
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePLGA/xylitol nanoparticles enhance antibiofilm activity via penetration into biofilm extracellular polymeric substances
Anam Anjuma,
Pooi-Yin Chungb and
Shiow-Fern Ng *a
*a
aCentre for Drug Delivery Research, Faculty of Pharmacy, University Kebangsaan Malaysia, 50300, Kuala Lumpur, Malaysia. E-mail: nsfern@gmail.com; Fax: +603 92897060; Tel: +603 92897977
bDepartment of Microbiology, International Medical University, 57000, Kuala Lumpur, Malaysia
First published on 7th May 2019
Abstract
Biofilms are gelatinous masses of microorganisms attached to wound surfaces. Previous studies suggest that biofilms generate resistance towards antibiotic treatments. It was reported that hydrogels containing xylitol and antibiotic combinations produced additive antibiofilm inhibition. However, hydrogel formulations lack specificity, due to which xylitol cannot penetrate into the biofilm matrix and gets easily degraded by bacterial beta lactamase enzymes. It was hypothesized that the incorporation of xylitol in PLGA (polylactic-co-glycolic acid) nanoparticles will enhance penetration into the EPS (extra polymeric substance) component of the biofilm matrix and potentially overcome the antibiotic resistance associated with the biofilms. The purpose of this study was to develop PLGA nanoparticles loaded with xylitol, which will enhance bacterial biofilm penetration. The nanoparticles were loaded with different amounts of xylitol (0.5–5% w/w) and characterized for physiochemical and drug release properties. The metabolic antibiofilm activity of the PLGA nanoparticles containing xylitol was demonstrated by an XTT assay using as references the cultures of Staphylococcus aureus (S. aureus) and Pseudomonas aeruginosa (P. aeruginosa) and the polymicrobial biofilms of both bacterial strains. Live/dead viability staining was also performed to investigate the viability ratio of bacterial cells present in the biofilms. The biofilm penetration study of the PLGA nanoparticles was assessed by combining the nanoparticles with conjugated concanavalin A (Con A)-fluorescein isothiocyanate (FITC) and by viewing using confocal laser scanning electron microscopy (CLSM). In conclusion, the PLGA nanoparticles loaded with xylitol were successfully developed and were found to promote the antibiofilm activity of xylitol in infected wounds.
1. Introduction
Biofilms can be defined as aggregations of microorganisms enclosed in self-produced EPS matrix. The study of chronic diabetic wound infections revealed the existence of polymicrobial bacterial communities on the wound surfaces.1 Bacterial biofilms interrupt the wound bed vascular supply and the host defense mechanism, resulting in wound chronicity, which becomes difficult to eradicate.2 Previous studies reported that biofilms showed resistance towards antimicrobials due to altered metabolism, substrate accessibility, permeability and EPS formation.3 For instance, Bjarnsholt and co-workers showed that P. aeruginosa biofilms produce a shelter effect, which ceases antibiotic activity, thus resulting in the inadequate eradication of wound infections.4 Antibiofilm strategies that are sought include debridement, eradication with chemical agents, and eradication by biological agents.5,42 Other approaches include inhibiting bacterial communication and signaling, also known as quorum sensing, and degeneration of EPS.5–7Xylitol is a five-carbon sugar alcohol found in various fruits and vegetables. Some studies on otitis media indicate that xylitol acts on polysaccharides present in the cell walls of Staphylococcus species and prevents biofilm adherence on the wound surface.8 Xylitol was also shown to inhibit the formation of glycocalyx in S. aureus bacteria.9 The inhibition of biofilm formation in P. aeruginosa was also demonstrated; it was also reported that xylitol shows more inhibition of the P. aeruginosa biofilm as compared to that of the S. aureus biofilm.10 Pathogenic bacteria excrete the sucrose metabolizing enzymes glycosyltransferase and fructosyltransferase that promote biofilm adhesion. Xylitol inhibits the formation of these enzymes by preventing the adhesion of bacteria on surfaces.11 Additionally, gentamicin in combination with xylitol was also found to be effective against the S. aureus and P. aeruginosa biofilms.12
In our previous study, two antibiofilm agents, namely, xylitol and EDTA along with the antibiotic gentamicin were loaded into an NaCMC hydrogel. It was found that the hydrogel produced additive antibiofilm inhibition but showed lack of specificity for the bacterial biofilms.13 Xylitol could not be effectively delivered into the biofilm matrix and thus, it could be degraded by bacterial beta lactamase enzymes, which resulted in less biofilm inhibition. PLGA-(polylactic-co-glycolic acid)-based nanoparticles as drug delivery carriers have shown potential activity owing to their sizes and related unique biodegradable properties.43 In a previous work, the sustained release of an antibiotic, ciprofloxacin, from PLGA nanoparticles was shown to be effective in the eradication of S. aureus biofilms.14 By loading an antibiofilm agent such as xylitol into the nanoparticles, one can expect improved delivery to the infected cells. Here, it was hypothesized that the incorporation of xylitol in PLGA nanoparticles will enhance the penetration into the biofilm EPS components within the biofilm matrix, thus potentially overcoming the biofilm-associated wound infection. Therefore, in this study, we aimed to develop PLGA nanoparticle formulations loaded with xylitol. The PLGA nanoparticles containing various xylitol amounts (0.5–5% w/w) were formulated and characterized in terms of particle size, zeta potential, polydispersity index, microscopy, entrapment efficiency, drug loading, infrared spectroscopy and drug release properties. Then, the metabolic antibiofilm testing of the PLGA nanoparticles was assessed using an XTT assay with the reference cultures of S. aureus and P. aeruginosa and polymicrobial biofilms of both species. Live/dead viability staining of PLGA nanoparticles was also performed to investigate the bacterial cell biofilm viability. Finally, the PLGA nanoparticle biofilm penetration studies were performed through combining with Con A-FITC, where Con A specifically binds to EPS in the biofilm matrix, and this was viewed under CLSM.
2. Materials and methods
2.1 Materials
PLGA (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50) (mol. wt 30000–60000 g mol−1), xylitol, polyvinyl alcohol (PVA), Pluronic F-125, PBS phosphate buffer saline (PBS), XTT salt, menadione and concanavalin A-FITC were purchased from Sigma-Aldrich Chemicals Co. Ltd. (St. Louis Missouri, USA). Acetonitrile, methanol and dichloromethane were purchased from J. T. Baker Avantor performance Materials, Inc (Malaysia). Tween 20 was purchased from Agilent Technologies, USA. Two biofilm-forming reference bacterial strains were used, i.e., Staphylococcus aureus (ATCC 6538) and Pseudomonas aeruginosa (ATCC 9027) and they were obtained from American Type Culture Collection (Erbetta, #23). A polymicrobial biofilm was formed by mixing equal ratios of both bacterial strains in the same culture. Mueller–Hinton agar (MHA) and Mueller–Hinton broth (MHB) were purchased from Becton, Dickinson and Company. Methanol, acetone and glacial acetic acid were purchased from R & M Chemicals Ltd (United Kingdom); ethanol was purchased from John Kollin Corporation (United Kingdom). A LIVE/DEAD™ BacLight™ Bacterial Viability Kit for microscopy and quantitative assays was purchased from Thermo-Fisher scientific, USA.
:50) (mol. wt 30000–60000 g mol−1), xylitol, polyvinyl alcohol (PVA), Pluronic F-125, PBS phosphate buffer saline (PBS), XTT salt, menadione and concanavalin A-FITC were purchased from Sigma-Aldrich Chemicals Co. Ltd. (St. Louis Missouri, USA). Acetonitrile, methanol and dichloromethane were purchased from J. T. Baker Avantor performance Materials, Inc (Malaysia). Tween 20 was purchased from Agilent Technologies, USA. Two biofilm-forming reference bacterial strains were used, i.e., Staphylococcus aureus (ATCC 6538) and Pseudomonas aeruginosa (ATCC 9027) and they were obtained from American Type Culture Collection (Erbetta, #23). A polymicrobial biofilm was formed by mixing equal ratios of both bacterial strains in the same culture. Mueller–Hinton agar (MHA) and Mueller–Hinton broth (MHB) were purchased from Becton, Dickinson and Company. Methanol, acetone and glacial acetic acid were purchased from R & M Chemicals Ltd (United Kingdom); ethanol was purchased from John Kollin Corporation (United Kingdom). A LIVE/DEAD™ BacLight™ Bacterial Viability Kit for microscopy and quantitative assays was purchased from Thermo-Fisher scientific, USA.
2.2 Formulation of PLGA/xylitol nanoparticles
The PLGA–xylitol nanoparticles were prepared by a solvent evaporation method using a sonicator and their composition is shown in Table 1. The PLGA nanoparticles were prepared using different xylitol concentrations (0.5%, 1%, 2%, 3%, 4% and 5% w/w) and the nanoparticles were named NpX1 to NpX6. Briefly, xylitol along with surfactant(s) was dissolved in distilled water, while PLGA was dissolved in acetone. The organic phase was added dropwise into the aqueous solution in a sonicator (Bransonic, USA), followed by evaporation by a rotary evaporator for 2 hours at 40 °C. The concentrated nanoparticles obtained were then placed in a −80 °C freezer for 18 h. The nanoparticles were then lyophilized using a freeze dryer (Scanvac coolsafe 110-4 system H031857, France) at −110 °C for 24 h.| Constituent | PLGA–xylitol nanoparticles | |||||
|---|---|---|---|---|---|---|
| NpX1 | NpX2 | NpX3 | NpX4 | NpX5 | NpX6 | |
| PLGA (%w/w) | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 |
| Xylitol (%w/w) | 0.5 | 1.0 | 2.0 | 3.0 | 4.0 | 5.0 |
| Pluronic F-125 (%w/w) | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 |
| Tween 20 (%w/w) | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 |
| PVA (%w/w) | — | — | — | — | — | — |
| Acetone (%w/w) | 3 | 3 | 3 | 3 | 3 | 3 |
| Distilled water q.s. | 100 | 100 | 100 | 100 | 100 | 100 |
2.3 Particle size, zeta potential and polydispersity index
Zetasizer ZS-90 (Malvern, UK) was used to measure the particle size, polydispersity index (PDI) and zeta potential of all nanoparticle dispersions. Twenty-five microliter (25 μL) of each nanoparticle dispersion was further diluted with distilled water. Particle size, polydispersity index (PDI) and zeta potential were measured using RI (refractive index) of 1.460 and viscosity (cP) of 0.8872 using distilled water as a dispersant at 25 °C. A disposable polystyrene quartz cuvette was filled with colloidal dispersion and placed inside the sample holder of the nano-sizer. The measurement was carried out in triplicates at 25 °C. The data was presented as mean ± standard deviation.2.4 Morphological analysis
Morphological analyses of nanoparticle dispersions were carried out using TEM (transmission electron microscopy) (FEI TECNAI G2 SPIRIT BIOTWIN, USA) at 120 kV. Freeze-dried nanoparticles were re-suspended in distilled water and sonicated for 10 min. A small amount of nanoparticle dispersion was placed on a glow discharged 400-mesh carbon-coated microscopic grid and stained using 0.1% phosphotungstic acid.2.5 Entrapment efficiency
Freeze-dried nanoparticles were dissolved in 25 mL distilled water and subjected to centrifugation at 9000 rpm for 30 min. Nanoparticles settled at the bottom of the centrifuge tube and free drug were quantified and analyzed by HPLC with a Shim-pack VP-ODS column (Shimadzu, Japan) using a deionized water and acetonitrile mixture (60:40) as eluent, with the column temperature set at 40 °C, flow rate of 1 mL min−1 and retention time of 5 min. The percentages of xylitol entrapped were calculated using eqn (1).
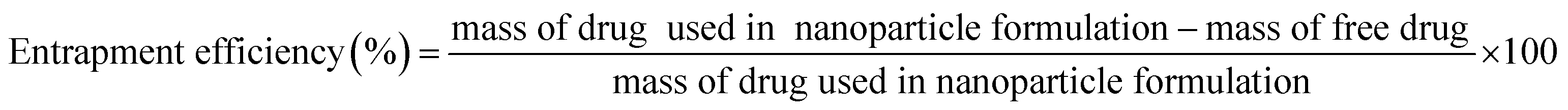 | (1) |
2.6 Fourier transform infrared spectroscopy (FTIR) analysis
Freeze-dried nanoparticles were analysed by FTIR using a PerkinElmer Spectrum 100 FT-IR spectrophotometer with PerkinElmer universal ATR (Attenuated Total Reflection) sampling assembly. Small amounts of freeze-dried nanoparticles were placed on an ATR crystal, and the maximum force was applied by using an ATR pressure clamp to allow for optimum contact with the samples. The FTIR spectra of different drug-loaded nanoparticles were recorded with a uniform resolution of 2 cm−1. The range selected was between 2000 cm−1 and 100 cm−1.2.7 XTT assay
Two bacterial strains were used in this study: Gram-positive cocci, i.e., S. aureus (ATCC 6538) and Gram-negative bacilli, i.e., P. aeruginosa (ATCC 9027), and their polymicrobial biofilm was formed by mixing equal ratios of both bacterial strains. The plates containing the bacterial inoculum were incubated at 37 °C for 24 h under static conditions. Planktonic (floating) bacteria were removed from the wells by rinsing with 100 μL of PBS and discarded. For the XTT assay, fresh XTT solutions were prepared by dissolving 4 mg of XTT in 10 mL of prewarmed (37 °C) PBS. The solution was mixed with 100 mL of a menadione stock solution containing 55 mg of menadione in 100 mL of acetone. Also, 100 mL of PBS and 100 mL of the XTT–menadione solution were added to each well. The plates were incubated in the dark for 5 h at 37 °C. Next, 100 mL of the supernatant was transferred to the wells of a new flat-bottom 96-well microtiter plate, and the absorbance was measured at 490 nm using a VersaMax™ ELISA Microplate Reader (Molecular Devices, USA). The percentage of biofilm inhibition was calculated using the following eqn (2):
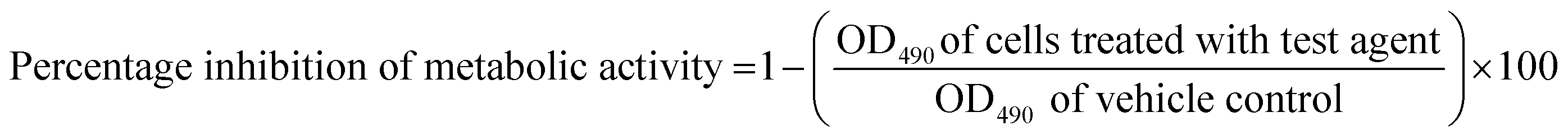 | (2) |
2.8 Drug release study
The nanoparticle in vitro drug release study was assessed using a dialysis tube method.15 To prepare the calibration curve, the stock solution was serially diluted using distilled water and yielded solutions of concentrations 12.5, 25, 50, 75, 100 and 125 mg mL−1 for linearity and lower limits of quantification assays. Five hundred microliter (500 μL) nanoparticle solutions were placed into a dialysis tubing cellulose acetate membrane (Sigma Aldrich, USA) with an average flat width of 33 mm and capacity of 110 mL ft−1. The dialysis tube was tied from its lower end and then, nanoparticles were poured into it. The other end of the tube was also tied and the tube was hanged into in a beaker containing 10 mL of PBS (phosphate buffer saline solution) of pH 7.4 and pH 4.5. The beakers were placed on a digital multi-point magnetic stirrer (WiseStir MS-MP8) and rotated at 100 rpm. After regular intervals, 0.5 mL of solution was withdrawn, diluted to 2 mL, and centrifuged at 40000 rpm at 18 °C for 60 min. The mobile phase was filtered through a 0.45 μm membrane filter and degassed via an ultrasonic water bath prior to use. An RP-HPLC auto sampler and a 2998 photodiode array detector, USA, were used for measuring the amount of drug released from the PLGA nanoparticles. The supernatants were analyzed by HPLC using Shim-pack VP-ODS as described above.
