same side of the pyrroline): cis (R2 and methyl group on opposite sides of the pyrrolines ring)] and ee values.
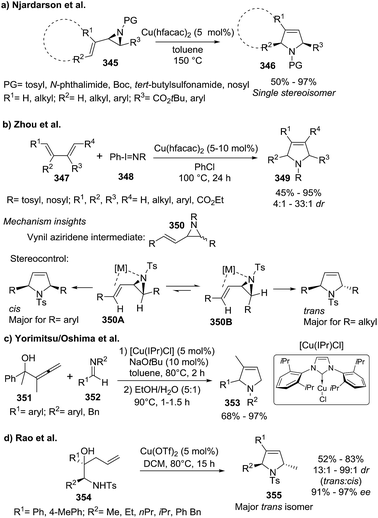 |
| | Scheme 115 Different synthetic approaches toward 3-pyrrolines via copper catalysis reported in 2010–2011. | |
Wang and Fang163 described subsequently the preparation of the highly substituted pyrrolines 357 from the alkynyl-substituted enamides 356 using Cu(II) catalysis. The key step in this synthesis is the preparation of the linear starting material 356 via a PhIO/Bu4NI-mediated oxidative cross-coupling. Cyclization of such enamides 356 in presence of copper salts led to different halogenated final products 357a–c (Scheme 116).
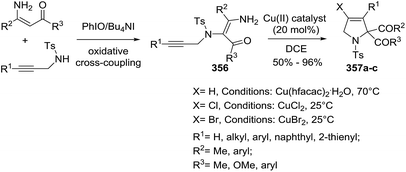 |
| | Scheme 116 Cu(II)-promoted cyclization of alkynyl-substituted enamides 356. | |
At the same time Tang et al.164 reported the construction of enantioenriched and highly substituted 3-pyrrolines 360 via asymmetric 1,3-dipolar cycloaddition the ethynyl ketones 358 to the azomethine ylides 359 (Scheme 117). The optimized reaction conditions used Cu(OAc)2·H2O with the FOXAP derivative L9 as the ligand. The pyrrolines were obtained in acceptable to excellent yields (48–99%) and high enantiomeric excess (84–98%).
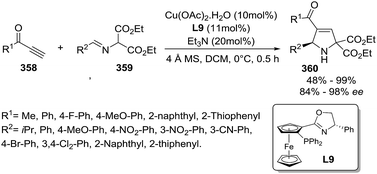 |
| | Scheme 117 Asymmetrical 1,3-dipolar cycloaddition of ethynyl ketone 358 to azomethine ylide 359 catalyzed by the complex Cu(OAc)2·H2O/FOXAP derivative L9. | |
2.3.2. Synthesis of 3-pyrrolines by iron catalysis. Schindler's group described the synthesis of chiral 3-aryl-3-pyrrolines 362 using iron(III)-catalyzed carbonyl-olefin metathesis taking advantages of commercially available amino acids as chiral pool reagents to access metathesis substrates 361 (Scheme 118).165 The optimized condition makes use of 0.5 equiv. of FeCl3 in dichloroethane at 0 °C for 1 h and then warmed to rt. The best results were obtained with carbonyl-olefin metathesis substrates 361 bearing prenyl-derived alkenes and electron-deficient sulfonamide protecting groups (R). This strategy provided 34 aryl-3-pyrrolines in yields up to 99% with complete stereoretention (up to 98% ee).
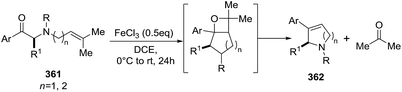 |
| | Scheme 118 3-Aryl-3-pyrrolines 362 via catalytic carbonyl-olefin metathesis. | |
2.3.3. Synthesis of 3-pyrrolines by gold catalysis. Lipshutz and Krause166 reported in 2011 the first example of 3-pyrroline scaffolds obtained by gold catalysis in micellar systems using PTS or TPGS-750-M as amphiphiles. The heterocycles 364 were efficiently produced from the α-functionalized amino allenes 363 via a cycloisomerization promoted by the air-stable aqueous gold(III) solution (Scheme 119a). Interestingly, the reaction presents a broad scope of substrates, and tolerates the TBS ether, ester, and sulfonamide functional groups. More recently Reissig et al.167 published an analogous transformation but using Au(I) catalysis. Starting from allenyl N-tosylamines 365, the 2,3-disubstituted pyrrolines 366 were obtained via 5-endo-trig cyclization (Scheme 119b). As previously discussed by the authors in preliminary results,168 this annulation could be also achieved by either Ag(I) catalysis or the use of a base such as KOtBu. However, the auric catalyst gave higher yields (94–100%) under mild reaction conditions, and with a more convenient work up (except for the 2-pyrrolyl pyrrolines, which required strongly basic conditions). A similar cycloisomerization was described as part of an interesting multicomponent protocol based on sequential use of metals (Cu/Rh/Au).169 This “three procedures/single work-up” methodology involved a final Au(I)-promoted cyclization of α-amino allenes analogous to those described above (see Subsection 2.3.11.: Synthesis of 3-pyrrolines by combined metal catalysis: Cu/Rh/Au, Scheme 138).
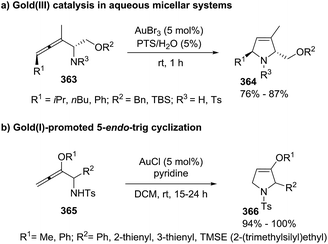 |
| | Scheme 119 Cyclization of allenyl amines and allenyl tosylamines. | |
Several strategies toward the gold-promoted generation of 3-pyrroline scaffolds were based on initial gold-alkyne coordination, taking advantage of the high alkynophilicity of this metal. For instance, Shi et al.59,170 developed a synthesis of 2,3-disubstituted 3-pyrrolines 368 by the gold(I)-promoted intramolecular cyclization of the functionalized 1,6-diynes 367 (Scheme 120). The use of the (tBu3P)AuCl/AgOTf catalytic system in the presence of water (1 equiv.) was optimal, providing the final cycloadducts in up to 88% yield. A plausible mechanism for this annulation, implicating two Au(I) cationic complexes with the two alkynes in 367, was proposed. Even though there is a broad scope of substrates, the terminal alkyne analogues compounds did not provide the pyrroline as a product, but instead the diketone “linear” products were produced.
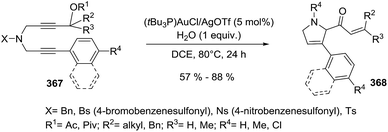 |
| | Scheme 120 Cyclization of 1,6-diynes 367 by Au(I) catalysis. | |
Xie, She et al.171 applied gold-catalyzed tandem 1,3-acyloxy rearrangement/intramolecular azacyclization to provide the pyrrolines 370 from γ-amino substituted propargylic esters 369 (Scheme 121a). The authors screened the catalytic system, solvent, and time. The use of AuPh3PCl/AgSbF6 in DCM (room temperature, 30 min) gave the best results. The proposed mechanism started with gold–alkyne coordination to generate via a 3,3-rearrangement an allenyl intermediate 371, on which an intramolecular SN2-type reaction occurred, followed by a final gold elimination leading to the pyrroline, and regenerating the gold catalytic species. The π–π interaction present in intermediate 371 explained the cis-stereochemistry (sole products with R1 and R4 on the same side of the pyrroline ring) observed in the 2,5-disubstituted pyrrolines 370. This cascade of transformations was applied efficiently to a total synthesis of natural alkaloid (±)-aphanorphine. Similarly, the Blanc and the Pale's groups172 developed a novel gold salt methodology to obtain spiro[isochroman-4,2′-pyrrolines] 373 in high yields from aryl-containing alkynylaziridines 372 (Scheme 121b). Similar to the previous example, a cascade reaction through the aminoallenylidene isochromane intermediate 374 leads to the products. In this case, a dual σ and π gold activation by double gold-coordination to the aziridine and alkynyl moieties, respectively has been proposed.
 |
| | Scheme 121 Au(I)-catalyzed cascade reactions involving allenyl intermediates. | |
2.3.4. Synthesis of 3-pyrrolines by niobium catalyst. Obora et al. developed an active in situ-generated niobium catalyst formed by niobium pentachloride, trimethylsilyl chloride, zinc, benzyl dichloride and THF as solvent.173 This Nb-based complex is an active ring-closing metathesis catalyst. 3-Pyrroline derivatives 376 were obtained in quantitative yields starting from N,N-diallyl-p-toluenesulfonamides 375 when heated at 60 °C for 2 h in presence of the Nb-based complex (Scheme 122).
