DOI:
10.1039/C8RA10636C
(Paper)
RSC Adv., 2019,
9, 8454-8462
A citric acid-assisted deposition strategy to synthesize mesoporous SiO2-confined highly dispersed LaMnO3 perovskite nanoparticles for n-butylamine catalytic oxidation†
Received
29th December 2018
, Accepted 5th March 2019
First published on
14th March 2019
Abstract
Catalytic oxidation can efficiently eliminate nitrogen-containing volatile organic compounds (NVOCs) and suppress the generation of toxic NOx in order to avoid secondary pollution. In this study, mesoporous SiO2-confined LaMnO3 perovskite nanoparticles with high dispersion were successfully prepared by a citric acid-assisted deposition method (LMO/SiO2-SD) and tested for the oxidation of n-butylamine. The method utilized the synergistic effect of abundant active hydroxyl groups existing on the SiO2 gel surface and citric acid, rendering the metal ions more uniformly scattered on the SiO2 surface. Strikingly, the LMO/SiO2-SD sample exhibited the optimum catalytic performance (T90 at 246 °C) and the highest N2 selectivity, which was mainly ascribed to its abundant surface acid sites, superior low-temperature reducibility and higher ratio of surface Mn4+ species. The apparent activation energy (Ea) for n-butylamine oxidation over LMO/SiO2-SD sample was 29.0 kJ mol−1. Furthermore, the reaction mechanism of n-butylamine oxidation was investigated by in situ FITR and a reasonable reaction route for n-butylamine oxidation over the LMO/SiO2-SD sample was proposed.
1 Introduction
Nitrogen-containing volatile organic compounds (NVOCs) have caused serious impacts on air pollution and public health, and have being a focus of attention in the field of air pollution prevention and treatment. The varied sources and varieties of NVOCs lead to disparate degrees of influence due to their different toxic, mutagenic and carcinogenic properties. Examples are amines, nitro compounds, nitrile and so on, which are odorous, highly toxic and would cause NOx secondary pollution and other problems by improper disposal.1–5 Compared to various developed technologies for NVOCs elimination,6 the catalytic oxidation method has always been judged as a highly efficient and promising alternative technology due to its superior advantages, such as lower temperature, lower energy consumption, no secondary pollution and so on. Within this eco-friendly method, NVOCs would be utterly decomposed into CO2, H2O and N2 at a relatively low reaction temperature. In addition, the yield of toxic product NOx would be strictly controlled in order to avoid secondary pollution.
With regard to the catalysts applied to NVOCs oxidation, precious metal (Pd, Ag, etc.) and transition metal (Cr, Mn, etc.) catalysts are commonly investigated by researchers due to their optimum catalytic performances.5–7 Although noble metal catalysts exhibited superior catalytic activities, toxicity resistance and better regeneration, their expensive cost, poor high thermal stability and scarce resources limit the large-scale application in industrial production. Perovskite-type metal oxides have always been considered as an alternative for noble metal catalysts due to their unique catalytic performances and good thermal stability for catalytic oxidation of VOCs.8–12 The application of conventional perovskites in catalytic oxidation were nonetheless currently limited due to low specific surface area and serious accumulation due to calcination of precursor material at high temperature. To improve the catalytic performances of perovskites, enormous methods were introduced, such as fabricating three-dimensionally ordered macroporous structure by PMMA-templating approach; dispersing nano-sized perovskites onto metal oxides support with larger specific surface area and so on.13–16 Numerous articles have substantiated that the supported nanoscale perovskites would enhance catalytic performances, but the supported perovskites prepared by conventional methods were poorly dispersed and crystallized, which would have an influence on stability and catalytic performances.17–19 The above triggered our interesting in the study of optimization of perovskite catalysts. In this work, mesoporous SiO2-confined perovskite nanoparticles with high dispersion was prepared by a novel avenue (citric acid-assisted deposition method). The figure shown in Scheme 1 depicts the facile preparation of the mesoporous SiO2 support and the ensuing synthesis of the LMO/SiO2-SD catalyst. The TEOS was firstly hydrolyzed to form SiO2 gel, and subsequently citric acid was coated on the surface of SiO2 gel. Ultimately, the synergistic effect of abundant active hydroxyl groups existed on SiO2 gel surface and citric acid, rendering the metal ions better and uniformly scattered on the SiO2 surface. As a contrast, LMO/SiO2 samples were also prepared by conventional citrate complexation impregnation and wetness impregnation methods, respectively. Catalytic performances were measured using the conversion of n-butylamine as a probe reaction for NVOCs abatement. Moreover, amounts of characterization techniques containing XRD, SEM, TEM, N2 sorption, NH3-TPD, H2-TPR, O2-TPD, XPS and in situ FTIR were employed to establish a constructive relationship between physicochemical properties and catalytic performances. Finally, a possible and reasonable catalytic reaction mechanism for n-butylamine oxidation was proposed.
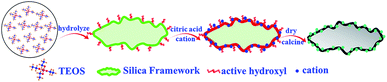 |
| | Scheme 1 Illustration of the preparation process of LMO/SiO2-SD catalyst. | |
2 Experimental
2.1 Catalyst preparation
Synthesis of SiO2 support.
In a typical preparation, the stoichiometric tetraethyl orthosilicate (TEOS), glacial acetic acid and ethanol were dissolved in distilled water. After homogeneous mixing, 9.0 g methanamide was added and stirring for 0.5 h, and then 70 g epoxypropane was added. The formed solution was heated at 60 °C with circulation reflux and aged overnight. The obtained sample was dried 100 °C and then calcined in a muffle furnace at 800 °C for 4 h.
Synthesis of LMO/SiO2-SD catalyst.
In a typical preparation, the stoichiometric tetraethyl orthosilicate (TEOS), glacial acetic acid and ethanol were dissolved in distilled water. After homogeneous mixing, 9.0 g methanamide was added and stirring for 0.5 h, and then 70 g epoxypropane was added. The formed solution was heated at 60 °C with circulation reflux and aged overnight. The mixture of stoichiometric citric acid and distilled water was gradually added to the above solution, and then stirred 2 h. Subsequently, the solution of nitrate containing La(NO3)3·6H2O and Mn(NO3)2 was added, and the formed mixture stirred for another 2 h to obtain viscous gel. The obtained viscous gel was dried at 100 °C and then calcined in a muffle furnace at 800 °C for 6 h.
