
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNovel cathepsin K inhibitors block osteoclasts in vitro and increase spinal bone density in zebrafish†
Si-tu
Xue‡
,
Ya-li
Wang‡
,
Xiao-wan
Han
,
Hong
Yi
,
Wei
Jiang
,
Shu-yi
Si
,
Hui-fang
Guo
* and
Zhuo-rong
Li
 *
*
Institute of Medicinal Biotechnology, Chinese Academy of Medical Science and Peking Union Medical College, Beijing 100050, China. E-mail: lizhuorong@imb.pumc.edu.cn; huifangguo@imb.pumc.edu.cn
First published on 14th March 2019
Abstract
Cathepsin K (Cat K) is a predominant cysteine protease and highly potent collagenase expressed in osteoclasts. Cat K inhibitors are anti-resorptive agents to treat osteoporosis. A novel scaffold of cathepsin K inhibitors, exemplified by lead compound 1x, was used as the template for designing and synthesizing a total of 61 derivatives that have not been reported before. An exploratory structure–activity relationship analysis identified the potent Cat K inhibitor A22, which displayed an IC50 value of 0.44 μM against Cat K. A22 was very specific for Cat K and caused a significantly higher in vitro inhibition of the enzyme as compared to that of lead compound 1x. A surface plasmon resonance analysis confirmed in vitro binding of A22 to Cat K. Molecular docking studies indicated several favourable interaction sites for A22 within the active pocket of Cat K. Furthermore, A22 also blocked active osteoclasts in vitro and increased spinal bone density in zebrafish, in which it showed an activity that was higher than that of the marketed therapeutic bone metabolizer etidronate disodium. A22 represents a very promising lead compound for the development of novel antiresorptive agents functioning as orthosteric inhibitors of Cat K.
1. Introduction
Osteoporosis (OP) is a systemic disease associated with a declining rigidity and mechanical stability of the bones.1 Current estimates indicate that over 200 million people worldwide suffer from this disease, causing more than 8.9 million fractures annually. In fact, an osteoporotic fracture is estimated to occur every 3 s, and one out of every three women and one in five men over 50 years of age will have an OP-related fracture in their lifetime.2,3 A better knowledge about the progression of bone metabolic disorder and the development of effective treatments will have important socio-economic and medical consequences. The most commonly prescribed OP medications include nitrogen-containing bisphosphonates, denosumab, raloxifene, hormone replacement therapy, and calcitonin.4 Although these therapies improve the quality of life in patients with OP, they have many side effects.5–7 Thus, the discovery of novel OP medications is a priority that would provide new options for OP patients and represent a major advancement in the treatment of OP.Bone health in adults depends on resorption that involves the continuous removal of old or damaged bone tissue by osteoclast (OC) cells, which is balanced by the deposition of new bone formed by osteoblast (OB) cells. As the skeleton ages, the action of the OCs outpaces that of the OBs, causing a shift toward excessive resorption and the development of chronic OP associated with reduced bone mineral density (BMD) and an increased risk of bone fractures.8,9 Cathepsin K (Cat K) is a lysosomal cysteine protease released by OCs, which cleaves the collagen matrix of bone tissue.10,11 Cat K knockout mice have been shown to develop osteopetrosis with elongated bones and vertebrae and abnormal joint morphology due to a deficit in matrix degradation,12 indicating the potential role of Cat K in OP and other bone diseases. In recent years, Cat K inhibitors have been intensively studied by pharmaceutical companies to find antiresorptive therapies for OP.13–15 The most advanced drug candidates include odanacatib,16,17 relacatib,18,19 balicatib20,21 and ONO-5334 (Fig. 1).22,23 Odanacatib and balicatib are reversible covalent Cat K inhibitors carrying a cyano group that can form a covalent bond with C25 of the active pocket in Cat K.
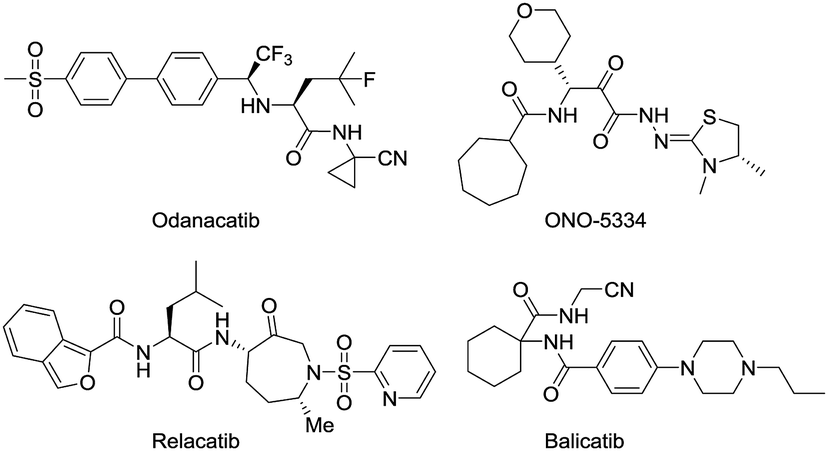 | ||
| Fig. 1 Structures of Cat K inhibitors. | ||
Odanacatib was once the most advanced drug candidate as a Cat K inhibitor that reduced fractures in postmenopausal women in a large multinational randomized, double-blinded phase III clinical study. However, odanicatib was also associated with an increased risk of cerebrovascular accidents and subsequently withdrawn from the regulatory approval process.
Thus, there is significant value in the study of Cat K because it is a major drug target for OP and related-bone disorders.
Our research team has been working on the development of anti-OP drugs for many years.24,25 Our recent efforts are focused on the discovery of Cat K inhibitors as potent antiresorptive agents. A virtual screen of our in-house compound library against the Cat K (PDB code: 4dmy, Fig. 2) active site identified compound 1q (Fig. 2). 1q had a higher CDOCKER score than the covalent Cat K inhibitor 14 which was reported by Alexander G. Dossetter and his colleagues (Fig. 2a).26 The orientation of 1q also showed a high fitness comparable to that of 14 (Fig. 2b). The scaffold of 1q has not been reported by others as a Cat K inhibitor. A search of our in-house compound library identified several structural analogues of 1q that were all tested for in vitro Cat K inhibiting effects using an enzymatic assay. However, 1q was not active against Cat K at 100 μM. Interestingly, we identified compound 1x (Fig. 2) with a scaffold of benzimidazole-2-substituted pyridine propyl ketene that inhibited Cat K at a concentration below 100 μM. Two other compounds (Fig. 3, 1o and 1p27) with a scaffold containing benzoxazole or benzothiazole did not inhibit Cat K. We also found that the carbonyl group in 1x scaffold is probably essential for the Cat K inhibiting activity because the switch to a hydroxyl group (see 1s and 1t27 in Fig. 3) abolished the activity.