 |
| | Scheme 122 Synthesis of 3-pyrrolines by in situ-generated niobium-based catalyst. | |
2.3.5. Synthesis of 3-pyrrolines promoted by magnesium. The rearrangement of vinylaziridines to pyrrolines is an exceptionally reliable methodology. The key for its success is the availability of the required highly substituted starting aziridine, and the selection of conditions to control reaction pathway. In this manner, De Kimpe's group174 developed a mild and efficient addition of vinylmagnesium bromide 378 to chiral α-chloro N-tert-butanesulfinyl ketimines (377) to produce in situ the vinylaziridines 380, leading exclusively to the 3-pyrrolines 381 via a SN2′ ring opening with C–N bond cleavage (Scheme 123). The crude reaction mixture contained a combination of dechlorinated imines (378), N-sulfinyl-2-aryl-2-vinylaziridenes intermediates (380), and final pyrrolines 381. However, recrystallization from ether or purification by silica chromatography induced the 2-sulfinyl-2-aryl-2-vinylaziridines rearrangement to give the 3-aryl-3-pyrrolines 381 in good yields. Careful analysis gave insight into the effect of the substitution pattern on both the aziridine ring as well as the alkene moiety. In particular, when the R2 substituent was different from hydrogen, the pyrroline was not obtained; the corresponding vinylaziridene was produced as the major product instead.
 |
| | Scheme 123 : Synthesis of 3-aryl-3-pyrrolines 381 from vinylmagnesium bromide (378) and chiral α-chloro N-tert-butanesulfinyl ketimines (377). | |
2.3.6. Synthesis of 3-pyrrolines by palladium catalysis. By analysis of the Pd-catalyzed transformations of (Z)-vinyl iodides, Tong's group175 discovered a domino process, combining Heck and aza-Michael reactions, to produce the 3-pyrroline. When tosylamide-substituted vinyl iodides 382 were used in combination with acrylic ester (and related structures 383) in the presence of catalytic Pd(OAc)2/PPh3 and base (2 equiv.), the domino reaction gave the substituted 3-pyrrolines 384 in 40–71% yields (Scheme 124). Addition of Bu4NBr increased the yield to 65%. Bridged derivatives (385 and 386) were obtained via synthetic modifications, initializing with ester hydrolysis and ester reduction. The proposed mechanism involves the oxidative addition of vinyl iodide 382 to Pd(0) to generate A, which undergo a Heck-type reaction with the acrylic ester leading to intermediate C (Scheme 124). Subsequently, spontaneous aza-Michael cyclization provides the desired N-tosyl pyrrolines.
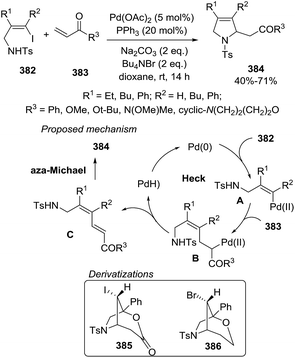 |
| | Scheme 124 Pd(0)-catalyzed domino Heck-aza-Michael reaction between (Z)-N-(3-iodoallyl)-tosylamides 382 and acrylic esters 383 and related. | |
Another strategy to access 2,5-dihydropyrroles, also based on a Pd-promoted cascade reaction, was developed by Sun et al.176 In this case the combination of metal and organic catalysis produced chiral 3-pyrrolines 390 from the readily available α,β-unsaturated aldehydes 387 and N-tosyl propargylamines 388 as the starting materials, via an iminium/enamide cascade (Scheme 125). The proposed mechanism integrates the two catalytic cycles. The overall cascade begins with the iminium activation of the aldehyde by the chiral amine 389, followed by an aza-Michael addition and final Pd-promoted carbocyclization, in which the metal coordinates simultaneously the enamide and the alkyne moieties. This cascade organo-metal cooperative catalysis procedure facilitates the production of 3-pyrrolines in acceptable to good yields (41–87%) and with excellent diastereo- (>16:1 (cis:trans) dr, only the major cis diastereomer is shown) and enantioselectivities (73 to > 99% ee). Although limited to the use of terminal alkynes, diverse enantiomerically enriched tri- and tetrasubstituted pyrrolines were produced, presenting a wide scope of substituents at R1 and R2 positions.
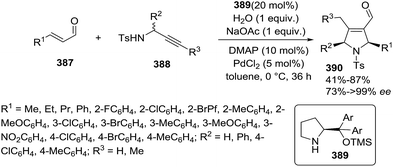 |
| | Scheme 125 Asymmetric Pd/amine-catalyzed synthesis of tri- and tetrasubstituted 3-pyrrolines 390. | |
The Pd-promoted cycloisomerization of enynes to 3-pyrrolines was exploited by Liang and coworkers (Scheme 126).177 The starting p-toluensulfonamides 1,6-enynes (391) underwent cycloisomerization in the presence of Pd(acac)2 to generate cyclopropylpalladium complexes, which were captured by the indoles 392 leading to the final 3-pyrrolines derivatives 393 with moderate yields (up to 74%). In the absence of the indole, an acetate anion from the reaction medium is incorporated, producing the truncated 3-pyrrolines 394.
 |
| | Scheme 126 Pd-catalyzed addition of indoles to hydroxyl 1,6-enynes. | |
The use of polystyrene-supported palladium (Pd@PS) nanoparticles (NPs) has been applied for the generation of fused 3-pyrrolines.178 This heterogeneous catalyst promotes a domino decarboxylative coupling-cyclization (DCC) from phenylpropiolic acid (396) and aminomethyl benzocycloheptene bromide 395, leading to a new class of bioactive 3-pyrroline derivatives 397 with acceptable to good yields (52–76%) (Scheme 127). The catalytic species was proven to be Pd(0) in heterogeneous form. Furthermore, the catalyst could be used up to five times without significant decreased activity.
 |
| | Scheme 127 Heterogeneous Pd-catalyzed tandem decarboxylative coupling-5-exo cyclization. | |
2.3.7. Synthesis of 3-pyrrolines by rhodium catalysis. A Rh-catalyzed multicomponent procedure was developed by Reddy et al.179 to obtain highly functionalized spirooxindolyl pyrrolines 401, which can be thought as hybrid drugs, as they present two active moeities (Scheme 128). The optimized multicomponent reaction required cyclic ketimines (398), dimethyl diazomalonate (399), and dimethyl acetylenedicarboxylate (400) in the presence of Rh2(OAc)2 (5 mol%) in benzene (80 °C, 15 min). The rhodium is proposed to catalyze the formation of azomethine ylides 402 from 398 and 399, which are subsequently trapped by the alkyne derivatives 400 present in the reaction mixture through Huisgen's cycloaddition, to generate the pyrroline. This methodology results in the remarkably efficient, single-step production of the complex spirocycles 401 with good to excellent yields (80–86%).
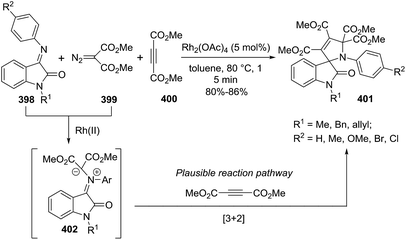 |
| | Scheme 128 Multicomponent reactions towards spirooxindolyl pyrroline catalyzed by rhodium. | |
2.3.8. Synthesis of 3-pyrrolines by ruthenium catalysis. Ru-catalyzed ring-closing metathesis (RCM) is one of the most efficient and broadly employed synthetic methods for the generation of various cyclic products.180 The application of this methodology towards the generation of 3-pyrrolines has been broadly explored, encompassing different aspects such as mechanistic studies with different ruthenium catalysts,181 an enyne version of the transformation,182 and the evaluation of the catalyst loading.183 Over the last years, this transformation has been improved and expanded. Most reports describe the Ru-promoted annulation of dialkenyl-substituted amines affording 3-pyrrolines whose substitution patterns were directly determined by the chemical structure achieved during the synthesis of such diolefinic starting materials. Diallyl sulfonamides are commonly used to study this RCM. For example, Perfetto et al.181 described the performance of new ruthenium NHC ligand (L10) complexes 405 (Scheme 129a). Kinetics studies showed that the complex catalysts syn-405a and syn-405b are remarkably efficient for the RCM of hindered olefins. Samec et al.184 expanded the substrate scope, as described by the synthesis of N-aryl, N-tosyl, and N-alkyl pyrrolines from diallylated amines 403, derived from the Pd-catalyzed reaction between allylic alcohols and the corresponding amines (Scheme 129b, R2 = H). The linear amine substrates 403 were transformed into the heterocycles 404 in good to excellent yields by RCM using (H2IMes)(PCy3)Cl2RuCHPh (406). The sequence of reactions exhibited high atom economy, with only water and ethane as side products. A one-pot version of the transformation, with the co-presence of the Pd and Ru metals, was successful. Some years later, Samec's group185 expanded the scope of this strategy towards unsymmetrical diallylated aromatic amines, which enabled RCM promoted by the same Ru catalyst to give 3-substituted 3-pyrrolines (Scheme 129b, R2 ≠ H). The dialkenyl containing amines were obtained by a two-step coupling sequence between aniline and the two corresponding allylic alcohols in presence of in situ-generated Pd[P(OPh)3]3. An expansion of this methodology was achieved by adding Fe(III) (as FeCl3·6H2O) during the cyclization step, leading via a two-step, one-pot procedure to the product of pyrroline oxidation, the pyrrole. In addition, Clayden et al.186 explored the incorporation of urea functionality at R1 position. Using N,N-diallylureas as suitable substrates for the RCM in presence of 5 mol% of Grubbs 1st generation catalyst, N-[(alkylamino)carbonyl]-2,5-dihydropyrroles were obtained.
