
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLong non coding RNA UCA1 contributes to the autophagy and survival of colorectal cancer cells via sponging miR-185-5p to up-regulate the WISP2/β-catenin pathway
Chao Liu,
Le Ji and
Xue Song *
*
Department of Gastroenterology, Affiliated Hospital of Yan'an University, No. 43 North Street, Yan'an, 716000, Shaanxi, China. E-mail: songxuedoctor@outlook.com; Tel: +86 09112881486
First published on 7th May 2019
Abstract
The estimated number of new cases of colorectal cancer (CRC) will increase to 140![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 250 in 2018 worldwide. The long non-coding RNA (lncRNA) urothelial carcinoma-associated 1 (UCA1) has recently been shown to be dysregulated in CRC, which plays an important role in the progression of CRC. However, the biological role and the underling mechanism of UCA1 in the carcinogenesis of CRC remain unclear. Herein, we found that UCA1 was aberrantly upregulated in two CRC cell lines (SW620 and HT29) compared to colorectal cell CCD-18Co. UCA1 knockdown inhibited the apoptosis, growth and autophagy of CRC cell lines in vitro. Furthermore, UCA1 could act as an endogenous sponge by directly interacting with miR-185-5p and downregulation miR-185-5p expression. In addition, UCA1 could reverse the inhibitory effect of miR-185-5p on the growth and autophagy of CRC cells, which might be involved in the derepression of member 1 (WNT1)-inducible signaling pathway protein 2 (WISP2, a target gene of miR-185-5p) expression and the activation of the WISP2/β-catenin signaling pathway. In vivo, the present study elucidates a novel UCA1-miR-185-5p-WISP2-Wnt/β-catenin axis in CRC, which may help us to understand the pathogenesis and the feasibility of lncRNA-directed diagnosis and therapy of CRC.
250 in 2018 worldwide. The long non-coding RNA (lncRNA) urothelial carcinoma-associated 1 (UCA1) has recently been shown to be dysregulated in CRC, which plays an important role in the progression of CRC. However, the biological role and the underling mechanism of UCA1 in the carcinogenesis of CRC remain unclear. Herein, we found that UCA1 was aberrantly upregulated in two CRC cell lines (SW620 and HT29) compared to colorectal cell CCD-18Co. UCA1 knockdown inhibited the apoptosis, growth and autophagy of CRC cell lines in vitro. Furthermore, UCA1 could act as an endogenous sponge by directly interacting with miR-185-5p and downregulation miR-185-5p expression. In addition, UCA1 could reverse the inhibitory effect of miR-185-5p on the growth and autophagy of CRC cells, which might be involved in the derepression of member 1 (WNT1)-inducible signaling pathway protein 2 (WISP2, a target gene of miR-185-5p) expression and the activation of the WISP2/β-catenin signaling pathway. In vivo, the present study elucidates a novel UCA1-miR-185-5p-WISP2-Wnt/β-catenin axis in CRC, which may help us to understand the pathogenesis and the feasibility of lncRNA-directed diagnosis and therapy of CRC.
Introduction
The number of cases of human colorectal cancer (CRC) is estimated to increase to 140250 and the estimated deaths to increase to 50630 in males and females worldwide in 2018.1 CRC is still a serious disease. More effort needs to be made to clarify the carcinogens and find out which factor is the most important in the development of CRC.2,3 DNA-designed chemistry and DNA nanotechnology are gradually maturing and are widely used in biomedicine,4,5 and might be effective strategies for CRC therapy.
Urothelial carcinoma-associated 1 (UCA1), one of the long non-coding RNA (lncRNA), has been proved to be significantly up-regulated in most tumor tissues and cancer cells.6–10 As reported, UCA1 promotes cell survival of CRC.6 However, the underlying mechanisms of how UCA1 contributes to CRC are still perplexing. Meanwhile, it is well-known that Wnt/β-catenin is a potential target in colon cancer.11 Abundance of articles found that UCA1 can interact with varies of microRNAs (miRNAs) to modulate cancer development, such as miR-204-5p,7 miR-143,8 miR-216b,12 miR-193a-3p,13 and so on. It is interesting that another research has reported miR-185-5p can inhibit the Wnt/β-catenin pathway and works as a tumor suppressor in CRC.14 Therefore, we speculated that UCA1 may interact with miR-185-5p to regulate CRC by WNT1-inducible signaling pathway protein 2 (WISP2)/β-catenin pathway.
In the present study, UCA1 was overexpressed in CRC cells and it contributed to CRC cells proliferation and autophagy inhibition. Importantly, this paper demonstrated that UCA1 might upregulate the expression of WISP2 through directly interacting and inhibiting the expression of miR-185-5p. Our present results provide a novel UCA1-miR-185-5p-WISP2-Wnt/β-catenin signaling pathway in regulation of CRC, shedding new light on the diagnosis and treatment for CRC.
Materials and methods
Cell culture
Normal colorectal cell CCD-18Co, normal epithelial cell HIEC-6, CRC-derived cells SW620 and HT29 were purchased from ATCC (Manassas, VA, USA). Cells were cultured in RPMI1640 (HyClone, UT, USA) or DMEM (HyClone) which was supplemented with 10% FBS (Gibco, NY, USA), 100 U ml−1 penicillin/streptomycin (HyClone) at 37 °C in a humidified incubator with 5% CO2.Reagents and transfections
The miR-185 inhibitor, sh-UCA1, and sh-UCA1 control sequences were purchased from the GenePharma Company (Shanghai, China). SW620 and HT29 cells were transfected with miR-185 inhibitor, sh-UCA1, sh-UCA1 control, or miR-185 inhibitor + sh-UCA1, separately. Lipofectamine2000 transfection reagent (Invitrogen, Carlsbad, CA, USA) was used for transfection according to the manufacturer's instructions.Luciferase reporter assay
To validate the predicted miRNA-binding sequence for UCA1, pmirGLO-UCA1-WT or pmirGLO-UCA1-MUT were transfected into HEK 293T cells with or without synthetic hsa-miR-185 mimic (GenePharma) by Lipofectamine 2000 (Invitrogen). HEK 293T cells (2.0 × 104) were plated in a 24-well plate and transfected with 0.05 μg hsa-miR-185 mimic. The wild type sequence of UCA1 (UCA1-wt) or its mutant sequence (UCA1-mut) was subcloned into the pMIR luciferase reporter and then co-transfected with miR-185-5p mimics or controls into 293T cells. Finally, luciferase signals were measured by the dual-luciferase reporter assay system (Promega, CA, USA) and normalized to Renilla activity.Cell counting kit-8 assay (CCK-8)
CCK-8 assay was performed to detect cell growth in our study. SW620 and HT29 cells were transfected with sh-UCA1 or sh-ctrl, then cells were cultured for 24 h. Cells were trypsinized and seeded into 96-well plates (1.5 × 103 cells in 200 μL). Then, the plate was incubated at 37 °C with 5% CO2. CCK-8 assay was performed for the nest continuous 4 days. Absorbance at 490 nm (OD490) was detected by using a microplate reader (BioTek Instruments, VT, USA).Q-PCR
miR-185-5p and UCA1 RNA were extracted from cultured CCD-18Co, HIEC-6, SW620, and HT29 cells by Trizol reagent (Invitrogen). miR-185-5p expression levels were determined by SYBR PrimeScript miRNA q-PCR kit (Takara Bio, Shiga, Japan). The relative miR-185-5p level was normalized to U6. The relative amount of UCA1 was normalized to GAPDH. q-PCR was performed according to the manufacturer's instructions of SYBR-Green (Applied Biosystems, Foster City, CA, USA) in a PCR amplifier (ABI Prism 7000; Applied Biosystems). Data were analyzed according to the comparative Ct method also referred to as the 2−ΔΔCt method.Western blot
Cellular and tissue proteins were extracted by RIPA lysis buffer (Beyotime, Shanghai, China), and then the protein was quantified by Bicinchoninic Acid (BCA) Kit (Solarbio, Beijing, China). The proteins were separated by SDS-PAGE and then transferred onto a PVDF membrane (Millipore Corp, Billerica, USA). 5% skim milk in TBST was used for 1 h blocking at 25 °C. After that, the membranes were incubated with specific primary antibodies against Ki-67 (ab15580; Abcam, Cambridge, UK), beclin1 (ab62557), p62 (ab56416), LC3B (ab48394), WISP2, (ab38317), transcription factor 4 (TCF-4; ab60727), β-catenin (ab32572) and GAPDH (ab8245) overnight at 4 °C, separately. The membranes were washed 10 min/3 times and then incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies (Life Technologies, Carlsbad, CA, USA) for 1 h at 25 °C. Finally, ECL reagent (GE Healthcare, Little Chalfont, UK) was used to detect the protein expression signals, and the blots were analyzed by ImageJ software (National Institutes of Health, Bethesda, USA).Immunofluorescence staining
SW620 cells were washed 3 times in PBS, and then fixed in 4% paraformaldehyde for 15 min at 25 °C and blocked with 3% BSA for 90 min. Next, samples were washed with PBS then treated with primary antibody against LC3 (Sigma, St. Louis., MO, USA) for 1 h at 25 °C. After that, samples were stained with DAPI solution for 3 min followed by incubation of secondary antibody for 30 min at 25 °C in dark. Fluorescence images were acquired by a confocal fluorescence microscope (Leica TCS SP8, Germany).Immunohistochemical staining
Specimens were incubated with 3% H2O2 for 10 min, and washed with PBS, then incubated with Ki67 and caspase-3 antibody at 4 °C overnight. Next, specimens were washed with PBS and then incubated at 25 °C for 30 min with secondary antibody, the samples were incubated in streptavidin–peroxidase for another 30 min. Finally, DAB (Sigma) solution was used. The final percentage of positive cells was calculated.Terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling (TUNEL) assay
Apoptosis was detected by TUNEL staining according to the manufacturer's instructions (UPSTATE, Lake Placid, NY). Brown staining within the nucleus represented TUNEL-positive apoptotic cells. The extent of apoptosis was calculated and expressed as a number of TUNEL-positive tubular epithelial cells from the same field.Animal experiments
6 week-old BALB/c athymic nude mice were purchased from Shanghai Institutes for Life Science, the Chinese Academy of Sciences (Shanghai, China). Mice were free to food and water ad libitum. SW620 cells (1 × 107 cells in 100 μl) transfected with sh-UCA1 or sh-RNA control were injected subcutaneously into the flanks of mice. Our animal studies have been approved by the Affiliated Hospital of Yan'an university ethics committee and the protocols have been performed in accordance with the ethical standards of Declaration of Helsinki. Tumor growth was determined every 5 d for 30 days. At the 30th day, mice were sacrificed.Statistical analysis
One-way ANOVA was used to analyze in vivo tumor volumes. The columns were presented as the mean ± SD, then evaluated with a Student's t-test, post hoc test or two-way ANOVA. Statistical significance was indicated by p values less than 0.05. All experiments were performed at least three in triplicate.Results
UCA1 knockdown suppressed the survival of SW620/HT29 cells
We found that the expression levels of UCA1 in SW620 (p < 0.001) and HT29 (p < 0.01) cells were significantly higher than those in normal CCD-18Co cells (Fig. 1A). UCA1 knockdown results shows that UCA1 expression levels are decreased in SW620 cells after sh-UCA1 transfection (p < 0.01, Fig. 1B). CCK-8 results showed that sh-UCA1 significantly inhibited the proliferation activity of SW620 and HT29 cells (p < 0.01, Fig. 1C and D). Protein expression level of caspase-3 was significantly elevated, while Ki67 was significantly downregulated after sh-UCA1 transfection (p < 0.001, Fig. 1E–G). As shown in Fig. 1H, the sh-UCA1 transfection decreased LC3-II levels (p < 0.001). The sh-UCA1 transfection decreased the expression of beclin1 (p < 0.001, Fig. 1H). The decreased LC3-II resulted in significantly decrease of LC3-II/LC3-I ratio (Fig. 1K). sh-UCA1 increased the expression of p62 (p < 0.001, Fig. 1J).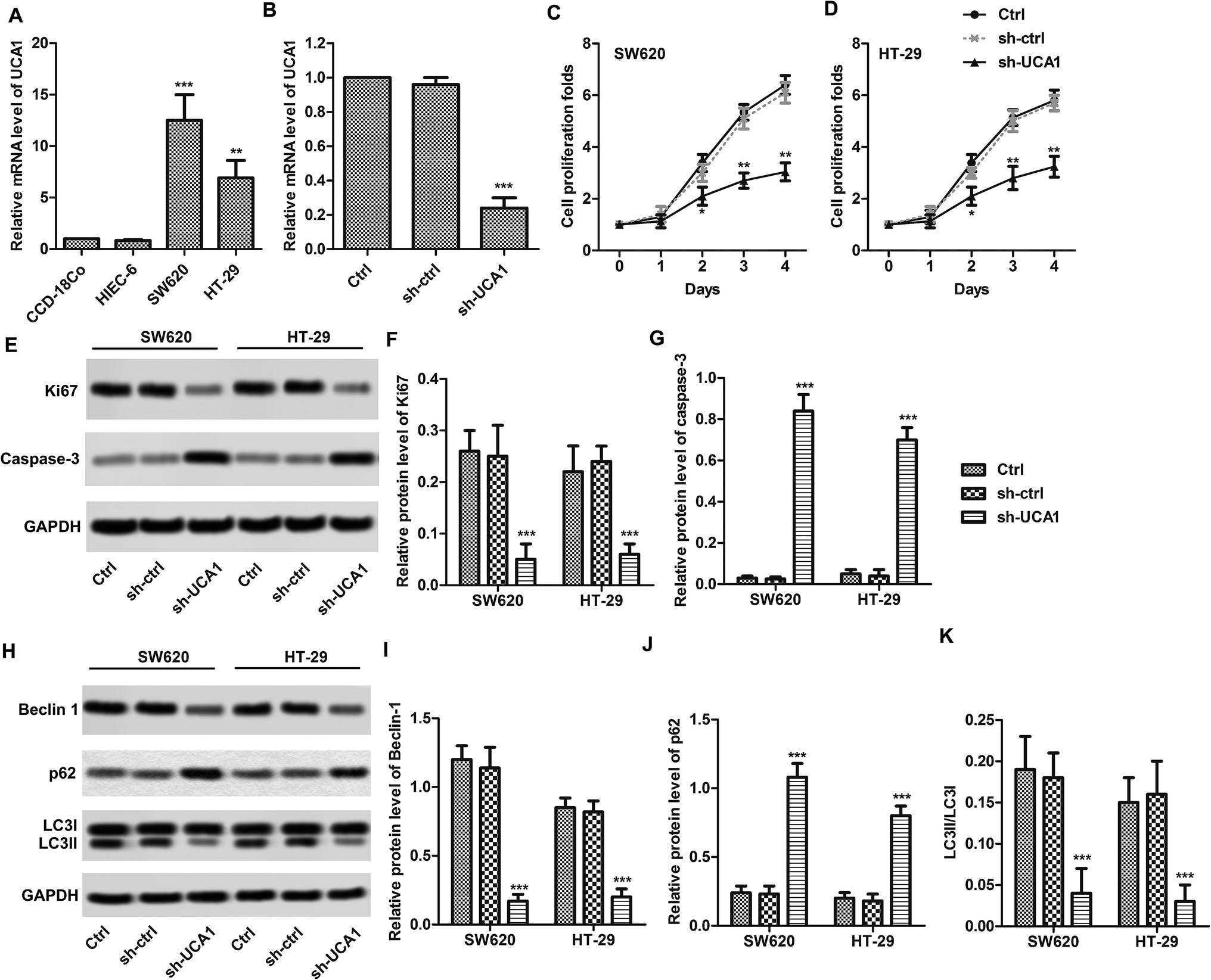 | ||
| Fig. 1 UCA1 regulated CRC cell proliferation, apoptosis and autophagy in vitro (A) UCA1 expression was upregulated in CRC cell lines (SW620 and HT29). UCA1 expression was detected in human normal colorectal cell CCD-18Co and normal epithelial cell HIEC-6 as well as CRC-derived cells SW620, HT29 by q-PCR. U6 was used as internal control. **p < 0.01, ***p < 0.001, compared with CCD-18Co group. (B) UCA1 was knockdown by shRNA. ***p < 0.001 compared with control group. (C and D) Cell proliferation was detected by CCK-8. **p < 0.01, compared with control group. (E–G) The relative expression levels of Ki67 and caspase-3 were detected by Western blot. ***p < 0.001 compared with control group. (H–K) The relative expression levels of autophagy markers were detected by Western blot. All data were represented as the mean ± SD from three independent experiments. ***p < 0.001 compared with control group. | ||
UCA1 directly interacted with miR-185-5p in SW620 and HT29 cells
As shown in Fig. 2A, UCA1 contains elements complementary to various miR-185-5p regions. MiR-185-5p expression was markedly downregulated (Fig. 2B) in SW620 (p < 0.001) and HT29 (p < 0.01) cells compared with CCD-18Co cells. The expression levels of miR-185-5p were measured in sh-UCA1 transfected SW620 and HT29 cells by q-PCR. Expectedly, the expression levels of miR-185-5p were markedly evaluated compared with the control group (p < 0.001, Fig. 2C). As shown in Fig. 2D, miR-185-5p reduced luciferase activity in cells transfected with UCA1-wt (p < 0.01). These results suggested that there existed direct interactions between miR-185-5p and UCA1.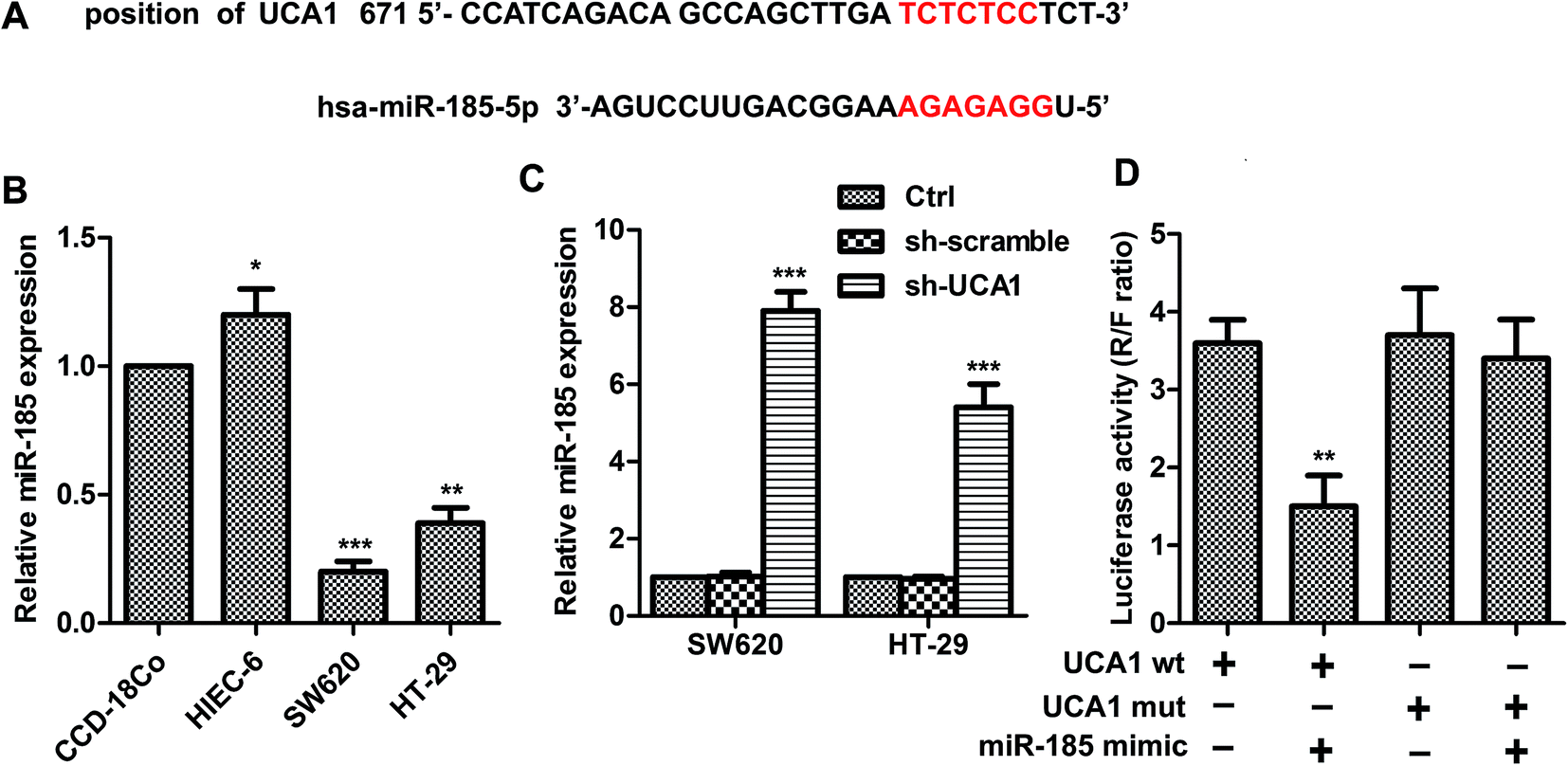 | ||
| Fig. 2 UCA1 can directly interact with miR-185-5p in CRC cell lines. (A) Predict binding sites between UCA1 and miR-185-5p. (B) miR-185-5p expression was detected in human normal colorectal cell CCD-18Co and normal epithelial cell HIEC-6 as well as CRC-derived cells by q-PCR. *p < 0.05, **p < 0.01, ***p < 0.001 compared with CCD-18Co group. (C) The expression of miR-185-5p mRNA level was detected in SW620 and HT29 after sh-UCA1 transfection by q-PCR. U6 was used as internal control. ***p < 0.001, compared with control group. (D) Luciferase activity was detected by luciferase reporter assay. HEK 293T cells were co-transfected with either 50 nM miR-185-5p mimics or NC oligos and 200 ng of pmirGLO-UCA1-wt or pmirGLO-UCA1-mut using Lipofectamien 2000. The relative firefly luciferase activity measured 24 hours after transfection and was normalized with renilla luciferase activity **p < 0.01 compared with UCA1-wt group. The data shown are the mean ± standard error of three individual experiments. | ||
UCA1 modulates WISP2 activity and autophagy through miR-185-5p
Bioinformatics prediction result (Fig. 3A) showed the 3′UTR of WISP2 has the complementary sequence to miR-185-5p. Expectedly, the mRNA expression level of WISP2 was significantly suppressed by sh-UCA1 in both SW620 and HT29 cells compared with the control (p < 0.001, Fig. 3B). In particular, the protein expression of WISP2 were significantly decreased by sh-UCA1, miR-185-5p inhibitor can partially restored the decrease of WISP2 protein level (p < 0.001, Fig. 3C and D). These results suggested that UCA1 promoted SW620 cell proliferation (Fig. 3E) and autophagy (Fig. 3F–I) through inactivating miR-185-5p.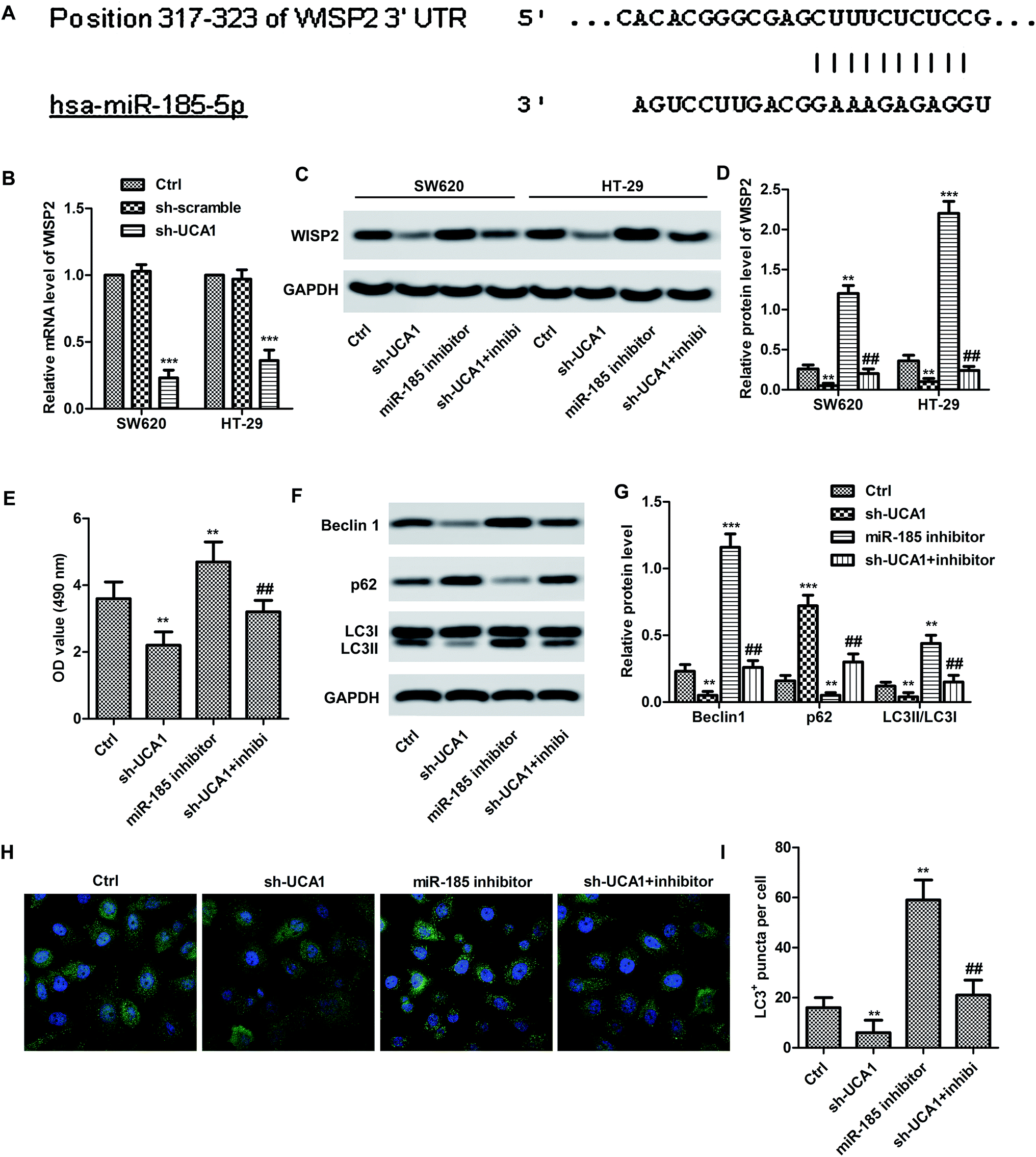 | ||
| Fig. 3 UCA1 upregulated WISP2 expression and promoted autophagy. (A) Result of bioinformatics analysis. WISP2 is a target of hsa-miR-185-5p. (B) Cells were divided into 4 group: Ctrl, sh-UCA1, miR-185 inhibitor, sh-UCA1 + inhibi. mRNA expression of WISP2 in SW620 and HT29 with or without transfected sh-UCA1. ***p < 0.001, compared with control group. (C and D) Protein expression of WISP2 in SW620 and HT29 with or without transfected sh-UCA1. A miR-185-5p inhibitor was transfected. **p < 0.01, ***p < 0.001 compared with control group. ##p < 0.01 compared with miR-185 inhibitor group. (E) The cell proliferation was detected by CCK-8 in SW620. (F and G) Relative expression of beclin1, P62 and LC3 I/II was detected by Western blot in SW620. **p < 0.01 compared with control group. ##p < 0.01 compared with miR-185 inhibitor group. (H and I) LC3+ puncta per cell was detected by immunofluorescence staining. The data shown are the mean ± standard error of three individual experiments. **p < 0.01, ***p < 0.001, compared with control group. ##p < 0.01, compared with sh-UCA1 group. | ||
UCA1 promoted tumor survival trough upregulating WISP2/β-catenin pathway in vivo
We therefore studied the effects of UCA1 in SW620 cell xenografted tumor mice in vivo. Consistent with the results in vitro, UCA1 knockdown markedly suppressed the tumor growth (volume) compared to the control (Fig. 4A). Obviously, UCA1 knockdown upregulated the mRNA level of miR-185-5p (Fig. 4B). Moreover, immunohistochemistry and TUNEL results showed that UCA1 knockdown increased the Ki67 expression but reduced the caspase-3 expression. In particular, the apoptosis rate was decreased by UCA1 knockdown (Fig. 4C–D). As shown in Fig. 4E and F, UCA1 knockdown resulted to autophagy inhibition. It is worth noting that UCA1 knockdown negatively regulated the expression of WISP2, β-catenin and TCF4, which indicated that UNA1 may activate the WISP2/β-catenin pathway to promote tumor cell survival.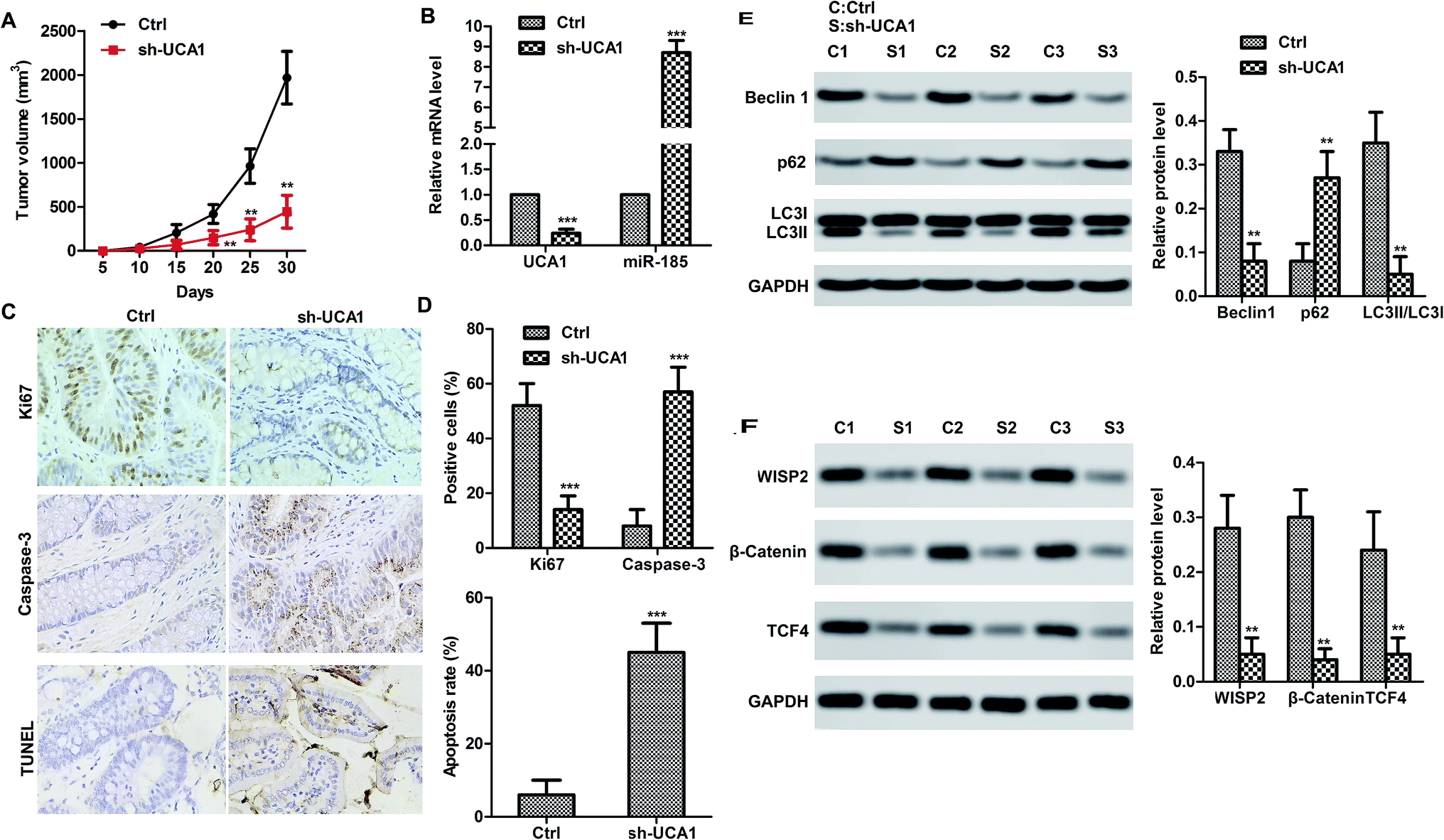 | ||
| Fig. 4 UCA1 contributed SW620 xenografted tumor survival in vivo. (A) sh-UCA1 decreased tumor volume. **p < 0.01 compared with control group. (B) UCA1 downregulated miR-185-5p mRNA level in vivo. (C and D) The expression of caspase-3 and Ki67 was detected by immunohistochemistry. Apoptosis rate was detected by TUNEL. The data shown are the mean ± standard error of three individual experiments. ***p < 0.001, compared with control group. (E) Relative expression of beclin1, P62 and LC3 I/II was detected by Western blot in vivo. (F) Relative expression of WISP2, β-catenin and TCF4 was detected by Western blot in vivo. The data shown are the mean ± standard error of three individual experiments. **p < 0.01 compared with control group. | ||
Discussion
Early diagnosis is meaningful to decrease the deaths of CRC.15 A lot of lncRNAs and miRNAs have been proved to be potential biomarkers in CRC.16 However, more biomarkers need to be found to apply to clinical CRC diagnosis or treatment.17 In present study, UCA1 and miR-185-5p may be effective biomarkers.A large number of researches reported that UCA1 might be biomarker of CRC18 and participated in proliferation, apoptosis and metastasis in CRC.6,17 Yu et al. pointed out UCA1 was overexpressed in CRC tissues and cell lines, including CaCO-2, LoVo, HCT116 and SW480 cells.6 Expectedly, we found that UCA1 was up-regulated CRC cell lines, including SW620 and HT29, as well as in SW620 cell xenografted tumor. As previously researches mentioned, UCA1 increased CRC cell proliferation and inhibited cell apoptosis.6,17 In present study, UCA1 knockdown significantly decreased cell proliferation folds and autophagy flux, while increased apoptosis in SW620 and HT29 cells. Our results demonstrated that UCA1 promoted CRC malignant progression. In contrast with UCA1, previous studies showed that miR-185-5p could suppress CRC development,14 including growth and invasion of colon cancer cells.19 It is worth noting that miR-185 showed decreased expression levels in human CRC cell lines, such as SW620, HT29 and LOVO.20 Consistent with the previous study, our findings suggested that there existed a negative correlation between UCA1 expression and miR-185-5p expression in CRC cell lines. Mounting evidences have identified that the UCA1 contributes to various cancers by interacting with miRNAs thus inhibit their expression.7,8,12,13 Additionally, Zhu et al. reported that lncRNA FOXD2-AS1 contributed to CRC proliferation through interacting with miR-185-5p.21 According to the previous study results, it is reasonable to speculate that UCA1 might interact with miR-185-5p. As expected, a direct bind was found in miR-185-5p elements and the 3′-UTR UCA1 RNA by luciferase assays. The regulatory mechanism between UCA1 and miR-185-5p is similar to aforementioned studies.7,8,12,13,21
To investigated inhibitory effect of UCA1 miR-185-5p, miR-185-5p inhibitor and/or sh-UCA1 were transfected into CRC cell lines. As a result, cell proliferation and autophagy were increased in miR-185-5p inhibitor treated CRC cells in vitro. Whereas, UCA1 knockdown could reverse the effect of miR-185-5p inhibitor on the proliferation and autophagy of CRC cells. Consistent with previous reports, our present study indicate that UCA1 may regulate CRC via inhibiting miR-185-5p expression.
WISP2 is a member of the CCN family of cysteine-rich and glycosylated signaling proteins, which involves in various developmental processes.22 A previous study demonstrated elevated expression of WISP2 in gastric cancer compared to normal tissues, which was associated with good prognosis in patients with gastric cancer.23 β-Catenin is the core component of the canonical Wnt signaling pathway, which plays key regulatory role in cell growth, proliferation, morphology, motility and organ development.24 T-cell factor-4 (TCF-4) was reported to promote initiation and progression of many cancers.25 Usually, TCF-4 binds to β-catenin and then transactivates Wnt target genes.26 It is notable that a previous study revealed that WISP2 was a direct target of miR-185-5p.14 Herein, we showed that the 3′UTR of WISP2 has the complementary sequence to miR-185-5p. The mRNA and protein levels of WISP2 were increased by miR-185-5p inhibitor but reduced by sh-UCA1. The data indicates that UCA1 upregulates WISP2 via miR-185-5p derepression, thus contributing to CRC development. Accumulating evidences exhibited that WISP2 played a crucial role in facilitating tumor growth and survival.27,28 In addition, our Western blot results comformed that UCA1 could promote CRC malignant progression through WISP2/β-catenin signaling pathway. Mounting evidences show that WISP2/β-catenin signal plays a crucial role in cell proliferation and tumorigenesis.29,30 Therefore, due to the important roles of WISP2/β-catenin signal in a variety of cancers, it is a promising therapeutic target in cancers.31 In the present study, sh-UCA1 obviously down-regulated the levels of β-catenin, TCF-4 and WISP2 in vivo, suggesting that UCA1 affected cell proliferation via activating WISP2/β-catenin pathway. Overall, we elucidate the UCA1-miR-185-5p-WISP2/β-catenin axis in CRC regulation.
Autophagy is a process that recycles cellular components via lysosomal degradation. Autophagy has been shown to play a vital role in CRC development and its manipulation is a therapeutic target. Autophagy was reported to be upregulated in various treatment strategies for CRC.32 Previous study investigated the expression and significance of three autophagy-related proteins, namely, beclin1, LC3, and mTOR, in the tumorigenesis and development of CRC. The expression of LC3 was positively correlated with beclin1 but negatively correlated with P62 in CRC tissues.33 In present study, UCA1 was positively correlated with beclin1 and LC3. The results indicated that UCA1 was positively correlated with tumor autophagy and survival.
Conclusions
In conclusion, our present work indicate that UCA1 may be an oncogene through promoting human CRC malignant progression. Importantly, our study reveals a novel UCA1-miR-185-5p-WISP2-Wnt/β-catenin regulatory axis in CRC.Conflicts of interest
There are no conflicts to declare.Abbreviations
| CRC | Colorectal cancer |
| WISP2 | WNT1-inducible signaling pathway protein 2 |
| lncRNA | Long non-coding RNAs |
| UCA1 | Urothelial carcinoma-associated 1 |
| CCK-8 | Cell counting kit-8 |
| TUNEL | Terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling |
| TCF-4 | T-cell factor-4 |
| RT-qPCR | Real-time polymerase chain reaction |
References
- R. L. Siegel, K. D. Miller and A. Jemal, Ca-Cancer J. Clin., 2018, 68, 7–30 CrossRef PubMed.
- M. D. Xu, P. Qi and X. Du, Mod. Pathol., 2014, 27, 1310–1320 CrossRef CAS PubMed.
- F. Jing, H. Jin, Y. Mao, Y. Li, Y. Ding, C. Fan and K. Chen, Tumor Biol., 2017, 39, 101042831770365 CrossRef PubMed.
- C. Ashworth, Nat. Rev. Mater., 2018, 3, 17092 CrossRef.
- Y. Tokura, S. Harvey, C. Chen, Y. Wu, D. Y. W. Ng and T. Weil, Bull. Chem. Soc. Jpn., 2017, 90, 967–1004 CrossRef.
- Y. Han, Y. N. Yang, H. H. Yuan, T. T. Zhang, H. Sui, X. L. Wei, L. Liu, P. Huang, W. J. Zhang and Y. X. Bai, Pathology, 2014, 46, 396–401 CrossRef CAS PubMed.
- Z. Bian, L. Jin, J. Zhang, Y. Yin, C. Quan, Y. Hu, Y. Feng, H. Liu, B. Fei, Y. Mao, L. Zhou, X. Qi, S. Huang, D. Hua, C. Xing and Z. Huang, Sci. Rep., 2016, 6, 23892 CrossRef CAS PubMed.
- Y. L. Tuo, X. M. Li and J. Luo, Eur. Rev. Med. Pharmacol. Sci., 2015, 19, 3403–3411 Search PubMed.
- H. M. Wang, J. H. Lu, W. Y. Chen and A. Q. Gu, Int. J. Clin. Exp. Med., 2015, 8, 11824–11830 CAS.
- Y. Fan, B. Shen, M. Tan, X. Mu, Y. Qin, F. Zhang and Y. Liu, FEBS J., 2014, 281, 1750–1758 CrossRef CAS PubMed.
- A. Sebio, M. Kahn and H. J. Lenz, Expert Opin. Ther. Targets, 2014, 18, 611–615 CrossRef CAS PubMed.
- F. Wang, H. Q. Ying, B. S. He, Y. Q. Pan, Q. W. Deng, H. L. Sun, J. Chen, X. Liu and S. K. Wang, Oncotarget, 2015, 6, 7899–7917 Search PubMed.
- W. Nie, H. J. Ge, X. Q. Yang, X. Sun, H. Huang, X. Tao, W. S. Chen and B. Li, Cancer Lett., 2016, 371, 99–106 CrossRef CAS PubMed.
- W. Dong-Xu, L. Jia and Z. Su-Juan, Indian J. Cancer, 2015, 52(suppl. 3), E182–E185 CrossRef PubMed.
- M. Boehler, V. Advani, C. J. Schwind, E. D. Wietfeldt, Y. Becker, B. Lewis, J. Rakinic and I. Hassan, J. Cancer Educ., 2011, 26, 147–152 CrossRef PubMed.
- Y. Yang, Y. Du, X. Liu and W. C. Cho, Adv. Exp. Med. Biol., 2016, 937, 19–51 CrossRef CAS PubMed.
- B. Ni, X. Yu, X. Guo, X. Fan, Z. Yang, P. Wu, Z. Yuan, Y. Deng, J. Wang, D. Chen and L. Wang, Internet J. Oncol., 2015, 47, 1329–1338 CrossRef CAS PubMed.
- H. H. Hong, L. K. Hou, X. Pan, C. Y. Wu, H. Huang, B. Li and W. Nie, Oncotarget, 2016, 7, 44442–44447 Search PubMed.
- Z. J. Lu, L. G. Lu, K. Z. Tao, D. F. Chen, Q. Xia, J. J. Weng, F. Zhu, X. P. Wang and P. Zheng, Mol. Med. Rep., 2014, 10, 2401–2408 CrossRef CAS PubMed.
- Z. Zhang, X. Liu, B. Feng, N. Liu, Q. Wu, Y. Han, Y. Nie, K. Wu, Y. Shi and D. Fan, Oncogene, 2015, 34, 4808–4820 CrossRef CAS PubMed.
- Y. Zhu, L. Qiao, Y. Zhou, N. Ma and C. Wang, Cancer Sci., 2018, 109(7), 2235–2242 CrossRef CAS PubMed.
- G. Dhar, S. Mehta, S. Banerjee, A. Gardner, B. M. McCarty, S. C. Mathur, D. R. Campbell, S. Kambhampati and S. K. Banerjee, Cancer Lett., 2007, 254, 63–70 CrossRef CAS PubMed.
- J. Ji, S. Jia, Y. Jia, K. Ji, R. Hargest and W. G. Jiang, Br. J. Cancer, 2015, 113, 921–933 CrossRef CAS PubMed.
- Z. M. Dai, S. Sun, C. Wang, H. Huang, X. Hu, Z. Zhang, Q. R. Lu and M. Qiu, J. Neurosci., 2014, 34, 8467–8473 CrossRef PubMed.
- Y. Tomimaru, H. Koga, H. Yano, S. de la Monte, J. R. Wands and M. Kim, Liver Int., 2013, 33, 1100–1112 CrossRef CAS PubMed.
- W. Gu, X. Li and J. Wang, Oncol. Rep., 2014, 31, 397–404 CrossRef CAS PubMed.
- S. C. Barreto, A. Ray and P. Ag Edgar, J. BUON, 2016, 21, 1359–1367 Search PubMed.
- L. F. Leal, L. M. Mermejo, L. Z. Ramalho, C. E. Martinelli Jr, J. A. Yunes, A. L. Seidinger, M. J. Mastellaro, I. A. Cardinalli, S. R. Brandalise, A. C. Moreira, L. G. Tone, C. A. Scrideli, M. Castro and S. R. Antonini, J. Clin. Endocrinol. Metab., 2011, 96, 3106–3114 CrossRef CAS PubMed.
- K. Li, Z. Y. Zhou, P. P. Ji and H. S. Luo, Oncol. Lett., 2016, 11, 3896–3900 CrossRef CAS PubMed.
- J. Ji, S. Jia, K. Ji and W. G. Jiang, Oncol. Rep., 2014, 31, 533–539 CrossRef CAS PubMed.
- M. Y. Wu, X. Xie, Z. K. Xu, L. Xie, Z. Chen, L. M. Shou, F. R. Gong, Y. F. Xie, W. Li and M. Tao, Oncol. Rep., 2014, 32, 513–522 CrossRef PubMed.
- K. Lai, M. C. Killingsworth and C. S. Lee, J. Clin. Pathol., 2014, 67, 854–858 CrossRef CAS PubMed.
- S. Wu, C. Sun, D. Tian, Y. Li, X. Gao, S. He and T. Li, Int. J. Clin. Exp. Pathol., 2015, 8, 3882–3891 CAS.
| This journal is © The Royal Society of Chemistry 2019 |