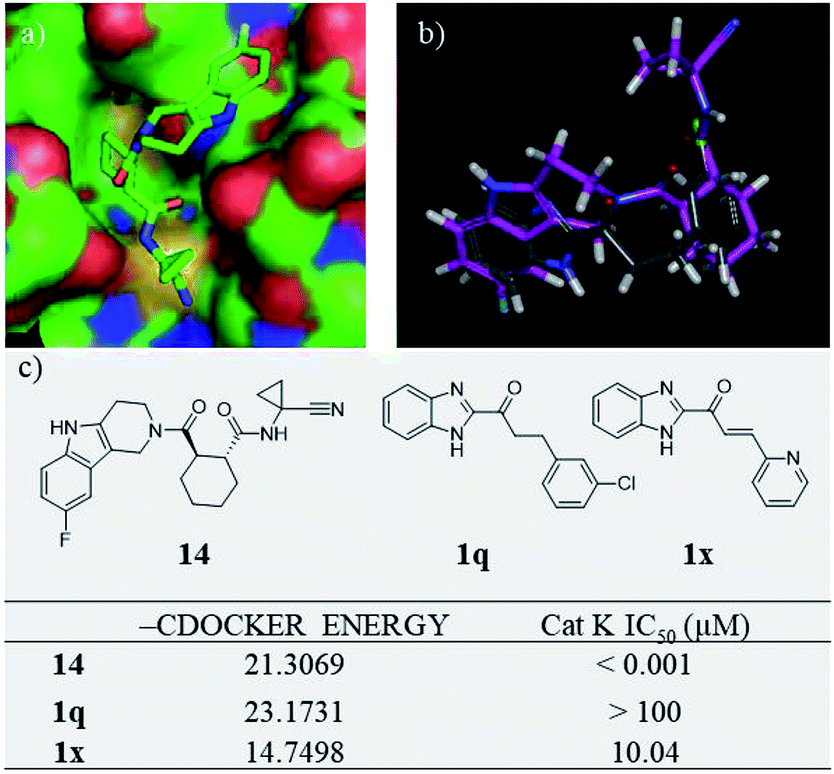 | ||
| Fig. 2 Virtual screen found Cat K inhibitor 1x. (a) The 3D model of Cat K inhibitor 14 in docking position (PDB code 4dmy; the picture was prepared with Pymol) in the active site of Cat K and (b) comparison of the docking position of 1q (grey) with 14 (purple) and (c) structures, CDOCKER scores and Cat K IC50s of 14, 1q and 1x. | ||
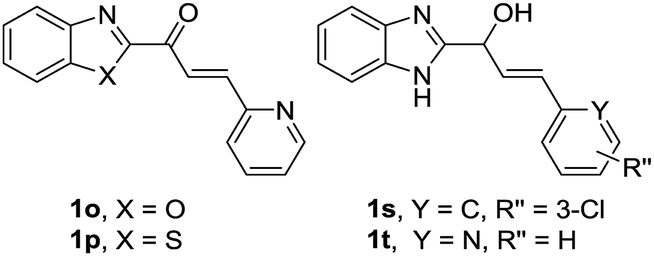 | ||
| Fig. 3 Inactive compounds against Cat K. | ||
The IC50 of compounds 1o, 1p, 1s, and 1t against Cat K were all above 100 μM.
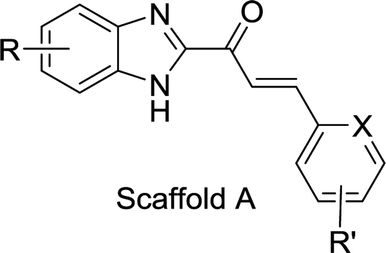
In this study, we synthesized derivatives based on the structure of 1x with the benzimidazole scaffold and explored their antiresorptive potential. In total, 61 compounds containing either benzimidazole-2-substituted phenyl or pyridine propyl ketenes were synthesized. The compounds were evaluated for their potential to inhibit the kinase activity of Cat K. Many derivatives with benzimidazole scaffold showed inhibitory activity at less than 10 μM against Cat K in vitro. Then, we tested the selectivity of 1x, A20, A22, and A32 (Table 1) against several other cathepsins (L, S and B) and found that these compounds were highly selective for Cat K. Modelling studies identified several favourable interactions of A22 with the active pocket of Cat K. Surface plasmon resonance (SPR) analysis confirmed the high affinity of A22 to Cat K. Compounds A20, A22 and A32 showed inhibitory activity toward OCs and also inhibited bone resorption caused by C-telopeptide (CTX-I) decrease observed in vitro. Moreover, A22 also increased spinal bone density in zebrafish.
