
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanism and stereoselectivity in NHC-catalyzed β-functionalization of saturated carboxylic ester†
Yan Li * and
Zhiqiang Zhang
* and
Zhiqiang Zhang
School of Chemical Engineering, University of Science and Technology Liaoning, Anshan 114051, P. R. China. E-mail: snow2007liyan@163.com; Fax: +86-412-5929627; Tel: +86-18741219506
First published on 6th March 2019
Abstract
To understand the mechanism and origin of the stereoselectivity of the [3 + 2] annulation reaction between a carboxylic ester and an isatin generating spirooxindole lactone catalyzed by N-heterocyclic carbene (NHC), density functional theory (DFT) calculations have been carried out. DFT results indicate that the catalytic cycle begins with the coupling of the catalyst with benzotriazole ester, followed by α-deprotonation to produce the enolate intermediate. The subsequent 1,4-proton transfer affords the homoenolate intermediate. The next crucial step is the stereoselective C–C bond formation. Then proton transfer takes place leading to the formation of the lactone intermediate. Finally, the elimination of the catalyst furnishes the final product. The presence of 1-hydroxybenzotriazole (HOBt) dramatically accelerates the proton transfer step. More importantly, HOBt has a non-negligible impact on stereoselective C–C bond formation, and the SR-configured product is the major stereoisomer of the annulation product, which is in good agreement with the experimental observations. The differential π⋯π stacking, C–H⋯π, lone pair (LP)⋯π and repulsion interactions are found to be responsible for the stereoselectivity. The obtained mechanistic insights should provide valuable information for understanding the important roles of the NHC catalyst and HOBt additive and be helpful for designing better NHC catalysts for this kind of reaction.
1. Introduction
As one of the most versatile and useful building blocks, saturated carbonyl compounds (e.g., esters and aldehydes) have been widely used in fields as diverse as pharmacology, chemistry, and materials science. Because of their great synthetic value, the functionalization of saturated carboxylic compounds has attracted considerable attention.1 However, the majority of the research work deals with either the carbonyl carbon atom or the α-carbon atom functionalization of saturated carbonyl compounds.2 Developing direct and efficient methods for the β-functionalization of these compounds is highly desirable and is still a challenge for chemists.3During the past few years, N-heterocyclic carbenes (NHCs) have become a hot topic of research due to their widely applications as ligands in organometallic catalysis4 and in Lewis base catalysis.5 More importantly, NHCs have received considerable attention because of their diverse applications in organocatalytic reactions with excellent efficiency, such as umpolung reaction, benzoin, Stetter, Mannich, Michael, cycloadditions, redox reactions, CO2 fixation, cooperative catalysis and cascade reactions.6 Indeed, many cycloaddition reactions using NHC catalysts affording various heterocycles have been reported, including the [2 + 2],7 [2 + 4],8 [3 + n] (n = 2, 3, 4),9 and [4 + n] (n = 2, 3)10 cycloadditions.
Recently, Xu et al. developed a catalytic protocol for the synthesis of spirooxindole lactones via β-functionalization of saturated carboxylic esters in tetrahydrofuran (THF) solvent at room temperature.11 It was found that the reaction of carboxylic esters with isatins could be effectively catalyzed by using NHC as catalyst and 1-hydroxybenzotriazole (HOBt) as additive. Additional experiments showed that HOBt is important in the current reaction because the yields and enantio- and diastereo-selectivities were higher in the presence of HOBt as compared with that in the absence of HOBt.