 |
| | Scheme 129 RCM promoted by Ru catalysts towards 3-pyrrolines. | |
Asymmetric syntheses of 3-pyrrolines via RCM promoted by ruthenium, in which the pyrroline stereochemistry is defined by the RCM substrates are also possible. For instance, Hong's and Wang's groups187 collaborated to explore the used of Zhan 1-B Ru catalyst to obtain the 2-substituted 3-pyrroline 409 as a single enantiomer (83–87% ee) from chiral N-diallyl p-toluensulfonamides 410, whose stereocenter was defined by the quinidine present in the previous reaction between 407 and 408 (Scheme 130a). Similarly, but with a different stereoselective approach, Kamimura's group188 reported the preparation of optically active 2,5-dihydropyrroles (412) from enantiomerically-enriched aza-Baylis–Hillman adducts (411, Scheme 130b). These key substrates 411 were generated via an optimized domino-reaction procedure from chiral sulfinimines and tert-butyl acrylate. The annulation step promoted by Grubbs' second generation catalyst proceeded smoothly to provide the desired pyrrolines 412 in high yields (92–100%) and conserving the initial enantiomeric excess (90–98% ee). In addition, the formal synthesis of the natural pyrrolizidine alkaloid (−)-trachelanthamidine 414 was proposed based on this methodology, to afford the immediate precursor 413.189 Another interesting application of chiral pyrrolines as key intermediates in the synthesis of natural products was achieved by Davies and coworkers.190 This group developed a diastereoselective lithium amide conjugation methodology that enabled the generation of enantiomerically pure β-amino esters 417 and 418, from lithium (R)-N-allyl-N-(α-methylbenzyl)-amides 416 and dienyl ester 415 (Scheme 130c). Subsequent ring closing metathesis of 417 and 418 promoted by Grubbs I catalyst provided the dihydropyrroles as the single diastereoisomers 419 and 420 (>99:1 dr). This lithium amide conjugate addition was applied as key stereodefining step during the synthesis of the natural alkaloids (+)-tetraponerine-1, -2, -5, and -6 (Scheme 130c).37 The dehydroxy derivative 319 was used for access to the tricyclic core of the target molecules, which were isolated enantiomerically pure and in acceptable overall yields from commercial starting materials.
 |
| | Scheme 130 Asymmetric synthesis of 3-pyrrolines and their applications for the generation of natural products. | |
2.3.9. Synthesis of 3-pyrrolines by scandium catalysis. As previously mentioned, aziridines are convenient substrates for the generation of pyrrolines scaffolds. For instance, these ring-strained three-membered cyclic amines (421) produce azomethine ylides via selective C–C heterolysis under the mild reaction conditions involving Sc(OTf)3 as the Lewis acid (Scheme 131).191 These in situ generated dipoles underwent 1,3-dipolar cycloadditions with various electron-rich alkynes (422) to afford the highly substituted 3-pyrrolines 423. This scandium-catalyzed [3 + 2] cycloaddition afforded the desired final heterocycles as a single regioisomer in good yields (65–91%) and with a diverse substitution pattern. Further synthetic modifications of the obtained 3-pyrroline core diversified the substitution, and allowed the transformation of these heterocycles into pyrroles, 1-pyrrolines, and pyrrolidines.
 |
| | Scheme 131 Sc(III)-catalyzed synthesis of highly substituted 3-pyrrolines from N-tosylaziridines and alkynes. | |
2.3.10. Synthesis of 3-pyrrolines by silver catalysis. The use of catalytic silver to produce 3-pyrrolines from amine-containing allenes has been broadly explored.45,168,192,193 Usually in these cases, diversely N-protected allenic amines 424 underwent AgNO3-catalyzed 5-endo-trig cyclization towards the 3-pyrrolines, involving a protodeargentation of the vinyl-silver intermediate 425 as the final step (hydroamination products 426, Scheme 132a). A novel modification of this approach incorporated different electrophiles, instead of the proton.194 This concept was probed successfully using N-chloro-succinimide (NCS) as the electrophilic species to obtain the halogenated 3-pyrrolines 428 from allene 427 (Scheme 132b). The annulation was achieved with catalytic [Ag(phe)OTf], as a silver catyalyst with increased air and moisture stability, and lutidine as the base. The cyclic products of this chloroamination were isolated in up to 94% yield. The proposed mechanism is analogous to the previously indicated mechanism. Ag-allene coordination (A) leads to the cationic silver complex B, followed by nitrogen intramolecular attack with corresponding cyclization (C), but with final chlorination by NCS instead of a protodemetallation (Scheme 133). The presence of base is essential, as the base neutralizes the charged species C and prevents protonation as a side reaction. Unfortunately, all attempts to use N-bromosuccinimide (NBS) failed to give bromodihydropyrroles. Nonetheless, the chloropyrrolines were able to provide different pyrrolines by substitution of chloride, as well as the corresponding chloro-pyrrole cores after oxidative conditions.
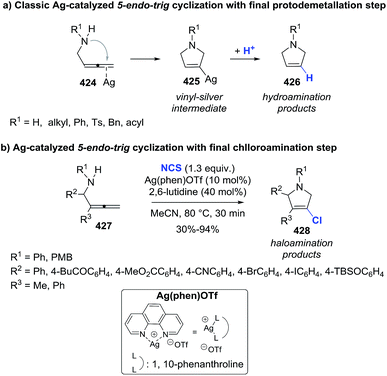 |
| | Scheme 132 Cyclization of allenic amines catalyzed by Ag(I). | |
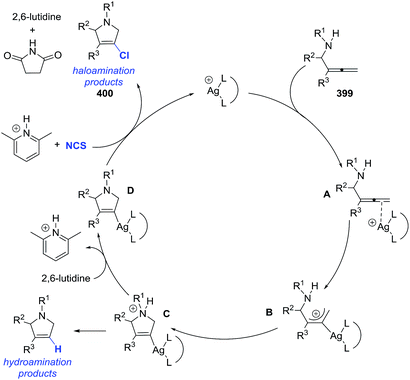 |
| | Scheme 133 Proposed mechanism for the formation of chloro-3-pyrrolines 428. | |
Based on the same strategy, the use of N-sulfonamide-substituted allenylamines 429 enabled the preparation of 2,5-disubstituted 3-pyrrolines 430 (Scheme 134).195 This cyclization proceeded smoothly with AgNO3 to afford the heterocycles in excellent yields (82–90%) and with high diastereoselectivity (>95:5 dr, only the major cis-diastereomer is shown).
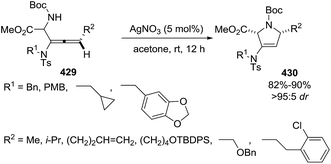 |
| | Scheme 134 AgNO3-catalyzed synthesis of 2,5-disubstituted 3-pyrrolines 430. | |
Harmata et al. treated a series of allenyl sulfonamides 431 with 2 mol% of AgF in acetonitrile at reflux to obtain the corresponding 3-sulfonyl-2,5-disubstituted-3-pyrrolines 432 with yields ranging from 78 to 99% (Scheme 135).196 The 5-endo-trig cyclization catalyzed by silver tolerates phenyl rings with electron-withdrawing and electron-donating groups, heteroaromatic moieties and aliphatic chains in position R3. Cyclic system like cyclopentyl and cyclohexyl groups in position R1 and R2 are rapidly transform in product indicating that there are no steric effects.