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compd. | X | R | R′ | IC50 | Compd. | X | R | R′ | IC50 |
| A1 | N | H | 3′-Cl | 26.15 | A33 | C | 5-Cl | 4′-CN | 2.61 |
| A2 | N | H | 4′-Cl | 2.47 | A34 | C | 4-Cl | 3′-CN | 6.19 |
| A3 | N | H | 5′-Cl | 8.48 | A35 | N | 5,6-Dichloro | H | 6.94 |
| A4 | N | H | 6′-Cl | 8.83 | A36 | C | 5,6-Dichloro | 3′-CN | 7.62 |
| A5 | N | H | 3′-Me | 9.51 | A37 | C | 5,6-Dichloro | 4′-CN | 5.57 |
| A6 | N | H | 6′-Me | 28.14 | A38 | N | 5-Br | H | 7.34 |
| A7 | N | H | 5′-Br | 6.12 | A39 | C | 5-Br | 3′-Cl | 6.67 |
| A8 | N | H | 6′-Br | 9.48 | A40 | C | 5-Br | 3′-CN | 2.34 |
| A9 | N | H | 3′-F | 3.75 | A41 | C | 5-Br | 4′-CN | 0.77 |
| A10 | N | H | 6′-OMe | 8.96 | A42 | N | 5-NO2 | H | 6.54 |
| A11 | C | H | 2′-CN | >100 | A43 | C | 5-NO2 | H | 8.19 |
| A12 | C | H | 3′-CN | 7.33 | A44 | C | 5-NO2 | 2′-Cl | 11.01 |
| A13 | C | H | 4′-CN | 9.16 | A45 | C | 5-NO2 | 3′-Cl | 10.70 |
| A14 | N | 5-Me | H | 25.86 | A46 | C | 5-NO2 | 4′-Cl | 26.11 |
| A15 | C | 5-Me | 2′-CN | >100 | A47 | C | 5-NO2 | 2′-Br | 11.32 |
| A16 | C | 5-Me | 3′-CN | 47.04 | A48 | C | 5-NO2 | 3′-Br | 9.70 |
| A17 | C | 5-Me | 4′-CN | 2.96 | A49 | C | 5-NO2 | 4′-Br | 15.90 |
| A18 | N | 5-Cl | H | 4.24 | A50 | C | 5-NO2 | 3′-Me | 25.41 |
| A19 | N | 5-Cl | 3′-Cl | 7.89 | A51 | C | 5-NO2 | 3′-CN | 3.14 |
| A20 | N | 5-Cl | 4′-Cl | 0.60 | A52 | C | 5-NO2 | 3′-OMe | 22.91 |
| A21 | N | 5-Cl | 5′-Cl | 4.73 | A53 | N | 5-NH2 | H | 4.29 |
| A22 | N | 5-Cl | 6′-Cl | 0.44 | A54 | C | 5-NH2 | H | 8.75 |
| A23 | N | 5-Cl | 3′-Me | 15.49 | A55 | C | 5-NH2 | 2′-Cl | 9.09 |
| A24 | N | 5-Cl | 5′-Br | 5.26 | A56 | C | 5-NH2 | 3′-Cl | 4.38 |
| A25 | N | 5-Cl | 3′-F | 2.82 | A57 | C | 5-NH2 | 4′-Cl | 6.40 |
| A26 | N | 5-Cl | 6′-OMe | 6.07 | A58 | C | 5-NH2 | 2′-Br | 18.77 |
| A27 | C | 5-Cl | 2′-Cl | >100 | A59 | C | 5-NH2 | 3′-Br | 4.11 |
| A28 | C | 5-Cl | 3′-Cl | 23.42 | A60 | C | 5-NH2 | 4′-Br | 4.99 |
| A29 | C | 5-Cl | 3′-Br | 3.06 | A61 | C | 5-NH2 | 3′-Me | 9.39 |
| A30 | C | 5-Cl | 3′-OMe | >100 | 1x | N | H | H | 10.04 |
| A31 | C | 5-Cl | 2′-CN | 51.14 | Odanacatib | 0.002 | |||
| A32 | C | 5-Cl | 3′-CN | 0.88 | |||||
2. Results and discussion
2.1 Chemistry
The search for potential Cat K inhibitors led to the identification of compound 1x with a scaffold containing benzimidazole, a carbonyl group, and a double bond that is required for in vitro Cat K inhibition. The design and synthesis of a series of 1x derivatives were performed as follows. The synthesis route of benzimidazole-2-substituted phenyl or pyridine propyl ketenes is described in Scheme 1. In total, 61 derivatives have been synthesized. There are different substituents on the two aromatic rings. In this round of synthesis, we only implanted substituents with smaller size, such as methyl, halogen or cyano groups. The detailed structures of the compounds are reported in Table 1.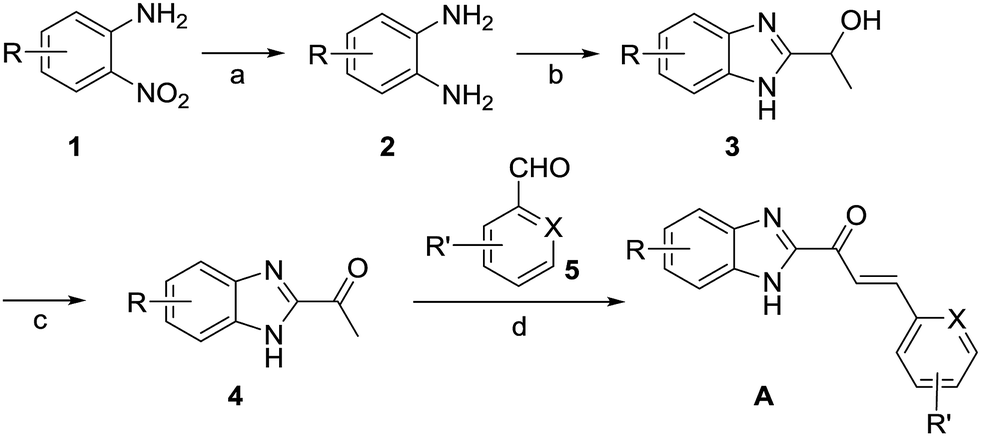 | ||
| Scheme 1 Synthesis route of derivatives of 1x started from 1 or 2. (a) H2, Pd/C, 30 °C; (b) 2-hydroxypropanoic acid, 4 M HCl, H2O, 90 °C; ammonia; (c) TEMPO, NaClO, KBr, CH3CN, r.t.; (d) NaOH, CH3CH2OH, r.t. | ||
2.2 Inhibition of Cat K in vitro and preliminary structure–activity relationships
The inhibitory activities of our focused compound library against Cat K are reported in Table 1 including odanacatib, which was used as a positive control. Importantly, many derivatives of scaffold A showed Cat K inhibitory effects at concentrations below 100 μM, which confirmed that this novel scaffold has a high potential to serve as a template for antiresorptive agents.The phenyl or pyridine substitution (such as A4 and A39) in the scaffold is favoured. The 2-substitution on the phenyl ring seems detrimental to Cat K inhibitory activities according to the IC50 data (>100 μM) of A11, A15, A27, and A30. The 5-halogen substitution on benzimidazole appears to be favourable for the activity. A20, A22, A32, and A41 showed inhibitory activities below 1 μM. Halogen and cyano group substitution on the terminal phenyl or pyridine ring may also contribute to the activity. The molecular docking calculations of A22 (Cat K IC50 = 0.44 μM, Fig. 4) in the Cat K active pocket also suggested that its imidazole H atom formed a hydrogen bond with the O atom of the backbone carbonyl group at N161. This could explain why compounds benzoxazole 1o and benzothiazole 1p did not show any Cat K inhibiting effect. They lack this key hydrogen bond donor in their scaffold which appears to be a necessity for this activity. The phenyl ring of benzimidazole was coplanar with the glycine shelf (G23) and generated a hydrophobic interaction. The hydrophobic interaction between benzimidazole and C25 also enhanced the affinity of A22 and Cat K. Thus, chlorine substitution on benzimidazole of A22 is beneficial to the affinity to Cat K.