However, the mechanism of the NHC-catalyzed [3 + 2] annulation reaction between carboxylic esters and isatins (Scheme 1) has not been studied theoretically until now. How does the catalytic reaction proceed? Which step is the stereocontrolling step? What role does the additive HOBt play? In this work, we will examine the NHC-catalyzed [3 + 2] annulation reaction between a carboxylic ester and an isatin using density functional theory calculations. We concentrate on the reaction mechanism as well as the origin of stereoselectivity. The objective of this work is to provide mechanistic insights for the catalyzed reaction shown in Scheme 1 and explain the enhanced stereoselectivity in the presence of HOBt in comparison to that in its absence.
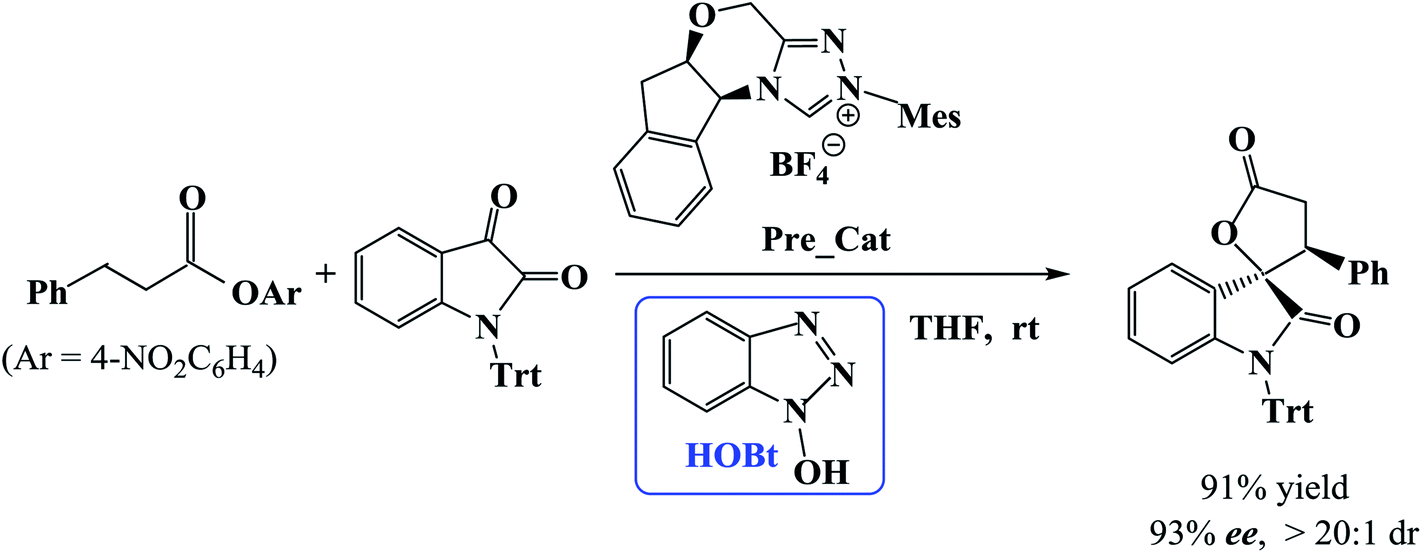 | ||
| Scheme 1 NHC-catalyzed [3 + 2] annulation reaction. | ||
2. Computational methods
Geometries of all the structures involved in the reaction were optimized using the M06-2X functional12 with the 6-31G(d) basis set. Previous studies have confirmed the reliability of the chosen M06-2X functional.13 Frequency calculations were performed in order to check the minimum has all real frequencies while the transition state has one and only one imaginary frequency and to obtain thermodynamic corrections at 298.15 K and 1 atm pressure. In order to confirm the connection of the transition states and the desired minima, intrinsic reaction coordinate (IRC) calculations were also performed.14 To account for solvent effect, single-point energy calculations were carried out at the M06-2X/6-311++G(d,p) level in the solvent phase modeled by the Cramer–Truhlar continuum solvation model SMD15 in tetrahydrofuran (THF, ε = 7.43). To assign the atomic charges, natural bond orbital (NBO) analyses were also carried out.16 All calculations were carried out with the Gaussian 09 software package.173. Results and discussion
3.1 Reaction mechanism
In this section, we present and discuss the detailed results for the [3 + 2] cycloaddition reaction between 4-nitrophenyl 3-phenylpropanoate and isatin depicted in Scheme 1. On the basis of our calculation results, the detailed mechanism for the whole catalytic cycle is presented in Scheme 2. Before considering the possible reaction pathways of the current reaction, we explore first the actual catalyst in the current reaction. As shown in Scheme 2, in the presence of cesium carbonate Cs2CO3, the catalyst precursor Pre_Cat will undergo deprotonation to form the active NHC catalyst and release bicarbonate anion HCO3−.18 Experimentally, 4-nitrophenyl 3-phenylpropanoate was used as one substrate (isatin as the other substrate) and 1-hydroxybenzotriazole (HOBt) as an additive. Computationally, we use the benzotriazole ester, depicted in Scheme 2 as one starting reactant because benzotriazole ester can be generated through the reaction of 4-nitrophenyl 3-phenylpropanoate with HOBt.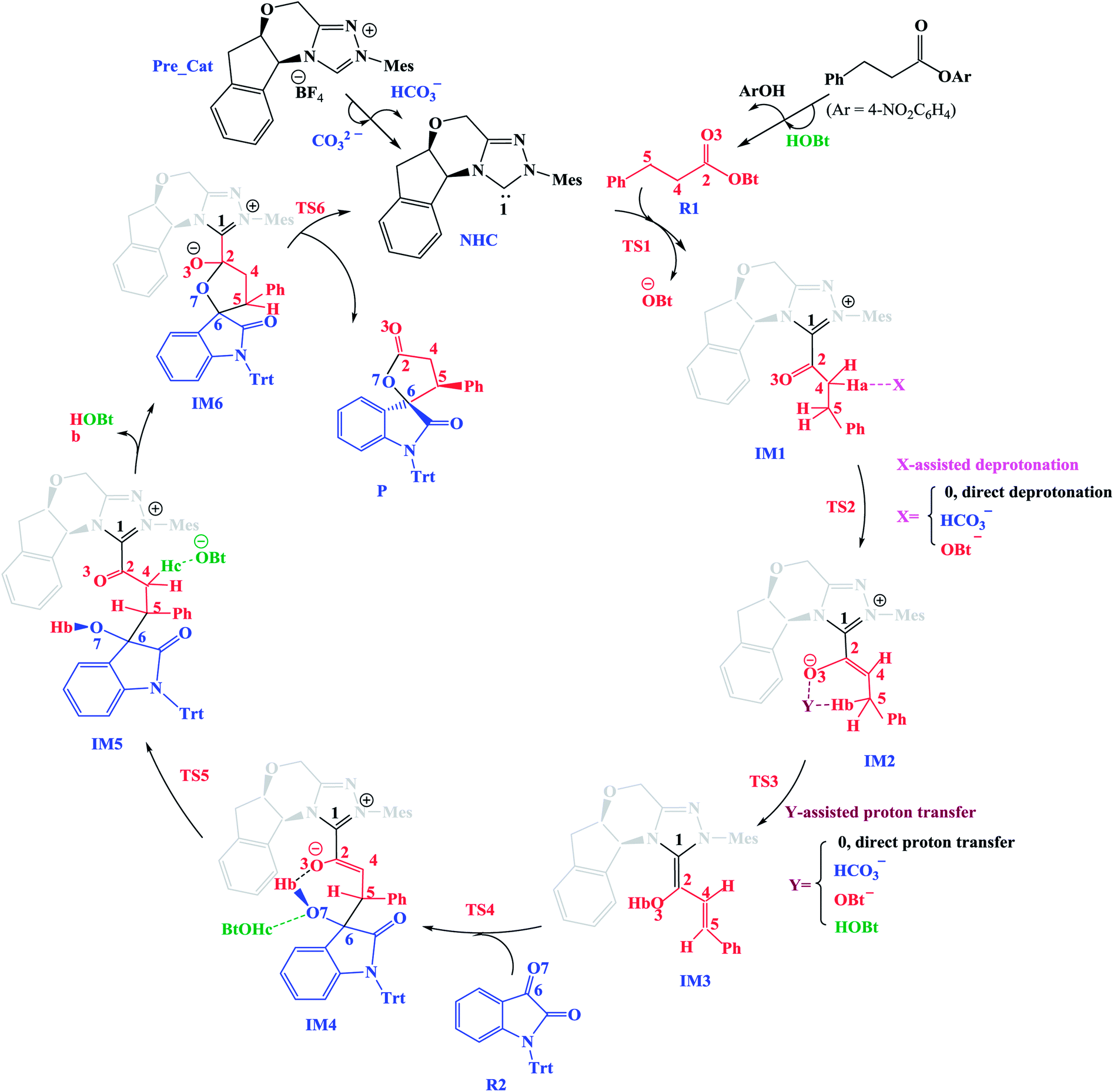 | ||
| Scheme 2 The key steps involved in the NHC-catalyzed [3 + 2] annulation reaction. | ||
As presented in Scheme 2, the catalytic reaction proceeds via six steps: (1) the additon of the active catalyst NHC to benzotriazole ester R1 to form the acyl azolium intermediate IM1 and release OBt−; (2) the deprotonation of the α-C–H in IM1 to generate the enolate intermediate IM2; (3) the proton transfer of IM2 leads to the homoenolate intermediate IM3; (4) the stereoselective carbon–carbon bond formation process which can be achieved from the addition of isatin R2 to IM3 leading to intermediate IM4; (5) the proton transfer followed by the intramolecular lactonization will eventually provide access to the five membered ring intermediate IM6, and (6) the elimination of the NHC catalyst yields the spirooxindole lactone product P with two chiral carbon atoms. The corresponding free energy profiles and optimized structures are shown in Fig. 1 (steps I–III) and 2 (steps IV–VI). In the following part, the reaction mechanism will be examined in greater detail.