Synthesis of LMO/SiO2-CI catalyst.
In a typical preparation, the stoichiometric La(NO3)3·6H2O, Mn(NO3)2 and citric acid were dissolved in distilled water. After homogeneous mixing, the support SiO2 were added and stirred for 6 h at 80 °C to form viscous gel. The obtained viscous gel was dried 100 °C and then calcined in a muffle furnace at 800 °C for 6 h.
Synthesis of LMO/SiO2-WI catalyst.
In a typical preparation, the stoichiometric La(NO3)3·6H2O and Mn(NO3)2 used as precursors were dissolved in distilled water. After homogeneous mixing, the support SiO2 were added and stayed for 12 h. The obtained precipitate dried at 100 °C and then calcined in a muffle furnace at 800 °C for 6 h.
2.2 Catalyst characterization
X-ray diffraction (XRD) patterns were recorded on a SmartLab X-ray Diffractometer (Rigaku Corporation, Japan) equipped with Cu Kα radiation (λ = 0.1541 nm) within 2θ range of 10–80° at the scanning step of 0.02°. Scanning electron microscopy (SEM) images were analyzed with a Hitachi S-3000 N scanning electron microscope. The high resolution TEM images (HRTEM) were recorded using a JEOL JEM-2100 electron microscope. The nitrogen adsorption and desorption isotherms were carried out on BEL SORP II apparatus. Before adsorption, all LMO/SiO2 samples were outgassed in vacuum at 200 °C. The surface area was determined by the BET theory and the pore size was calculated by the BJH method. Temperature-programmed desorption of NH3 (NH3-TPD) was investigated by AutoChemII 2920 apparatus. Prior to each test, the catalyst (50 mg, 40–60 meshes) was pretreated under high purified He at 200 °C for 1 h, then cooled down to 50 °C, and turned the flow of 10 vol% NH3/He into the system with a flow rate of 30 mL min−1 for 1 h. After that, the catalyst was flushed with He to remove physically adsorbed NH3 on the catalyst surface. Finally, the system was heated to 500 °C at a heating rate of 10 °C min−1, and the desorption profiles were recorded under He flow. Temperature-programmed reduction of H2 (H2-TPR) was carried on the same instrument. Firstly, the catalysts were pretreated under Ar flow at 200 °C for 1 h. Then the temperature was cooled to 50 °C, the catalyst was heated from 50 °C to 900 °C under 10 vol% H2/Ar flow. The consumption of H2 was detected by thermal conductivity detector (TSD). Temperature programmed desorption of O2 (O2-TPD) experiment was also determined by AutoChemII 2920 analyzer. After pretreated 50 mg of catalyst at 200 °C for 1 h under He flow, the sample was exposed to 4 vol% O2/He at 50 °C for 1 h. After the catalyst was purged with helium until stabilization of the instrument baseline, the sample was heated (10 °C min−1) to 800 °C. The amount of desorbed ammonia was measured using a thermal conductivity detector (TSD). The X-ray photoelectron spectroscopy (XPS) spectra were performed using a PHI 5000 Versa Probe high performance electron spectrometer. All binding energies (BE) were calibrated using C 1s (BE = 284.8 eV) as a standard.
2.3
In situ FTIR studies
In situ FTIR spectra were recorded by IS50 infrared spectrometer equipped with an in situ diffuse reflectance pool containing CaF2 window and MCT detector. The real reaction temperature was controlled by the programmed temperature controller precisely. All spectra were recorded by accumulating 32 scans at a spectra resolution of 4 cm−1. Prior to data collection, the catalyst would be pretreated with 20 vol% O2/He at 300 °C for 1 h in order to remove the contaminants and then cooled down to 50 °C. In this process, spectra of the fresh catalyst surface were collected per 50 °C and employed as the background. Reaction was carried out by exposing catalyst to a stream of flow composed of 1000 ppm n-butylamine, 20% O2 and He as balance gas. In situ FTIR spectra were collected from 50 to 300 °C for every 50 °C where a steady state was reached.
2.4 Catalytic evaluation
The measurements of catalytic activities were carried out in a fixed bed quartz reactor at atmospheric pressure for the complete oxidation of n-butylamine. Quantitative catalyst (1.0 g, 16–40 mesh) was diluted with 1.0 g of quartz sands and positioned at the middle of the fixed bed quartz reactor. For the measurements of catalytic performances, the total gas flow was kept at 250 mL min−1, and concentrations of the feed components were controlled as follows: 1000 ppm n-butylamine + 20% O2 + He (balance). The reactants and products were analyzed by VARIO industrial flue gas analyzer (MRU Corporation, Germany) and an on-line GC equipped with FID detector. The n-butylamine conversion (xNVOCs) and the selectivity of N2 (SN2) were defined, respectively, as:
| xNVOCs = (cin − cout)/cin × 100%; |
where, cin and cout represent the n-butylamine concentration of inlet and outlet, respectively; YN2 represent the yield of N2.
3 Results and discussion
3.1 Catalyst characterization
XRD results.
The crystal structure of LMO/SiO2 samples were examined by XRD, and the corresponding patterns with 2θ range of 10–80° were displayed in Fig. 1. For all LMO/SiO2 samples, the diffraction peaks at around 2θ = 23°, 33°, 40°, 46°, 53°, 58°, 69° and 78° can be observed, indicating the existence of rhombohedral perovskite-type crystalline phase (PDF 50-0299). Furthermore, one can see that both LMO/SiO2-SD and LMO/SiO2-CI samples exhibited well single perovskite structure, and no reflections belonging to other impurities such as La2O3 and MnOx crystalline phase were observed, which revealed that citric acid had the better metal ion complexing action. Differently, for LMO/SiO2-WI sample, the coexistence of La2O3 phase (PDF 74-2430) and LaMnO3 perovskite phase (PDF 50-0299) was demonstrated from the corresponding XRD patterns. La2O3 impurity in LMO/SiO2-WI sample was attributed to the surface enrichment of La species without well fabricating LaMnO3 with Mn species during the calcination process.12
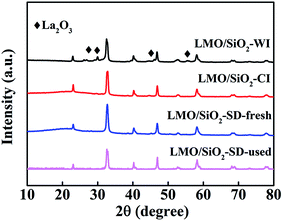 |
| | Fig. 1 XRD patterns of LMO/SiO2 catalysts. | |
SEM and TEM results.
The spatial morphological structure of three LMO/SiO2 samples were further examined by SEM. As shown in Fig. 2, both LMO/SiO2-SD and LMO/SiO2-CI samples featured a rough stacked pore structure with quantities of nanoparticles. Noticeably, the nanoparticles and channels generated of LMO/SiO2-SD were uniform and well-distributed, which might be ascribed to the spontaneously uniform deposition arose from the interaction between abundant active hydroxyl groups existed on SiO2 gel surface and La, Mn metal ions, forming LaMnO3 nanoparticles homogeneously embedded in the framework of SiO2 aerogel. For LMO/SiO2-CI sample, majority of nanoparticles had gathered, which was consistent with its larger crystallite size. In contrast, LMO/SiO2-WI sample exhibited a smooth sheet surface structure with only trace amount of nanoparticles.
 |
| | Fig. 2 SEM and TEM images of LMO/SiO2-WI (a and d), LMO/SiO2-CI (b and e) and LMO/SiO2-SD (c and f). | |
TEM characterization was conducted out to study the structure for all LMO/SiO2 samples. In Fig. 2(d–f), LaMnO3 perovskite was deposited onto the SiO2 support as larger nanoparticles of 15–30 nm diameters, which was similar microtopography to SEM results. The observed 0.387 nm, 0.221 nm and 0.234 nm lattice fringe were ascribed to the (012), (006) and (113) crystallographic planes of LaMnO3 (PDF 50-0299), respectively. This result was in good accordance with the XRD results of LMO/SiO2 samples. It could be explicitly from all samples that the layered sheet-like structures assigned to LaMnO3 was decorated by the black spots. When the LMO/SiO2 sample was prepared by citric acid-assisted deposition method, the black spots covered the surface of SiO2 homogeneously. On the contrary, the black spots existed on the LMO/SiO2-WI and LMO/SiO2-CI samples obviously exhibited the pulverous aggregation and accumulation.
N2 adsorption–desorption results.
The N2 adsorption–desorption isotherms and pore-size distributions of LMO/SiO2 samples were plotted in Fig. S1.† It could be obviously seen that the isotherms of all samples (Fig. S1(a)†) exhibited the typical IV type with an apparent type H2 hysteresis loop in the relative pressure (p/p0) range of 0.4–0.9 as defined by IUPAC, implying the existence of mesopore structure in LMO/SiO2 samples, which could be substantiated by Fig. S1(b).† The agglomeration or compaction of the nanoparticle building blocks led to the formation of mesopore structure of these samples. The pore size distribution curves (Fig. S1(b)†) suggested that the pore diameter of LMO/SiO2 samples were distributed predominantly in the range of 4–5 nm, which matched well with the isotherms. The surface area of LMO/SiO2 samples was calculated by BET equation, and the pore volume and pore diameter LMO/SiO2 samples were obtained by BJH model (Table 1). As seen clearly in Table 1, the LMO/SiO2-WI sample exhibited the largest surface area (300 m2 g−1), pore volume (0.39 cm3 g−1) and pore diameter (5.03 nm), LMO/SiO2-SD sample followed and corresponding values were (253 m2 g−1, 0.36 cm3 g−1 and 4.31 nm), and the LMO/SiO2-CI sample exhibited the lowest surface area (205 m2 g−1), pore volume (0.31 cm3 g−1) and pore diameter (4.02 nm). Combining with the results of XRD, SEM and TEM, one can find that the LMO/SiO2-SD sample might expose more active sites, which might result from the synergistic effect of the citric acid-assisted deposition method.
Table 1 Physico-chemical properties of LMO/SiO2 catalysts
| Catalysts |
SSAa (m2 g−1) |
V
p
b (cm3 g−1) |
D
p
b (nm) |
Acid amount (mmol gcat−1) |
H2 consumption (mmol gcat−1) |
O2 desorption (mmol gcat−1) |
| A |
B |
1st peak |
2nd peak |
3rd peak |
Oα |
Oβ |
|
SSA: calculated by BET method.
Vp and Dp: obtained by BJH method.
|
| LMO/SiO2-WI |
300 |
0.39 |
5.03 |
0.038 |
0.019 |
0.202 |
0.052 |
0.120 |
0.228 |
0.121 |
| LMO/SiO2-CI |
205 |
0.31 |
4.02 |
0.039 |
0.022 |
0.347 |
0.249 |
— |
0.096 |
0.125 |
| LMO/SiO2-SD |
253 |
0.36 |
4.31 |
0.042 |
0.027 |
0.254 |
0.196 |
0.259 |
0.122 |
0.137 |
NH3-TPD results.
The acidity of LMO/SiO2 samples was investigated by NH3-TPD technique. As is well known, the acidity of samples is another essential factor to affect the catalytic performance for n-butylamine oxidation,20,21 the adsorption and activation of n-butylamine on the sample surface was a vital step in the reaction, meanwhile the amount of surface acid sites and acid strength would alter its adsorption. As illustrated in Fig. 3, all LMO/SiO2 samples mainly presented two broad NH3 desorption peaks ranged from 200 to 500 °C, assigned to the weak-strength acid sites and the medium-strength acid sites, respectively.22 Strikingly, the LMO/SiO2-SD sample displayed higher peak-temperature of desorption peaks, about 280 and 370 °C, indicating its relatively stronger acidity. The corresponding acid amounts of LMO/SiO2 samples were listed in Table 1. It could be easily seen that LMO/SiO2-SD sample exhibited the most amount of weak acid sites (0.042 mmol gcat−1) and medium-strength acid sites (0.027 mmol gcat−1), LMO/SiO2-CI sample followed and corresponding values were 0.039 mmol gcat−1 and 0.022 mmol gcat−1, and the LMO/SiO2-WI sample exhibited the lowest amount of weak acid sites (0.038 mmol gcat−1) and medium-strength acid sites (0.019 mmol gcat−1). Combined with the analyses of previous characterization results, the presence of more acidic sites on the surface could be attributed to the homogeneous and well-distributed nanoscale pore structure of LMO/SiO2-SD sample.
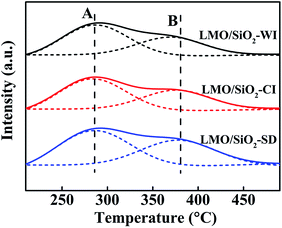 |
| | Fig. 3 NH3-TPD curves of LMO/SiO2 catalysts. | |
Redox properties.
The redox properties of all LMO/SiO2 samples were characterized by H2-TPR experiments (Fig. 4). Since La3+ is unreducible under the H2-TPR conditions adopted in the present study, the observed reduction peaks could be assigned to the reduction of Mnn+ species in LMO/SiO2 samples.23 For all LMO/SiO2 samples, two main reduction peaks were observed, the first one within temperature range of 250–600 °C, which could be attributed to the reduction of Mn4+ into Mn3+,24,25 and the latter one above 600 °C were due to the reduction of Mn3+ into Mn2+,26 whereas the reduction of Mn2+ into Mn0 hardly occurred in the reduction process of MnOx.27 It could be easily seen that the reduction peak temperature of LMO/SiO2-SD sample was significantly lower than LMO/SiO2-CI and LMO/SiO2-WI samples, indicating that LMO/SiO2-SD sample exhibited an excellent reducibility behaviour among all LMO/SiO2 samples. Meanwhile the H2 consumption related to each reduction region calculated through quantitative integration of the corresponding H2-TPR peaks were listed in Table 1. The H2 consumption of LMO/SiO2-SD sample was the highest (0.709 mmol gcat−1), LMO/SiO2-CI sample followed (0.596 mmol gcat−1), and LMO/SiO2-WI sample exhibited the lowest H2 consumption (0.374 mmol gcat−1). It was known from the H2-TPR experiments that the LMO/SiO2-SD sample revealed the conspicuous low temperature redox capability compared to other LMO/SiO2-CI and LMO/SiO2-WI samples, which could be related to the better surface oxygen mobility of LMO/SiO2-SD sample.28
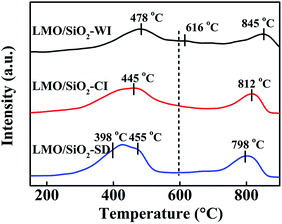 |
| | Fig. 4 H2-TPR curves of LMO/SiO2 catalysts. | |
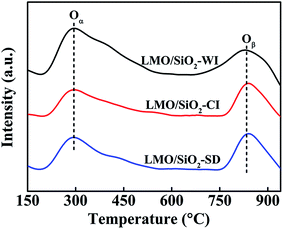
O2-TPD measurements were performed in order to investigate the surface and bulk oxygen species as well as their mobility of the perovskite oxides and corresponding oxygen desorption curves of LMO/SiO2 samples were depicted in Fig. 5. Apparently, all LMO/SiO2 samples showed two main oxygen desorption regions. The first region (Oα) ranging from 200 to 550 °C could be attributed to the surface adsorbed molecular oxygen and/or oxygen species migrating via surface oxygen vacancies, which mainly related to the physical and chemical properties of B-site metal.20,29,30 Another region ranging from 750–900 °C could be assigned to the desorption of lattice oxygen, denoted as Oβ,31–33 which mainly related to the ability of B-site metal ions to be reduced. Strikingly, the LMO/SiO2-SD sample exhibited the lowest peak-temperature of desorption peaks, indicating its better oxygen mobility. The corresponding oxygen desorption amounts were further analyzed by quantitative integration of O2-TPD curve (Table 1). The desorption amounts of LMO/SiO2-SD sample were 0.122 and 0.137 mmol gcat−1, respectively, which were remarkably higher than that of LMO/SiO2-CI samples (0.096 and 0.125 mmol gcat−1).
 |
| | Fig. 5 O2-TPD curves of LMO/SiO2 catalysts. | |
XPS results.
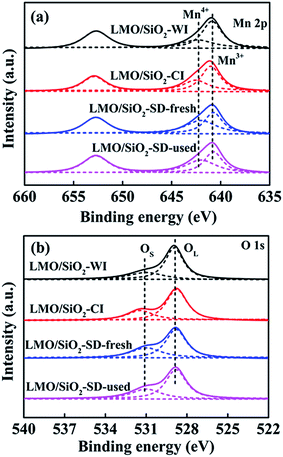
All LMO/SiO2 samples were subjected to XPS characterization analysis for investigating the atomic concentrations and chemical states of major elements (Fig. 6). Two main asymmetrical peaks assigned to Mn 2p3/2 and Mn 2p1/2 of LMO/SiO2 samples were observed, ranging from 640.8 eV to 652.7 eV. Relevant references reported that Mn4+ and Mn3+ species coexisted in LaMnO3 perovskite-type samples.27,34–36 After a peak linear fitting deconvolution, the Mn 2p3/2 XPS spectra could be decomposed into tow peaks, appeared at binding energies of 642.4 ± 0.2 eV and 640.8 ± 0.2 eV assignable to Mn4+ peak and Mn3+ peak, respectively.37,38 The relative percentages of manganese species were calculated by quantitative analysis of the corresponding characteristic peaks and ultimate results were listed in Table S1.† A significant distinction existed in Mn4+/(Mn4+ + Mn3+) ratio between the different LMO/SiO2 samples and followed the sequence LMO/SiO2-SD (41.64%) > LMO/SiO2-CI (34.54%) > LMO/SiO2-WI (29.47%). Based on which, the citric acid-assisted deposition method is conducive to obtaining a sample containing abundant surface concentration of Mn4+ species.
 |
| | Fig. 6 (a) Mn 2p and (b) O 1s XPS spectra of LMO/SiO2 catalysts. | |
As previously reported,41,42 the surface chemisorbed oxygen (OS) species were more active than the lattice oxygen (OL) species due to their higher mobility, therefore OS species played a more important role in catalytic oxidation reaction. It can be seen from Fig. 6(b), the O 1s peak could be split into two peaks: surface chemisorbed oxygen (531.2 ± 0.2 eV, denoted as OS) and lattice oxygen (529.0 ± 0.2 eV, denoted as OL).39,40 The OS/(OS + OL) ratios of all LMO/SiO2 samples were calculated by quantitative analysis of the corresponding characteristic peaks of OS and OL, the OS/(OS + OL) ratio of LMO/SiO2-SD sample calculated from deconvoluted O 1s XPS spectrum was 38.72%, which was larger than that of LMO/SiO2-CI (32.16%) and LMO/SiO2-WI (27.74%) samples. Further investigating the H2-TPR, O2-TPD and XPS results, higher ratio of surface Mn4+ species and luxuriant OS species of LMO/SiO2-SD sample could be attributed to the synergic effect of the citric acid-assisted deposition method, which can contribute to the conspicuous low temperature redox capability and better surface oxygen mobility.
3.2 Catalytic evaluation
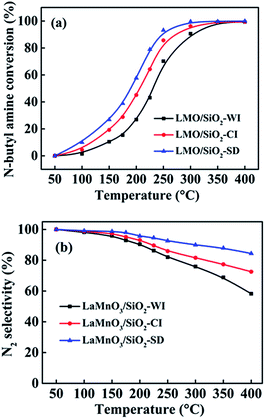
The catalytic performances of all LMO/SiO2 samples for n-butylamine oxidation were tested within the temperature range from 50 to 400 °C (Fig. 7). Apparently, the conversion of n-butylamine showed a monotonic increasing tendency with the increase of reaction temperature, and a complete conversion of n-butylamine could be ultimately achieved below 400 °C over all LMO/SiO2 samples. The sequence in terms of catalytic performance over LMO/SiO2 samples followed by LMO/SiO2-SD > LMO/SiO2-CI > LMO/SiO2-WI, revealing that SD method can be capable of improving the catalytic performance for the complete oxidation of n-butylamine. T10, T50 and T90 (representing the temperature at 10%, 50% and 90% n-butylamine conversion) were respectively listed in Table 2. Among all LMO/SiO2 samples, LMO/SiO2-SD sample displayed the optimum catalytic oxidation performance for n-butylamine with the T10, T50 and T90 values of 95, 188 and 246 °C, respectively, which were lower than that of LMO/SiO2-CI (116, 206 and 273 °C) and LMO/SiO2-WI (146, 230 and 305 °C) samples.
 |
| | Fig. 7 Catalytic oxidation performances of n-butylamine over LMO/SiO2 catalysts: (a) n-butylamine conversion (b) N2 selectivity. | |
Table 2 Catalytic activities and Ea of LMO/SiO2 catalysts
| Catalysts |
Catalytic activities |
R
a/(mmol m−2 s−1) |
E
a (kJ mol−1) |
|
T
10 (°C) |
T
50 (°C) |
T
90 (°C) |
|
Calculated from the active test at 250 °C.
|
| LMO/SiO2-WI |
146 |
230 |
305 |
4.35 × 10−7 |
34.7 |
| LMO/SiO2-CI |
116 |
206 |
273 |
7.77 × 10−7 |
32.5 |
| LMO/SiO2-SD |
95 |
188 |
246 |
6.85 × 10−7 |
29.0 |
To deeply explore the n-butylamine catalytic oxidation reaction, a comprehensive identification of reaction products was carried out. At 400 °C or above, n-butylamine was entirely disintegrated into CO2, N2, NOx and so on. As shown in Fig. 7(b), the results of N2 selectivity over LMO/SiO2 samples were observed and LMO/SiO2-SD sample displayed the highest N2 selectivity. With the reaction temperature increasing, the N2 selectivity over all LMO/SiO2 samples showed an obvious decline, which could be ascribed to the more products of NOx at higher temperature. Moreover, to highlight the optimum catalytic performance of LMO/SiO2-SD sample for n-butylamine catalytic oxidation, their activities were compared based on specific activity (R). It was found that LMO/SiO2-SD sample possessed higher specific activity (6.85 × 10−7 mmol m−2 s−1), indicating that more active sites existed on the unit specific surface area of sample. Based on the catalytic performances and characterization results, one can find that LMO/SiO2-SD sample showed the optimum catalytic performance, which was assigned to its better dispersity, abundant surface acid sites, superior low-temperature reducibility, high ratio of surface Mn4+ species and more available surface adsorbed oxygen species. Among them, acid sites of LMO/SiO2-SD sample played an essential role in reaction. By which, n-butylamine could be adsorbed efficiently and gone through further oxidation.
The apparent activation energy (Ea) for n-butylamine oxidation over all LMO/SiO2 samples with a first-order Arrhenius equation: r = −kc = (−A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−Ea/RT))c, in which r, k, A, Ea represent the reaction rate (mol s−1), the rate constant (s−1), the pre-exponential factor and the apparent activation energy (kJ mol−1), respectively.36–38 The Arrhenius plots for n-butylamine catalytic oxidation over all the LMO/SiO2 samples showed well-line relationship (Fig. S2†). In addition, the Ea value values were calculated and summarized in Table 2. As listed in Table 2, the sequence of Ea values followed by LMO/SiO2-SD (29 kJ mol−1) < LMO/SiO2-CI (32.5 kJ mol−1) < LMO/SiO2-WI (34.7 kJ mol−1), suggesting that LMO/SiO2-SD sample was beneficial to the proceeding of n-butylamine catalytic oxidation, which was in good accordance with its excellent catalytic performance.
exp(−Ea/RT))c, in which r, k, A, Ea represent the reaction rate (mol s−1), the rate constant (s−1), the pre-exponential factor and the apparent activation energy (kJ mol−1), respectively.36–38 The Arrhenius plots for n-butylamine catalytic oxidation over all the LMO/SiO2 samples showed well-line relationship (Fig. S2†). In addition, the Ea value values were calculated and summarized in Table 2. As listed in Table 2, the sequence of Ea values followed by LMO/SiO2-SD (29 kJ mol−1) < LMO/SiO2-CI (32.5 kJ mol−1) < LMO/SiO2-WI (34.7 kJ mol−1), suggesting that LMO/SiO2-SD sample was beneficial to the proceeding of n-butylamine catalytic oxidation, which was in good accordance with its excellent catalytic performance.
In the literature reported previously,5,6,21 the temperature of n-butylamine complete conversion over the reported catalysts was about 280 °C. Apparently, the conversion of n-butylamine over the LMO/SiO2-SD catalyst showed a monotonic increasing tendency with the increase of reaction temperature, and the temperature of n-butylamine complete conversion was similar to other reported catalysts. Moreover, the conversion of LMO/SiO2-SD and reported catalysts showed no noticeable fluctuation with the increase of reaction temperature, which indicated that the LMO/SiO2-SD catalyst in this study had an advantage in catalytic oxidation of n-butylamine.
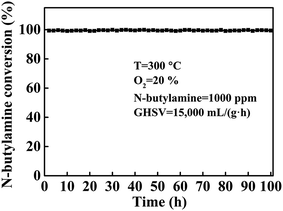
To assess catalyst fatigue, the stability of LMO/SiO2-SD has been studied. A durability test for the oxidation of n-butylamine over catalyst LMO/SiO2-SD was carried out for 100 h at the reaction temperature of 300 °C, and the curves of n-butylamine conversion versus the time on stream was shown in Fig. 8. Obviously, complete conversion of n-butylamine was always observed during the entire reaction time and no deactivation was observed, indicating that the LMO/SiO2-SD sample showed the stable catalytic performance of n-butylamine. To investigate the fate of the catalyst after reaction, the XRD and XPS analyses of the LMO/SiO2-SD catalyst after stability test were further carried out. From Fig. 1, the XRD analysis revealed that no phase transformations were observed, indicating its superior structural stability. Meanwhile, the XPS result (Table S1†) showed that the used LMO/SiO2-SD catalyst displayed the similar atomic concentrations to the fresh catalyst, and the chemical states of major elements only showed a slightly drop than that of the fresh catalyst, which was in good accordance with the XRD result. All of the above characterization results demonstrated that the LMO/SiO2-SD catalyst possessed the excellent stability.
 |
| | Fig. 8 Catalytic stability of LMO/SiO2-SD catalyst. | |
3.3
In situ FTIR
To further investigate reaction mechanism of n-butylamine catalytic oxidation over all LMO/SiO2 samples, we employed in situ FITR under 1000 ppm n-butylamine, 20% O2 and He conditions to analysis intermediate products at different temperature intervals and established a constructive relationship between samples' physicochemical properties and catalytic performances (Fig. 9). As shown in Fig. 9, when the reaction temperature was 50 °C, the induced n-butylamine adsorbed on the surface of LMO/SiO2-SD sample, and occurred four adsorption peaks. Four intense bands belong to n-butylamine adsorbed over the LMO/SiO2-SD catalyst at 1286 cm−1 (vC–N), 1462 cm−1 (δCH2), 1545 cm−1 (δCH3) and 1659 cm−1 (δN–H) were observed.43–47 However, these adsorption peaks would gradually weakened and vanished during the temperature-programmed surface reaction, and emerged three new intense bands located at 2347 cm−1, 1724 cm−1 and 1596 cm−1, which could be assigned to the formation of CO2, δ(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and NH3 adsorbed on at 2347 cm−1, 1724 cm−1 and 1596 cm−1, which could be assigned to the formation of CO2, δ(CO) and NH3 adsorbed on the sample surface, respectively.39,40 Meanwhile, the relative intensity of CO2 adsorption peak enhanced with the increase of reaction temperature, suggesting the increase of CO2 formation. However, the other two intense bands of δ(CO) and NH3 fade away with reaction temperature increasing, which could be ascribed to the generation of CO2, H2O, N2 and so on during the reaction process. When the reaction temperature reached to 300 °C, novel intense bands at 1557 cm−1 and 1334 cm−1 respectively ascribed to bidentate nitrate and cis-N2O22− species were observed, since more formed NOx absorbed on the surface of sample with reaction temperature rising.48,49 The in situ FTIR spectra of other LMO/SiO2-CI and LMO/SiO2-WI samples were similar to that of LMO/SiO2-SD sample (Fig. S3†), merely the adsorption peaks appeared slightly shift.
O) and NH3 adsorbed on at 2347 cm−1, 1724 cm−1 and 1596 cm−1, which could be assigned to the formation of CO2, δ(CO) and NH3 adsorbed on the sample surface, respectively.39,40 Meanwhile, the relative intensity of CO2 adsorption peak enhanced with the increase of reaction temperature, suggesting the increase of CO2 formation. However, the other two intense bands of δ(CO) and NH3 fade away with reaction temperature increasing, which could be ascribed to the generation of CO2, H2O, N2 and so on during the reaction process. When the reaction temperature reached to 300 °C, novel intense bands at 1557 cm−1 and 1334 cm−1 respectively ascribed to bidentate nitrate and cis-N2O22− species were observed, since more formed NOx absorbed on the surface of sample with reaction temperature rising.48,49 The in situ FTIR spectra of other LMO/SiO2-CI and LMO/SiO2-WI samples were similar to that of LMO/SiO2-SD sample (Fig. S3†), merely the adsorption peaks appeared slightly shift.
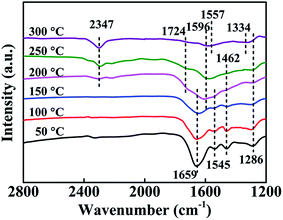 |
| | Fig. 9
In situ FTIR spectra of catalytic oxidation of n-butylamine over LMO/SiO2-SD catalyst. | |
Based on the results of in situ FTIR, the reaction pathway for catalytic oxidation of n-butylamine over LMO/SiO2 samples was proposed, as shown in Scheme 2. There was a condensation route over the all samples: the n-butylamine was firstly induced and adsorbed on the surface of sample, forming the adsorbed n-butylamine, which was consecutively split C–N bond and produced –NH2 and butyl. On the one hand, the –NH2 species over sample might be adsorbed and oxidized to generate NOx, which might be ascribed to the deep oxidation of NH3 in channels of samples.21,22 Subsequently, the formed NOx reacted with another –NH2 to produce non-toxic N2 and H2O. On the other hand, the –NH2 species might also form NH3 via hydrogenation reaction and subsequently generated non-toxic N2 and H2O by means of NH3–SCO. The butyl over the LMO/SiO2-SD sample might be oxidized by active oxidation species to generate intermediate species, and then were oxidized to produce CO2 and H2O terminally.
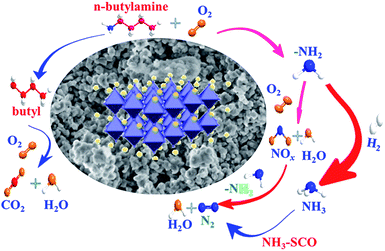 |
| | Scheme 2 Reaction mechanism for catalytic oxidation of n-butylamine over LMO/SiO2 catalysts. | |
4 Conclusions
In summary, citric acid-assisted deposition method utilized the synergistic effect of abundant active hydroxyl groups existed on SiO2 gel surface and citric acid to prepare the mesoporous SiO2-confined LaMnO3 perovskite nanoparticles with high dispersion. The LMO/SiO2-SD sample showed the optimum catalytic performance (T90 at 246 °C) and the highest N2 selectivity for n-butylamine oxidation compared with other LMO/SiO2-CI and LMO/SiO2-WI samples, which was mainly assigned to its abundant surface acid sites, superior low-temperature reducibility and high ratio of Mn4+ species. Moreover, acid sites of LMO/SiO2-SD sample played an essential role in reaction. By which, n-butylamine could be adsorbed efficiently and gone through further oxidation. In addition, the LMO/SiO2-SD sample possessed the lowest Ea value (29 kJ mol−1), proving its excellent catalytic activity. In addition, one can find that the LMO/SiO2-SD with high dispersion exhibited advantage in controlling NOx production. Based on analysis, one reason for this phenomenon is that the high dispersion can decrease the reaction temperature and then reduce the NOx production. It is predictable that highly dispersed perovskite nano-catalysts will draw more attentions and play a vital role in catalytic oxidation of NVOCs.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Key Research and Development Program (Grant No. 2017YFC0210903), the National Natural Science Foundation of China (Grant No. 21306089), the State Key Laboratory of Materials-Oriented Chemical Engineering (Grant ZK201610, ZK201703), and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
Notes and references
- Z. Jiang, C. He, N. F. Dummer, J. Shi, M. Tian, C. Ma, Z. Hao, S. H. Taylor, M. Ma and Z. Shen, Appl. Catal., B, 2018, 226, 220–233 CrossRef CAS.
- S. Cimino, S. Colonna, S. De Rossi, M. Faticanti, L. Lisi, I. Pettiti and P. Porta, J. Catal., 2002, 205, 309–317 CrossRef CAS.
- R. Spinicci, M. Faticanti, P. Marini, S. De Rossi and P. Porta, J. Mol. Catal. A: Chem., 2003, 197, 147–155 CrossRef CAS.
- Z.-J. Sui, L. Vradman, I. Reizner, M. V. Landau and M. Herskowitz, Catal. Commun., 2011, 12, 1437–1441 CrossRef CAS.
- M. Ma, H. Huang, C. Chen, Q. Zhu, L. Yue, R. Albilali and C. He, Mol. Catal., 2018, 455, 192–203 CrossRef CAS.
- Z. Shi, Q. Huang, P. Yang and R. Zhou, J. Porous Mater., 2015, 22, 739–747 CrossRef CAS.
- T. Nanba, S. Masukawa, J. Uchisawa and A. Obuchi, J. Catal., 2008, 259, 250–259 CrossRef CAS.
- L. Wang, C. Wang, H. Xie, W. Zhan, Y. Guo and Y. Guo, Catal. Today, 2018, 327, 190–195 CrossRef.
- X. Wang, J. Zuo, Y. Luo and L. Jiang, Appl. Surf. Sci., 2017, 396, 95–101 CrossRef CAS.
- Y. Chai, Y. Fu, H. Feng, W. Kong, C. Yuan, B. Pan, J. Zhang and Y. Sun, ChemCatChem, 2018, 10, 2078–2086 CrossRef CAS.
- J. Chen, M. Shen, X. Wang, J. Wang, Y. Su and Z. Zhao, Catal. Commun., 2013, 37, 105–108 CrossRef CAS.
- C. Zhang, Y. Guo, Y. Guo, G. Lu, A. Boreave, L. Retailleau, A. Baylet and A. Giroir-Fendler, Appl. Catal., B, 2014, 148, 490–498 CrossRef.
- Q. Ding, H. Xian, Y. Tan, N. Tsubakiet and X. Li, Catal. Sci. Technol., 2013, 3, 1493–1496 RSC.
- H. Arandiyan, H. Dai, J. Deng, Y. Wang, H. Sun, S. Xie, B. Bai, Y. Liu, K. Ji and J. Li, J. Phys. Chem. C, 2014, 118, 14913–14928 CrossRef CAS.
- X. Li, H. Dai, J. Deng, Y. Liu, Z. Zhao, Y. Wang, H. Yang and C. Au, Appl. Catal., A, 2013, 458, 11–20 CrossRef CAS.
- R. Lima, M. Batista, M. Wallau, E. Sanches and E. A. Urquieta-González, Appl. Catal., B, 2009, 90, 441–450 CrossRef.