 | ||
| Fig. 4 Molecular docking calculations of the binding mode of A22 with Cat K (PDB code: 4dmy). (a) and (b) The calculated interactions of A22 (grey) with amino acids (green) in the active pocket of Cat K. (c) Overlay of the crystal structures of Cat L (PDB code: 6ezp), Cat S (PDB code: 4bs5) and Cat B (PDB code: 6ay2) with Cat K. N161 (green) of Cat K located at a loop region where the conformations of the Cat L, S, B and K are different. Compared to the orientation of Y193 (purple) of Cat S, Y67 (green) of Cat K has much less clash with pyridine ring of A22. (d) Backbone carbonyl groups of N161 of Cat K, N286 (purple) of Cat S and D162 (yellow) of Cat L. | ||
2.3 Selectivity of the inhibition of cathepsins in vitro
We compared the inhibitory activities of compounds 1x, A20, A22, and A32 against Cat K, Cat L, Cat S, and Cat B (Table 2). Compounds 1x, A20, A22, and A32 showed an inhibitory effects against the cathepsins, especially toward Cat K and Cat L. The order of the inhibitory activities of the four test compounds against the cathepsins were Cat K > Cat L > Cat S > Cat B. The selectivity of A22 for Cat K over Cat S and Cat B was 106 and 168 times, respectively. According to the docking result, the hydrogen bond between A22 and N161 of Cat K probably played a key role of the selectivity of A22. N161 of Cat K was in a loop region where the conformation of Cat B was highly different from Cat L, S, and K (Fig. 4c). Different from the loops of Cat K, L and S, it was a β-sheet of Cat B in this region. There was no asparagine here in Cat B. The change of asparagine to histidine and glycine distorted the binding pocket, which explained well that A22 showed much weaker inhibitory activity against Cat B. Similar to their counterpart N161 in Cat K, D162 of Cat L and N286 of Cat S (Fig. 4d) also can form hydrogen bond with A22. A22 showed high inhibiting effect against Cat L. But the higher clash of Y193 of Cat S to A22 may alleviate the interaction (Fig. 4c).| Compd. | IC50 (μM) | |||
|---|---|---|---|---|
| Cat K | Cat L | Cat S | Cat B | |
| 1x | 10.04 | 15.82 | >100 | >100 |
| A20 | 0.60 | 1.64 | 24.73 | 47.73 |
| A22 | 0.44 | 2.62 | 46.72 | 73.85 |
| A32 | 0.88 | 1.79 | 25.34 | 101.76 |
| Odanacatib | 0.002 | 8.46 | 0.013 | 0.013 |
As compared to the inhibitory activities of 1x, the positive control odanacatib had a higher activity against Cat K and Cat L, but a lower activity against Cat S and Cat B. The selectivity of odanacatib for Cat K over Cat S and Cat B was 6.5 times for both enzymes. The order of the inhibitory activity of odanacatib against the cathepsins was Cat K > Cat S = Cat B > Cat L. These results indicated that A22 and odanacatib differed in their selectivity toward cathepsin family proteins and, therefore, may differ in their activity and toxicity. Therefore, A22 has the potential to be novel antiresorptive agents.
2.4 Antiresorptive effects in vitro
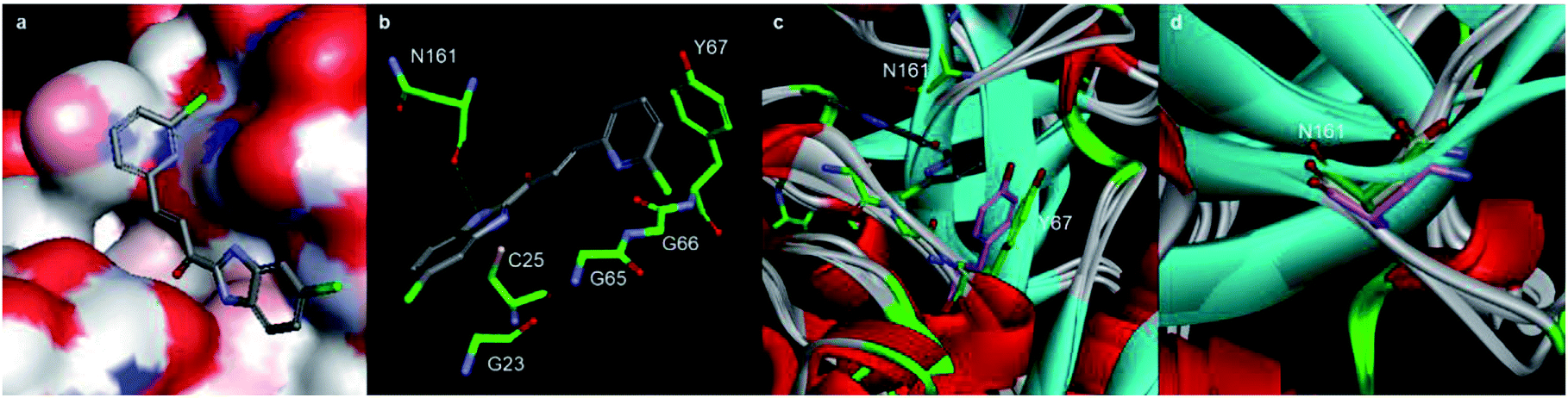
Type I collagen comprises about 95% of the entire collagen content of bone.28 During skeletal renewal, type I collagen is degraded, releasing short peptide fragments into the bloodstream. The bone resorption process can be studied in vitro by culturing bone cells on bone sections or dentin sheets. During the process of bone resorption by OCs, the type I collagen C-terminal peptide (CTX-I) is degraded into fragments and released. In this experiment, the concentration of CTX-I released into the cell culture medium by bone degradation can be detected and quantified by ELISA. The in vitro antiresorptive effect of our compounds was tested in RANKL-induced RAW264.7 cells. Osteoclastic features can be induced in RAW264.7 by RANKL with high efficiency and reproducibility.29 We detected the CTX-I26,30,31 concentration released into the culture medium from RANKL-induced RAW264.7 cells by ELISA.Importantly, RAW264.7 cells released much less CTX-I in the presence of A20, A22, A32, 1×, and positive control odanacatib, as compared to that released by untreated RAW264.7 cells. The concentrations of all experimental compounds and positive control drugs were 5 μM. The antiresorptive effects of all the experimental compounds was similar to that of odanacatib, especially the activity of A20 was much stronger than that of the control drug. And also the efficacy of all the test compounds was significantly better than that of 1x on RAW264.7 cells (Fig. 5A).