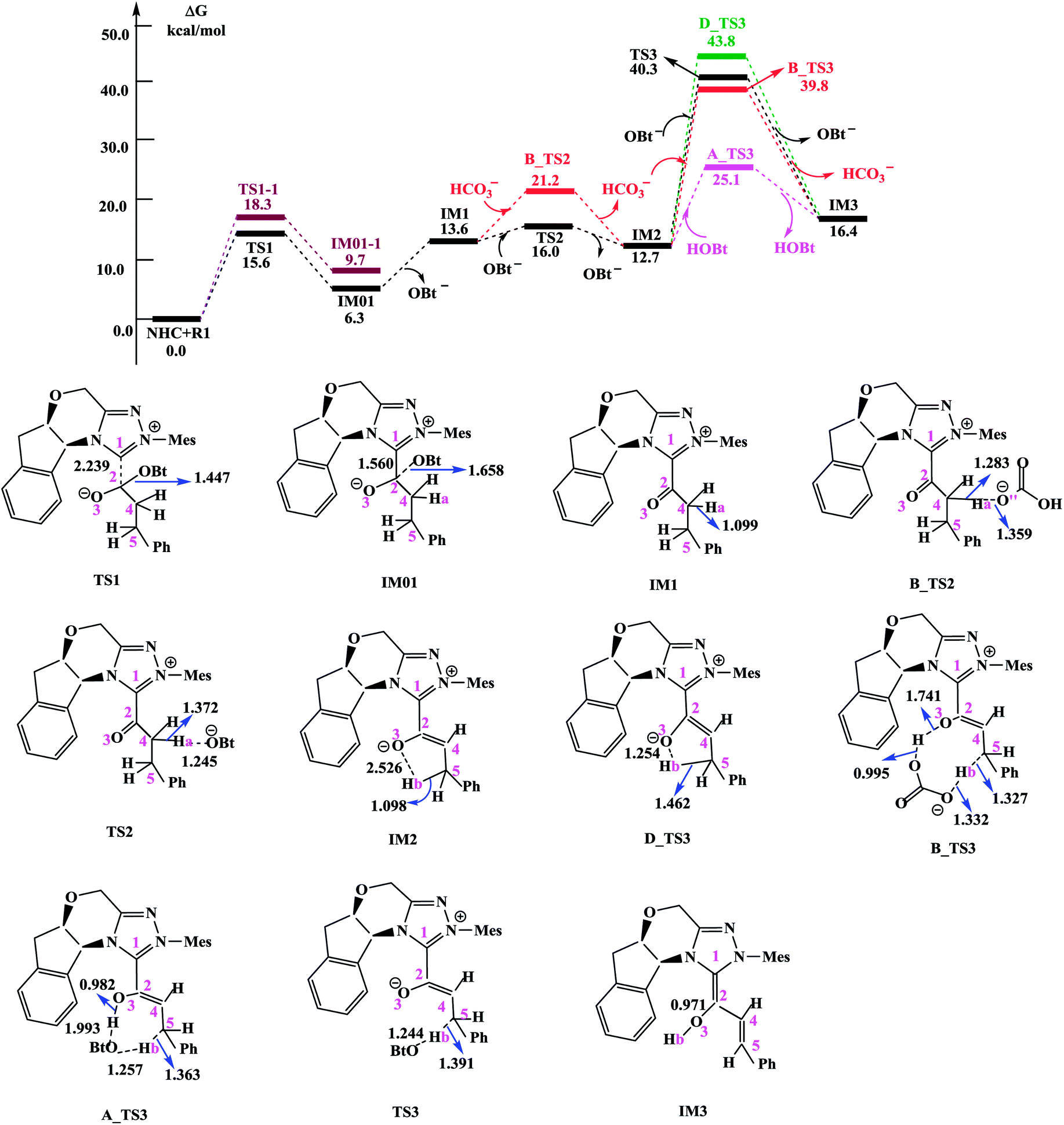 | ||
| Fig. 1 Free energy profile obtained at SMD(THF)/M06-2X/6-311++G(d,p) level (kcal mol−1) and optimized structures for steps I–III. Bond lengths are given in Å. | ||
Scheme 3 shows the NBO charge analysis results. As can be seen from Scheme 3, for Cat + R1 → IM01 process, the positive charge value on the C1 atom increases from 0.148e to 0.414e, while that on the C2 atom decreases from 0.840e to −0.041e. Furthermore, the negative charge value on the N atom decreases from −0.489e to −0.415e, while that of the carbonyl oxygen O3 atom increases from −0.543e to −0.909e. These results indicate that the electron density is transferred from Cat to R1 during the process of Cat + R1 → IM01.
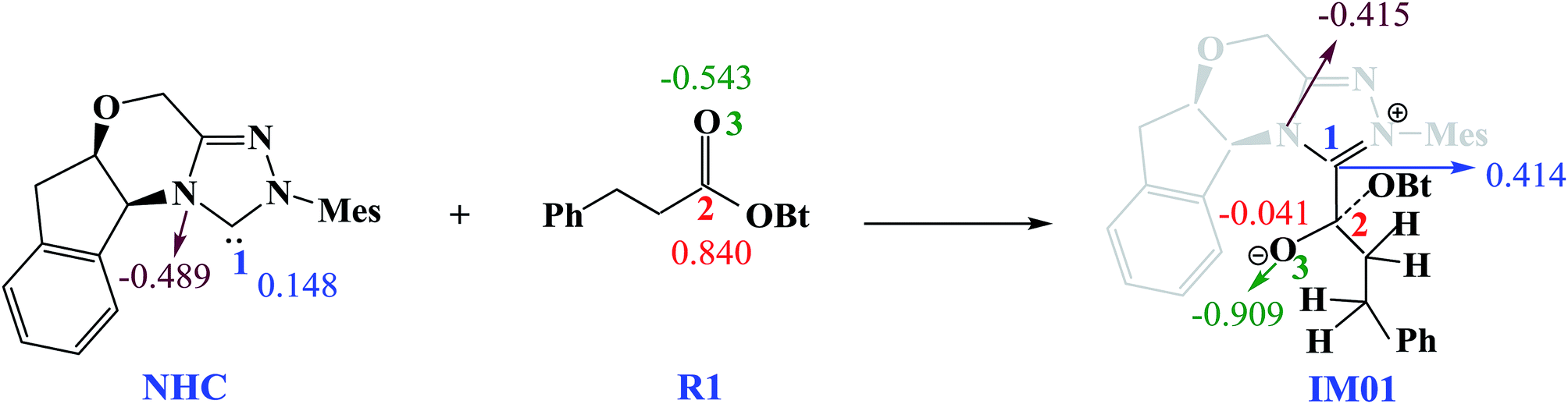 | ||
| Scheme 3 The NBO charges of selected atoms in NHC, R1 and IM01 (units of e). | ||
(1) Direct deprotonation. For direct deprotonation, we can not locate the corresponding transition state despite lots of attempts. We performed relax potential energy scan for the C4–Ha bond. The corresponding pointwise potential curve was provided in Fig. S1 in the ESI.† Fig. S1† clearly shows that the energy keeps increasing when the Ha atom getting farther from the C4 atom of IM1. The high energy barrier estimated for direct deprotonation process (∼70 kcal mol−1), indicates that the direct deprotonation pathway is kinetically unfeasible. In view of this, a mediator assisted deprotonation, facilitated by the previously formed OBt− (or HCO3−) has been examined.
(2) OBt− assisted deprotonation. The anion OBt− generated in the first step can remove the hydrogen atom (Ha) in IM1 via the transition state TS2 to generate intermediate IM2. The calculated free energy barrier is 2.4 kcal mol−1 for TS2 with respect to IM1, demonstrating that the OBt− mediated deprotonation of α-C–H is a fast process under the experimental conditions. During this process (IM1 → TS2 → IM2), the distance of C4–Ha bond is elongated from 1.099 Å to 1.372 Å (IM1 → TS2), whereas the distance of Ha–O bond is shortened from 1.245 Å, to 0.973 Å (TS2 → IM2 + OBt). These results indicate that the deprotonation process is completed. The transformation from IM1 to IM2 is found to be slightly exergonic with a reaction energy of −0.9 kcal mol−1.