2.9 Biofilm penetration study
Con A was bound to nanoparticles by the method adopted by Weissenboeck A. and Bogner E. in 2004.15 A concentration of 50 μg mL−1 of Con-A conjugated with FITC was dissolved in PBS. Freeze-dried nanoparticles were allowed to incubate with the Con-A-FITC solution for 3 h at room temperature. The labeled nanoparticles were then centrifuged and the nanoparticles settled at the bottom of the tube were then purified by repeated washings with PBS. The fluorescein Con A-FITC-labeled PLGA nanoparticles were again lyophilized and stored at 4 °C until use.Ligand-bound nanoparticles penetration study was done by pouring 100 μL of 0.5 McFarland standard (approximate cell density 1 × 108/mL) bacterial inoculum were placed in six wells flat bottom plates and allowed to incubate for 24 h under static conditions16 and observed under CLSM (Leica TCS SP5 II) at different time intervals of 5 min, 10 min, 15 min, 20 min, 25 min and 30 min using a 20× oil immersion lens. For each sample, images were obtained from three different positions and analysed by an NIS element image viewer 4.20.
2.10 Live/dead biofilm viability assay
The bacterial biofilm viability test was performed by a ready-to-use, dual staining kit named BacLight™ live dead bacterial viability kit. It consists of two florescent dyes that are SYTO9 and propidium iodide (PI).17 The principle of both stains depends on the cell membrane integrity of bacterial cells in the biofilm. For Live/dead staining, both bacterial strains P. aeruginosa and S. aureus and their polymicrobial biofilm were grown as described previously. The inoculum was then treated with a nanoparticle formulation NpX6, a 5% xylitol solution and blank nanoparticles and allowed to incubate for 72 ± 2 h. The test also included untreated biofilm cells incubated with PBS only. Following incubation, non-adherent cells were washed thrice with PBS and then stained using SYTO9 and PI. The biofilm was first stained using 50 μg mL−1 (2.5 μM) SYTO9 and allowed to stand for 15 min at room temperature, followed by staining with 50 μL (15 μM) PI. The mixture was allowed to stand for 15 min at room temperature. Visualization by a confocal laser electron microscope showed green stains, which represented live cells, while red stains represented dead cells. The method used in this assay was previously described by Stiefel et al. (2015).182.11 Statistical analysis
Statistical analysis of data was carried out by one-way ANOVA (one way analysis of variance) followed by Tukey's comparison test and Student t-test. All data were presented as mean ± standard deviation; p < 0.05 was set as significant difference within the group tested.3. Results and discussion
3.1 Particle size, zeta potential, polydispersity, morphology and entrapment efficiency
The mean particle sizes of the six nanoparticle dispersions ranged between 106.8 and 140.8 nm (Table 2). It was observed that an increase in xylitol concentration from 0.5% (NpX1) to 2% (NpX3) resulted in decrease in the mean particle size from 140.8 nm to 106.8 nm. Thereafter, with the rise in xylitol concentration from 3% to 5%, the nanoparticle size increased slightly to 124.7 (NpX4) nm and remained stable at 127 nm (NpX5 and NpX6). It has been reported in the literature that the particle sizes of most polymeric nanoparticles for wound applications range from 100 to 300 nm.19–22 For example, a study demonstrated that the mean particle size of PLGA nanoparticles containing VEGF for promoting the healing of non-diabetic and diabetic wounds was 203 ± 9 nm.22 Another study showed that the sustained release of curcumin, a topical wound healing agent, from PLGA significantly improved the wound healing activity in vivo and the PLGA nanoparticles possessed a mean particle size of 176.5 ± 7.0 nm.21 It was also reported that if the nanoparticle size ranges from 300 to 500 nm, they can rapidly enter the liver, the spleen, and the bone marrow and produce toxicity; thus, nanoparticles with sizes ranging between 100 and 300 nm are considered appropriate for topical treatment applications.23 The TEM images (Fig. 1) of the nanoparticles show a fine spherical shape with an outer smooth surface. The images also confirm that the nanoparticles do not show aggregation. It was also observed that the concentration of xylitol did not affect the shape of the nanoparticles.| Formulations | Xylitol concentration | Particle size (nm) | Polydispersity index | Zeta potential (mV) | Entrapment efficiency (%) |
|---|---|---|---|---|---|
| a Statistically significant (p < 0.0001) as compared to other nanoparticle formulations. | |||||
| NpX1 | 0.5% | 140.8 ± 34.8 | 0.36 ± 0.09 | −12.29 ± 3.57 | 63.6 ± 0.2 |
| NpX2 | 1% | 134.7 ± 25.8 | 0.41 ± 0.22 | −15.06 ± 5.81 | 73.1 ± 3.3 |
| NpX3 | 2% | 106.8 ± 35.1a | 0.39 ± 0.15 | −18.52 ± 1.49 | 82.1 ± 0.1 |
| NpX4 | 3% | 124.7 ± 20.4 | 0.33 ± 0.08 | −34.05 ± 3.68 | 86.4 ± 2.2 |
| NpX5 | 4% | 127.1 ± 6.56 | 0.29 ± 0.06 | −33.83 ± 4.13 | 87.6 ± 0.01 |
| NpX6 | 5% | 127.3 ± 20.6 | 0.52 ± 0.18 | −33.41 ± 3.29 | 89.7 ± 0.7a |
 | ||
| Fig. 1 Transmission electron microscopy morphological analysis of the nanoparticle formulations NpX1–NpX6. | ||
The PDI values of all nanoparticle formulations fall in the acceptable range of 0.3–0.5, indicating narrow size distribution. For stable nanoparticles, the zeta potential usually lies below −30 mV or above +30 mV. The zeta potential data showed that only NpX4, NpX5 and NpX6 produced stable nanoformulations with zeta potentials above +30 mV. The zeta potential is represented by the interfacial potential between the attached layer formed on the surface of the nanoparticles and the solvent used. This quantitative analysis is used to determine the repulsive interactions between the nanoparticles and the solvent. Higher zeta potential values indicate high stability of the nanoparticles in the formulation. We hypothesized that the xylitol concentration of 4% and above introduced steric repulsions, which would enhance the PLGA nanoparticle stability.
The entrapment efficiencies of PLGA nanoparticle formulations are also shown in Table 2. In this study, the concentration of polymers remained constant; therefore, the entrapment mainly depends on the concentration of xylitol. The xylitol entrapment efficiency was found to increase from 63.6 to 86.4% as the xylitol concentration increased from 0.5 to 3%. The entrapment reached a plateau at approximately 86–89% as the xylitol concentrations were increased from 4 to 5%. The highest xylitol entrapment was found with the NpX6 nanoparticle formulation at 89.7% (p < 0.05). A higher entrapment percentage indicates a low amount of xylitol wasted during nanoparticle formation.
3.2 FTIR (Fourier transform infrared spectroscopy) analysis
The FTIR spectra of the nanoparticles were compared with those of pure xylitol powder, PLGA polymer and blank nanoparticles (Fig. 2a) to ascertain notable trends with increasing drug concentrations. Fig. 2b presents the results of the FTIR analysis of the xylitol nanoparticles. The stretching bands occurring in the range of 1760–1750 cm−1 represent the stretching vibrations of the carbonyl groups present in PLGA.24 The stretching band vibrations between 1300 and 1150 cm−1 are due to the presence of asymmetric and symmetric C–C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)–O functional groups that represent esters. The peaks obtained between 900 cm−1 and 600 cm−1 are characteristic of alkanes and alkynes. These stretching vibrations indicated the presence of the PLGA polymer in the nanoparticles. The stretching vibrations in the range of 400–190 cm−1 indicate the presence of xylitol in the PLGA nanoparticles. The nanoparticles showed a combination of the stretching vibrations of xylitol and PLGA. The bands occurring in all six nanoformulations followed the same pattern, indicating the presence of similar functional groups, and no chemical interactions were observed. With an increase in the xylitol concentration (NpX1–NpX6), the absorption band obtained at 400–390 cm−1 became more prominent and sharp, indicating the encapsulation of more xylitol inside the PLGA core. The FTIR result revealed that increasing the xylitol concentration did not affect the nanoparticle properties, which indicated compatibility between the drug and the polymer;25 therefore, it can be inferred that the FTIR curve obtained is similar to that of the pure drug and polymer, confirming that there was no interaction between PLGA and xylitol. Hence, it can be concluded that xylitol was successfully loaded in the PLGA polymer and there was no chemical interaction between the drug and the polymer.