- M. Alifanti, M. Florea and V. I. Parvulescu, Appl. Catal., B, 2007, 70, 400–405 CrossRef CAS.
- J. Zhang, X. Weng, Z. Wu, Y. Liu and H. Wang, Appl. Catal., B, 2012, 126, 231–238 CrossRef CAS.
- J. Deng, L. Zhang, H. Dai and C. Au, Appl. Catal., A, 2009, 352, 43–49 CrossRef CAS.
- C. Zhang, C. Wang, S. Gil, A. Boreave, L. Retailleau, Y. Guo, J. L. Valverde and A. Giroir-Fendler, Appl. Catal., B, 2017, 201, 552–560 CrossRef CAS.
- Q. Huang, S. Zuo and R. Zhou, Appl. Catal., B, 2010, 95, 327–334 CrossRef CAS.
- P. Li, X. Hu, L. Zhang, H. Dai and L. Zhang, Nanoscale, 2011, 3, 974–976 RSC.
- H. Arandiyan, H. Dai, J. Deng, Y. Wang, H. Sun, S. Xie, B. Bai, Y. Liu, K. Ji and J. Li, J. Phys. Chem. C, 2014, 118, 14913–14928 CrossRef CAS.
- Y. Lu, Q. Dai and X. Wang, Catal. Commun., 2014, 54, 114–117 CrossRef CAS.
- Y. Liu, H. Dai, Y. Du, J. Deng, L. Zhang and Z. Zhao, Appl. Catal., B, 2012, 119, 20–31 CrossRef.
- A. Kaddouri, S. Ifrah and P. Gelin, Catal. Lett., 2007, 119, 237–244 CrossRef CAS.
- C. Zhang, C. Wang, W. Zhan, Y. Guo, Y. Guo, G. Lu, A. Baylet and A. Giroir-Fendler, Appl. Catal., B, 2013, 129, 509–516 CrossRef CAS.
- J. Yang, L. Li, X. Yang, S. Song, J. Li, F. Jing and W. Chu, Catal. Today, 2018, 327, 19–27 CrossRef.
- J. Zhu, Z. Zhao, D. Xiao, J. Li, X. Yang and Y. Wu, J. Mol. Catal. A: Chem., 2005, 238, 35–40 CrossRef CAS.
- B. P. Barbero, J. A. Gamboa and L. E. Cadús, Appl. Catal., B, 2006, 65, 21–30 CrossRef CAS.
- H. Ziaei-Azad, A. Khodadadi, P. Esmaeilnejad-Ahranjani and Y. Mortazavi, Appl. Catal., B, 2011, 102, 62–70 CrossRef CAS.
- M. Pena and J. Fierro, Chem. Rev., 2001, 39, 1981–2017 CrossRef.
- J. Quiroz, J.-M. Giraudon, A. Gervasini, C. Dujardin, C. Lancelot, M. Trentesaux and J.-F. Lamonier, ACS Catal., 2015, 5, 2260–2269 CrossRef CAS.
- M. Alifanti, J. Kirchnerova and B. Delmon, Appl. Catal., A, 2003, 245, 231–244 CrossRef CAS.
- R. Hammami, S. B. Aïssa and H. Batis, Appl. Catal., A, 2009, 353, 145–153 CrossRef CAS.
- C. Zhang, C. Wang, W. Hua, Y. Guo, G. Lu, S. Gil and A. Giroir-Fendler, Appl. Catal., B, 2016, 186, 173–183 CrossRef CAS.
- B. Kucharczyk and W. Tylus, Catal. Lett., 2007, 115, 122–132 CrossRef CAS.
- S. Cimino, M. P. Casaletto, L. Lisi and G. Russo, Appl. Catal., A, 2007, 327, 238–246 CrossRef CAS.
- X. Yao, F. Gao, Q. Yu, L. Qi, C. Tang, L. Dong and Y. Chen, Catal. Sci. Technol., 2013, 3, 1355 RSC.
- J. Liu, X. Li, Q. Zhao, J. Ke, H. Xiao, X. Lv, S. Liu, M. Tadé and S. Wang, Appl. Catal., B, 2017, 200, 297–308 CrossRef CAS.
- T. Boningari, P. R. Ettireddy, A. Somogyvari, Y. Liu, A. Vorontsov, C. A. MSDonald and P. G. Smirniotis, J. Catal., 2015, 325, 145–155 CrossRef CAS.
- P. Sun, R.-t. Guo, S.-m. Liu, S.-x. Wang, W.-g. Pan and M.-y. Li, Appl. Catal., A, 2017, 531, 129–138 CrossRef CAS.
- S. Benard, A. Baylet, P. Vernoux, J. L. Valverde and A. Giroir-Fendler, Catal. Commun., 2013, 36, 63–66 CrossRef CAS.
- A. Baylet, C. Capdeillayre, L. Retailleau, J. L. Valverde, P. Vernoux and A. Giroir-Fendler, Appl. Catal., B, 2011, 102, 180–189 CrossRef CAS.
- B. de Rivas, R. López-Fonseca, C. Jiménez-González and J. I. Gutiérrez-Ortiz, Chem. Eng. J., 2012, 184, 184–192 CrossRef CAS.
- Z. Wu, B. Jiang, Y. Liu, H. Wang and R. Jin, Environ. Sci. Technol., 2007, 16, 5812 CrossRef.
- L. Chen, J. Li and M. Ge, Environ. Sci. Technol., 2010, 24, 9590–9596 CrossRef PubMed.
- F. Liu, H. He, Y. Ding and C. Zhang, Appl. Catal., B, 2009, 93, 194–204 CrossRef CAS.
- M. Machida, M. Uto, D. Kurogi and T. Kijima, J. Mater. Chem., 2001, 11, 900–904 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Complementary characterisation and catalytic data. See DOI: 10.1039/c8ra10636c |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Zuliang
Tao
,
Minghong
Wang
and
Xu
Qiao
*
*,
Zuliang
Tao
,
Minghong
Wang
and
Xu
Qiao
*










