
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSingle-molecule study on conformational dynamics of M.HhaI†
Shanshan Heab,
Chen Yangab,
Sijia Pengc,
Chunlai Chen c and
Xin Sheng Zhao*ab
c and
Xin Sheng Zhao*ab
aBeijing National Laboratory for Molecular Sciences, State Key Laboratory for Structural Chemistry of Unstable and Stable Species, Department of Chemical Biology, College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, China. E-mail: zhaoxs@pku.edu.cn
bBiomedical Pioneering Innovation Center (BIOPIC), Peking University, Beijing 100871, China
cSchool of Life Sciences, Tsinghua-Peking Joint Center for Life Sciences, Beijing Advanced Innovation Center for Structural Biology, Tsinghua University, Beijing 100084, China
First published on 13th May 2019
Abstract
We found that apo DNA methyltransferase M.HhaI under the physiological salt concentration does not possess the structure characterized by X-ray crystallography; instead, it interchanges between prefolded and unfolded states. Only after binding to the substrate, it transforms into a crystal-structure-like state. Flipping rates of its catalytic loop were directly measured.
DNA methylation is an essential epigenetic mark, which plays regulatory roles in cell development and disease.1 A thorough understanding of the DNA methylation mechanism is important to exploit epigenetics and also to guide drug design. Bacterial DNA methyltransferase M.HhaI is the first DNA methyltransferase whose structure was determined using X-ray crystallography.2 It recognizes and methylates the inner cytosine of GCGC target sites in duplex DNA in the presence of cofactor S-adenosylmethionine (AdoMet), which transforms into S-adenosylhomocysteine (AdoHcy) after the reaction. Structural studies can provide valuable clues to understand how the enzyme executes its catalytic function. By comparing the crystal structures of M.HhaI with and without DNA, an essential catalytic loop (residues 80–100) in M.HhaI is observed to flip from the open to closed state along with a 180° turn of the target base. These motions adjust the target base into a proper position for methylation and have been under intensive studies.3–6 The flipping motion of the catalytic loop is an essential part of the catalytic process, but its kinetic information was only inferred indirectly and its actual rates remain to be determined.4–6
Apart from the catalytic loop reorganization, M.HhaI may experience global rearrangement upon substrate binding. Molecular dynamics (MD) simulations showed the anti-correlated motions of residues separated by 3 nm, indicating domain compression during catalysis.7 NMR studies suggested that M.HhaI is a dynamic protein8 and that there are dramatic chemical shift perturbations reaching many distal sites upon DNA binding.9 Due to the lack of direct access to tertiary structures, they could not differentiate whether these distal changes result from structural adjustments from the crystal structure at the local level or concerted, global structural changes.
To answer all the above questions, in this study, we used single-molecule techniques, i.e., single-molecule fluorescence resonance energy transfer (smFRET) and fluorescence correlation spectroscopy (FCS) to study the conformational dynamics of M.HhaI. smFRET, well known as ‘spectroscopic ruler’, which has been successfully used to study many fundamental biological mechanisms10 and has long-term progress and applications in structural biology.11 FCS can detect the dynamics of transition under equilibrium condition.12 With time resolution as high as nanoseconds, it can detect a process that is too fast to be studied by ensemble methods. By applying smFRET and FCS techniques, we directly measured the flipping rates of the catalytic loop and revealed the global conformational reorganizations of M.HhaI. Surprisingly, we found that M.HhaI alone (apo M.HhaI) under the physiological salt concentration did not possess the structure characterized by X-ray crystallography. Instead, it interchanges between prefolded and unfolded states. Only after it binds to the substrate, M.HhaI turns into its crystal-structure-like state.