 | ||
| Fig. 5 (A) CTX-I detection of test compounds in the bone absorption process; (B) Effects on OC differentiation by test compounds in vitro. Three replicates were tested in each group. **Differences from the control group with probability p < 0.05. ***Differences from the control group with probability p < 0.001. The p value is calculated by Tukey–Kramer multiple comparisons test. The difference is considered statistically significant when probability p value is less than 0.05 and statistically highly significant when probability p value is less than 0.001. | ||
2.5 Inhibition of osteoclast (OC) cells
The inhibition of OCs in vitro was tested by counting the numbers of tartrate-resistant acid phosphatase (TRAP) positive cells. In this experiment, the RANKL-induced RAW264.7 cells were identified by OC-specific TRAP staining specific. The RANKL-induced RAW264.7 cells were incubated with the test compounds added at 20 μg mL−1. After 72 h of culture, the TRAP staining was performed and the TRAP + cells were counted. As compared to the growth of the untreated control OCs, the growth of the OCs incubated with the test compounds (20 μg mL−1) was reduced. The OC growth inhibition of A20, A22, and A32 was 31.3%, 33.3%, and 42.2%, respectively (Fig. 5B).2.6 Binding affinity to Cat K tested by SPR
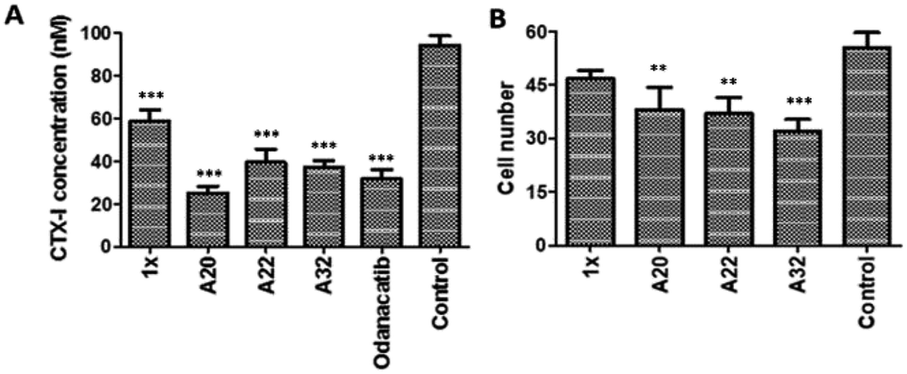
To determine whether the active compounds can bind to Cat K, real-time binding analysis by SPR (Biacore 8K) was performed using Cat K and different dilutions of A20 and A22 (Fig. 6). The two compounds showed moderately strong affinities to Cat K. The calculated affinities (KD) of A20 and A22 to Cat K were 53.2 ± 43.7 μM and 17.2 ± 5.6 μM, respectively. Moderately strong affinities implied that the compounds may show antiresorptive efficacy in vivo via inhibition of Cat K. | ||
Fig. 6 Binding curves of A20 and A22 dilutions. The affinities (KD) were calibrated with the steady state affinity 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding model using the evaluation software of Biacore 8K. :1 binding model using the evaluation software of Biacore 8K. | ||
2.7 OP therapeutic activity in zebrafish
To probe into the in vivo activities, we chose A22 as a pilot compound and evaluated its activity in zebrafish32,33 because A22 was the most potent Cat K inhibitor in vitro among our focused library compounds and showed high selectivity and affinity to Cat K. Etidronate disodium was used as a positive control drug. Etidronate disodium is a bone resorption inhibitor and now mainly used as an antiosteoporotic medication.34 Its action mechanism involves the hydroxyapatite crystal growth inhibition according to the in vitro chemisorption onto the crystal surface.35 Prednisone is a glucocorticoid medication and often used to suppress the immune system and relieve inflammation. The use of prednisone can induce bone loss.36–38 In our study, we use prednisone to set up a zebrafish bone loss model.Our dosing pilot study showed at the concentration of 1.00 μM, A22 didn't show obvious toxicity to the 30 zerafish in the group. We then tested OP therapeutic activity in zebrafish of A22 at concentrations of 0.11, 0.33, and 1.00 μM. Fluorescent chromophore calcein, which binds specifically to calcified skeletal structures, was used to indicate zebrafish spine bone density.39 Fluorescence intensities were enhanced as compared to that of the model group, indicating that A22 treatment improved bone density at tested concentrations. A22 also showed dose–response relationship in OP zebrafish. At the highest concentration group, A22 exhibited higher activity than etidronate disodium (ED) (Fig. 7). The OP therapeutic effect in zebrafish bone loss model showed that A22 is a promising anti-OP agent.