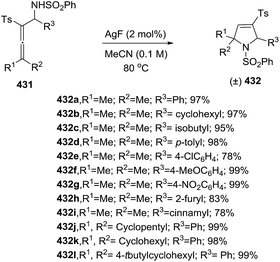 |
| | Scheme 135 Synthesis of 3-sulfonyl-2,5-disubstituted-3-pyrrolines 432 catalyzed by silver fluoride. | |
Wang et al.197 reported a novel generation of 2,5-dihydropyrroles 434 from ketopropargylamines 433 via a 5-endo-dig cyclization promoted by Ag(I) through a Conia-ene-type reaction (Scheme 136). This report represents the first example of transition metal-catalysis of Conia-ene reactions employing mono-carbonyl groups as starting materials. The reaction was compatible with a broad scope of functional groups, and with both terminal and internal alkynes. The reaction proceeded efficiently with tosyl and nosyl groups in R2 position. However, the reaction failed if the N-substituent was alkyl, aryl, or acyl. The final pyrrolines were obtained in acceptable to excellent yields (49–94%). Some of the pyrrolines were oxidized successfully to the corresponding pyrroles.
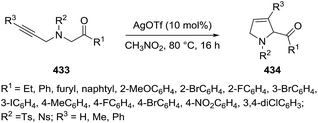 |
| | Scheme 136 Silver(I)-catalyzed Conia-ene type reaction. | |
Reissig et al. published the synthesis of 1,2,3,5-tetrasubstituted 3-pyrrolines 437 from a mixture of pro-cis and pro-trans allenylamines (435 and 436) using silver nitrate as catalyst at room temperature (Scheme 137).198 The cis and trans 3-pyrrolines (cis-437 and trans-437) were obtained in good yields. However, the silver-catalyzed cyclization was not stereoespecific since silver nitrate would allow a configurational isomerization of axially chiral allenylamines 435 and 436.
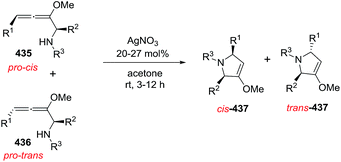 |
| | Scheme 137 Silver-catalyzed cyclization to give polysubstituted 3-pyrrolines 437. | |
2.3.11. Synthesis of 3-pyrrolines by combined metal catalysis: Cu/Rh/Au. Miura, Murakami and coworkers169 reported a one-pot, multistep-pathway to 3-pyrrolines 441 from terminal alkynes 438, sulfonyl azides 439, and propargyl alcohols 440. The relay action of a set of three metals (copper, rhodium and gold: Scheme 138a) achieved three sequential reactions culminating with a single final work-up and purification. The plausible mechanism uses initial Cu(I)-catalyzed 1,3-dipolar cycloaddition between alkyne 438 and azide 439 to provide the triazole 442a, which equilibrates with the α-diazo imine 442a′ (Scheme 138b). Intermediate 442a′ reacts with Rh(II) leading to the α-imine rhodium carbene complex A, which reacts with the propargyl alcohol 440 to generate the zwitterionic intermediate B. Rhodium release and a proton shift give the (Z)-isomer of C, which at 100 °C undergoes a spontaneous Claisen-type rearrangement to give the α-allenyl-α-aminoketone 443. The final step is a gold-catalyzed cycloisomerization to the 3-pyrroline 441, in a similar manner to the examples described previously in the gold subsection (see Subsection 2.3.3.: Synthesis of 3-pyrrolines by gold catalysis, Scheme 119). In addition, the application of sequential independent second Rh-catalyzed and third Au-catalyzed procedures (each step with its adequate work-up) was evaluated, using the corresponding triazoles 442 as starting materials in combination with propargylic alcohols 440 or 1-ethynylcyclohexan-1-ol 444.
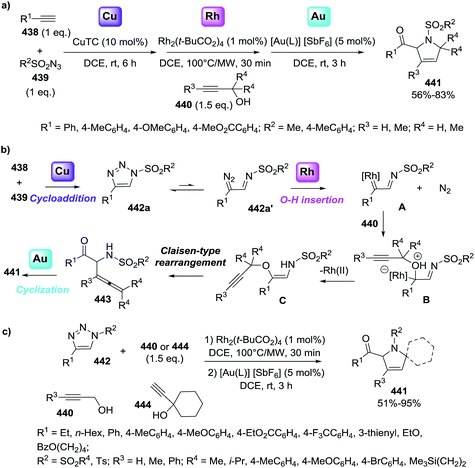 |
| | Scheme 138 Sequential one-pot synthesis of 3-pyrrolines catalyzed by three different metals (Cu, Rh and Au). (a) Three transformations with a single, final work-up. (b) Plausible mechanism. (c) Evaluation of the sequential independent Rh-catalyzed and Au-catalyzed reactions. | |
3. Conclusions
In this review, we describe the latest advances in the transition metal-catalyzed or -mediated synthesis of the three classes of pyrrolines, illustrating the most important aspects of each synthetic method. These advances are classified according to the metal involved in the formation of the pyrroline ring. It is worth noting that a comprehensive and up-to-date compilation on the existing methodologies for the synthesis of the three classes of pyrrolines is missing in the literature. Transition metal-catalyzed reactions are no longer a niche of the organometallic chemist but have entered the mainstream synthesis of heterocycles and complex natural products. Efficiency, mild reaction conditions, and tolerance of a wide variety of functional groups are key characteristics of many of these metal-mediated syntheses. There are many articles encompassing a variety of methodologies that use metals in the synthesis of the pyrroline ring. The most common metals used are copper, gold, silver, palladium, rhodium and ruthenium. A popular methodology is the palladium-catalyzed synthesis of pyrrolines from alkene-tethered oxime esters, which allows decorating the pyrroline ring with different functional groups. Another methodology that we would like to highlight is the use of N-sulfonyl-1,2,3-triazoles as precursors of α-imino rhodium carbene (azavinylcarbenes), which can react with a variety of olefins to produce enantioenriched pyrrolines if chiral rhodium catalysts are used. The metal-catalyzed rearrangement of vinylaziridines to pyrrolines is another exceptionally reliable methodology. Metals such as scandium and nickel have been used lately with excellent results. For example, the scandium-catalyzed synthesis of multi-substituted pyrrolines through sequential reactions has generated complex heterocycles. Nickel catalysts have the advantage that they do not suffer β-H-elimination in cross-coupling reactions, which contrast with the situation met with those based on palladium.
The intention of this review is to inspire further development of the metal-catalyzed synthesis of nitrogen heterocycles, using the pyrrolines as a particular example. Future developments in the transition metal-catalyzed synthesis of pyrrolines will probably involve the adoption of greener reaction conditions, using easily-accessible and less expensive metal catalysts. An increased effort should be devoted to the design of new metal-catalyzed visible-light photo-induced reactions leading to pyrrolines and other nitrogen heterocycles. Apart from being conceptually appealing, these processes are usually efficient, enantioselective and green.
The presence of the pyrroline ring in many bioactive compounds and the remarkable breadth of their reactivity justified the development of new synthetic methods for their construction. Newer approaches to the synthesis of multi-substituted pyrrolines and catalytic asymmetric methodologies for the synthesis of pyrrolines, will be required in the future to further develop these heterocycles for materials and for medicinal chemistry.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors gratefully acknowledge Dr Jed F. Fisher, University of Notre Dame, for critical discussions of the manuscript. The research activities of this laboratory are supported by Agencia Nacional de Promoción Científica y Tecnológica (PICT-2015-2449 and PICT-2016-1069) and Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET, Argentina).
References
- V. C. Clark, C. J. Raxworthy, V. Rakotomalala, P. Sierwald and B. L. Fisher, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 11617 CrossRef CAS PubMed.
- K. L. Rinehart, J. Kobayashi, G. C. Harbour, J. Gilmore, M. Mascal, T. G. Holt, L. S. Shield and F. Lafargue, J. Am. Chem. Soc., 1987, 109, 3378–3387 CrossRef CAS.
- C. Marti and E. M. Carreira, J. Am. Chem. Soc., 2005, 127, 11505–11515 CrossRef CAS PubMed.
- D. Tsukamoto, M. Shibano, R. Okamoto and G. Kusano, Chem. Pharm. Bull., 2001, 49, 492–496 CrossRef CAS PubMed.
- A. Adams and N. De Kimpe, Chem. Rev., 2006, 106, 2299–2319 CrossRef CAS PubMed.
- T.-C. Huang, C.-S. Teng, J.-L. Chang, H.-S. Chuang, C.-T. Ho and M.-L. Wu, J. Agric. Food Chem., 2008, 56, 7399–7404 CrossRef CAS PubMed.
- C. B. Cui, H. Kakeya and H. Osada, J. Antibiot., 1996, 49, 832–835 CrossRef CAS PubMed.
- L. H. Hurley and R. Petrusek, Nature, 1979, 282, 529–531 CrossRef CAS PubMed.
- M. Ozawa, T. Etoh, M. Hayashi, K. Komiyama, A. Kishida and A. Ohsaki, Bioorg. Med. Chem. Lett., 2009, 19, 234–236 CrossRef CAS PubMed.
- The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic, San Diego, 2003 Search PubMed.
- S. Tyroller, W. Zwickenpflug and E. Richter, J. Agric. Food Chem., 2002, 50, 4909–4915 CrossRef CAS PubMed.
- D. Bacos, J. J. Basselier, J. P. Celerler, C. Lange, E. Marx, G. Lhommet, P. Escoubas, M. Lemaire and J. L. Clement, Tetrahedron Lett., 1988, 29, 3061–3064 CrossRef CAS.
- S. Castellano, H. D. G. Fiji, S. S. Kinderman, M. Watanabe, P. de Leon, F. Tamanoi and O. Kwon, J. Am. Chem. Soc., 2007, 129, 5843–5845 CrossRef CAS PubMed.
- S. Schann, V. Bruban, K. Pompermayer, J. Feldman, B. Pfeiffer, P. Renard, E. Scalbert, P. Bousquet and J.-D. Ehrhardt, J. Med. Chem., 2001, 44, 1588–1593 CrossRef CAS PubMed.
- J.-B. Behr, M. S. M. Pearson, C. Bello, P. Vogel and R. Plantier-Royon, Tetrahedron: Asymmetry, 2008, 19, 1829–1832 CrossRef CAS.
- I. V. Magedov, G. Luchetti, N. M. Evdokimov, M. Manpadi, W. F. A. Steelant, S. Van slambrouck, P. Tongwa, M. Y. Antipin and A. Kornienko, Bioorg. Med. Chem. Lett., 2008, 18, 1392–1396 CrossRef CAS PubMed.
- Q.-Y. Mou, J. Chen, Y.-C. Zhu, D.-H. Zhou, Z.-Q. Chi and Y.-Q. Long, Bioorg. Med. Chem. Lett., 2002, 12, 2287–2290 CrossRef PubMed.
- D. Rondeau, P. Gill, M. Chan, K. Curry and W. D. Lubell, Bioorg. Med. Chem. Lett., 2000, 10, 771–773 CrossRef CAS PubMed.
- C. D. Cox, P. J. Coleman, M. J. Breslin, D. B. Whitman, R. M. Garbaccio, M. E. Fraley, C. A. Buser, E. S. Walsh, K. Hamilton, M. D. Schaber, R. B. Lobell, W. Tao, J. P. Davide, R. E. Diehl, M. T. Abrams, V. J. South, H. E. Huber, M. Torrent, T. Prueksaritanont, C. Li, D. E. Slaughter, E. Mahan, C. Fernandez-Metzler, Y. Yan, L. C. Kuo, N. E. Kohl and G. D. Hartman, J. Med. Chem., 2008, 51, 4239–4252 CrossRef CAS PubMed.
- S. Peddibhotla and J. J. Tepe, J. Am. Chem. Soc., 2004, 126, 12776–12777 CrossRef CAS PubMed.
- D. Imbri, N. Netz, M. Kucukdisli, L. M. Kammer, P. Jung, A. Kretzschmann and T. Opatz, J. Org. Chem., 2014, 79, 11750–11758 CrossRef CAS PubMed.
- X. F. Bai, L. Li, Z. Xu, Z. J. Zheng, C. G. Xia, Y. M. Cui and L. W. Xu, Chem.–Eur. J., 2016, 22, 10399–10404 CrossRef CAS PubMed.
- M. K. Majhail, P. M. Ylioja and M. C. Willis, Chem.–Eur. J., 2016, 22, 7879–7884 CrossRef CAS PubMed.
- G. Dannhardt and W. Kiefer, Arch. Pharm., 2001, 334, 183–188 CrossRef CAS PubMed.
- B. B. Snider and B. J. Neubert, Org. Lett., 2005, 7, 2715–2718 CrossRef CAS PubMed.
- F. A. Davis, N. Theddu and R. Edupuganti, Org. Lett., 2010, 12, 4118–4121 CrossRef CAS PubMed.
- V. B. Reddy Iska, V. Verdolino, O. Wiest and P. Helquist, J. Org. Chem., 2010, 75, 1325–1328 CrossRef PubMed.
- M. M. Nebe, M. Kucukdisli and T. Opatz, J. Org. Chem., 2016, 81, 4112–4121 CrossRef CAS PubMed.
- J. M. Humphrey, Y. Liao, A. Ali, T. Rein, Y.-L. Wong, H.-J. Chen, A. K. Courtney and S. F. Martin, J. Am. Chem. Soc., 2002, 124, 8584–8592 CrossRef CAS PubMed.
- R. Martin, A. Jager, M. Bohl, S. Richter, R. Fedorov, D. J. Manstein, H. O. Gutzeit and H. J. Knolker, Angew. Chem., Int. Ed., 2009, 48, 8042–8046 CrossRef CAS PubMed.
- J. Wegner, S. V. Ley, A. Kirschning, A.-L. Hansen, J. Montenegro Garcia and I. R. Baxendale, Org. Lett., 2012, 14, 696–699 CrossRef CAS PubMed.
- H. Zhang and D. P. Curran, J. Am. Chem. Soc., 2011, 133, 10376–10378 CrossRef CAS PubMed.
- T. Ritthiwigrom, A. C. Willis and S. G. Pyne, J. Org. Chem., 2010, 75, 815–824 CrossRef CAS PubMed.
- S. Kaden and H.-U. Reissig, Org. Lett., 2006, 8, 4763–4766 CrossRef CAS PubMed.
- V. Dhand, J. A. Draper, J. Moore and R. Britton, Org. Lett., 2013, 15, 1914–1917 CrossRef CAS PubMed.
- A. L. L. Garcia, M. J. S. Carpes, A. C. B. M. de Oca, M. A. G. dos Santos, C. C. Santana and C. R. D. Correia, J. Org. Chem., 2005, 70, 1050–1053 CrossRef CAS PubMed.
- S. G. Davies, A. M. Fletcher, I. T. T. Houlsby, P. M. Roberts and J. E. Thomson, J. Org. Chem., 2017, 82, 6689–6702 CrossRef CAS PubMed.
- F. Bellina and R. Rossi, Tetrahedron, 2006, 62, 7213–7256 CrossRef CAS.
- M. G. A. Shvekhgeimer, Chem. Heterocycl. Compd., 2003, 39, 405–448 CrossRef CAS.
- P. N. D. Singh, R. F. Klima, S. Muthukrishnan, R. S. Murthy, J. Sankaranarayanan, H. M. Stahlecker, B. Patel and A. D. Gudmundsdóttir, Tetrahedron Lett., 2005, 46, 4213–4217 CrossRef CAS.
- S. Asghari and M. Qandalee, Synth. Commun., 2010, 40, 2172–2177 CrossRef CAS.
- E. Elamparuthi, S. Sarathkumar, S. Girija and V. Anbazhagan, Tetrahedron Lett., 2014, 55, 3992–3995 CrossRef CAS.
- A. Claesson, C. Sahlberg and K. Luthman, Acta Chem. Scand., 1979, 33, 309–310 CrossRef.
- R. K. Dieter and H. Yu, Org. Lett., 2001, 3, 3855–3858 CrossRef CAS PubMed.
- R. K. Dieter, N. Chen and V. K. Gore, J. Org. Chem., 2006, 71, 8755–8760 CrossRef CAS PubMed.
- S. Mangelinckx, N. Giubellina and N. De Kimpe, Chem. Rev., 2004, 104, 2353–2400 CrossRef CAS PubMed.
- B. Cui, J. Ren and Z. W. Wang, J. Org. Chem., 2014, 79, 790–796 CrossRef CAS PubMed.
- P. Pandit, N. Chatterjee and D. K. Maiti, Chem. Commun., 2011, 47, 1285–1287 RSC.
- P. A. Wender and D. Strand, J. Am. Chem. Soc., 2009, 131, 7528–7529 CrossRef CAS PubMed.