O)–O functional groups that represent esters. The peaks obtained between 900 cm−1 and 600 cm−1 are characteristic of alkanes and alkynes. These stretching vibrations indicated the presence of the PLGA polymer in the nanoparticles. The stretching vibrations in the range of 400–190 cm−1 indicate the presence of xylitol in the PLGA nanoparticles. The nanoparticles showed a combination of the stretching vibrations of xylitol and PLGA. The bands occurring in all six nanoformulations followed the same pattern, indicating the presence of similar functional groups, and no chemical interactions were observed. With an increase in the xylitol concentration (NpX1–NpX6), the absorption band obtained at 400–390 cm−1 became more prominent and sharp, indicating the encapsulation of more xylitol inside the PLGA core. The FTIR result revealed that increasing the xylitol concentration did not affect the nanoparticle properties, which indicated compatibility between the drug and the polymer;25 therefore, it can be inferred that the FTIR curve obtained is similar to that of the pure drug and polymer, confirming that there was no interaction between PLGA and xylitol. Hence, it can be concluded that xylitol was successfully loaded in the PLGA polymer and there was no chemical interaction between the drug and the polymer.
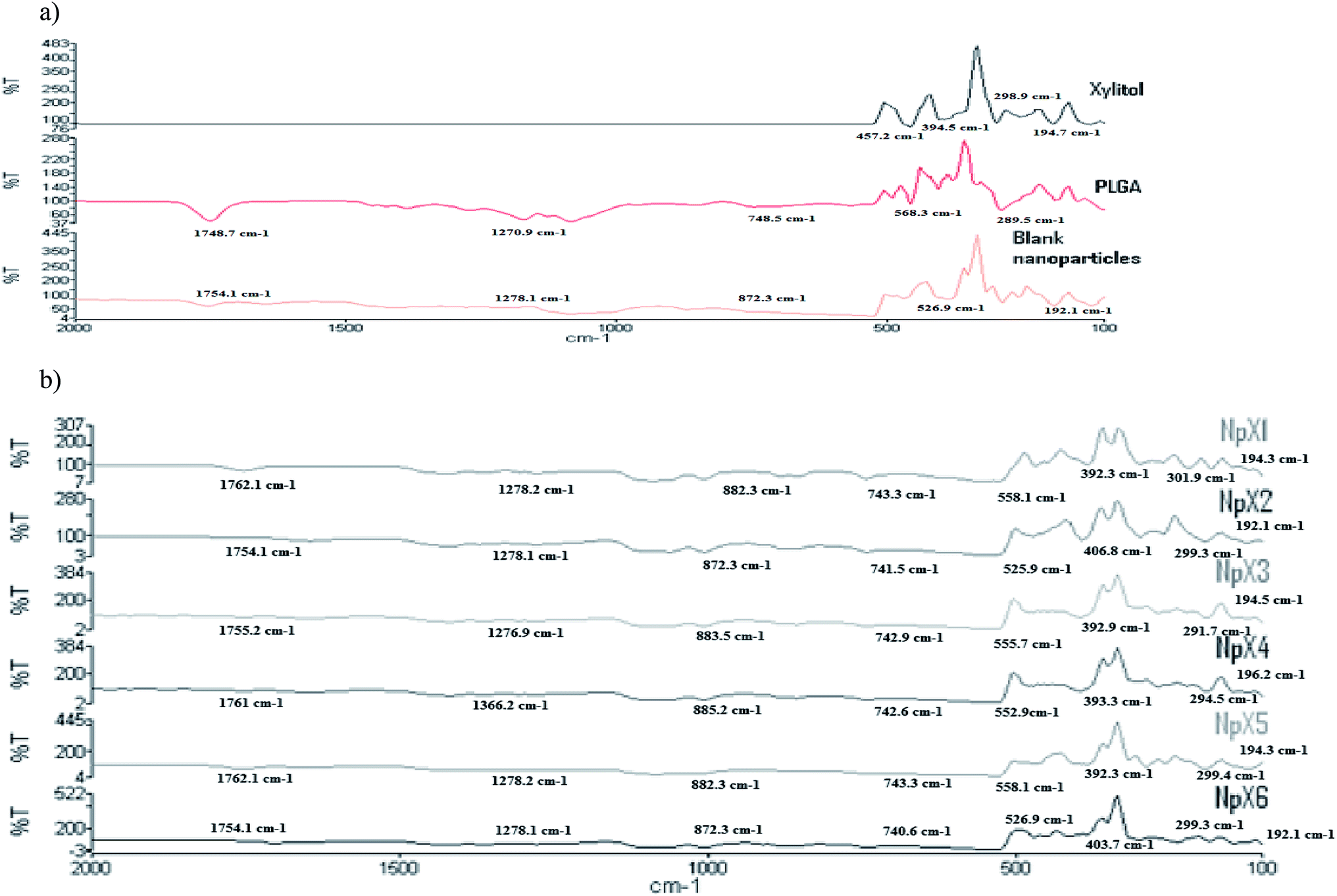 | ||
| Fig. 2 FTIR results for (a) pure xylitol, PLGA and blank nanoparticles and (b) nanoparticle formulations having different xylitol concentrations (NpX1–NpX6). | ||
3.3 XTT assay
An XTT assay involves the measurement of the metabolic activity of the bacterial biofilms, representing the living bacterial cells in a biofilm community. The XTT assay is preferred as it is simple, efficient and non-invasive as compared to other quantification methods.26 S. aureus and P. aeruginosa were chosen because the biofilms of these two strains are the most common for biofilm-associated chronic wound infections.27,44 Additionally, the co-infection of S. aureus and P. aeruginosa has both intrinsic and acquired antibiotic resistance in patients, which can delay wound healing.28The XTT results obtained from the S. aureus biofilms are shown in Fig. 3a. Overall, the xylitol nanoparticles showed significantly higher inhibition (53–69%) compared to the xylitol solutions (35–59%), with NpX6 having the most prominent biofilm inhibition (69%). The results obtained from the XTT assay against S. aureus confirmed that the xylitol nanoparticles have better antibiofilm activities compared to the xylitol solutions. Similar results were obtained for P. aeruginosa biofilms but to a lower extent (Fig. 3b). The PLGA nanoparticles also demonstrated the ability to decrease polymicrobial biofilm formation, which is a more realistic representation of clinical biofilms as in wounds, the biofilms are normally present with multispecies microorganisms. One study demonstrated that a xylitol solution (3–18%) is an efficient antibiofilm agent as well and it shows concentration-dependent antibiofilm activity.29 Other studies also reported that xylitol showed positive antibiofilm efficacy when in combination with other antibiofilm agents such as lactoferrin.30,31 Comparatively, our study showed that the antibiofilm effect can be observed when xylitol is loaded in nanoparticulate systems. Our preliminary data showed that NpX6 (5% xylitol concentration) exhibited the most promising antibiofilm activity.30 This study showed that xylitol formulated as PLGA nanoparticles can enhance the xylitol antibiofilm activities. From our data, 1% xylitol as nanoparticles showed no significant difference in antibiofilm inhibition to 5% xylitol solution, which indicated that PLGA nanoparticles are able to provide protection to xylitol from bacterial enzymatic degradation.32 These findings confirmed that PLGA nanoparticles can effectively eradicate S. aureus and P. aeruginosa biofilms.
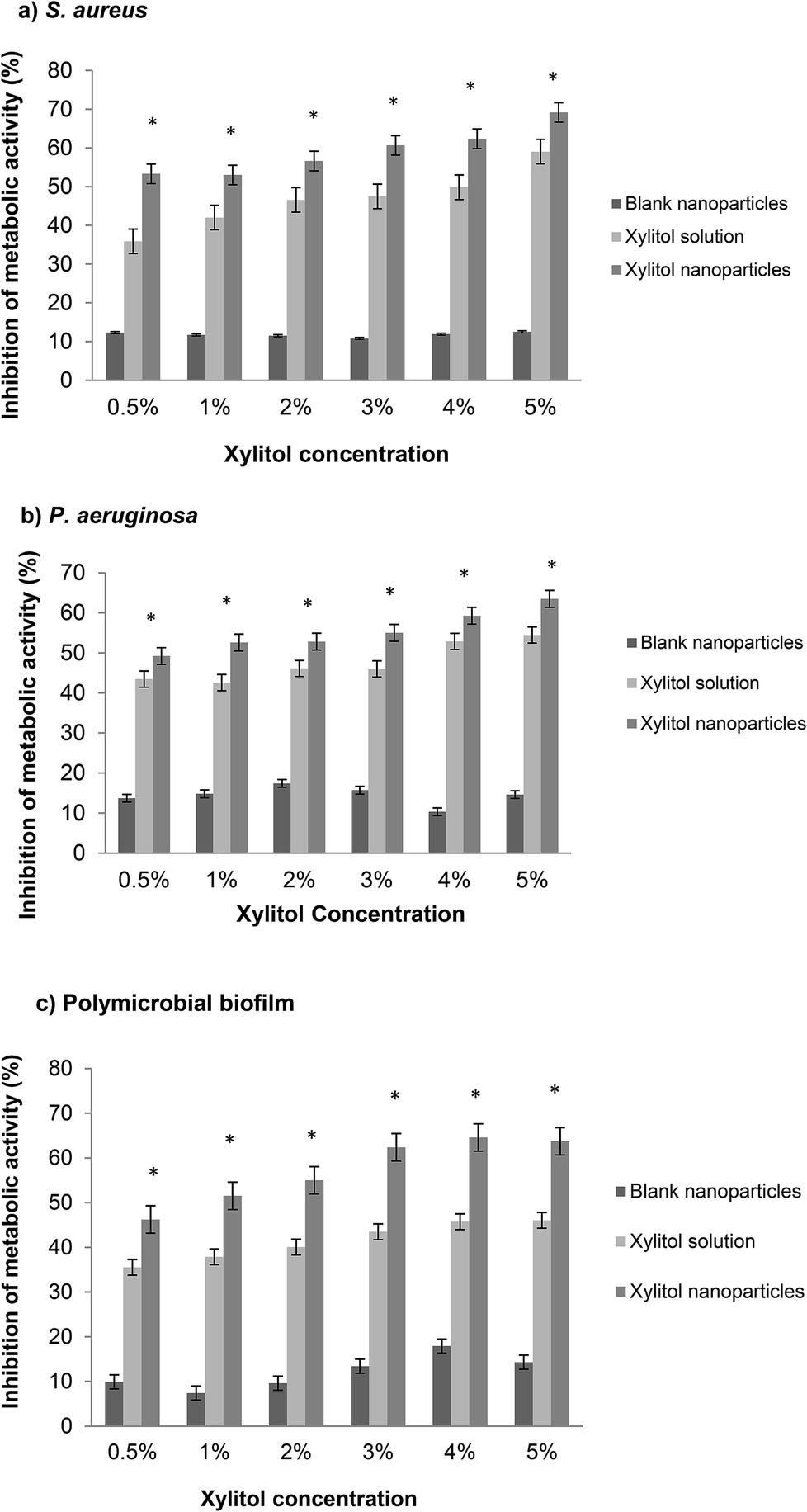 | ||
| Fig. 3 XTT assay showing the percentage of inhibition of the metabolic activity of xylitol solutions and nanoparticles NpX1–NpX6 against (a) S. aureus, (b) P. aeruginosa and (c) polymicrobial biofilms. *p < 0.002 is statistically significant as compared to xylitol solutions of the same concentrations; n = 6. | ||
3.4 Drug release study
The percentage of the cumulative amount of xylitol released (%) as a function of time (h) is shown in Fig. 4. The release profiles of the PLGA nanoparticles with different xylitol concentrations in pH7.4 PBS are presented in Fig. 4a. All nanoparticle formulations show good release profiles for 72h. However, it was also observed that an increase in drug concentration did not affect the release characteristic of nanoparticles after 72 h, in which 85 to 95% of drugs were released from the PLGA matrix in all the nanoparticle formulations (NpX1–NpX6). Many factors affect the release of xylitol from PLGA nanoparticles, and these include polymer–drug interactions, drug–drug interactions and water absorption that can influence the rate of PLGA drug diffusion and degradation.33 The release was controlled by the rate of PLGA degradation, which is a three-step mechanism. The first step includes a random chain breakage, causing rapid drug release, which is followed by breakage to oligomers; finally, the oligomers are broken into monomers and cause polymer solubilization.33 A similar trend was observed in this study. The rapid elution of the drug in the first hour is due to the presence of the drug on the surface layers of the nanoparticles, followed by a continuous increase of drug release till 8 h, indicating PLGA degradation and formation of oligomers; after 8 h, the drug released steadily till 72 h and showed slow conversation of oligomers into monomers.
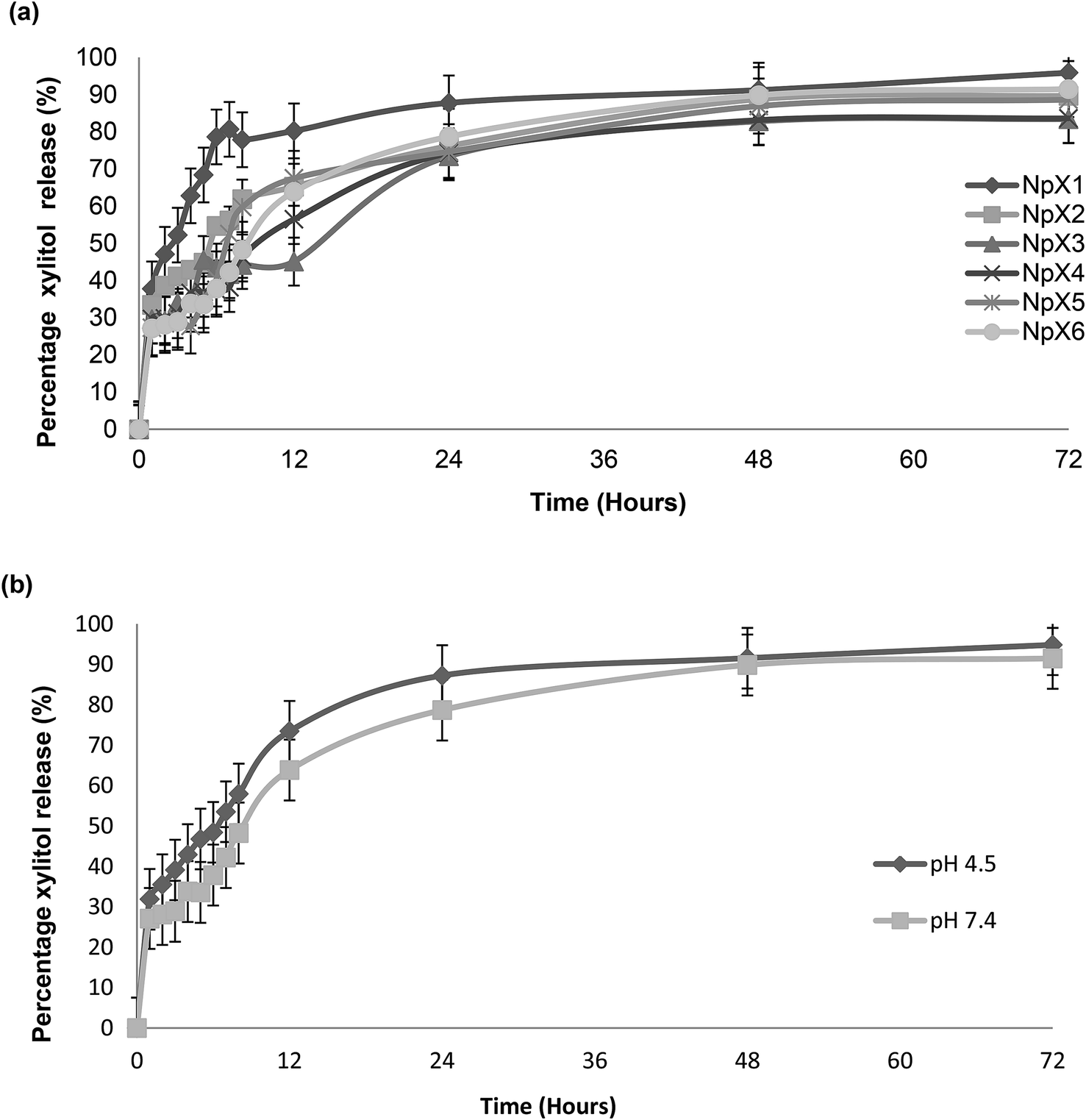 | ||
| Fig. 4 (a) Percentage of xylitol release of NpX1–NpX6. (b) Percentage of xylitol release of NpX6 at pH 4.5 and 7.4 (*statistically significant p < 0.03 at pH 4.5 as compared to the release at pH 7.4). | ||
The PLGA nanoparticles having the highest concentration of an antibiofilm agent (NpX6) were also assessed to obtain the release profile at pH 4.5 along with the release profile at pH 7.4 (Fig. 4b). The nanoparticles showed a higher drug release at pH 4.5 compared to that at pH 7.4. However, the patterns of release were similar and followed the first order kinetics. This result indicated the pH-dependent nature of the PLGA polymer as expected due to the higher solubility of PLGA at acidic pH. The proposed mechanism includes faster polymer degradation, promoting surface pitting and pore formation. It was also reported elsewhere that degradation occurred in a more homogeneous pattern at acidic pH as compared to that at alkaline pH.34 Autocatalysis of PLGA occurs as a result of the generation of acidic oligomer units, and these oligomers play an important role in PLGA degradation, resulting in the formation of channels through which drug release occurs.34 Acidic pH catalyzes the breakage of the ester linkage of the polymer backbone, enhancing polymer erosion and drug release from it.35 This mechanism was explained by a change in the morphological analysis observed at different time intervals at both acidic and alkaline pH.34–36 The effect of pH has been reported to play an important role in the wound healing process by influencing the activities of tissue inhibitors, metalloproteinases and fibroblasts; proliferation of keratinocytes and microorganisms; and immunological responses.29 The change in pH alters the patient's defense mechanisms, thereby inhibiting wound microorganism invasion and proliferation. The pH change disturbs the activity of antimicrobials and biofilm formation and affects treatment efficacy and wound management strategies.30 It became quite clear that pH plays an important role in both healing and treatment of chronic and acute wounds. Hence, a change in pH is usually very significant for wound healing and biofilm formation and can affect treatment efficacy and wound management strategies; it was measured that the pH of chronic wounds is in the range of 7.15–8.9 for both acute and chronic wounds.30 It was also stated that both acute and chronic wounds having an alkaline pH showed lower rates of healing than wounds having a pH closer to neutral that ranges between 7 and 8. As the wound progresses, the pH changes from a neutral to an acidic environment in the range from 5 to 4.29 Hence, pH has an important impact on the activity of antibiofilm agents.
3.5 Biofilm penetration study
The penetration of nanoparticles into bacterial biofilms is highly dependent on the surface characteristics of the nanoparticles. The surface properties of nanoparticles can be enhanced by adding a ligand onto the nanoparticle surface.37 Con A is a type of lectin representing a marker for glycoconjugates that are the major components of the biofilm EPS, and it binds selectively to a-glucopyranosyl molecules such as glucans. NpX6 was chosen to conduct this study because NpX6 nanoparticle formulations showed the highest drug entrapment as well as the highest biofilm inhibition in the XTT assay. When NpX6 nanoparticles combined with Con A-FITC entered the biofilms, Con A bound to the biofilm EPS matrix and green fluorescence was produced. Fig. 5a shows the S. aureus biofilm penetration of nanoparticles up to 30 min. As the time increased, the intensity of the green fluorescence also increased. A similar trend was observed in the P. aeruginosa biofilm under CLSM, as shown in Fig. 5b. The green fluorescence proved that the NpX6 nanoparticles have successfully entered both Gram-positive and Gram-negative bacterial biofilms. The polymicrobial biofilm of both bacteria also showed higher penetration of NpX6 nanoparticles with denser and higher intensity of green fluorescence, as shown in Fig. 5c.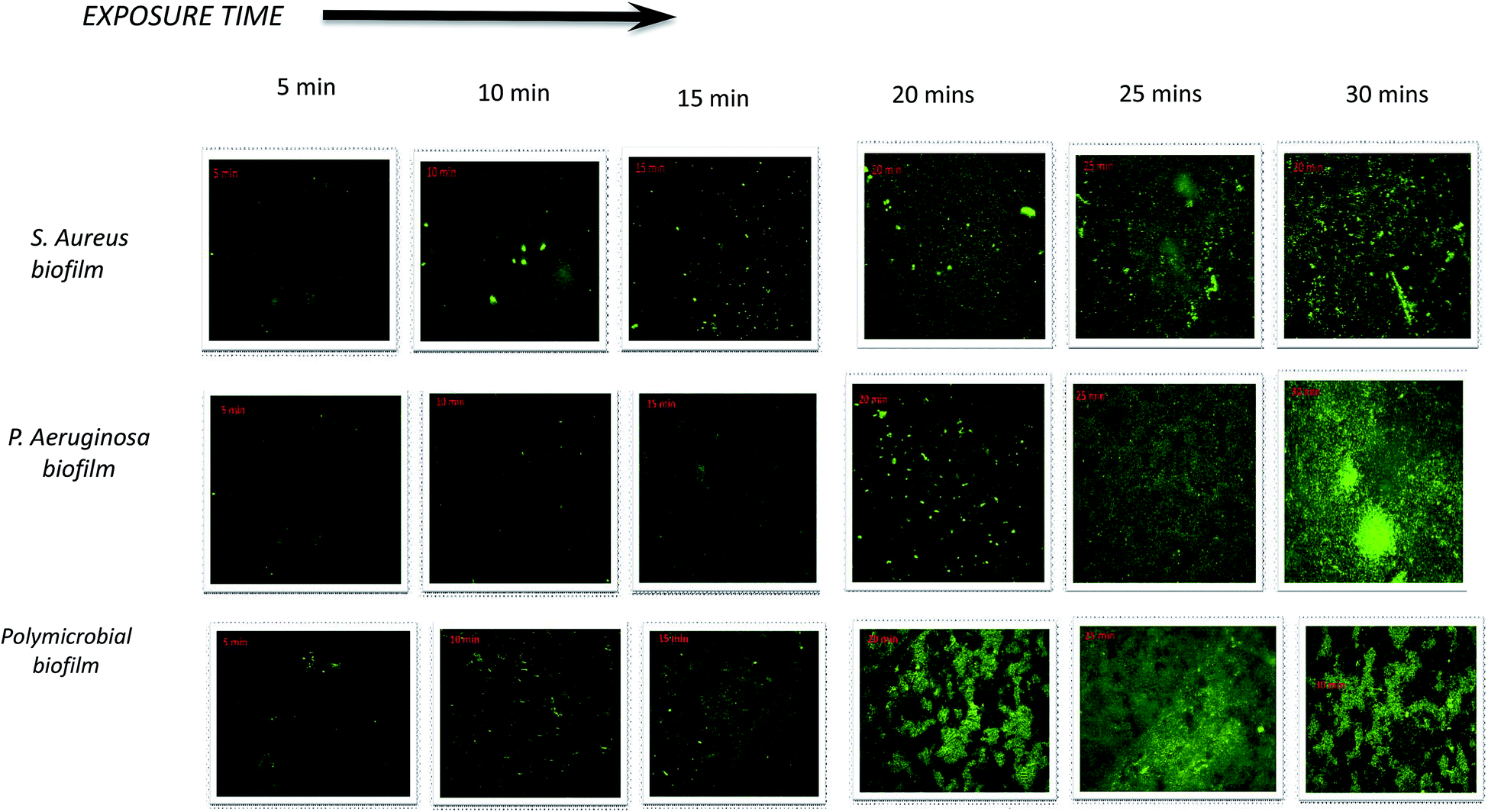 | ||
| Fig. 5 Confocal fluorescence images of the S aureus biofilm ligand with Con A FITC to investigate the penetration into bacterial biofilms. Time-dependent penetration of NpX6 nanoparticles, Staphylococcus aureus, Pseudomonas aeruginosa and polymicrobial biofilms; images obtained after 5 min, 10 min, 15 min, 20 min, 25 min and 30 min. | ||
The ability of nanoparticles to attach and penetrate into biofilm cells depends on their physiochemical properties such as size, morphology, surface charge and composition16. Con A, a lectin, is commonly used as a ligand binding to different nano-based drug delivery systems against bacterial cells and biofilm matrices. It is the most preferred because it binds to the mannose residues of glycoproteins present in bacterial cell membranes and biofilm matrices. Con A FITC produces green fluorescence upon activation, which is usually related to its binding to the glycoproteins present in the bacterial cells.38 Con A FITC binds specifically at the α-Man and α-Glc target specific sites of the glycoproteins in the S. aureus biofilm.39 The CLSM images obtained show that the nanoparticles actively penetrate into the biofilm matrix as compared to the xylitol solution and hence facilitate the release of the drug inside the biofilm matrix; this promotes the antibiofilm activity of xylitol. It also explained the results obtained from the XTT assay.
3.6 Live/dead biofilm viability assay
The bacterial viability assay was performed using BacLight™ (LIVE/DEAD Bacterial Viability Kit), which consists of two fluorescent stains: SYTO 9 and PI. The bacterial live/dead viability assay indicates the assessment of membrane integrity. The live/dead viability images for S. aureus, P. aeruginosa and the polymicrobial biofilm are shown in Fig. 6. The green stains in the CLSM images represent living biofilm cells with intact membranes, while the red cells represent dead biofilm cells with damaged cell membranes.18 Visually, the xylitol solution showed higher red stain intensity compared to untreated and blank nanoparticle samples, which indicated a higher number of dead cells, while NpX6 showed higher intensity of red staining compared to the xylitol solution. The live/dead viability obtained from the CLSM images indicated that the nanoparticles showed enhanced penetration into bacterial biofilms and higher dead cell content compared to a xylitol solution of the same concentration. Higher intensity red staining seen with NpX6 nanoparticles indicated that the xylitol nanoparticles were able to penetrate into the biofilm due to their small size. Nanoparticles facilitate xylitol penetration into the biofilm, while the xylitol solution can partially penetrate into the biofilm matrix due to rapid enzymatic degradation.31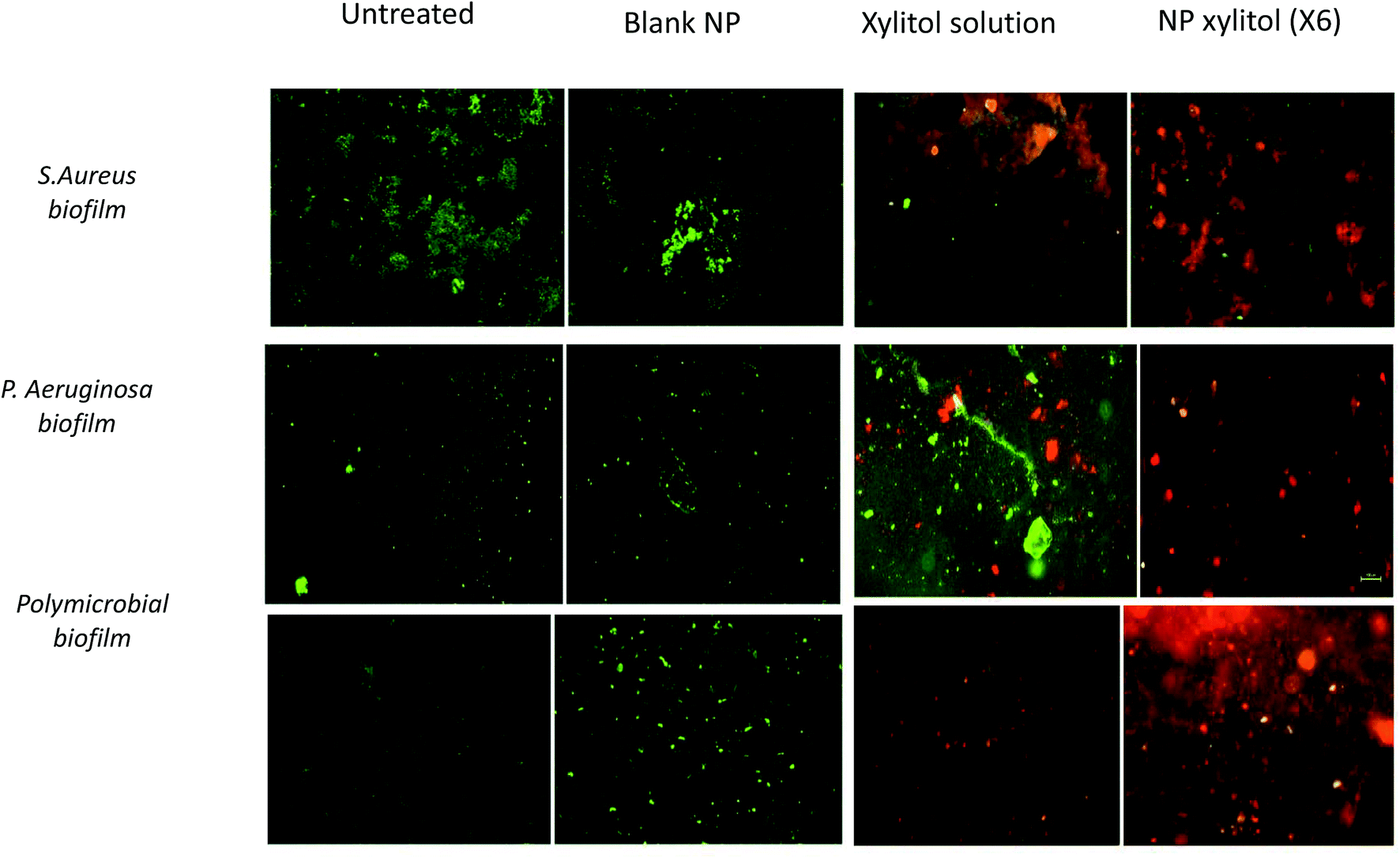 | ||
| Fig. 6 CLSM images of S aureus, P aeruginosa and polymicrobial biofilm with green for the SYTO9 signal and red for the PI signal; merged images are shown with untreated samples, blank nanoparticles, xylitol solution and NpX6 nanoparticles. | ||
4. Conclusion
Most chronic wounds contain biofilms that perpetuate the inflammatory phase of wound healing. The biofilm matrix provides resistance to biocides, host defenses, and antibiotic penetration.40 It was suggested that addressing the biofilm in the first 48h of formation provides the most effective therapeutic window.41 Effective treatments for these wound infections include debridement, but it is only beneficial for a short duration. A treatment that provides both antimicrobial and antibiofilm activities will serve as an ideal treatment regimen for chronic wound infections. In this study, PLGA nanoparticle-based delivery systems were designed and evaluated. It was demonstrated that PLGA nanoparticles containing xylitol successfully penetrated into the EPS matrix. In conclusion, PLGA nanoparticles containing xylitol promote antibiofilm activity due to their unique physiochemical properties and enhanced penetration in biofilm EPS. It can be considered as a new strategy in chronic wound management. We hypothesize that the PLGA nanoparticles can passively target the EPS matrix for the delivery of xylitol to induce the dispersion of biofilms. Further investigations using animal model and clinical wound isolates are warranted to conclude the efficacy against biofilm-associated chronic wound infection.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors would like to thank the Ministry of Energy, Science, Technology, Environment & Climate Change Malaysia (MESTECC) Grant no. 02-01-02-SF1228 and the Faculty of Pharmacy, Universiti Kebangsaan Malaysia, for the research infrastructure and facility support.References
- G. A. James, E. Swogger, R. Wolcott, E. d. Pulcini, P. Secor, J. Sestrich, J. W. Costerton and P. S. Stewart, Wound Repair. Regen., 2008, 16, 37–44 CrossRef
.
- A. R. Siddiqui and J. M. Bernstein, Clin. Dermatol., 2010, 28, 519–526 CrossRef PubMed
- S. L. Percival, K. E. Hill, D. W. Williams, S. J. Hooper, D. W. Thomas and J. W. Costerton, Wound Repair. Regen., 2012, 20, 647–657 CrossRef
- T. Bjarnsholt, K. Kirketerp-Møller, P. Ø. Jensen, K. G. Madsen, R. Phipps, K. Krogfelt, N. Høiby and M. Givskov, Wound Repair. Regen., 2008, 16, 2–10 CrossRef PubMed
- M. C. B. Ammons, Recent Pat. Anti-Infect. Drug Discovery, 2010, 5, 10–17 CrossRef CAS
- M. S. Blackledge, R. J. Worthington and C. Melander, Curr. Opin. Pharmacol., 2013, 13, 699–706 CrossRef CAS PubMed
- P. Y. Chung and Y. S. Toh, Pathog. Dis., 2014, 70, 231–239 CrossRef CAS
- T. Bapiainen, R. Sormunen, T. Kaijalainen, T. Kontiokari and M. Uhari, J. Antimicrob. Chemother., 2004, 54, 225–228 CrossRef
- N. P. Truong, M. R. Whittaker, C. W. Mak and T. P. Davis, Expert Opin. Drug Delivery, 2015, 12, 129–142 CrossRef CAS PubMed
- K. Masako, I. Hideyuki, O. Shigeyuki and I. Zenro, Attack on biofilm of Staphylococcus aureus without antibiotics, J. Dermatol. Sci., 2005, 38, 197–205 CrossRef CAS PubMed
- S. E. Dowd, Y. Sun, E. Smith, J. P. Kennedy, C. E. Jones and R. Wolcott, J. Wound Care, 2009, 18(508), 510–512 Search PubMed
- S.-F. Ng and H.-L. Leow, Drug Dev. Ind. Pharm., 2015, 41, 1902–1909 CrossRef CAS PubMed
- A. Anjum, C. H. Sim and S. F. Ng, AAPS PharmSciTech, 2017, 3, 1–12 Search PubMed
- N. Thomas, C. Thorn, K. Richter, B. Thierry and C. Prestidge, J. Pharm. Sci., 2016, 105, 3115–3122 CrossRef CAS PubMed
- A. Weissenboeck, E. Bogner, M. Wirth and F. Gabor, Pharm. Res., 2004, 21, 1917–1923 CrossRef CAS
- T. Wang, J. Bai, X. Jiang and G. U. Nienhaus, ACS Nano, 2012, 6, 1251–1259 CrossRef CAS PubMed
- S. Leuko, A. Legat, S. Fendrihan and H. Stan-Lotter, Appl. Environ. Microbiol., 2004, 70, 6884–6886 CrossRef CAS PubMed
- P. Stiefel, S. Schmidt-Emrich, K. Maniura-Weber and Q. Ren, BMC Microbiol., 2015, 1, 36 CrossRef PubMed
- H. Nurhasni, J. Cao, M. Choi, I. Kim, B. L. Lee, Y. Jung and J. W. Yoo, Int. J. Nanomed., 2015, 10, 3065–3080 CAS
- G. Baier, A. Cavallaro, K. Friedemann, B. Müller, G. Glasser, K. Vasilev and K. Landfester, Nanomedicine, 2014, 10, 131–139 CrossRef CAS PubMed
- K. K. Chereddy, R. Coco, P. B. Memvanga, B. Ucakar, A. des Rieux, G. Vandermeulen and V. Préat, J. Controlled Release, 2013, 171, 208–215 CrossRef CAS PubMed
- K. K. Chereddy, A. Lopes, S. Koussoroplis, V. Payen, C. Moia, H. Zhu, P. Sonveaux, P. Carmeliet, A. des Rieux, G. Vandermeulen and V. Préat, Nanomedicine, 2015, 11, 1975–1984 CrossRef CAS PubMed
- M. Gaumet, A. Vargas, R. Gurny and F. Delie, Eur J Pharm Biopharm, 2008, 69, 1–9 CrossRef CAS PubMed
- C. D. Erbetta, R. J. Alves, J. M. Resende, R. F. Freitas and R. G. de Sousa, J. Biomater. Nanobiotechnol., 2012, 2, 208 CrossRef
- H. Gupta, M. Aqil, R. K. Khar, A. Ali, A. Bhatnagar and G. Mittal, Nanomedicine, 2010, 6, 324–333 CrossRef CAS PubMed
- D. M. Kuhn, M. Balkis, J. Chandra, P. K. Mukherjee and M. A. Ghannoum, J. Clin. Microbiol., 2003, 41, 506–508 CrossRef CAS PubMed
- R. Serra, R. Grande, L. Butrico, A. Rossi, U. F. Settimio, B. Caroleo, B. Amato, L. Gallelli and S. de Franciscis, Expert Rev. Anti-Infect. Ther., 2015, 13, 605–613 CrossRef CAS PubMed
- B. Gilchrist, Nursing Times, 1994, 90, 55–56 CAS
- S. Radmerikhi, E. Azul, K. R. Fajardo and B. Formantes, J. Restor. Dent., 2013, 3, 95 CrossRef
- M. C. Ammons, G. A. Ward Ls Fau - James and G. A. James, Int. Wound J., 2011, 8, 268–273 CrossRef PubMed
- M. C. Ammons, L. S. Ward, S. Dowd and G. A. James, Int. J. Antimicrob. Agents, 2011, 37, 316–323 CrossRef CAS PubMed
- R. S. Kadam, D. W. A. Bourne and U. B. Kompella, Drug Metab. Dispos., 2012, 40, 1380–1388 CrossRef CAS PubMed
- F. Alexis, Polym. Int., 2005, 54, 36–46 CrossRef CAS
- B. S. Zolnik and D. J. Burgess, J. Controlled Release, 2007, 122, 338–344 CrossRef CAS PubMed
- X. S. Wu and N. Wang, J. Biomater. Sci., Polym. Ed., 2001, 12(1), 21–34 CrossRef CAS PubMed
- J. Shen and D. J. Burgess, Int. J. Pharm., 2012, 422, 341–348 CrossRef CAS PubMed
- Y. Zhao, Y. Tian, Y. Cui, W. Liu, W. Ma and X. Jiang, J. Am. Chem. Soc., 2010, 132, 12349–12356 CrossRef CAS PubMed
- M. M. Baum, A. Kainović, T. Keeffe, R. Pandita and K. McDonald, BMC Microbiol., 2009, 9, 103 CrossRef PubMed
- P. Anastasiadis, K. D. A. Mojica, J. S. Allen and M. L. Matter, J. Nanobiotechnol., 2014, 12, 24 CrossRef PubMed
- P. S. Stewart, J. A. G. L. Fau - Diemer and J. A. Diemer, J. Appl. Microbiol., 1998, 85, 495–500 CrossRef CAS PubMed
- R. D. Wolcott, G. Rumbaugh Kp Fau - James, G. James G Fau - Schultz, P. S. G. Fau - Phillips, Q. Phillips P Fau - Yang, C. Yang Q Fau - Watters, P. S. Watters C Fau - Stewart, S. E. Stewart Ps Fau - Dowd and S. E. Dowd, J. Wound Care, 2010, 19, 45–5342 CrossRef CAS PubMed
- C. Martin, W. L. Low, A. Gupta, M. C. Amin, I. Radecka, S. T. Britland, P. Raj and K. M. Kenward, Curr. Pharm. Des., 2015, 21, 43–66 CrossRef CAS PubMed
- R. H. Ansary, M. M. Rahman, N. Mohamad, T. M. Arrif, A. Z. A. Latif, H. Katas, W. Nik and M. B. Awang, Polymers, 2017, 9, 485 CrossRef PubMed
- C. Kong, C. F. Chee, K. Richter, N. Thomas, N. Abd Rahman and S. Nathan, Sci. Rep., 2018, 8, 2758 CrossRef PubMed
| This journal is © The Royal Society of Chemistry 2019 |