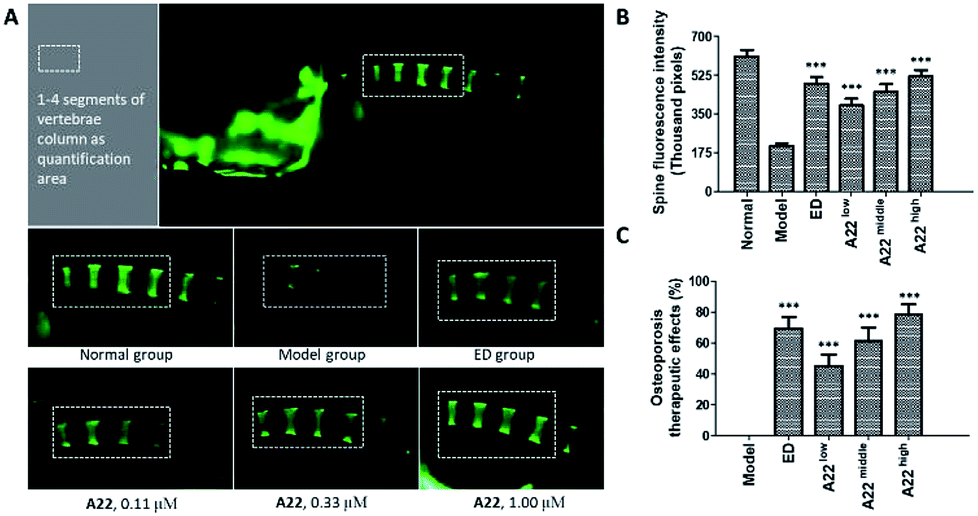 | ||
| Fig. 7 Therapeutic effect of A22 in OP zebrafish. (A) Zebrafish spine fluorescence intensity data were collected within the white rectangle area. A22 was administered at a dose of 0.11 μM, 0.33 μM, 1.00 μM, and etidronate disodium (ED) was administered at a dose of 600 μM; (B) spine fluorescence intensity data were calculated based on values obtained from 30 fish each group. (C) A22 therapeutic effect on zebrafish OP. ***Differences from the model group with probability p < 0.001. The p value is calculated using Dunnett's T test and differences with probability p < 0.05 are considered statistically significant. | ||
3. Experimental section
3.1 Chemistry
1-(5-Chloro-1H-benzo[d]imidazol-2-yl)ethan-1-ol (3, 1.97 g, 0.01 mol), TEMPO (0.016 g, 0.1 mmol) and KBr (0.12 g, 1 mmol) were added to a 100 mL three-necked round bottom bottle. Then, 20 mL of acetrile and 20 mL of water were added. An 8% aqueous sodium hypochlorite (13.96 g, 0.015 mol) solution was added dropwise within 20 min while keeping the temperature of the mixture between 10 °C and 15 °C. The mixture was stirred for 30 min until full conversion (as monitored by TLC). The mixture was extracted by ethyl acetate (50 mL × 3). The organic phases were combined and washed with 60 mL of 10% sodium thiosulfate and 60 mL of brine. The organic layer was then separated, dried over anhydrous Na2SO4 and evaporated to recover 1-(5-chloro-1H-benzo[d]imidazol-2-yl)ethan-1-one (4, yield 71%). mp 156.1–157.4 °C; MS (ESI) m/z: 195 [M + H]+.
1-(5-Chloro-1H-benzo[d]imidazol-2-yl)ethan-1-one (4, 607.2 mg, 3.1 mmol) and ethanol (10 mL) were added to a 25 mL three-necked round bottom bottle. The resulting mixture was stirred until 4 was fully dissolved. NaOH (0.12 g, 3.12 mmol) was then added and then stirred for another 15 min. 6-Chloropicolinaldehyde (0.44 g, 3.12 mmol) was added. The reaction was stirred for 2 h until full conversion (as monitored by TLC). The reaction was quenched with 5 mL of water while stirring. A yellow solid precipitated from the solution. Filtration was performed to collect the solid and wash it with water. The solid was first dried under air in a culture dish and then separated by flash column chromatography (PE:EA = 10:1). (E)-1-(5-chloro-1H-benzo[d]imidazol-2-yl)-3-(6-chloropyridin-2-yl)prop-2-en-1-one (A22, yield 54%) was obtained as yellow solid. mp 251.4–252.7 °C; 1H NMR (400 MHz, d6-DMSO) δ 13.68 (s, 1H), 8.39 (d, J = 15.7 Hz, 1H), 7.99–7.93 (m, 2H), 7.88 (d, J = 15.7 Hz, 1H), 7.84 (d, J = 7.5 Hz, 1H), 7.58 (d, J = 7.6 Hz, 2H), 7.43–7.34 (m, 1H); 13C NMR (101 MHz, d6-DMSO) δ 181.26, 153.43, 151.18, 141.46, 134.07, 130.78, 126.19, 125.80, 124.38, 123.28, 121.01, 114.84, 112.88; HRMS calcd for C15H10ON3Cl2+ [M + H]+ 318.01954, found 318.01942.
3.2 Molecular docking calculations
CDOCKER of Discovery Studio 4.5 was used to carry out the docking simulation of the compounds to the protein. X-ray crystal structure of Cat K (PDB code 4dmy) was used for the docking studies. The ligand was extracted from the catalytic active site of Cat K, which was defined as the docking site for binding. Both the receptor and the compounds were employed using the CHARMm force field before docking. A higher CDOCKER score (i.e. –DOCKER _ENERGY) indicates a more favourable affinity. Using the docking calculations of CDOCKER, we also obtained the exact pose of the ligand in 4dmy.3.3 Enzymatic activity assays in vitro
Quenched fluorescent resonance energy transfer (QFRET) technology was used to test the inhibitory activity of the compounds against Cat K (ENZO BML-SE553) catalysing the cleavage of the synthetic peptide Z-Phe-Arg-AMC. Compounds were tested at eight concentrations (diluted from 200 μg mL−1, three replicates per dilution). To each test sample well on a 96-well plate, 2 μL of test compound were mixed with 25 μL of Cat K (0.3 ng mL−1). In control wells, DMSO was added instead of the compounds. In blank wells, DMSO was added instead of compounds and buffer was added instead of Cat K solution. The plate was incubated for 15 min at 37 °C. The fluorescence (F1) was determined at a wavelength of 355 nm excitation and 460 nm emission. Then, 25 μL of 20 μM of Z-Phe-Arg-AMC was then added and the plate was incubated for 1 h at 37 °C. Then, the fluorescence (F2) was measured again at a wavelength of 355 nm excitation and 460 nm emission. The calculation was performed as follows: inhibition (%) = [control (F2–F1) − sample (F2–F1)]/[control (F2–F1) − blank (F2–F1)] × 100%. IC50 was calculated using SPSS.The Cat L (Sigma, C6854), Cat S (Sigma, SRP6297), and Cat B (Sigma, C8571) inhibitory assays were carried out using a similar protocol, except that the substrates for Cat S and Cat B were replaced with Ac-Lys-Gln-Lys-Leu-Arg-AMC and Z-Arg-Arg-AMC, respectively.