(3) HCO3− assisted deprotonation. In another possibility, the previously formed HCO3− was used as promoter. The energy barrier calculated for the HCO3− assisted deprotonation is 7.6 kcal mol−1 via B_TS2. As shown in Fig. 1, the distance of C4–Ha bond increases from 1.099 Å in IM1 to 1.283 Å in B_TS2, while that of Ha–O′′ bond decreases from 1.359 Å in IM1 to 0.968 Å in H2CO3.
Taken together, the mediators (BtO− and HCO3−) promote the deprotonation process. The most favored proton transfer pathway is found to be that from IM1 to IM2 via TS2 with the assistance of OBt−.
For direct 1,4-proton transfer, migration of the Hb atom connected to C5 atom to O3 atom via the corresponding five-membered ring transition state D_TS3 with a free energy barrier of 31.1 kcal mol−1 (43.8 kcal mol−1 with respect to reactants) (Fig. 1). Therefore, direct 1,4-proton transfer in intermediate IM2 is a kinetically inaccessible process under the experimental conditions.
We also calculated the HCO3− and OBt− assisted proton transfer. The corresponding energy profiles are also presented in Fig. 1. However, the high activation energies of B_TS3 (39.8 kcal mol−1 relative to reactants, associated with the HCO3− mediated proton transfer) and TS3 (40.3 kcal mol−1 relative to reactants, associated with the OBt− mediated proton transfer) indicate that the mediator (HCO3− and BtO−) has minimal effects on the proton transfer event. Thus these pathways are also kinetically unfavorable, and can be ruled out.
Experimentally, HOBt was used as an additive, promoting us to investigate the HOBt assisted proton transfer process. According to our calculation results, the 1,4-proton transfer proceeds via the seven-membered ring transition state A_TS3 leading to the formation of the homoenolate intermediate IM3 in the presence of HOBt (Fig. 1). Computational results show that the transition state for this process (A_TS3 (25.1 kcal mol−1)) is much lower in energy than those of the unassisted and the BtO− and HCO3− assisted proton transfer processes (i.e., D_TS3 (43.8 kcal mol−1), B_TS3 (39.8 kcal mol−1) and TS3 (40.3 kcal mol−1)). As can be seen from Fig. 1, the Hb atom migrates from C5 to O, at the same time, the H atom transfers from O to O3 via the seven-membered ring transition state A_TS3. The bond distances of C5–Hb, Hb–O, O–H and O3–H in A_TS3 are 1.363, 1.257, 1.993 and 0.982 Å, respectively, indicating the HOBt mediated 1,4-proton transfer occurs in a concerted manner but not synchronous.
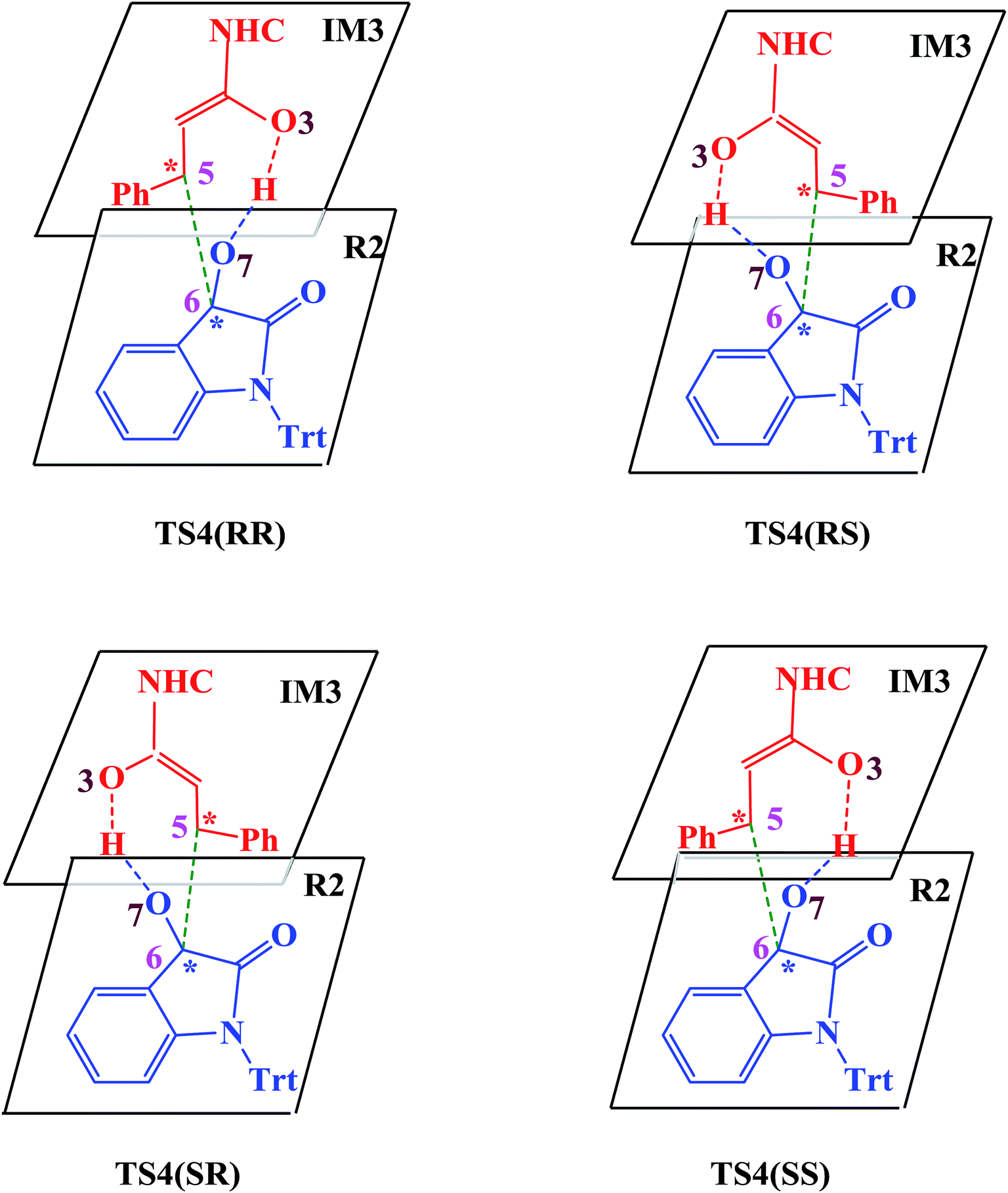 | ||
| Scheme 4 Four attack modes involved in the stereoselectivity determining step (the hydrogen atoms are omitted). | ||
In the absence of HOBt, the activation energy barriers of this step via transition states D_TS4(RR&RS&SR&SS) are 13.2/20.2/14.0/17.9 kcal mol−1 (29.6/36.6/30.4/34.3 kcal mol−1 with respect to reactants), as shown in Fig. S2.† Considering that the activation energy barrier of D_TS4(RR) is lower than those of D_TS4(RS&SR&SS), the reaction pathway associated with D_TS4(RR) should be more favorable than the other pathways. Therefore, the dominant product in the absence of HOBt should be the RR configuration product. This is inconsistent with the experimental results. The experimentally observed major product without the use of HOBt was the SR-stereoisomer of the cycloaddition product.11 Considering this inconsistency, more experimental studies are expected to be necessary to better understand the stereoselectivity associated with the reaction, depicted in Scheme 1 in the absence of HOBt.
Since HOBt was used as an additive in experiment,11 it might assist in this C–C bond formation. We thus have included an explicit molecule of HOBt in this step to form intermediate IM4. Fig. 2 shows that the free energy barriers of this step in the presence of HOBt via transition states TS4(RR&RS&SR&SS) are 9.8/10.2/7.9/12.1 kcal mol−1 (26.2/26.6/24.3/28.5 kcal mol−1 relative to reactants), which is found to be lower than the transition states in the absence of HOBt (for details see the ESI, Fig. S2†). To understand this result, we examined the non and HOBt assisted transition state (D_TS4 and TS4) structures (shown in Fig. S3†). As depicted in Fig. S3,† additional interactions such as O–H⋯O and O–H⋯N are present in the HOBt-assisted transition state compared to the unassisted transition state. These interactions are depicted as follows: O–H⋯O (a, b, c and d) and O–H⋯N (e). Moreover, Fig. 2 clearly shows that the energy barrier of TS4(SR) is remarkably lower than those of other configuration transition states (i.e., TS4(RR), TS4(RS) and TS4(SS)), for simplicity of presentation, we discuss the reaction pathways starting from IM4(SR) here (as shown in Fig. 2). Results for the less favorable pathways which feature RR/RS/SS-configuration are provided in the ESI (Fig. S4†). Intrinsic reaction coordinate (IRC) results for transition state TS4(SR) reveal that the C5–C6 bond formation occurs along with the Hb atom migration from O3 to O7, leading to intermediate IM4(SR). From TS4(SR) to IM4(SR), the distance of C5–C6 bond changes from 2.502 Å to 1.570 Å, at the same time, the distance of O3–Hb bond increases from 0.975 Å to 1.426 Å, while the Hb–O7 bond shortens from 2.136 Å to 1.051 Å. The above structural analysis indicates that the C5–C6 bond formation is accompanied by the transformation of the Hb atom (from O3 to O7).
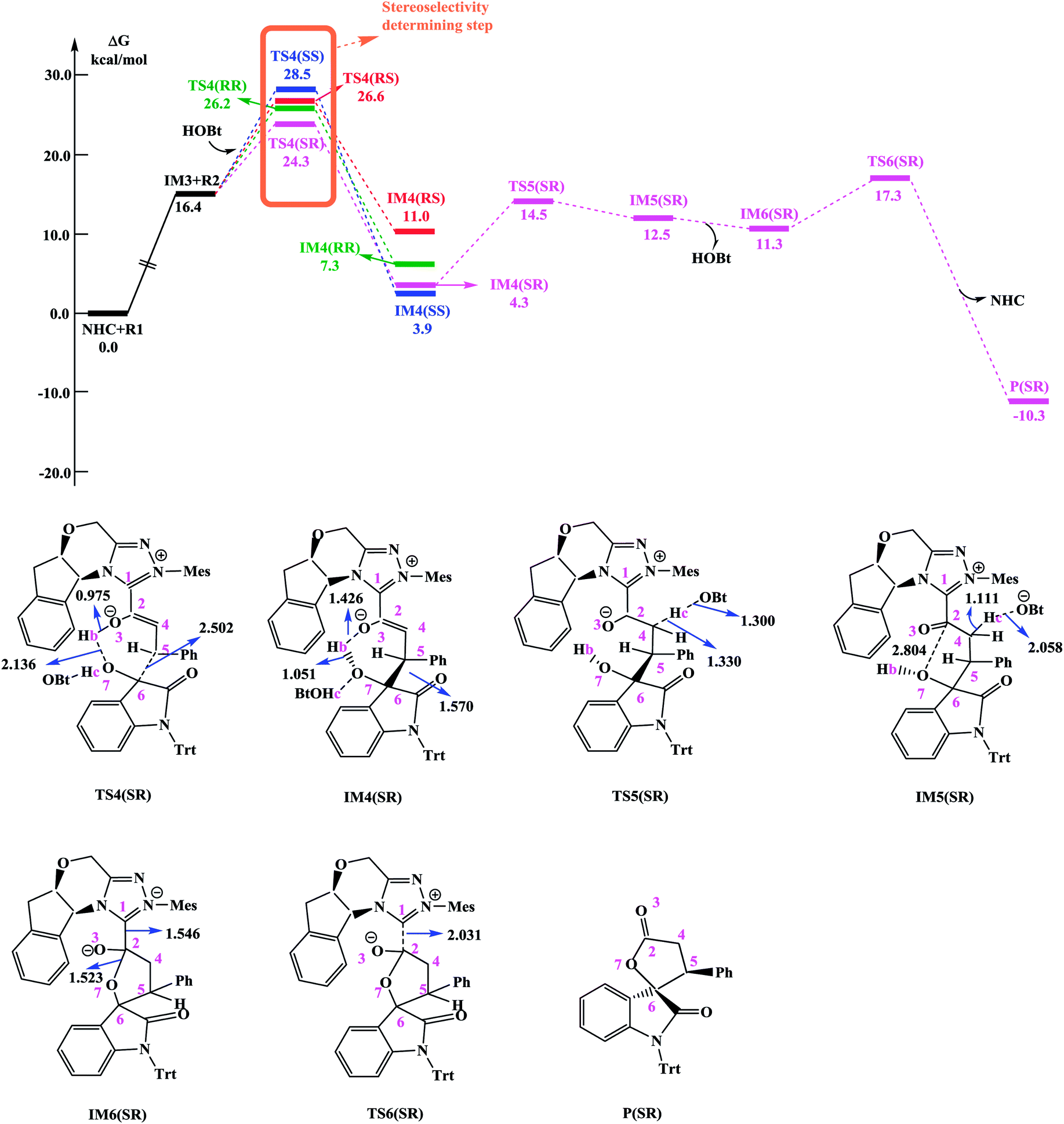 | ||
| Fig. 2 Free energy profile obtained at SMD(THF)/M06-2X/6-311++G(d,p) level (kcal mol−1) and optimized structures for steps IV–VI. Bbond lengths are given in Å. | ||
3.2 Origin of selectivity
As shown in Fig. 2 and discussed above in step IV, the addition of the homoenolate intermediate IM3 to isatin R2 leading to the formation of C–C bond is the stereoselectivity-determining step. Consequently, transition states TS4(RR), TS4(RS), TS4(SR) and TS4(SS) are found to be responsible for determining the stereoselectivity of the entire reaction. The activation energy barriers for generating the RR/RS/SR/SS configuration products via TS4(RR/RS/SR/SS) are calculated to be 26.2/26.6/24.3/28.5 kcal mol−1 (the energy difference between TS4(RR/RS/SR/SS) and reactants). Obviously, the SR-configurational product P(SR) is the major product, agrees well with the experimental observations. Based on the Curtin–Hammett Principle,20 the product distribution is determined by the standard free energy difference of the stereoselectivity-determining transition states. The energy difference between TS4(SR) and TS4(RS), which responsible for enantioselectivity, is 2.3 kcal mol−1 corresponding to an enantiomeric excess of 96%. The computed enantioselectivity is in good agreement with the experimental reported value (93%). Similarly, the energy difference between TS4(SR) and TS4(RR), which is related to diastereoselectivity, is 1.9 kcal mol−1. This energy difference corresponds to a diastereomeric ratio of 92%, which is in good agreement with the experimentally observed de of 91% (diastereomeric ratio > 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1).
:1).
Furthermore, in order to better understand the origin of stereoselectivity of the NHC-catalyzed [3 + 2] annulation reaction, we performed the noncovalent interaction (NCI) analysis21 which is demonstrated to be able to distinguish the strong interactions as well as the repulsion interactions. NCI analysis results for the stereoselectivity-determining transition states TS3(RR), TS3(RS), TS3(SR) and TS3(SS) are presented in Fig. 3. As depicted in Fig. 3, it is clear that a large green cloud between the π-systems of the NHC-bounded homoenolate (IM3-part) and HOBt is observed in TS3(SR), but absent in the other transition states. In addition, the repulsion interactions exist in TS3(RR), TS3(RS) and TS3(SS). Moreover, a number of noncovalent interactions including π⋯π interactions, lone pair (LP)⋯π interactions and C–H⋯π interactions are identified in TS3(SR). Therefore, TS3(SR) is more stable than the other transition states (TS3(RR), TS3(RS) and TS3(SS)), which is in good agreement with the experimentally observed enantioselectivity.
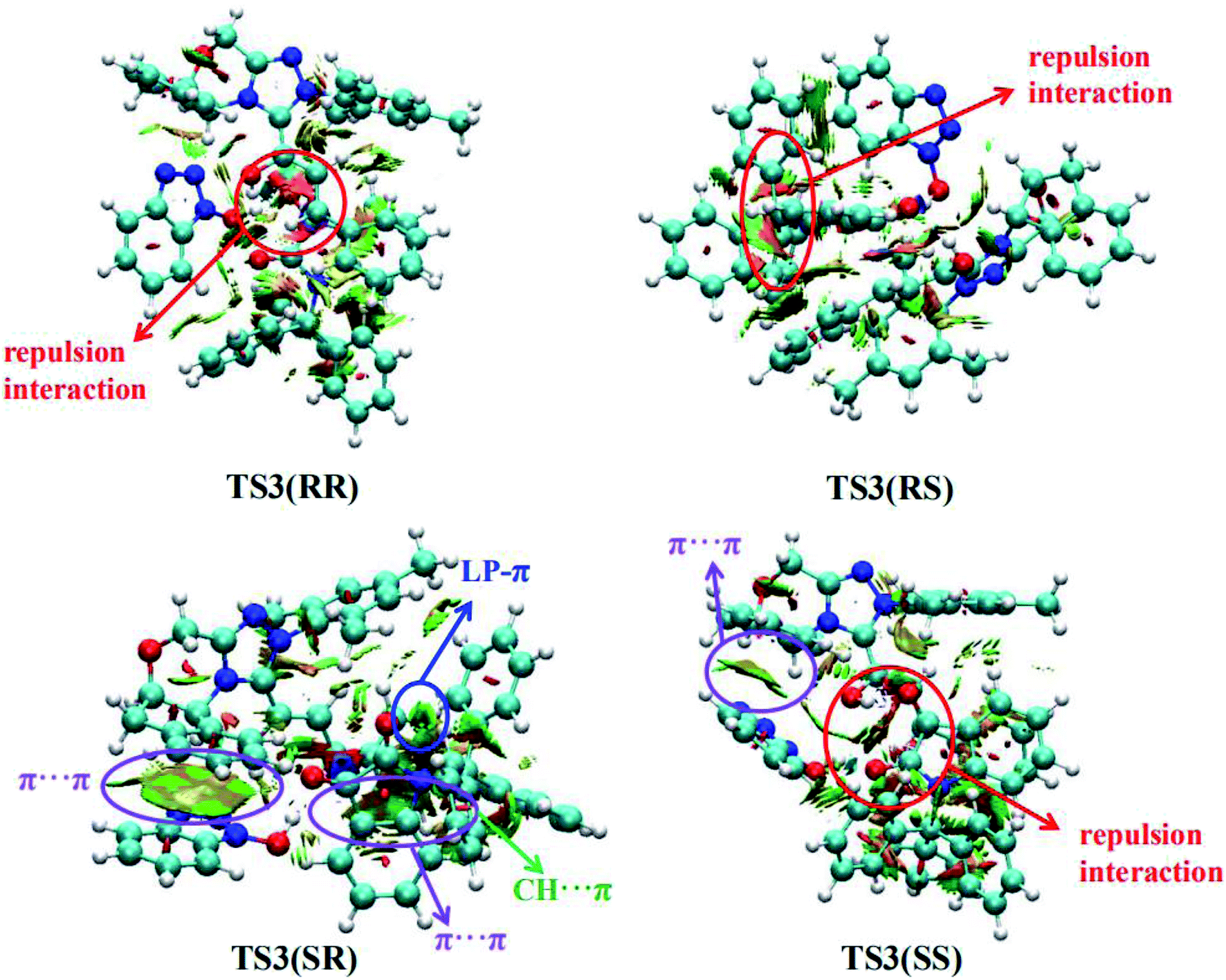 | ||
| Fig. 3 Noncovalent interactions analysis (blue, strong interaction; green, weak interaction; and red, strong repulsion) for stereoselective transition states TS3(RR), TS3(RS), TS3(SR) and TS3(SS). | ||
3.3 The nature of catalyst
In this section, global reactivity indexes (GRIs) analysis was carried out in order to understand the role of the NHC catalyst. The results are presented in Table 1. In GRI analysis, the molecular global electrophilicity character represented by the electrophilicity index ω, can be derived from the following expression, ω = (μ2/2η) (eV),22 where μ and η are chemical potential and chemical hardness, respectively. μ and η can be calculated from the one-electron energies of the highest occupied molecular orbital (HOMO) and the lower unoccupied molecular orbital (LUMO), εH and εL with the expressions μ ≈ (εH − εL)/2 and η ≈ (εL − εH), respectively.23 Within the Kohn–Sham scheme,24 the empirical (relative) nucleophilicity index N25 introduced by Domingo et al. was written as N = εH(Nu) − εH(TCE). The nucleophilicity scale was referenced to tetracyanoethylene (TCE). Table 1 shows that from R1 to IM01, the electronic chemical potential increases (−4.10 eV vs. −3.46 eV), which is consistent with what we found in NBO charge analysis, i.e., the charge goes from NHC to R1. Table 1 also shows that the coupling of NHC catalyst with carboxylic esters R1 strengthens its nucleophilicity (the nucleophilicities of IM2 (4.96 eV) and IM3 (5.40 eV) are larger than that of R1, N = 2.67 eV), but weakens its electrophilicity (the electrophilicity of IM2 (IM3) is smaller than that of R1, 0.67 (0.79) eV vs. 1.10 eV). The GRI results suggest that the coordination of NHC to R1 makes the nucleophilic addition to R2 easier.| Species | η | μ | ω | N |
|---|---|---|---|---|
| R1 | 7.66 | −4.10 | 1.10 | 2.67 |
| IM01 | 7.05 | −3.46 | 0.85 | 3.61 |
| IM2 | 5.71 | −2.77 | 0.67 | 4.96 |
| IM3 | 4.85 | −2.77 | 0.79 | 5.40 |
4. Conclusion
The reaction mechanism of the NHC-catalyzed cycloaddition reaction between carboxylic esters and isatins generating spirooxindole lactones with high enantio- and diastereo-selectivities has been investigated by using density functional theory (DFT) calculations at SMD(THF)/M06-2X/6-311++G(d,p) level. The role of the additive HOBt is also examined in detail. The current computations reveal that the catalytic cycle comprises six steps: the NHC catalyst first attacks carboxylic ester, followed by α–C–H deprotonation gives the enolate intermediate. Subsequently, proton transfer occurs leading to the homoenolate intermediate and the next step is the stereoselective C–C bond formation. The fifth step is the generation of lactone intermediate and finally liberation of catalyst gives the cycloaddition product. DFT results reveal that the additive HOBt plays a critical role both in the proton transfer and in the stereoselective C–C formation. The activation barriers both for the proton transfer and for the stereoselective C–C bond formation are much lower in the presence of HOBt. The computed stereoselectivity in the presence of HOBt agrees very good with the experimentally reported enantioselectivity (computed 96%, experimental 93%) as well as diastereoselectivity (computed 92%, experimental 91%). The origin of stereoselectivity is traced to differential π⋯π stacking, C–H⋯π, lone pair (LP)⋯π and repulsion interactions that exist in the stereoselective-determining C–C bond formation transition state. These results should provide helpful insights for understanding the detailed reaction mechanism and the significant role of the HOBt additive.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (No. 21403024), and National Supercomputing Center in Shenzhen.References
- (a) L. Wang, J. Xiao and T. P. Loh, ChemCatChem, 2014, 6, 1183–1185 CAS; (b) K. J. R. Murauski, A. A. Jaworskia and K. A. Scheidt, Chem. Soc. Rev., 2018, 47, 1773–1782 RSC; (c) D. F. Chen, T. C. K. Chu and T. Rovis, J. Am. Chem. Soc., 2017, 139, 14897–14900 CrossRef CAS PubMed.
- (a) Z. G. Hajos and D. R. Parrish, J. Org. Chem., 1974, 39, 1615–1621 CrossRef CAS; (b) B. List, R. A. Lerner and C. F. III Barbas, J. Am. Chem. Soc., 2000, 122, 2395–2396 CrossRef CAS; (c) W. Notz, F. Tanaka and C. F. III Barbas, Acc. Chem. Res., 2004, 37, 580–591 CrossRef CAS PubMed; (d) S. Mukherjee, J. W. Yang, S. Hoffmann and B. List, Chem. Rev., 2007, 107, 5471–5569 CrossRef CAS PubMed; (e) A. J. B. Watson, D. W. C. Macmillan, Y. Wang, L. Deng, S. Shirakawa and K. Maruoka, in Catalytic Asymmetric Synthesis, ed. I. Ojima, John Wiley & Sons, Hoboken, NJ, 3rd edn, 2010, ch. 2, pp. 37–117 Search PubMed.
- (a) G. Yan and A. J. Borah, Org. Chem. Front., 2014, 1, 838–842 RSC; (b) Z. Huang and G. Dong, Tetrahedron Lett., 2014, 55, 5869–5889 CrossRef CAS.
- (a) F. Glorius, N-heterocyclic carbene in catalysis – an introduction, Springer-Verlag, Berlin, Heidelberg, 2006, vol. 21, p. 1 Search PubMed; (b) S. P. Nolan, Acc. Chem. Res., 2010, 44, 91–100 CrossRef PubMed; (c) D. Zhang and G. F. Zi, Chem. Soc. Rev., 2015, 44, 1898–1921 RSC; (d) C. Chen, M. H. Kim and S. H. Hong, Org. Chem. Front., 2015, 2, 241–247 RSC.
- (a) D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606–5655 CrossRef CAS PubMed; (b) F. Glorius and K. Hirano, Nucleophilic carbenes as organocatalysts, Springer-Verlag, Berlin, Heidelberg, 2008, vol. 2, p. 159 Search PubMed; (c) J. L. Moore and T. Rovis, Top. Curr. Chem., 2010, 291, 77–144 CrossRef CAS PubMed; (d) D. Enders and T. Balensiefer, Acc. Chem. Res., 2004, 37, 534–541 CrossRef CAS PubMed; (e) V. Nair, S. Vellalath and B. P. Babu, Chem. Soc. Rev., 2008, 37, 2691–26981 RSC; (f) M. Fèvre, J. Pinaud, Y. Gnanou, J. Vignolle and D. Taton, Chem. Soc. Rev., 2013, 42, 2142–2172 RSC.
- (a) M. He, J. R. Struble and J. W. Bode, J. Am. Chem. Soc., 2006, 128, 8418–8420 CrossRef CAS PubMed; (b) J. J. Song, Z. Tan, J. T. Reeves, N. K. Yee and C. H. Senanayake, Org. Lett., 2007, 9, 1013–1016 CrossRef CAS PubMed; (c) D. A. DiRocco, K. M. Oberg, D. M. Dalton and T. Rovis, J. Am. Chem. Soc., 2009, 131, 10872–10874 CrossRef CAS PubMed; (d) Y. Kayaki, M. Yamamoto and T. Ikariya, Angew. Chem., Int. Ed., 2009, 48, 4194–4197 CrossRef CAS PubMed; (e) D. Enders, A. Grossmann, J. Fronert and G. Raabe, Chem. Commun., 2010, 46, 6282–6284 RSC; (f) J. Kaeobamrung, M. C. Kozlowski and J. W. Bode, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20661–20665 CrossRef CAS PubMed; (g) D. E. A. Raup, B. Cardinal-David, D. Holte and K. A. Scheidt, Nat. Chem., 2010, 2, 766–771 CrossRef CAS PubMed; (h) Q. Kang and Y. Zhang, Org. Biomol. Chem., 2011, 9, 6715–6719 RSC; (i) S. J. Ryan, L. Candish and D. W. Lupton, J. Am. Chem. Soc., 2011, 133, 4694–4697 CrossRef CAS PubMed; (j) T. Jousseaume, N. E. Wurz and F. Glorius, Angew. Chem., Int. Ed., 2011, 50, 1410–1414 CrossRef CAS PubMed; (k) D. A. DiRocco and T. Rovis, J. Am. Chem. Soc., 2011, 133, 10402–10405 CrossRef CAS PubMed; (l) Z. Fu, J. Xu, T. Zhu, W. W. Y. Leong and Y. R. Chi, Nat. Chem., 2013, 5, 835–839 CrossRef CAS PubMed; (m) J. Chen and Y. Huang, Nat. Commun., 2014, 5(1–8), 3437 CrossRef PubMed; (n) Z. Jin, S. Chen, Y. Wang, P. Zheng, S. Yang and Y. R. Chi, Angew. Chem., Int. Ed., 2014, 53, 13506–13509 CrossRef CAS PubMed; (o) V. Yatham, J. M. Neudörfl, N. E. Schlörer and A. Berkessel, Chem. Sci., 2015, 6, 3706–3710 RSC; (p) B. S. Li, Y. Wang, Z. Jin, P. Zheng, R. Ganguly and Y. R. Chi, Nat. Commun., 2015, 6, 6207–6211 CrossRef PubMed; (q) C. Guo, M. Fleige, D. Janssen-Müller, C. G. Daniliuc and F. Glorius, Nat. Chem., 2015, 7, 842–848 CrossRef CAS PubMed; (r) K. Yamashita, S. Hase, Y. Kayaki and T. Ikariya, Org. Lett., 2015, 17, 2334–2337 CrossRef CAS PubMed; (s) Y. Nakano and D. W. Lupton, Angew. Chem., Int. Ed., 2016, 55, 3135–3139 CrossRef CAS PubMed; (t) C. Zhao, F. Li and J. Wang, Angew. Chem., Int. Ed., 2016, 55, 1820–1824 CrossRef CAS PubMed; (u) S. R. Yetra, S. Mondal, S. Mukherjee, R. G. Gonnade and A. T. Biju, Angew. Chem., Int. Ed., 2016, 55, 268–272 CrossRef CAS PubMed.
- (a) X. N. Wang, P. L. Shao, H. Lv and S. Ye, Org. Lett., 2009, 11, 4029–4032 CrossRef CAS PubMed; (b) X. L. Huang, X. Y. Chen and S. Ye, J. Org. Chem., 2009, 74, 7585–7587 CrossRef CAS PubMed; (c) J. Douglas, J. E. Taylor, G. Churchill, A. M. X. Slawin and A. D. Smith, J. Org. Chem., 2013, 78, 3925–3938 CrossRef CAS PubMed; (d) X. N. Wang, L. T. Shen and S. Ye, Org. Lett., 2011, 13, 6382–6385 CrossRef CAS PubMed; (e) T. Wang, X. L. Huang and S. Ye, Org. Biomol. Chem., 2010, 8, 5007–5010 RSC; (f) H. M. Zhang, Z. H. Gao and S. Ye, Org. Lett., 2014, 16, 3079–3081 CrossRef CAS PubMed.
- (a) X. Q. Fang, X. K. Chen and Y. R. Chi, Org. Lett., 2011, 13, 4708–4711 CrossRef CAS PubMed; (b) L. M. Fang, F. Wang, P. J. Chua, Y. B. Lv, L. J. Zhong and G. F. Zhong, Org. Lett., 2012, 14, 2894–2897 CrossRef PubMed.
- (a) H. Lu, J. Y. Liu, C. G. Li, J. B. Lin, Y. M. Liang and P. F. Xu, Chem. Commun., 2015, 51, 4473–4476 RSC; (b) S. H. Hu, B. Y. Wang, Y. Zhang, W. F. Tang, M. Y. Fang, T. Lu and D. Du, Org. Biomol. Chem., 2015, 13, 4661–4667 RSC; (c) S. R. Yetra, S. Mondal, E. Suresh and A. T. Biju, Org. Lett., 2015, 17, 1417–1420 CrossRef CAS PubMed; (d) J. D. Tessier, E. A. O'Bryan, T. B. H. Schroeder, D. T. Cohen and K. A. Scheidt, Angew. Chem., Int. Ed., 2012, 124, 5047–5051 CrossRef; (e) Z. Q. Liang, Z. H. Guo, W. Q. Jia and S. Ye, Chem.–Eur. J., 2015, 21, 1868–1872 CrossRef CAS PubMed; (f) C. Guo, M. Schedler, C. G. Daniliuc and F. Glorius, Angew. Chem., Int. Ed., 2014, 53, 10232–10236 CrossRef CAS PubMed; (g) A. G. Kravina, J. Mahatthananchai and J. W. Bode, Angew. Chem., Int. Ed., 2012, 51, 9433–9436 CrossRef CAS PubMed; (h) Y. Zhang, Y. Y. Lu, W. F. Tang, T. Lu and D. Du, Org. Biomol. Chem., 2014, 12, 3009–3015 RSC.
- (a) J. Izquierdo, A. Orue and K. A. Scheidt, J. Am. Chem. Soc., 2013, 135, 10634–10637 CrossRef CAS PubMed; (b) Y. W. Xie, Y. L. Que, T. J. Li, L. Zhu, C. X. Yu and C. S. Yao, Org. Biomol. Chem., 2015, 13, 1829–1835 RSC; (c) H. Lv, W. Q. Jia, L. H. Sun and S. Ye, Angew. Chem., Int. Ed., 2013, 125, 8769–8772 CrossRef.
- F. Xu, S. R. Yuan, M. Z. Miao and Z. K. Chen, J. Org. Chem., 2016, 81, 11454–11460 CrossRef PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2007, 120, 215–241 Search PubMed.
- (a) H. Xue, D. Jiang, H. Jiang, C. W. Kee, H. Hirao, T. Nishimura, M. W. Wong and C. H. Tan, J. Org. Chem., 2015, 80, 5745–5752 CrossRef CAS PubMed; (b) M. W. Wong and A. M. E. Ng, Aust. J. Chem., 2014, 67, 1100–1109 CrossRef CAS; (c) H. Yang and M. W. Wong, J. Am. Chem. Soc., 2013, 135, 5808–5818 CrossRef CAS PubMed; (d) E. H. Krenske, E. C. Davison, I. T. Forbes, J. A. Warner, A. L. Smith, A. B. Holmes and K. N. Houk, J. Am. Chem. Soc., 2012, 134, 2434–2441 CrossRef CAS PubMed; (e) H. Yang and M. W. Wong, J. Org. Chem., 2011, 76, 7399–7405 CrossRef CAS PubMed; (f) Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS PubMed.
- C. Gonzalez and H. B. Gonzalez, J. Phys. Chem., 1990, 94, 5523–5527 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- (a) E. D. Glendening, A. E. Reed, J. E. Carpenter and F. Weinhold, NBO Version 3.1, Theoretical Chemistry Institute, University of Wisconsin, Madison, WI, 1996 Search PubMed; (b) A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS; (c) J. P. Foster and F. Weinhold, J. Am. Chem. Soc., 1980, 102, 7211–7218 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 (Revision D.01), Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- (a) Q. Zhang, H. Z. Yu and Y. Fu, Org. Chem. Front., 2014, 1, 614–624 RSC; (b) J. Mahatthananchai and J. W. Bode, Acc. Chem. Res., 2013, 47, 696–707 CrossRef PubMed.
- (a) Y. Wang, D. Wei, Y. Wang, W. Zhang and M. Tang, ACS Catal., 2016, 6, 279–289 CrossRef CAS; (b) Y. Wang, D. Wei and M. Tang, J. Org. Chem., 2017, 82, 13043–13050 CrossRef CAS PubMed; (c) Y. Wang, Y. Qiao, D. Wei and M. Tang, Org. Chem. Front., 2017, 4, 1987–1998 RSC; (d) Y. Wang, D. Wei and W. Zhang, ChemCatChem, 2018, 10, 338–360 CrossRef CAS.
- D. Y. Curtin, Rec. Chem. Prog., 1954, 15, 111–128 CAS.
- T. Lu and F. W. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- R. G. Parr, L. V. Szentpály and S. Liu, J. Am. Chem. Soc., 1999, 121, 1922–1924 CrossRef CAS.
- (a) R. G. Parr and W. Yang, Density Functional Theory of Atoms and Molecules, Oxford University Press, New York, 1989 Search PubMed; (b) R. G. Parr and R. G. Pearson, J. Am. Chem. Soc., 1983, 105, 7512–7516 CrossRef CAS.
- W. S. Kohn and L. J. Sham, Phys Rev., 1965, 140, 1133–1138 CrossRef.
- L. R. Domingo, E. Chamorro and P. Pérez, J. Org. Chem., 2008, 73, 4615–4624 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra10262g |
| This journal is © The Royal Society of Chemistry 2019 |