- Y.-Q. Fang and E. N. Jacobsen, J. Am. Chem. Soc., 2008, 130, 5660–5661 CrossRef CAS PubMed.
- C. E. Henry, Q. Xu, Y. C. Fan, T. J. Martin, L. Belding, T. Dudding and O. Kwon, J. Am. Chem. Soc., 2014, 136, 11890–11893 CrossRef CAS PubMed.
- P. J. Campos, A. Soldevilla, D. Sampedro and M. A. Rodríguez, Tetrahedron Lett., 2002, 43, 8811–8813 CrossRef CAS.
- P. J. Campos, A. Soldevilla, D. Sampedro and M. A. Rodríguez, Org. Lett., 2001, 3, 4087–4089 CrossRef CAS PubMed.
- A. Soldevilla, D. Sampedro, P. J. Campos and M. A. Rodríguez, J. Org. Chem., 2005, 70, 6976–6979 CrossRef CAS PubMed.
- R. S. Atkinson and C. W. Rees, Chem. Commun., 1967, 1232 RSC.
- T. Hudlicky and J. W. Reed, Angew. Chem., Int. Ed., 2010, 49, 4864–4876 CrossRef CAS PubMed.
- Y. J. Liang, D. W. Dong, Y. M. Lu, Y. Wang, W. Pan, Y. Y. Chai and Q. Liu, Synthesis, 2006, 3301–3304 CAS.
- G. Sathishkannan and K. Srinivasan, Org. Lett., 2011, 13, 6002–6005 CrossRef CAS PubMed.
- D. H. Zhang, Z. Zhang and M. Shi, Chem. Commun., 2012, 48, 10271–10279 RSC.
- R. Miyauchi, C. Ono, T. Ohnuki and Y. Shibad, Appl. Environ. Microbiol., 2016, 82, 6414–6422 CrossRef CAS PubMed.
- E. Tsujii, M. Muroi, N. Shiragami and A. Takatsuki, Biochem. Biophys. Res. Commun., 1996, 220, 459–466 CrossRef CAS PubMed.
- M. X. Zhao, H. K. Zhu, T. L. Dai and M. Shi, J. Org. Chem., 2015, 80, 11330–11338 CrossRef CAS PubMed.
- T. J. Hagen, A. A. Bergmanis, S. W. Kramer, K. F. Fok, A. E. Schmelzer, B. S. Pitzele, L. Swenton, G. M. Jerome, C. M. Kornmeier, W. M. Moore, L. F. Branson, J. R. Connor, P. T. Manning, M. G. Currie and E. A. Hallinan, J. Med. Chem., 1998, 41, 3675–3683 CrossRef CAS PubMed.
- V. Vaillancourt and S. M. K. Sheehan, PCT Int. Appl. WO2013169622A1, 2013.
- D. Aicher, A. Wiehe, C. B. W. Stark, and V. Albrechr, US Pat., Appl. US20130041307A1, 2013.
- D. Aicher, V. Albrecht, B. Gitter, C. B. W. Stark, and A. Wiehe, PCT Int. Appl. WO2013015774A1, 2013.
- D. Aicher, S. Gräfe, C. B. W. Stark and A. Wiehe, Bioorg. Med. Chem. Lett., 2011, 21, 5808–5811 CrossRef CAS PubMed.
- D. Aicher, A. Wiehe, C. B. W. Stark, V. Albrecht and S. Grafe, PCT Int. Appl. WO2012012809A2, 2012.
- R. M. Rosser and D. J. Faulkner, J. Org. Chem., 1984, 49, 5157–5160 CrossRef CAS.
- E. T. Newcomb, P. C. Knutson, B. A. Pedersen and E. M. Ferreira, J. Am. Chem. Soc., 2016, 138, 108–111 CrossRef CAS PubMed.
- S. Sanjaya, S. H. Chua and S. Chiba, Synlett, 2012, 1657–1661 CAS.
- W. Debrouwer, T. S. A. Heugebaert, K. Van Hecke and C. V. Stevens, J. Org. Chem., 2013, 78, 8232–8241 CrossRef CAS PubMed.
- X.-H. Ouyang, R.-J. Song, Y. Liu, M. Hu and J.-H. Li, Org. Lett., 2015, 17, 6038–6041 CrossRef CAS PubMed.
- J. Duan, Y. Cheng, R. Li and P. Li, Org. Chem. Front., 2016, 3, 1614–1618 RSC.
- S.-P. Jiang, Y.-T. Su, K.-Q. Liu, Q.-H. Wu and G.-W. Wang, Chem. Commun., 2015, 51, 6548–6551 RSC.
- B. Liu, Z.-M. Zhang, B. Xu, S. Xu, H.-H. Wu, Y. Liu and J. Zhang, Org. Chem. Front., 2017, 4, 1772–1776 RSC.
- A. Faulkner, N. J. Race, J. S. Scott and J. F. Bower, Chem. Sci., 2014, 5, 2416–2421 RSC.
- T. Saegusa, Y. Ito, H. Kinoshita and S. Tomita, J. Org. Chem., 1971, 36, 3316–3323 CrossRef CAS.
- S. Padilla, J. Adrio and J. C. Carretero, J. Org. Chem., 2012, 77, 4161–4166 CrossRef CAS PubMed.
- K. Goutham, N. Mangina, S. Suresh, P. Raghavaiah and G. V. Karunakar, Org. Biomol. Chem., 2014, 12, 2869–2873 RSC.
- N. S. Medran, M. Villalba, E. G. Mata and S. A. Testero, Eur. J. Org. Chem., 2016, 3757–3764 CrossRef CAS.
- A. D. Melhado, G. W. Amarante, Z. J. Wang, M. Luparia and F. D. Toste, J. Am. Chem. Soc., 2011, 133, 3517–3527 CrossRef CAS PubMed.
- K. Guo, H. Zhang, S. Cao, C. Gu, H. Zhou, J. Li and Y. Zhu, Org. Lett., 2018, 20, 2261–2264 CrossRef CAS PubMed.
- S.-H. Cai, J. H. Xie, S. J. Song, L. Ye, C. Feng and T. P. Loh, ACS Catal., 2016, 6, 5571–5574 CrossRef CAS.
- S.-H. Cai, D.-X. Wang, L. Ye, Z.-Y. Liu, C. Feng and T.-P. Loh, Adv. Synth. Catal., 2018, 360, 1262–1266 CrossRef CAS.
- H. Jiang and A. Studer, Angew. Chem., Int. Ed., 2017, 56, 12273–12276 CrossRef CAS PubMed.
- C. Hua, K. Q. Vuong, M. Bhadbhade and B. A. Messerle, Organometallics, 2012, 31, 1790–1800 CrossRef CAS.
- H. Kawai, T. Kitayama, E. Tokunaga, T. Matsumoto, H. Sato, M. Shiro and N. Shibata, Chem. Commun., 2012, 48, 4067–4069 RSC.
- H. Kawai, Z. Yuan, T. Kitayama, E. Tokunaga and N. Shibata, Angew. Chem., Int. Ed., 2013, 52, 5575–5579 CrossRef CAS PubMed.
- H. B. Yang and N. Selander, Chem.–Eur. J., 2017, 23, 1779–1783 CrossRef CAS PubMed.
- M. M. Jackman, Y. Cai and S. L. Castle, Synthesis, 2017, 49, 1785–1795 CrossRef CAS.
- T. Shimbayashi, K. Okamoto and K. Ohe, Chem.–Asian J., 2018, 13, 395–399 CrossRef CAS PubMed.
- M. Bingham, C. Moutrille and S. Z. Zard, Heterocycles, 2014, 88, 953–960 CrossRef CAS.
- H. B. Yang, S. R. Pathipati and N. Selander, ACS Catal., 2017, 7, 8441–8445 CrossRef CAS.
- L. Wang and C. Wang, Org. Chem. Front., 2018, 5, 3476–3482 RSC.
- A. Faulkner, J. S. Scott and J. F. Bower, J. Am. Chem. Soc., 2015, 137, 7224–7230 CrossRef CAS PubMed.
- A. Faulkner, J. S. Scott and J. F. Bower, Chem. Commun., 2013, 49, 1521–1523 RSC.
- N. J. Race and J. F. Bower, Org. Lett., 2013, 15, 4616–4619 CrossRef CAS PubMed.
- N. J. Race, A. Faulkner, G. Fumagalli, T. Yamauchi, J. S. Scott, M. Ryden-Landergren, H. A. Sparkes and J. F. Bower, Chem. Sci., 2017, 8, 1981–1985 RSC.
- X. Bao, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2017, 56, 9577–9581 CrossRef CAS PubMed.
- Z. Shi, M. Suri and F. Glorius, Angew. Chem., Int. Ed., 2013, 52, 4892–4896 CrossRef CAS PubMed.
- C. Chen, L. Hou, M. Cheng, J. Su and X. Tong, Angew. Chem., Int. Ed., 2015, 54, 3092–3096 CrossRef CAS PubMed.
- D.-Y. Li, S. Liu, S. Chen, A. Wang, X.-P. Zhu and P.-N. Liu, ACS Catal., 2018, 8, 6407–6412 CrossRef CAS.
- F. Alonso, I. P. Beletskaya and M. Yus, Chem. Rev., 2004, 104, 3079–3160 CrossRef CAS PubMed.
- T. E. Müller, K. C. Hultzsch, M. Yus, F. Foubelo and M. Tada, Chem. Rev., 2008, 108, 3795–3892 CrossRef PubMed.
- X. Huang, X. Li, X. Xie, K. Harms, R. Riedel and E. Meggers, Nat. Commun., 2017, 8, 2245 CrossRef PubMed.
- V. Kanchupalli and S. Katukojvala, Angew. Chem., Int. Ed., 2018, 57, 5433–5437 CrossRef CAS PubMed.
- D. Chao, T.-X. Liu, N. Ma, P. Zhang, Z. Fu, J. Ma, Q. Liu, F. Zhang, Z. Zhang and G. Zhang, Chem. Commun., 2016, 52, 982–985 RSC.
- Z.-P. Wang, S. Xiang, P.-L. Shao and Y. He, J. Org. Chem., 2018, 83, 10995–11007 CrossRef CAS PubMed.
- A. Mondal and C. Mukhopadhyay, Eur. J. Org. Chem., 2017, 6299–6313 CrossRef CAS.
- J. J. Badillo, C. J. A. Ribeiro, M. M. Olmstead and A. K. Franz, Org. Lett., 2014, 16, 6270–6273 CrossRef CAS PubMed.
- X. Zhu and S. Chiba, Chem. Commun., 2016, 52, 2473–2476 RSC.
- J. N. Zhu, L. L. Chen, R. X. Zhou, B. Li, Z. Y. Shao and S. Y. Zhao, Org. Lett., 2017, 19, 6044–6047 CrossRef CAS PubMed.
- K. Liu, C. Zhu, J. Min, S. Peng, G. Xu and J. Sun, Angew. Chem., Int. Ed., 2015, 54, 12962–12967 CrossRef CAS PubMed.
- M. Yoshida, A. Kobayashi, A. Nakayama and K. Namba, Tetrahedron, 2016, 72, 2544–2551 CrossRef CAS.
- R. Wang, Y. OuYang, C. Xu, N. Yi, J. Jiang, W. Deng, Z. Zeng and J. Xiang, Org. Biomol. Chem., 2017, 15, 796–800 RSC.
- S.-P. Jiang, Q.-H. Wu and G.-W. Wang, J. Org. Chem., 2017, 82, 10823–10829 CrossRef CAS PubMed.
- B. Xu, Z.-M. Zhang, L. Zhou and J. Zhang, Org. Lett., 2018, 20, 2716–2719 CrossRef CAS PubMed.
- X. Shi, X. Chen, M. Wang, X. Zhang and X. Fan, J. Org. Chem., 2018, 83, 6524–6533 CrossRef CAS PubMed.
- B. Ritzen, G. J. J. Richelle, L. Brocken, F. L. van Delft and F. Rutjes, Synlett, 2014, 25, 270–274 CAS.
- D. Monge, K. L. Jensen, P. T. Franke, L. Lykke and K. A. Jørgensen, Chem.–Eur. J., 2010, 16, 9478–9484 CrossRef CAS PubMed.
- Y. F. Yu, C. Shu, B. Zhou, J. Q. Li, J. M. Zhou and L. W. Ye, Chem. Commun., 2015, 51, 2126–2129 RSC.
- Z.-S. Wang, T.-D. Tan, C.-M. Wang, D.-Q. Yuan, T. Zhang, P. Zhu, C. Zhu, J.-M. Zhou and L.-W. Ye, Chem. Commun., 2017, 53, 6848–6851 RSC.
- L. M. Joyce, A. C. Willis, C. J. T. Hyland and S. G. Pyne, Aust. J. Chem., 2018, 71, 682–689 CrossRef CAS.
- M. C. Chung, Y. H. Chan, W. J. Chang and D. R. Hou, Org. Biomol. Chem., 2017, 15, 3783–3790 RSC.
- C. R. Liu, B. H. Zhu, J. C. Zheng, X. L. Sun, Z. W. Xie and Y. Tang, Chem. Commun., 2011, 47, 1342–1344 RSC.
- B. Sun, Q. Ma, Y. Wang, Y. Zhao, P. Liao and X. Bi, Eur. J. Org. Chem., 2014, 2014, 7552–7555 CrossRef CAS.
- O. El-Sepelgy, A. Brzozowska, J. Sklyaruk, Y. K. Jang, V. Zubar and M. Rueping, Org. Lett., 2018, 20, 696–699 CrossRef CAS PubMed.
- T. Y. Lin, H. H. Wu, J. J. Feng and J. L. Zhang, Org. Lett., 2017, 19, 6526–6529 CrossRef CAS PubMed.
- J.-C. Zeng, H. Xu, R.-L. Huang, F. Yu and Z. Zheng, Tetrahedron Lett., 2018, 59, 1576–1580 CrossRef CAS.
- M. C. Martin, D. V. Patil and S. France, J. Org. Chem., 2014, 79, 3030–3039 CrossRef CAS PubMed.
- W. Zhou, G. An, G. Zhang, J. Han and Y. Pan, Org. Biomol. Chem., 2011, 9, 5833–5837 RSC.
- J. Peng, Y. Zhao, J. Zhou, Y. Ding and C. Chen, Synthesis, 2014, 46, 2051–2056 CrossRef CAS.
- B. Jiang, F. F. Meng, Q. J. Liang, Y. H. Xu and T. P. Loh, Org. Lett., 2017, 19, 914–917 CrossRef CAS PubMed.
- A. K. A. Persson and J.-E. Bäckvall, Angew. Chem., Int. Ed., 2010, 49, 4624–4627 CrossRef CAS PubMed.
- K. Okamoto, T. Oda, S. Kohigashi and K. Ohe, Angew. Chem., Int. Ed., 2011, 50, 11470–11473 CrossRef CAS PubMed.
- M. Yoshida, K. Kinoshita and K. Namba, Org. Biomol. Chem., 2014, 12, 2394–2403 RSC.
- P. V. Santhini, G. Nimisha, J. John, E. Suresh, R. L. Varma and K. V. Radhakrishnan, Chem. Commun., 2017, 53, 1848–1851 RSC.
- T. Zheng, D.-S. Shan, B. Jin and R.-F. Peng, Org. Biomol. Chem., 2018, 16, 8845–8853 RSC.
- Y. Y. Zhang, Y. Wei, X. Y. Tang and M. Shi, Chem. Commun., 2017, 53, 5966–5969 RSC.
- H. Kusama, Y. Karibe, R. Imai, Y. Onizawa, H. Yamabe and N. Iwasawa, Chem.–Eur. J., 2011, 17, 4839–4848 CrossRef CAS PubMed.
- S. Kim, J. Mo, J. Kim, T. Ryu and P. H. Lee, Asian J. Org. Chem., 2014, 3, 926–931 CrossRef CAS.
- T. Miura, T. Tanaka, K. Hiraga, S. G. Stewart and M. Murakami, J. Am. Chem. Soc., 2013, 135, 13652–13655 CrossRef CAS PubMed.
- S. W. Kwok, L. Zhang, N. P. Grimster and V. V. Fokin, Angew. Chem., Int. Ed., 2014, 53, 3452–3456 CrossRef CAS PubMed.
- X. Ma, L. Liu, J. Wang, X. Xi, X. Xie and H. Wang, J. Org. Chem., 2018, 83, 14518–14526 CrossRef CAS PubMed.
- H. Shang, Y. H. Wang, Y. Tian, J. Feng and Y. F. Tang, Angew. Chem., Int. Ed., 2014, 53, 5662–5666 CrossRef CAS PubMed.
- X. Zheng, B. Cao and X. Zhang, Tetrahedron Lett., 2014, 55, 4489–4491 CrossRef CAS.
- J.-J. Feng, T.-Y. Lin, C.-Z. Zhu, H. Wang, H.-H. Wu and J. Zhang, J. Am. Chem. Soc., 2016, 138, 2178–2181 CrossRef CAS PubMed.
- C. E. Kim, Y. Park, S. Park and P. H. Lee, Adv. Synth. Catal., 2015, 357, 210–220 CrossRef CAS.
- B. Alcaide, P. Almendros, S. Cembellin, T. M. del Campo and G. Palop, Chem.–Eur. J., 2017, 23, 13754–13759 CrossRef CAS PubMed.
- M. K. Ghorai and D. P. Tiwari, J. Org. Chem., 2013, 78, 2617–2625 CrossRef CAS PubMed.
- Y. Xia, X. Liu, H. Zheng, L. Lin and X. Feng, Angew. Chem., Int. Ed., 2015, 54, 227–230 CrossRef CAS PubMed.
- M. Schlegel, P. Coburger and C. Schneider, Chem.–Eur. J., 2018, 24, 14207–14212 CrossRef CAS PubMed.
- C. Arroniz, A. Gil-Gonzalez, V. Semak, C. Escolano, J. Bosch and M. Amat, Eur. J. Org. Chem., 2011, 3755–3760 CrossRef CAS.
- J. Song, C. Guo, P.-H. Chen, J. Yu, S.-W. Luo and L.-Z. Gong, Chem.–Eur. J., 2011, 17, 7786–7790 CrossRef CAS PubMed.
- W. K. Anderson and A. S. Milowsky, J. Med. Chem., 1987, 30, 2144–2147 CrossRef CAS PubMed.
- G. Bhaskar, Y. Arun, C. Balachandran, C. Saikumar and P. T. Perumal, Eur. J. Med. Chem., 2012, 51, 79–91 CrossRef CAS PubMed.
- W. Shi, Y. Duan, Y. Qian, M. Li, L. Yang and W. Hu, Bioorg. Med. Chem. Lett., 2010, 20, 3592–3595 CrossRef CAS PubMed.
- M. Brichacek, M. Navarro Villalobos, A. Plichta and J. T. Njardarson, Org. Lett., 2011, 13, 1110–1113 CrossRef CAS PubMed.
- Q. Wu, J. Hu, X. Ren and J. Zhou, Chem.–Eur. J., 2011, 17, 11553–11558 CrossRef CAS PubMed.
- M. Sai, H. Yorimitsu and K. Oshima, Angew. Chem., Int. Ed., 2011, 50, 3294–3298 CrossRef CAS PubMed.
- W. Rao, P. Kothandaraman, C. B. Koh and P. W. H. Chan, Adv. Synth. Catal., 2010, 352, 2521–2530 CrossRef CAS.
- C. Zheng, Y. Wang and R. Fan, Org. Lett., 2015, 17, 916–919 CrossRef CAS PubMed.
- F. F. Tang, W. L. Yang, X. X. Yu and W. P. Deng, Catal. Sci. Technol., 2015, 5, 3568–3575 RSC.
- E. J. Groso, A. N. Golonka, R. A. Harding, B. W. Alexander, T. M. Sodano and C. S. Schindler, ACS Catal., 2018, 2006–2011 CrossRef CAS PubMed.
- S. R. K. Minkler, B. H. Lipshutz and N. Krause, Angew. Chem., Int. Ed., 2011, 50, 7820–7823 CrossRef CAS PubMed.
- G. M. O. Amombo, O. Floge, S. K. D. Kalai, S. Schoder, U. Warzok and H. U. Reissig, Eur. J. Org. Chem., 2017, 1965–1972 CrossRef.
- M. O. Amombo, A. Hausherr and H.-U. Reissig, Synlett, 1999, 1999, 1871–1874 CrossRef.
- T. Miura, T. Tanaka, K. Matsumoto and M. Murakami, Chem.–Eur. J., 2014, 20, 16078–16082 CrossRef CAS PubMed.
- D.-H. Zhang, L.-F. Yao, Y. Wei and M. Shi, Angew. Chem., Int. Ed., 2011, 50, 2583–2587 CrossRef CAS PubMed.
- Z. Wang, H. Zheng, J. Yang, X. Xie and X. She, Adv. Synth. Catal., 2015, 357, 2082–2088 CrossRef CAS.
- N. Kern, A. Blanc, J. M. Weibel and P. Pale, Chem. Commun., 2011, 47, 6665–6667 RSC.
- M. Fuji, J. Chiwata, M. Ozaki, S. Aratani and Y. Obora, ACS Omega, 2018, 3, 8865–8873 CrossRef CAS.
- E. Leemans, F. Colpaert, S. Mangelinckx, S. De Brabandere, B. Denolf and N. De Kimpe, Synlett, 2011, 674–678 CAS.
- P. Wu, H. Liu and X. Tong, Tetrahedron Lett., 2012, 53, 4673–4675 CrossRef CAS.
- W. Sun, G. Zhu, L. Hong and R. Wang, Chem.–Eur. J., 2011, 17, 13958–13962 CrossRef CAS PubMed.
- M.-J. Zhong, H.-T. Zhu, P. Gao, Y.-F. Qiu and Y.-M. Liang, RSC Adv., 2014, 4, 8914–8917 RSC.
- C. B. Reddy, R. Bharti, S. Kumar and P. Das, RSC Adv., 2016, 6, 71117–71121 RSC.
- T. Rajasekaran, G. Karthik, B. Sridhar and B. V. S. Reddy, Org. Lett., 2013, 15, 1512–1515 CrossRef CAS PubMed.
- A. Deiters and S. F. Martin, Chem. Rev., 2004, 104, 2199–2238 CrossRef CAS PubMed.
- A. Perfetto, C. Costabile, P. Longo, V. Bertolasi and F. Grisi, Chem.–Eur. J., 2013, 19, 10492–10496 CrossRef CAS PubMed.
- Q. Yang, H. Alper and W.-J. Xiao, Org. Lett., 2007, 9, 769–771 CrossRef CAS PubMed.
- K. M. Kuhn, T. M. Champagne, S. H. Hong, W.-H. Wei, A. Nickel, C. W. Lee, S. C. Virgil, R. H. Grubbs and R. L. Pederson, Org. Lett., 2010, 12, 984–987 CrossRef CAS PubMed.
- S. Sawadjoon and J. S. M. Samec, Org. Biomol. Chem., 2011, 9, 2548–2554 RSC.
- A. Bunrit, S. Sawadjoon, S. Tsupova, P. J. R. Sjoberg and J. S. M. Samec, J. Org. Chem., 2016, 81, 1450–1460 CrossRef CAS PubMed.
- M. B. Tait, S. Butterworth and J. Clayden, Org. Lett., 2015, 17, 1236–1239 CrossRef CAS PubMed.
- W. Sun, X. Ma, L. Hong and R. Wang, J. Org. Chem., 2011, 76, 7826–7833 CrossRef CAS PubMed.
- S. Ishikawa, F. Noguchi and A. Kamimura, J. Org. Chem., 2010, 75, 3578–3586 CrossRef CAS PubMed.
- Y. Nagao, W.-M. Dai, M. Ochiai and M. Shiro, Tetrahedron, 1990, 46, 6361–6380 CrossRef CAS.
- K. Csatayova, S. G. Davies, A. L. A. Figuccia, A. M. Fletcher, J. G. Ford, J. A. Lee, P. M. Roberts, B. G. Saward, H. Song and J. E. Thomson, Tetrahedron, 2015, 71, 9131–9142 CrossRef CAS.
- L. Li and J. Zhang, Org. Lett., 2011, 13, 5940–5943 CrossRef CAS PubMed.
- R. K. Dieter, N. Chen, H. Yu, L. E. Nice and V. K. Gore, J. Org. Chem., 2005, 70, 2109–2119 CrossRef CAS PubMed.
- B. Mitasev and K. M. Brummond, Synlett, 2006, 2006, 3100–3104 CrossRef.
- M. Sai and S. Matsubara, Org. Lett., 2011, 13, 4676–4679 CrossRef CAS PubMed.
- J. Brioche, C. Meyer and J. Cossy, Org. Lett., 2013, 15, 1626–1629 CrossRef CAS PubMed.
- R. R. Tata, C. Fu, S. P. Kelley and M. Harmata, Org. Lett., 2018, 20, 5723–5726 CrossRef CAS PubMed.
- S. S. K. Boominathan, W. P. Hu, G. C. Senadi and J. J. Wang, Adv. Synth. Catal., 2013, 355, 3570–3574 CrossRef CAS.
- A. Hausherr and H.-U. Reissig, Eur. J. Org. Chem., 2018, 2018, 4071–4080 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*