First, we probed the global conformational changes of M.HhaI upon binding DNA using smFRET. For the convenience of dye labelling, the Cys81 of M.HhaI was mutated to serine in global conformational studies. The other natural cysteines in M.HhaI were proven to be inert to the dye labelling. We doubly mutated selected sites on M.HhaI into cysteine and labelled them with donor (AF555) and acceptor (AF647) fluorophores in a random manner. We designed 13 mutants with FRET pairs distributed over all the domains of M.HhaI (Fig. S1†), which are termed by the number of labelled sites as X-X (e.g., 90-210 means dyes are labelled at Q90C and K210C sites). The circular dichroism (CD) spectra showed that mutations and dye labelling did not disturb the structure of M.HhaI (Fig. S2†). smFRET experiments were performed for the 13 mutants with M.HhaI in two conditions: (1) apo state, i.e., ∼40 pM M.HhaI in the T50 buffer (50 mM Tris–HCl, 50 mM NaCl, 1 mM EDTA, pH 7.5); and (2) DNA bound state, i.e., ∼40 pM M.HhaI with 2 μM SP_DNA (a specific DNA substrate) and 200 μM AdoMet in the T50 buffer.
The experiments were conducted on a home-built inversed confocal fluorescence microscope.13 The ratio of the detected photon number in the acceptor channel over that in the acceptor and donor channels together was measured and is termed as apparent FRET efficiency (Eapp). The Eapp distribution histograms of the mutants in two conditions are presented in Fig. 1A and S3.† The Eapp peak at around 0.12 was caused by M.HhaI molecules labelled with only a bright donor (no acceptor or acceptor having been bleached), and was ignored in the analysis. The peaks at higher Eapp were attributed to the effective FRET signal. All the mutants of M.HhaI in the apo state exhibited similar high Eapp peaks, suggesting that apo M.HhaI stays in a collapsed conformation. After binding with DNA, lower Eapp peaks appeared. To compare with the crystal structure, the true FRET efficiency (E) for mutants in the DNA bound state was calculated by rectifying the influence of crosstalk between the donor and acceptor channels, detection efficiency, and background photons in two channels. We plotted E of the DNA bound M.HhaI with the distance (d) provided by the crystal structure (Fig. 1B). The E–d curve showed clear anti-correlation and can be fitted well via a theoretical equation, suggesting that the DNA bound M.HhaI stays in a folded state characterized by the crystal structure. Urea denaturation experiments of 129-210 and 142-210 mutants supported these assignments. With the increase in the urea concentration, DNA bound M.HhaI showed the denaturation pattern of folded proteins, while apo M.HhaI showed the denaturation pattern of denatured proteins (Fig. S4†).
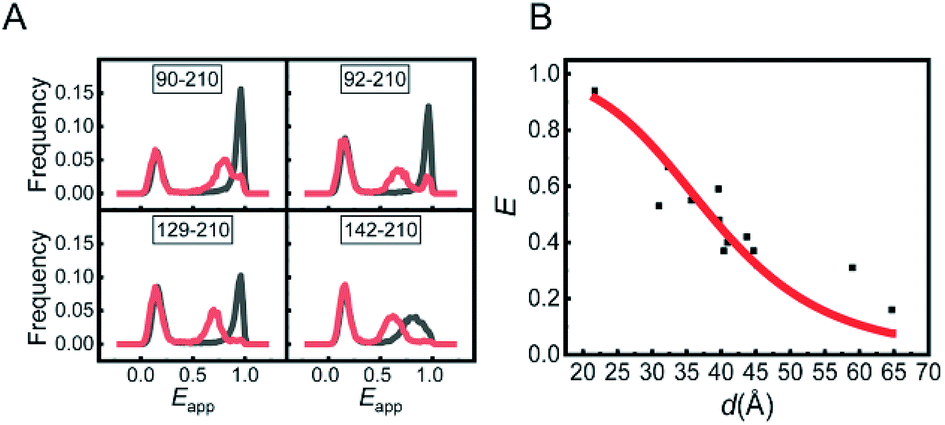 | ||
| Fig. 1 (A) FRET histograms of several representative mutants in apo M.HhaI (gray) and in DNA bound M.HhaI (red) in the T50 buffer. (B) Corrected FRET efficiency of all the mutants in DNA bound state vs. the distance measured from the crystal structure (5MHT). Red line is the fitting curve obtained using the theoretical equation: E = R06/(R06 + (d + l)6), where R0 = 51 Å is the Förster distance of the FRET pair of AF555 and AF647, and l = 12 Å is the equivalent length of linkers and dyes. | ||
This global conformational regulation upon DNA substrate binding was not expected from the crystal structure, nor has it been directly observed before. We also performed smFRET experiments on the binary complex of M.HhaI and AdoMet. AdoMet did not change the overall collapsed conformation of M.HhaI (Fig. S5†). We speculated that the different appearance of apo M.HhaI in our smFRET and crystallographic studies was due to different experimental conditions. A high salt concentration will make the folded state more stable. During crystallization, M.HhaI as well as the salts in buffer were highly concentrated. To confirm this, we conducted smFRET experiments at high salt concentrations using the T1000 buffer (50 mM Tris–HCl, 1000 mM NaCl, 1 mM EDTA, pH 7.5). Indeed, new and lower Eapp peaks appeared in apo M.HhaI (Fig. S6†). At such high salt concentrations, the affinity between M.HhaI and DNA was too weak to form a complex. To compare apo M.HhaI with DNA bound M.HhaI in the T1000 buffer, we connected the 142-210 mutant (without C81S mutation) with the DNA substrate by a covalent bond. The 142-210 mutant was doubly labelled after it reacted with F-DNA, which is a derivative of SP_DNA with the hydrogen on carbon 5 of target cytosine substituted by fluorine in the presence of AdoMet. Due to the substitution of fluorine, the covalent bond between the Cys81 of M.HhaI and the carbon 6 of target cytosine could not be cleaved after methyl transfer and was used for the crystallographic study of the M.HhaI and DNA complex.2,14 We termed the covalently bound complex of M.HhaI and DNA as 142-210_F-DNAMe. As a control, we observed that in both the covalently bound and non-covalently bound complexes, M.HhaI exhibited the same folded conformation in the T50 buffer (Fig. S7†). Then, in the T1000 buffer, we found the same Eapp peaks for the apo 142-210 mutant and the covalently bound complex of M.HhaI and DNA (both 142-210_F-DNAMe and 142-210_F-DNAMe·AdoHcy) (Fig. 2A), indicating that at a high salt concentration apo M.HhaI indeed adopts the structure as that determined by X-ray crystallography. The results on other mutants reached the same conclusion. Since our experimental condition of low salt is closer to the real physiological condition, we believe that our observation more realistically reflects the status of M.HhaI in vivo.
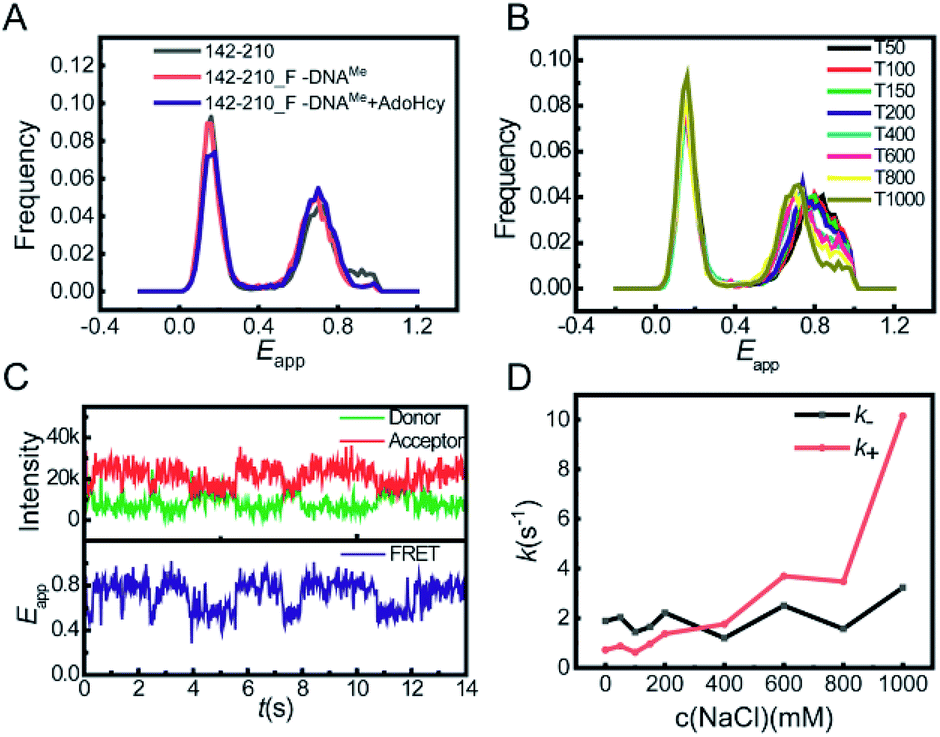 | ||
| Fig. 2 (A) FRET histograms of apo 142-210 (gray), 142-210_F-DNAMe (red) and 142-210_F-DNAMe with AdoHcy (blue) in the T1000 buffer. (B) FRET histograms of apo 142-210 at different concentrations of NaCl. Tn means that in the Tris buffer the concentration of NaCl is n mM. (C) Single-molecule fluorescence trace of apo 142-210 (upper panel) and corresponding Eapp trace (lower panel) in T50 buffer. (D) The calculated kinetic parameters k+ (from unfolded to prefolded, red) and k− (from prefolded to unfolded, black) for the transition between unfolded and prefolded states of apo M.HhaI at different salt concentrations. | ||
Careful scrutinization of the smFRET histogram revealed that apo M.HhaI in the T50 buffer has two FRET components. We assigned the species with lower Eapp as the prefolded state (the tertiary structure is partially maintained) because it behaves more or less like a folded protein but is much less stable than the M.HhaI–DNA complex under urea denaturation (Fig. S8†). The one with higher Eapp was assigned as the unfolded state (the tertiary structure is mostly destroyed) because it behaves as a typical denatured protein under urea denaturation (Fig. S4†). Due to partially maintaining the tertiary structure, the prefolded state is more extended than the unfolded state such that its Eapp is lower. As shown in Fig. 2B, with the increase in salt concentration, the proportion of the prefolded state increased and its Eapp peak shifted toward that of 142-210_F-DNAMe in the T1000 buffer (Fig. S9†), suggesting that the partially folded prefolded state of apo M.HhaI under low salt concentration converted into a fully folded state at high salt concentration because a high salt concentration stabilizes the folded crystal structure.
To capture the transition between the two states, we tracked the smFRET trace on the surface immobilized apo 142-210 mutant under a total internal reflection fluorescence microscope (TIRFM).15 Photon traces of donor and acceptor were recorded individually for each molecule (Fig. 2C, upper panel), from which Eapp–time trace was calculated (Fig. 2C, lower panel). We then quantified the transition rates between the unfolded and prefolded states of apo M.HhaI from T0 (50 mM Tris–HCl, 1 mM EDTA, pH 7.5) to T1000 buffers by statistical analysis on the fluorescence traces. Transition from the unfolded state to prefolded state was designated as the forward reaction. As the results show in Fig. 2D, with the increase in the salt concentration, k+ increases significantly while k− hardly changes. Overall, the global conformational transition of M.HhaI happens at the time scale of several hundred milliseconds. It is possible that M.HhaI prefers to bind DNA substrates in the prefolded state, from which it is easier to adjust into the folded conformation compared to starting from the unfolded state. The transition between the prefolded and unfolded states may serve as an additional regulation step for the substrate binding process.
Thus, by examining the global conformational dynamics of M.HhaI, we observed three conformational states of M.HhaI, i.e., the unfolded state and prefolded state for apo M.HhaI (predominant in the physiological salt concentration), and the folded state for M.HhaI, which exists in the complex with DNA and apo M.HhaI at a high salt concentration. The conformational transition of apo M.HhaI and the conformational reorganization upon DNA binding on the global scale may play a critical role in the substrate recognition process.
Next, we focused on the conformational changes of the catalytic loop during the catalytic process. First, we performed confocal-based smFRET experiments on the 90-210 mutant. In this study, we reserved Cys81 by blocking its accessibility with a DNA substrate during dye labelling (see the ESI† for details). To learn about the pre-catalytic steps, smFRET measurements were performed in the following four situations: (1) M.HhaI with SP_DNA, (2) M.HhaI with SP_DNA in the presence of AdoHcy, (3) M.HhaI with NS_DNA (nonspecific DNA substrate), and (4) M.HhaI with NS_DNA in the presence of AdoHcy. For the former two situations, the experiments were conducted in the T50 buffer. To improve the affinity between M.HhaI and NS_DNA, the experiments for the last two situations were conducted in the T0 buffer. The results showed that when the substrate is SP_DNA, the catalytic loop stays open (Eapp = 0.54, calculated d = 50 Å) in the binary complex of M.HhaI and SP_DNA. The loop then closes (Eapp = 0.84, calculated d = 37 Å) after forming the ternary complex of M.HhaI, SP_DNA and AdoHcy (Fig. 3A). When the substrate is NS_DNA, the loop always stays open whether there is AdoHcy or not (Fig. 3B). To the best of our knowledge, the conformations of catalytic loop in the first and last two situations have not been directly observed before. Our results directly showed that the catalytic loop stays open during the target searching process and only closes when both the specific substrate and cofactor exist. This observation is essential to fully elucidate the mechanism of catalytic DNA methylation.
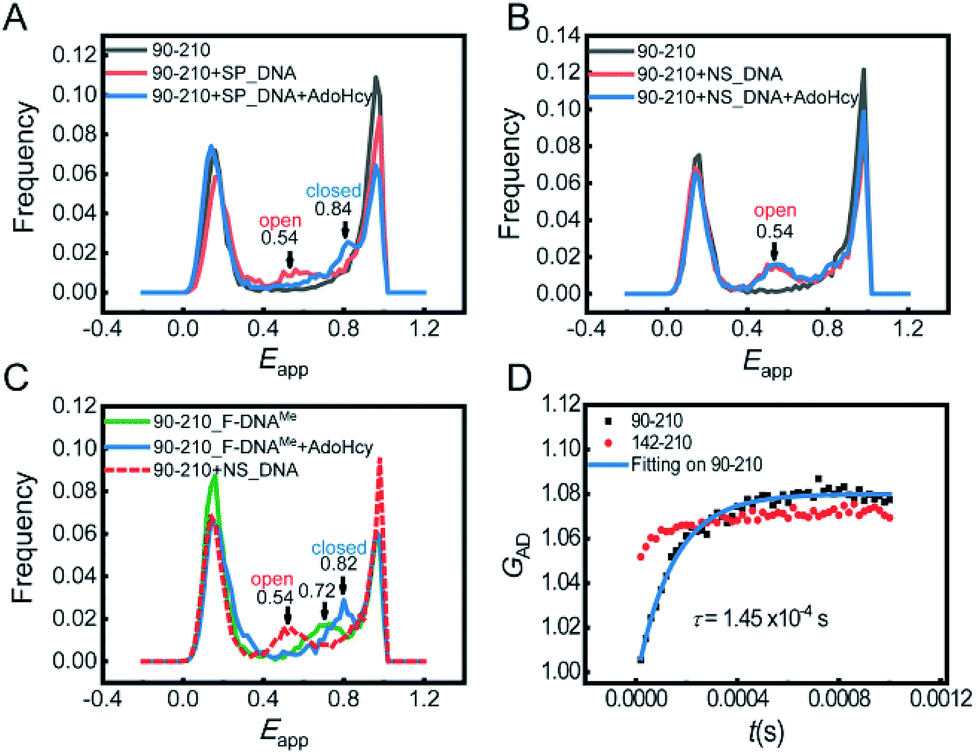 | ||
| Fig. 3 (A) FRET histograms of 90-210 (gray) that bind with specific DNA (labelled as SP_DNA) in the presence (blue) and absence of AdoHcy (red). (B) FRET histograms of 90-210 (gray), that bind with nonspecific DNA (labelled as NS_DNA) in the presence (blue) and absence of AdoHcy (red). (C) FRET histograms of binary complex of 90-210_F-DNAMe (green) and ternary complex of 90-210_F-DNAMe·AdoHcy (blue). 90-210 binding with nonspecific DNA is displayed (dashed line) as reference for the open state of loop. (D) The second order cross correlation curve GAD of 90-210 (black) in the T1000 buffer and corresponding fitting curve (blue line). The GAD of 142-210 is shown as control (red). | ||
To learn the post-catalytic steps, we detected the conformational changes of the loop during product release using F-DNA. The substitution of hydrogen for fluorine in the target cytosine pauses the reaction at the step right after methyl transfer, making the system with F-DNA a good model to study the post-catalytic step. smFRET measurements were conducted on the complex of 90-210_F-DNAMe with and without AdoHcy. In the ternary complex with AdoHcy, the catalytic loop stays in a fully closed state (Eapp = 0.82), while the loop stays in a half-open state (Eapp = 0.72) in the binary complex 90-210_F-DNAMe (without a cofactor) (Fig. 3C). To find out whether the single FRET peak represents a single half-open state or is due to the fast interchange between the open and closed states, we conducted the FRET-FCS experiment on a home-built scanning confocal microscope.16 No dynamic relaxation process was observed in 90-210_F-DNAMe (Fig. S10†), confirming that the FRET peak signals a single half-open state. This should be an important intermediate during the product release process that has not be recognized before. It suggests that immediately after the release of the product cofactor AdoHcy, the catalytic loop will spontaneously move out toward the open state and be ready to release the methylated base.
In previous kinetic studies, the rate of catalytic loop flipping was concealed by the base flipping such that the true loop flipping rates can only be estimated.5,6 Here, we were able to directly detect the dynamics of the catalytic loop by using the FRET-FCS technique with and without the substrate under equilibrium conditions. We measured the relaxation rate of the loop motion of apo M.HhaI in the T1000 buffer, where we had proven that apo M.HhaI stays in a folded state. Though the loop of apo M.HhaI showed a similar Eapp peak to that of 90-210_F-DNAMe in the T1000 buffer (Fig. S11†), they have very different dynamic behaviours. A clear anti-correlation was detected in the cross correlation curve (GAD) for apo 90-210, proving the existence of dynamic motion of the catalytic loop (Fig. 3D). As a control, the anti-correlation in the 142-210 mutant was much weaker, which may indicate that when the catalytic loop flips, other parts of M.HhaI make slight conformational adjustment as well. By including the recently developed third-order FRET-FCS calculation,17 we determined that kopen = (5.3 ± 0.3) × 103 s−1 and kclose = (1.6 ± 0.1) × 103 s−1. These values are faster than the rates estimated before5,6 but land in the region suggested by the NMR studies on global dynamic motions.8 The Eapp corresponding to the loop in closed and open states were calculated to be Eclosedapp = 0.77 ± 0.09 and Eopenapp = 0.44 ± 0.07. Because the smFRET experiment was performed on free molecules in solution, while the FRET-FCS experiment was performed on immobilized molecules on surface with different apparatuses, Eapp has a systematic deviation. After correction, the true FRET efficiencies are consistent with the assignment on the loop open and closed states in the smFRET measurement, suggesting that the catalytic loop in apo M.HhaI swings in a broad range. The intrinsically high dynamic behaviour of the loop enables it to react quickly to the target recognition and base flipping and to reorganize concertedly with these processes. In contrast, on binding with DNA, the motion of loop is restrained to a smaller scale and exhibited in the binary complex of M.HhaI and SP_DNA as well as in the ternary complex of M.HhaI, SP_DNA and AdoHcy, where weaker anti-correlation was observed (Fig. S12†). Their calculated Eapp from the second- and third-order FCS curves confirmed that the motion represents local fluctuation around the potential minima of the open and closed states, respectively.
In summary, we have used single-molecule fluorescence techniques, smFRET and FCS, to study the conformational dynamics of M.HhaI on two scales. On the global scale, we found that under the physiological salt concentration, apo M.HhaI does not possess the structure characterized by X-ray crystallography; instead, it interchanges between unfolded and prefolded states. It undergoes global conformational reorganizations to form a fully folded structure after the substrate recognition. On the local scale, we observed the conformational changes of catalytic loop during the catalytic process and found that the loop closes only when both the correct substrate and the cofactor are in correct positions. The rates of the loop flipping were directly quantified. These results provided new insights into the dynamic mechanisms of the catalytic DNA methylation. In addition, our results emphasized the fact that certain proteins in aqueous solutions may adopt conformations different from their crystal structures. Therefore, the crystal data should be cautiously used.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21233002 and 21521003).Notes and references
- C. Luo, P. Hajkova and J. R. Ecker, Science, 2018, 361, 1336–1340 CrossRef CAS PubMed.
- S. Klimasauskas, S. Kumar, R. J. Roberts and X. Cheng, Cell, 1994, 76, 357–369 CrossRef CAS PubMed.
- N. Huang, N. K. Banavali and A. D. Mackerell, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 68–73 CrossRef CAS PubMed.
- R. A. Estabrook and N. O. Reich, J. Biol. Chem., 2006, 281, 37205–37214 CrossRef CAS PubMed.
- D. M. Matje, D. F. Coughlin, B. A. Connolly, F. W. Dahlquist and N. O. Reich, Biochemistry, 2011, 50, 1465–1473 CrossRef CAS PubMed.
- R. Gerasimaitė, E. Merkienė and S. Klimašauskas, Nucleic Acids Res., 2011, 39, 3771–3780 CrossRef PubMed.
- R. A. Estabrook, J. Luo, M. M. Purdy, V. Sharma, P. Weakliem, T. C. Bruice and N. O. Reich, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 994–999 CrossRef CAS PubMed.
- H. Zhou, W. Shatz, M. M. Purdy, N. Fera, F. W. Dahlquist and N. O. Reich, Biochemistry, 2007, 46, 7261–7268 CrossRef CAS PubMed.
- H. Zhou, M. M. Purdy, F. W. Dahlquist and N. O. Reich, Biochemistry, 2009, 48, 7807–7816 CrossRef CAS PubMed.
- R. Roy, S. Hohng and T. Ha, Nat. Methods, 2008, 5, 507–516 CrossRef CAS PubMed.
- S. Kalinin, T. Peulen, S. Sindbert, P. J. Rothwell, S. Berger, T. Restle, R. S. Goody, H. Gohlke and C. A. Seidel, Nat. Methods, 2012, 9, 1218–1225 CrossRef CAS PubMed.
- O. Krichevsky and G. Bonnet, Rep. Prog. Phys., 2002, 65, 251–297 CrossRef CAS.
- G. Li, C. He, P. Bu, H. Bi, S. Pan, R. Sun and X. S. Zhao, ACS Chem. Biol., 2018, 13, 1082–1089 CrossRef CAS PubMed.
- D. V. Santi, C. E. Garrett and P. J. Barr, Cell, 1983, 33, 9–10 CrossRef CAS PubMed.
- S. Peng, R. Sun, W. Wang and C. Chen, Angew. Chem., Int. Ed., 2017, 56, 6882–6885 CrossRef CAS PubMed.
- H. Bi, Y. Yin, B. Pan, G. Li and X. S. Zhao, J. Phys. Chem. Lett., 2016, 7, 1865–1871 CrossRef CAS PubMed.
- L. Meng, S. He and X. S. Zhao, J. Phys. Chem. B, 2017, 121, 11262–11272 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra00021f |
| This journal is © The Royal Society of Chemistry 2019 |