3.4 The inhibition of TRAP in RANKL-induced OC
RAW264.7 cells were seeded into a sterile 96-well plate at a density of 1 × 102 cells per well. To the DMEM culture medium, 10 ng mL−1 RANKL was added to induce the differentiation in RAW264.7 cells. Test compounds were added at 20 μg mL−1; each compound was tested in triplicate. Wells without a compound were used as blanks. The cells were incubated for 72 h at 37 °C in 5% CO2. Then, the culture medium was removed. The residues in the wells were fixed with fixative solution (25.5% citrate solution + 66.3% acetone + 8.2% of 37% formaldehyde) for 30 s and washed with deionized water. Then mixing solution (naphthol AS-BI phosphate solution and tartrate solution) was added and the plate was incubated at 37 °C in the dark. After a final washing step with deionized water, the TRAP-positive cells were counted.3.5 CTX-I concentration
RAW264.7 cells were seeded into a sterile 12-well plates with bovine femur slices at a density of 1 × 105 cells per well. To the DMEM culture medium, 50 ng mL−1 RANKL was added to induce the differentiation in RAW264.7 cells. Test compounds were added at a concentration of 5 μM. The cells were cultured for five days at 37 °C under 5% CO2. The culture supernatants were collected by centrifugation for analysing the CTX-I concentrations by ELISA.3.6 Real-time binding to Cat K and analysed by SPR
SPR experiments were carried out using a Biacore 8K (GE Healthcare) at 25 °C. Cat K (ENZO BML-SE553) was preconditioned in pH 5.0 sodium acetate and then immobilized to a CM7 chip by amine coupling. Running buffer was 50 mM NaOAc (pH 5.5), 0.01% P20, 2 mM EDTA, 100 μM DTT. When the affinities of compounds were tested, 5% DMSO was added. Ten concentrations of each compound were used. A20 was diluted from 100 μM by two-fold serial dilution. A22 was diluted from 50 μM by two-fold serial dilution. Compounds were injected to the surface of the protein-coupled chip channels at the flow rate of 30 μL min−1. The affinities (KD) were calibrated with the steady state affinity 1:1 binding model with the evaluation software of Biacore 8K. The affinities of A20 and A22 were tested twice and five times, respectively.
3.7 OP therapeutic effects in zebrafish B
OP was induced in zebrafish at the age of 2 dpf (day-post-fertilization) by treating normal wild-type AB zebrafish with prednisone for 96 h. Zebrafish larvae were randomly added to wells in 6-well plates. There were 30 fish per well. Etidronate disodium and different concentrations of A22 were dissolved by water and then added to different wells to target concentration. The zebrafish larvae were incubated with the compounds for 96 h at 28 °C.Zebrafish larvae aged at 7 dpf was dyed with 0.05% calcein solution for 15 min. Then it was washed with water for 15 min on the shaking table (repeated three times). The zebrafish larvae were fixed with methyl cellulose. The spine fluorescent intensity (SFI) images of the calcein-stained vertebrate column were captured with AZ100 fluorescence microscopy (Nikon). The integral optical density of 1 to 4 segments of vertebrae was measured by Image J analysis software. OP therapeutic effects (%) = (sample SFI – model SFI)/(normal SFI – model SFI) × 100%.
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Institute of Medicinal Biotechnology, Chinese Academy of Medical Science and Peking Union Medical College and approved by the Animal Ethics Committee of Institute of Medicinal Biotechnology.
4. Conclusions
In this study, a total of 61 derivatives were designed and synthesized with a shared scaffold that has not been reported elsewhere as Cat K inhibitors and anti-osteoporosis agents. Several derivatives showed an IC50 value below 10 μM against Cat K. We rigorously analysed the structure–activity relationship. A20, A22, and A32 had a significantly higher inhibitory activity against Cat K in vitro than that of the lead compound 1x. Real-time binding analysis by SPR and molecular docking calculations confirmed the binding of A22 to Cat K in vitro. A22 improved spinal bone density in zebrafish with induced OP at an efficacy that was higher than that of the marketed therapeutic bone metabolizer etidronate disodium. Hence, we believe that A22 represents a novel lead compound for antiresorptive drugs that will be examined for further improvements in structure-based drug design, therapeutic efficacy, and pharmacokinetics.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the grants from the CAMS Innovation Fund for Medical Sciences (CIFMS, 2017-I2M-3-019) and the National Natural Science Foundation of China (81473097, 81402800).Notes and references
- S. Tanaka, Am. J. Nephrol., 2007, 27, 466–478 CrossRef.
- E. Hernlund, A. Svedbom, M. Ivergard, J. Compston, C. Cooper, J. Stenmark, E. V. McCloskey, B. Jönsson and J. A. Kanis, Arch. Osteoporos., 2013, 8, 1–115 Search PubMed.
- O. Ström, F. Borgström and J. A. Kanis, Arch. Osteoporos., 2011, 6, 59–155 CrossRef.
- Z. Amso, J. Cornish and M. A. Brimble, Med. Res. Rev., 2016, 36, 579–640 CrossRef.
- S. Fujiwara, E. Hamaya, M. Sato, P. Graham-Clarke, J. A. Flynn and R. Burge, Clin. Interventions Aging, 2014, 5, 1879–1893 Search PubMed.
- B. R. Bhavnani and F. Z. Stanczyk, J. Steroid Biochem. Mol. Biol., 2014, 142, 16–29 CrossRef CAS PubMed.
- S. Sanders and S. A. Geraci, South. Med. J., 2013, 106, 698–706 CrossRef CAS.
- G. Mazziotti, J. Bilezikian, E. Canalis, D. Cocchi and A. Giustina, Endocrine, 2012, 41, 58–69 CrossRef CAS.
- J. A. Kanis, E. V. McCloskey, H. Johansson, A. Oden, L. J. Melton and N. Khaltaev, Bone, 2008, 42, 467–475 CrossRef CAS.
- A. G. Costa, N. E. Cusano, B. C. Silva, S. Cremers and J. P. Bilezikian, Nat. Rev. Rheumatol., 2011, 7, 447–456 CrossRef CAS.
- M. Novinec and B. Lenarčič, Biol. Chem., 2013, 394, 1163–1179 CAS.
- M. Gowen, F. Lazner, R. Dodds, R. Kapadia, J. Field, M. Tavaria, I. Bertoncello, F. Drake, S. Zavarselk, I. Tellis, P. Hertzog, C. Debouck and I. Kola, J. Bone Miner. Res., 1999, 14, 1654–1663 CrossRef CAS.
- M. T. Drake, B. L. Clarke, M. J. Oursler and S. Khosla, Endocr. Rev., 2017, 38, 325–350 CrossRef.
- J. Wijkmans and J. Gossen, Expert Opin. Ther. Pat., 2011, 21, 1611–1629 CrossRef CAS.
- M. Frizler, M. Stirnberg, M. T. Sisay and M. Gütschow, Curr. Top. Med. Chem., 2010, 10, 294–322 CrossRef CAS.
- J. Y. Gauthier, N. Chauret, W. Cromlish, S. Desmarais, L. T. Duong, J. P. Falgueyret, D. B. Kimmel, S. Lamontagne, S. Léger, T. LeRiche, C. S. Li, F. Massé, D. J. McKay, D. A. Nicoll-Griffith, R. M. Oballa, J. T. Palmer, M. D. Percival, D. Riendeau, J. Robichaud, G. A. Rodan, S. B. Rodan, C. Seto, M. Thérien, V. L. Truong, M. C. Venuti, G. Wesolowski, R. N. Young, R. Zamboni and W. C. Black, Bioorg. Med. Chem. Lett., 2008, 18, 923–928 CrossRef CAS.
- H. Xie, G. Chen and R. N. Young, J. Med. Chem., 2017, 60, 7012–7028 CrossRef CAS.
- S. Kumar, L. Dare, J. A. Vasko-Moser, I. E. James, S. M. Blake, D. J. Rickard, S. M. Hwang, T. Tomaszek, D. S. Yamashita, R. W. Marquis, H. Oh, J. U. Jeong, D. F. Veber, M. Gowen, M. W. Lark and G. Stroup, Bone, 2007, 40, 122–131 CrossRef CAS PubMed.
- D. S. Yamashita, R. W. Marquis, R. Xie, S. D. Nidamarthy, H. J. Oh, J. U. Jeong, K. F. Erhard, K. W. Ward, T. J. Roethke, B. R. Smith, H. Y. Cheng, X. Geng, F. Lin, P. H. Offen, B. Wang, N. Nevins, M. S. Head, R. C. Haltiwanger, S. A. Narducci, L. M. Liable-Sands, B. Zhao, W. W. Smith, C. A. Janson, E. Gao, T. Tomaszek, M. McQueney, I. E. James, C. J. Gress, D. L. Zembryki, M. W. Lark and D. F. Veber, J. Med. Chem., 2006, 49, 1597–1612 CrossRef CAS PubMed.
- J. P. Falgueyret, S. Desmarais, R. Oballa, W. C. Black, W. Cromlish, K. Khougaz, S. Lamontagne, F. Massé, D. Riendeau, S. Toulmond and M. D. Percival, J. Med. Chem., 2005, 48, 7535–7543 CrossRef CAS.
- C. Jerome, M. Missbach and R. B. Gamse, Osteoporosis Int., 2012, 23, 339–349 CrossRef CAS.
- S. Nagase, M. Ohyama, Y. Hashimoto, M. Small, J. Sharpe, J. Manako, T. Kuwayama and S. Deacon, J. Bone Miner. Metab., 2015, 33, 93–100 CrossRef CAS.
- Y. Ochi, H. Yamada, H. Mori, Y. Nakanishi, S. Nishikawa, R. Kayasuga, N. Kawada, A. Kunishige, Y. Hashimoto, M. Tanaka, M. Sugitani and K. Kawabata, Bone, 2011, 49, 1351–1356 CrossRef CAS.
- H. F. Guo, H. Y. Shao, Z. Y. Yang, S. T. Xue, X. Li, Z. Y. Liu, X. B. He, J. D. Jiang, Y. Q. Zhang, S. Y. Si and Z. R. Li, J. Med. Chem., 2010, 53, 1819–1829 CrossRef CAS PubMed.
- S. T. Xue, H. F. Guo, M. J. Liu, J. Jin, D. H. Ju, Z. Y. Liu and Z. R. Li, Eur. J. Med. Chem., 2015, 96, 151–161 CrossRef CAS.
- A. G. Dossetter, H. Beeley, J. Bowyer, C. R. Cook, J. J. Crawford, J. E. Finlayson, N. M. Heron, C. Heyes, A. J. Highton, J. A. Hudson, A. Jestel, P. W. Kenny, S. Krapp, S. Martin, P. A. MacFaul, T. M. McGuire, P. M. Gutierrez, A. D. Morley, J. J. Morris, K. M. Page, L. R. Ribeiro, H. Sawney, S. Steinbacher, C. Smith and M. Vickers, J. Med. Chem., 2012, 55, 6363–6374 CrossRef CAS.
- L. T. Wu, Z. Jiang, J. J. Shen, H. Yi, Y. C. Zhan, M. Q. Sha, Z. Wang, S. T. Xue and Z. R. Li, Eur. J. Med. Chem., 2016, 114, 328–336 CrossRef CAS.
- C. Niyibizi and D. R. Eyre, Eur. J. Biochem., 1994, 224, 943–950 CrossRef CAS.
- V. G. Palacios, L. J. Robinson, C. W. Borysenko, T. Lehmann, S. E. Kalla and H. C. Blair, J. Biol. Chem., 2005, 280, 13720–13727 CrossRef.
- P. Garnero, M. Ferreras, M. A. Karsdal, R. Nicamhlaoibh, J. Risteli, O. Borel, P. Qvist, P. D. Delmas, N. T. Foged and J. M. Delaissé, J. Bone Miner. Res., 2003, 18, 859–867 CrossRef CAS PubMed.
- M. Herrmann and M. J. Seibel, Clin. Chim. Acta, 2008, 393, 57–75 CrossRef CAS PubMed.
- A. Fleming, M. Sato and P. Goldsmith, J. Biomol. Screening, 2005, 10, 823–831 CrossRef CAS PubMed.
- T. P. Barros, W. K. Alderton, H. M. Reynolds, A. G. Roach and S. Berghmans, Br. J. Pharmacol., 2008, 154, 1400–1413 CrossRef CAS.
- D. E. Padgett, K. G. Holley, M. Cummings, A. G. Rosenberg, D. R. Sumner, D. Conterato and J. O. Galante, J. Arthroplasty., 2003, 18, 677–686 CrossRef.
- B. J. Thomas and H. C. Amstutz, Hip, 1987, 59–69 CAS.
- F. N. Ton, S. C. Gunawardene, H. Lee and R. M. Neer, J. Bone Miner. Res., 2005, 20, 464–470 CrossRef CAS.
- L. Buckley and M. B. Humphrey, Glucocorticoid-Induced Osteoporosis, N. Engl. J. Med., 2018, 379, 2547–2556 CrossRef.
- H. Wang, T. Feng, D. Guo, M. Zhang, L. Chen and Y. Zhou, Molecules, 2018, 23, E2343 CrossRef.
- S. J. Du, V. Frenkel, G. Kindschi and Y. Zohar, Dev. Biol., 2001, 238, 239–246 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra10338k |
| ‡ The authors contribute equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |