DOI:
10.1039/C9RA00135B
(Paper)
RSC Adv., 2019,
9, 6658-6680
Efficient electroluminescent hybridized local and charge-transfer host materials with small singlet–triplet splitting to enhance exciton utilization efficiency: excited state transition configuration†
Received
7th January 2019
, Accepted 18th February 2019
First published on 26th February 2019
Abstract
A series of efficient electroluminescent materials with dual carrier transport properties shows enhanced singlet exciton utilization (ηs) due to small singlet–triplet splitting (ΔEST). The strong orbital-coupling transitions of N-(4-(1-(1-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)-4,5-diphenyl-1H-imidazol-2-yl)naphthalen-4-yl)phenyl)-N-phenyl benzenamine (DDPB) exhibit deep blue emission at 435 nm (CIEy, 0.07) with an external quantum efficiency of 2.01%. The electroluminescent efficiencies of 2-(1-(9H-carbazol-9-yl)naphthalen-4-yl)-1-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)-1H-phenanthro[9,10-d]imidazole (CDDPI) (L – 3992 cd m−2; ηex – 3.01%; ηc – 2.56 cd A−1; ηp – 2.12 lm W−1) are higher than those of the N-(4-(1-(1-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)-H-phenanthro[9,10-d]imidazole-2-yl)naphthalen-4-yl)phenyl)-N-phenylbenzenamine (DBDPA) based device (L – 3015 cd m−2; ηex – 2.85%; ηc – 2.01 cd A−1; ηp – 1.92 lm W−1). The blue emissive materials CDDPI and DBDPA are used as a host to construct green and red phosphorescent OLEDs: the green device based on CDDPI:Ir(ppy)3 exhibits higher efficiencies (L – 8812 cd m−2; ηex – 19.0%; ηc – 27.5 cd A−1; ηp – 33.0 lm W−1) at 2.7 V and the red device based on CDDPI:Ir(MQ)2(acac) exhibits a maximum luminance of 39![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 661 cd m−2 with excellent EL efficiencies [ηex – 19.2%; ηc – 27.9 cd A−1; ηp – 29.2 lm W−1; CIE (0.64, 0.34)] compared with those of the DBDPA:Ir(MQ)2(acac) based device [L – 37621 cd m−2; ηex – 18.5%; ηc – 25.2 cd A−1; ηp – 25.8 lm W−1; CIE (0.64, 0.34)].
661 cd m−2 with excellent EL efficiencies [ηex – 19.2%; ηc – 27.9 cd A−1; ηp – 29.2 lm W−1; CIE (0.64, 0.34)] compared with those of the DBDPA:Ir(MQ)2(acac) based device [L – 37621 cd m−2; ηex – 18.5%; ηc – 25.2 cd A−1; ηp – 25.8 lm W−1; CIE (0.64, 0.34)].
1. Introduction
The development of highly efficient blue emissive materials with balanced carrier injection in organic light-emitting devices (OLEDs) remains a challenging task.1–5 The blue emitters with higher energy gap (Eg) result in low electron affinities and the lowering of the device efficiency.6–9 Though the non-doped blue device based on dipyrenylbenzene (CIE: 0.15, 0.11) and anthracene derivative (CIE: 0.14, 0.12) exhibit high external quantum efficiencies (ηex) of 5.2% and 5.3%, respectively,10,11 their power (ηp) and current (ηc) efficiencies are low due to the wide band-gap (Eg). The blue device (CIE: 0.15, 0.16) with oligoquinoline emissive material shows high ηp and ηex values of 4.3 lm W−1 and 6.6%, respectively.12 The simple structured device based on bis(phenanthroimidazolyl)biphenyl derivative exhibits higher performances (CIEy-0.15; ηex: 6.31%) than the multi-layered one.13,14 Electroluminescent materials with thermally activated delayed fluorescence (TADF),15 hybridized local and charge-transfer (HLCT)16 and triplet–triplet annihilation (TTA: ηint – 62.5%)17–20 have enhanced the internal quantum efficiency (ηint).21–25 Constructing donor–spacer–acceptor (D–π–A) molecules is one of the widely used techniques to improve the charge injection and carrier transportation of OLED materials.26 In some D–π–A based OLEDs, the triplet excitons have been fully employed and excellent performances were obtained.27,28 D–π–A compounds usually possess weakly bound charge-transfer (CT) excitons which facilitate a reverse intersystem crossing (RISC) process in the OLEDs. However, it is rather rarely reported that the fluorescent OLEDs based on D–π–A molecular architecture exhibit high efficiency and favourable colour-purity. Hence, the molecular design is aimed to overcome the following remarks: (i) the D–π–A molecules are more suitable to design narrow-band-gap materials due to significant decrease in excited state energy of the CT state from donor to acceptor or more delocalized π–π* state between weak donor and weak acceptor;26b,29,30 (ii) CT state as an emissive state always leads to broadened photoluminescence (PL) and electroluminescence (EL) spectra which is unfavourable for high colour-purity;28 (iii) the CT state usually exhibits a low efficiency fluorescence which is attributed to the nature of the forbidden transition induced by the spatial separation between the hole and electron wave functions. On the contrary, the locally excited (LE) state is suited to producing high-efficiency fluorescence radiation due to the large orbital overlap. However, the CT state can provide a RISC channel that improves exciton-utilizing efficiency (ηs) in fluorescent OLEDs through a very small energy splitting between singlet and triplet states which has been proven to be an effective way to utilize triplet exciton energy in fluorescent OLEDs.27,28 Considering the above issues, if CT and LE states could be reasonably combined into one D–π–A compound, it would be possible to further greatly improve the efficiency of fluorescent OLEDs. That is to say, the low-lying LE state determines the efficient fluorescence radiation, wide band-gap and colour-purity, while the high-lying CT state is responsible for the triplet exciton utilization through the RISC process. Thus, this golden combination is surely beneficial to maximize the EL efficiency of OLEDs, and it can be a novel strategy to design emitters with high efficiency (ηPL and high ηs) and good colour-purity by taking advantage of the D–A structure.
The donor–spacer–acceptor (D–π–A) compounds with HLCT emissive state exhibit high exciton utilization efficiency (ηS) attributed by hot exciton mechanism.31–33 The external quantum efficiency (ηEQE) and exciton utilization efficiency (ηS) have been calculated by: ηEQE = ηout × ηIQE = ηout × ηrec × ηγ × ΦPL; ηs = ηout × ηrec × ηPL, respectively.34 The LE dominated HLCT provides high radiative rate (kr) which results high ηPL whereas the CT dominated HLCT is responsible for small singlet–triplet energy splitting (ΔEST).35–37 Designing D–π–A molecules is an effective strategy for balancing the carrier transport in the device due to their bipolar ability,38 however, the assembly of donor and acceptor units extends the π-conjugation and it is not beneficial for blue emission. Thus, it is difficult to achieve deep-blue emission with CIEy of 0.06: reduced conjugation,39,40 twisted conformation41 and linkage modification42 are the strategies adopted to achieve blue emission. Hence, it is aimed to design D–π–A geometry with side capping of bulky dihydrobenzodioxin in phenanthrimidazole core; expected to enhance the color purity, thermal stability, high quantum efficiency, bipolar properties and high exciton utilizing efficiency.43–53 A blue emissive material with balanced carrier transport characteristics and high triplet energy (ET) may be employed as host for green and red phosphorescent emitters.54 The efficient host for green and red phosphors exhibit low efficiency when they used as blue OLEDs.55 Therefore, it is still a challenging task to achieve efficient full color OLEDs with blue emissive material. Inspired by this, herein, we report multi-functional organic OLED materials namely, N-(4-(1-(1-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)-4,5-diphenyl-1H-imidazol-2-yl)naphthalen-4-yl)phenyl)-N-phenylbenzenamine (DDBP) obtained by single bond fission of phenanthrene moiety of N-(4-(1-(1-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)-H-phenanthro[9,10-d]imidazole-2-yl)naphthalen-4-yl)phenyl)-N-phenylbenzenamine (DBDPA) and 2-(1-(9H-carbazol-9-yl)naphthalen-4-yl)-1-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)-1H-phenanthro[9,10-d]imidazole (CDDPI) used as (i) emitters in blue OLEDs and (ii) host for green and red OLEDs. The strong orbital-coupling transitions of DDPB exhibit deep blue emission at 435 nm with CIEy 0.07 and maximum external quantum efficiency of 2.01%. The electroluminescent efficiencies of CDDPI (L – 3992 cd m−2; ηex – 3.01%; ηc – 2.56 cd A−1; ηp – 2.12 lm W−1) are higher than DBDPA based device (L – 3015 cd m−2; ηex – 2.85%; ηc – 2.01 cd A−1; ηp – 1.92 lm W−1). The blue emissive materials, CDDPI and DBDPA are used as a host to construct green and red phosphorescent OLEDs. The green device with CDDPI:Ir(ppy)3 exhibits maximum luminance of 8812 cd m−2, maximum ηc and ηp of 27.5 cd A−1 and 33.0 lm W−1, respectively and red device based on CDDPI:Ir(MQ)2(acac) exhibits excellent EL efficiencies [L – 39661 cd m−2;ηex – 19.2%; ηc – 27.9 cd A−1; ηp – 29.2 lm W−1; CIE (0.64, 0.34)].
2. Experimental
2.1. Synthesis of host dihydrobenzodioxin phenanthroimidazoles
The schematic synthetic route for the emissive and or host materials namely, N-(4-(1-(1-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)-4,5-diphenyl-1H-imidazol-2-yl)naphthalen-4-yl)phenyl)-N-phenylbenzenamine (DDPB), N-(4-(1-(1-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)-H-phenanthro[9,10-d]imidazole-2-yl)naphthalen-4-yl)phenyl)-N-phenylbenzenamine (DBDPA) and 2-(1-(9H-carbazol-9-yl)naphthalen-4-yl)-1-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)-1H-phenanthro[9,10-d]imidazole (CDDPI) are displayed in Scheme 1. Benzil (1 mmol) for BDBD or phenanthrene-9,10-dione (1 mmol) for BDBPI, 4-bromo-1-naphthaldehyde (1 mmol), 1,4-benzodioxane-6-amine (1 mmol) and ammonium acetate (1 mmol), all in 25 mL acetic acid was refluxed (120 °C; 24 h) and the crude 2-(1-bromonaphthalen-4-yl)-1-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)-4,5-diphenyl-1H-imidazole (BDBD) or 2-(1-bromonaphthalen-4-yl)-1-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)-1H-phenanthro[9,10-d]imidazole (BDBPI) was column chromatographed (hexane:ethyl acetate) and the pure sample was used for the synthesis of DDPB, DBDPA and CDDPI, respectively.
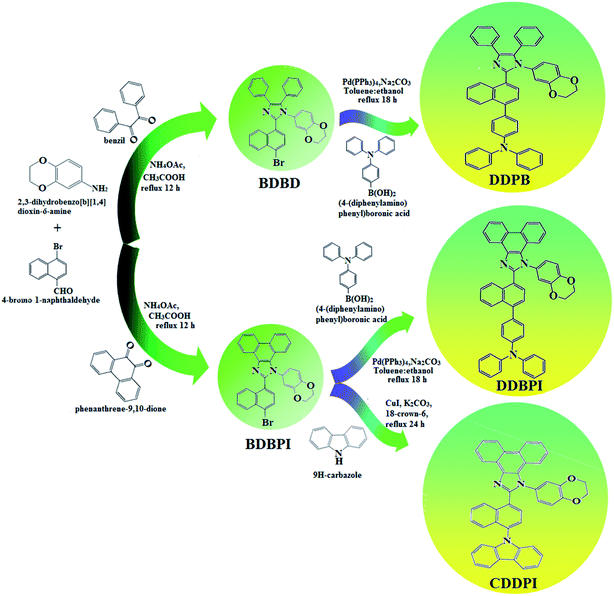 |
| | Scheme 1 Synthetic routes for BDBD, BDBPI, DDPB, DBDPA and CDDPI. | |
2.1.1. N-(4-(1-(1-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)-4,5-diphenyl-1H-imidazol-2-yl)naphthalen-4-yl)phenyl)-N-phenylbenzenamine (DDPB). 2-(1-Bromonaphthalen-4-yl)-1-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)-4,5-diphenyl-1H-imidazole (BDBD) (4.5 mmol), Pd(PPh3)4 (0.25 mmol) and 4-(diphenylamino)phenylboronic acid (7.5 mmol), all in aqueous Na2CO3 (15 mL) in toluene:ethanol (20:15 mL) was refluxed under N2 stream for 24 h. The dichloromethane extract was distilled off to get white DDPB. Yield 60%. Anal. calcd: C51H37N3O2: C, 84.62; H, 5.15; N, 5.81. Found: C, 84.59; H, 5.10; N, 5.77. 400 MHz 1H NMR (CDCl3): δ 4.35–4.38 (m, 4H), 6.46 (d, J = 8.2 Hz, 2H), 6.62–6.70 (m, 9H), 7.01 (t, 4H), 7.23–7.32 (m, 10H), 7.48 (d, J = 8.4 Hz, 4H), 7.60–7.67 (m, 4H) (Fig. S1†). 100 MHz 13C NMR (CDCl3): δ 64.21, 101.52, 114.71, 115.87, 122.62, 122.82, 125.32, 127.54, 128.24, 128.86, 131.08, 131.84, 133.15, 133.28, 134.86, 136.58, 141.45, 144.65, 147.61 (Fig. S2†). MALDI TOF MS: m/z. 723.29 [M+] (Fig. S7†). Calcd: 723.25.
2.1.2. N-(4-(1-(1-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)-H-phenanthro[9,10-d]imidazole-2-yl)naphthalen-4-yl)phenyl)-N-phenylbenzenamine (DBDPA). 2-(1-Bromonaphthalen-4-yl)-1-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)-1H-phenanthro[9,10-d]imidazole (BDBPI) (4.5 mmol), Pd(PPh3)4 (0.25 mmol), 4-(diphenylamino) phenylboronic acid (7.5 mmol) and aqueous Na2CO3 (15 mL), all in toluene:ethanol (20:15 mL) was refluxed for 2 days under N2 stream. The dichloromethane extract was distilled off to get DBDPA. Yield 52%. Anal. calcd: C51H35N3O2: C, 84.86; H, 4.89; N, 5.82. Found: C, 84.83; H, 4.87; N, 5.80. 400 MHz 1H NMR (CDCl3): δ 4.36–4.39 (m, 4H), 6.46 (d, J = 8.8 Hz, 2H), 6.61–6.69 (m, 9H), 7.12 (t, 4H), 7.24–7.34 (m, 4H), 7.61 (d, J = 8.8 Hz, 4H), 7.82–7.88 (m, 4H), 8.12 (d, J = 8.4 Hz, 2H), 8.93 (d, J = 8.0 Hz, 2H) (Fig. S3†). 100 MHz 13C NMR (CDCl3): δ 64.41, 101.70, 114.81, 115.86, 122.35, 122.61, 125.29, 126.41, 126.54, 126.72, 127.64, 128.41, 129.76, 129.81, 131.54, 133.45, 134.98, 136.86, 140.08, 141.25, 147.75, 149.51 (Fig. S4†). MALDI TOF MS: m/z. 721.27 [M+] (Fig. S7†). Calcd: 721.21.
2.1.3. 2-(1-(9H-Carbazol-9-yl)naphthalen-4-yl)-1-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)-1H-phenanthro[9,10-d]imidazole (CDDPI). A mixture of 2-(1-bromonaphthalen-4-yl)-1-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)-1H-phenanthro[9,10-d]imidazole (BDBPI) (4.5 mmol), 9H-carbazole (7.5 mmol), CuI (10.0 mg, 0.05 mmol), 18-crown-6 (13.2 mg, 0.05 mmol), and K2CO3 (0.83 g, 6.0 mmol) in tetrahydro-1,3-dimethylpyrimidin-2(1H)-one (2.0 mL) was refluxed in nitrogen atmosphere for 18 h. The solvent was distilled off and the pure CDDPI was used further analysis. Yield 58%. Mp 305 °C. Anal. calcd: C45H29N3O2: C, 83.96; H, 4.54; N, 6.53. Found: C, 83.82; H, 4.47; N, 6.40. 400 MHz 1H NMR (CDCl3): δ 4.41–4.50 (m, 4H), 6.16 (d, J = 8.2 Hz, 3H), 6.62–6.70 (m, 6H), 7.14 (t, 4H), 7.22–7.36 (m, 4H), 7.68 (d, J = 8.8 Hz, 4H), 8.22 (d, J = 8.4 Hz, 2H), 8.82 (d, J = 8.0 Hz, 2H) (Fig. S5†). 100 MHz 13C NMR (CDCl3): δ 64.25, 101.61, 111.15, 114.71, 115.88, 119.51, 120.35, 121.21, 122.25, 123.51, 125.54, 126.72, 126.84, 127.65, 128.31, 129.76, 130.51, 131.54, 132.85, 133.98, 135.86, 139.56, 146.08, 147.25, 149.46 (Fig. S6†). MALDI TOF MS: m/z. 643.17 [M+] (Fig. S7†). Calcd: 643.23.
2.2. Measurements and general methods
All reagents used for designing the manuscript are purchased from Sigma-Aldrich and NMR was recorded with 400 MHz spectrometer (Bruker). Agilent (LCMS VL SD) spectrometry was employed to analyze the mass of the blue emitters and or host materials. UV-vis absorption was measured on a Perkin-Elmer Lambda 35 (solution) and Lambda 35 spectrophotometer with integrated sphere (RSA-PE-20) instrument (film). PerkinElmer LS55 fluorescence spectrometer was employed to analyze the emission properties. Thermogravimetric analysis (TGA) and differential scanning calorimetric (DSC) were recorded with PerkinElmer thermal analysis system and NETZSCH-DSC-204, respectively. Time correlated single photon counting (TCSPC) spectrometer (Horiba Fluorocube-01-NL lifetime system and nano LED is excitation source with TBX-PS is detector: DAS6 software and χ2 – 0.8–1.2) was employed to examine the decay time of the emitters. The absolute PLQY was determined with fluorescence spectrometer Model-F7100. Cyclic voltammetry (CV) was recorded with potentiostat CHI 630A electrochemical analyzer with 100 mV s−1 scan (Ag/Ag+-reference electrode, platinum electrode-working electrode and platinum wire-counter electrode, ferrocene-internal standard HOMO-4.80 eV and 0.1 M tetrabutylammonium perchlorate-supporting electrolyte) and the HOMO [EHOMO = −(Eox + 4.8 eV)] and LUMO energies [ELUMO = EHOMO − 1239/λonset] of the blue emitters were calculated.
2.3. Computational details
For theoretical calculation, ground state (DFT)/excited state (TD-DFT) geometrical properties were optimized by employing Gaussian 09 program.56 Multifunctional wavefunction analyzer (Multiwfn)57 was used to know the nature of electronic transition of excited states and natural transition orbitals (NTOs).
2.4. Fabrication of devices
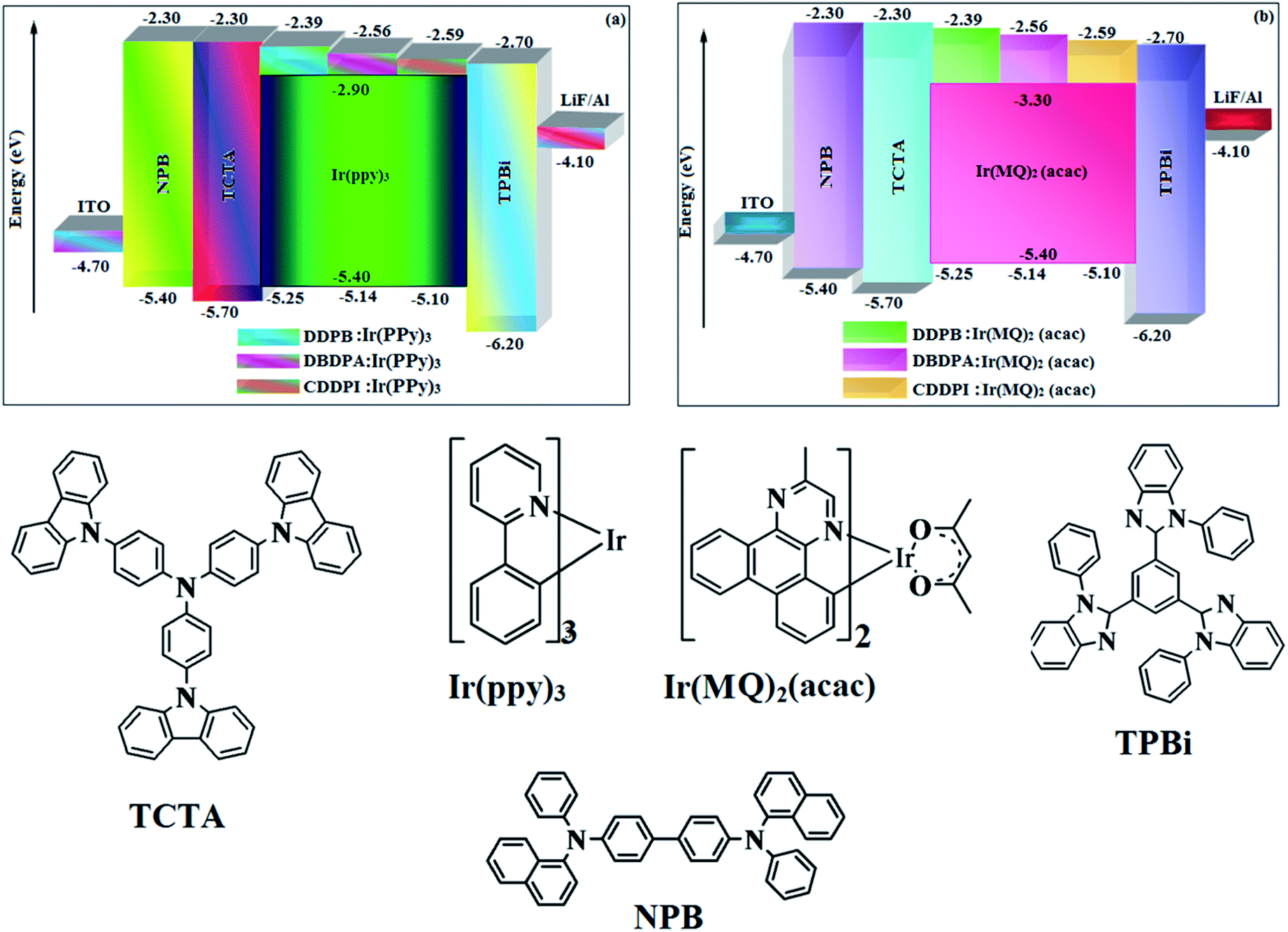
2.4.1. Device fabrication and measurement. Devices with configuration of (i) [ITO/NPB (70 nm)/DDPB/DBDPA/CDDPI (100 nm)/TPBI (20 nm)/LiF (0.5 nm)/Al], (ii) hole-only device: [ITO/HATCN (10 nm)/NPB (20 nm)/DBDPA/CDDPI (60 nm)/NPB (20 nm)/Al (100 nm)], (iii) electron-only device: [ITO/TPBi (10 nm)/DBDPA/CDDPI (60 nm)/TPBi (10 nm)/LiF (1 nm)/Al (100 nm)], (iv) green device: [ITO/NPB (40 nm)/TCTA (5 nm)/DBDPA (30 nm): 5 wt% Ir(ppy)3/CDDPI (30 nm): 5 wt% Ir(ppy)3/TPBI (50 nm)/LiF (1 nm)/Al (100 nm)] and (v) red device: [ITO/NPB (40 nm)/TCTA (5 nm)/DBDPA (30 nm): 8 wt% Ir(MQ)2(acac)/CDDPI (30 nm): 8 wt% Ir(MQ)2(acac)/TPBI (50 nm)/LiF (1 nm)/Al (100 nm)] were fabricated on pre-cleaned ITO-coated glass substrates with resistance of 20 Ω sq−1. Current density–voltage characteristics were measured with Keithley 2400 power source. The EL spectra and CIE coordinates were recorded with spectrometer (USB-650-VIS-NIR, Ocean Optics, Inc, USA).
3. Results and discussion
3.1. Potential energy scan (PES) and HLCT character
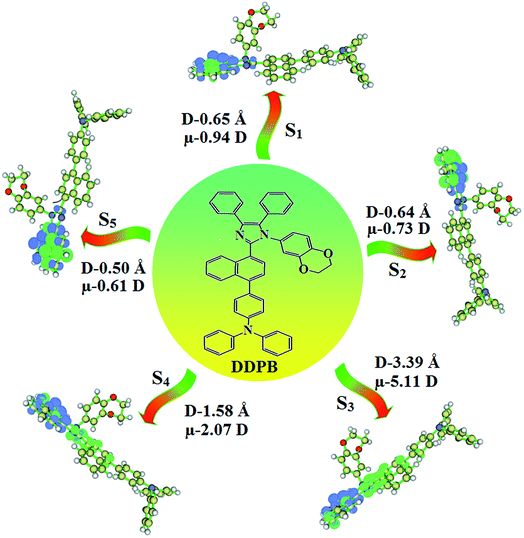
The ground state (S0) and excited state (S1) geometries of D–π–A compounds, DBDPA, CDDPI and DDPB were optimized with DFT/B3LYP/6-31G(d,p) and TD-DFT/B3LYP/6-31G(d,p) methods using Gaussian-09 (Fig. 1). The non-coplanar geometry of DDPB, DBDPA and CDDPI was confirmed by DFT studies. The naphthyl linkage adopts planar geometry and the dihydrobenzodioxin at imidazole nitrogen is perpendicular about 77.6° (DDPB), 87° (DBDPA) and 80° (CDDPI) with respect to their corresponding imidazole plane due to the interaction between aryl rings and repulsion between adjacent hydrogen atoms. Theoretical studies confirmed that DDPB exist in fully twisting geometry whereas DBDPA and CDDPI exist in partially twisting molecular structure, respectively with rigid phenanthro[9,10-d]imidazole plane.58 The intramolecular charge migration was detailed by potential energy surface scan of DDPB, DBDPA and CDDPI. The twist angles (θ°), namely, (i) C–θ° (θ1) (between phenanthrimidazole plane and naphthyl at imidazole carbon), (ii) N–θ° (θ2) (between phenanthrimidazole plane and dihydrobenzodioxin at imidazole nitrogen) and (iii) NA–θ° (θ3) (between naphthyl and phenyl group TPA for DDPB and DBDPA:naphthyl and carbazole for DDPB) imparts a major role in HOMO–LUMO π-electron overlap (Fig. 2).58 Because of the steric interaction of neighboring hydrogen atoms, the dihydrobenzodioxin at imidazole nitrogen is perpendicular to imidazole fragment and the conjugation was restricted. Because of the stronger repulsion between the neighboring hydrogen atoms of phenyl ring in DBDPA and CDDPI, the ground state twist angle (θ1) of DBDPA (88°) and CDDPI (73°) is higher when compared with DDPB (62°), however, increased excited state twist angle (θ1) of DDPB (108°), DBDPA (98°) and CDDPI (82°) was obtained.59
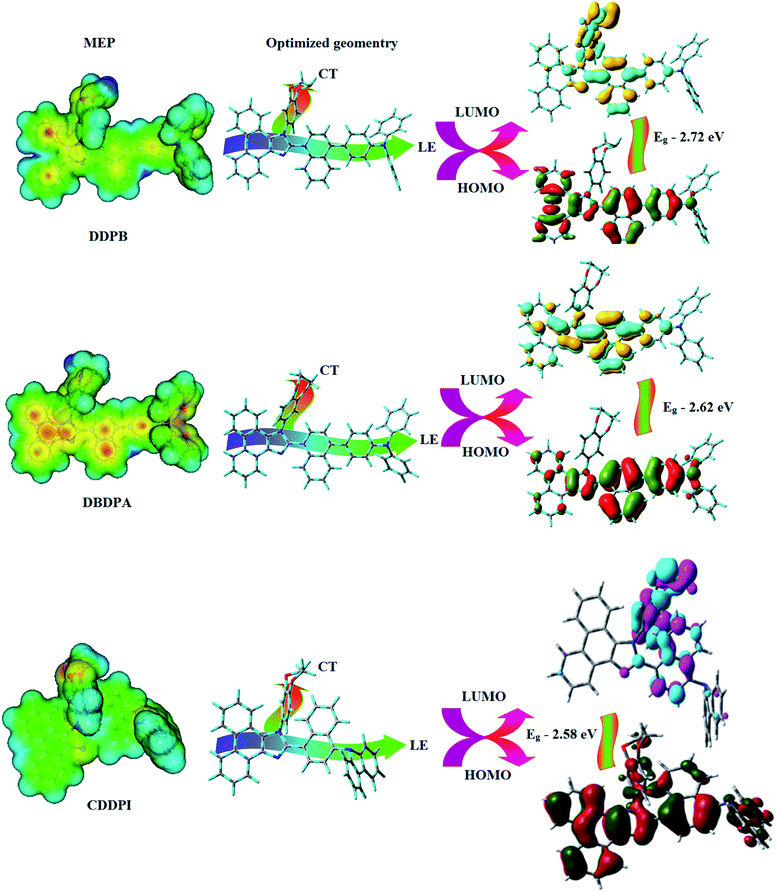 |
| | Fig. 1 HOMO, LUMO contour maps and molecular electrostatic potential (ESP) surface of DDPB, DBDPA and CDDPI. | |
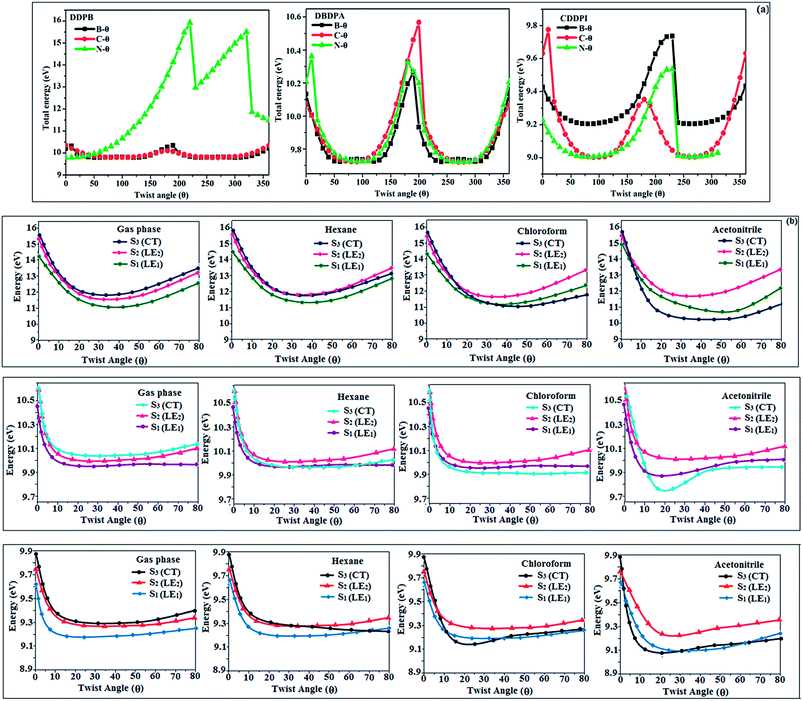 |
| | Fig. 2 (a) Potential energy scan (PES) at different twist angles C–θ, B–θ and N–θ of DDPB, DBDPA and CDDPI; (b) potential energy scan (PES) of excited states of DDPB, DBDPA and CDDPI with increasing solvent polarity. | |
The TPA moiety in DDPB and DBDPA and Cz moiety in CDDPI was twisted with dihedral angle of 130.8°, 148.2° and 159.1°, respectively.60 The larger twist angle of CDDPI when compared with DDPB and DBDPA is due to the stronger repulsion between the two adjacent hydrogen atoms in carbazole and naphthyl spacer as a result of stronger rigidity of Cz than TPA. The excited state twist angle (θ2) of DDPB, DBDPA and CDDPI are increased to 79.1°, 89.0° and 91.0°, respectively when compared with ground state twist angle (θ2). Similarly smaller increased twist angle (θ1) was obtained for CDDPI (173°) when compared with DDPB (108.1°) and DBDPA (98.1°). The elongated bond length (R1) of DDPB (0.06), DBDPA (0.03 Å) and CDDPI (0.02 Å) was calculated from S0 to S1. The smaller change in geometry of Cz in CDDPI from ground state (S0) to excited state (S1) was observed than that of TPA unit in DBDPA. This may facilitate the suppression of non-radiation for the enhancement of ηPL.60 From the potential energy surfaces of the twisted geometry of CDDPI at ground and excimer at excited states (Fig. 2) reveal that the CDDPI needs very small relaxation energy to form excimer of excited state corresponding to slightly increased interplanar separation of carbazole moieties from the linker fragment (1.4680 Å to 1.4880 Å, 36.6° to 38.6°, Fig. S8†) and then only small energy needs to return to an equilibrium geometry at ground state. At this point, the CDDPI with rigid geometry from initial state to excimer indicating that the minimized non-radiative energy dissipation contributes to the enhanced emission. Hence, the lifetime of excitons are increased due to the suppression of non-radiative pathways. Since the non-radiative pathway of CDDPI was blocked effectively, CDDPI shows higher photoluminescence efficiency (ηPL).61 The molecule shows twisted structure which is essential for separating HOMO and LUMO distribution effectively which can also achieve fully excition utilization (ηs) through efficient upconversion of non-radiative triplets to radiative singlets. The overlap between HOMO and LUMO is extremely small for CDDPI, ensuring the small ΔEST of the molecule. Meanwhile, the slight HOMO–LUMO overlap also ensures fairish radiative decay from intramolecular CT excitons. Moreover, large steric hindrances of carbazole group, naphthyl and phenanthrimidazole core lead to a rigid structure of CDDPI compound, which can limit torsional flexibility, thus preventing non-radiative transition62
This orthogonal dihedral angle confirmed the non-coplanar twisting conformation of DDPB, DBDPA and DBDPA which suppresses the red shift and harvested high external quantum efficiency (ηex) in film by restraining intermolecular interaction.63–66 Fig. 2 shows that the relative energy of DDPB is higher than that of DBDPA and CDDPI. The twist angles (θ2 and θ3) in D-A linkage of DDPB, DBDPA and CDDPI could be the origin for CT and LE intercross. In DDPB, DBDPA and CDDPI, the S1 state remained unchanged with increasing solvent polarity whereas the S3 state decreased to intercross with S1 state at moderate polarity and to be much lower than S1 state at high polarity (Fig. 2).
3.2. Thermal properties and electrochemical properties
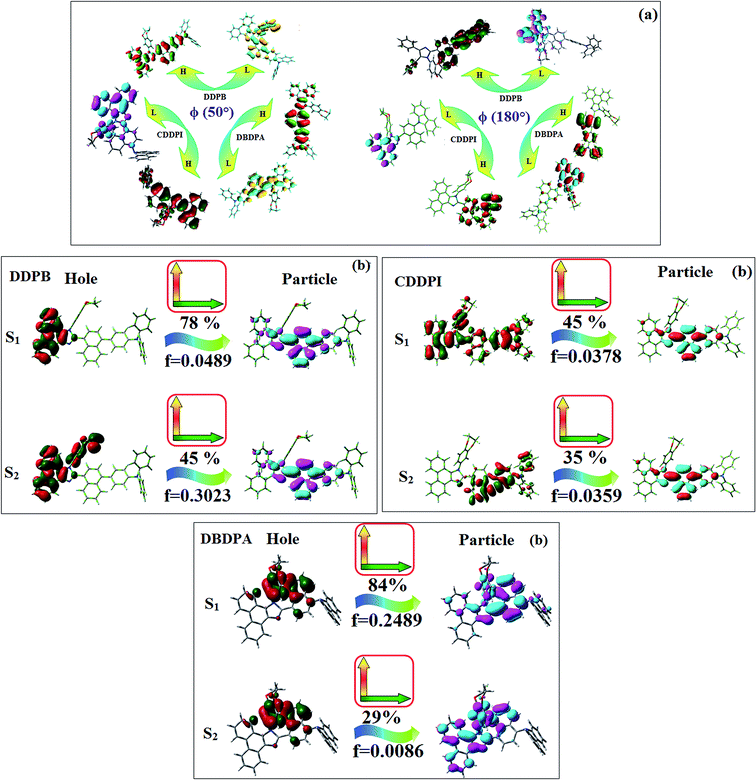
The incorporation of highly rigid bulky moiety at imidazole carbon and side capping at imidazole nitrogen enlarged the size and improved their thermal stability (Td5 & Tg) which is required for efficient devices (Table 1). Among the donor–spacer–acceptor compounds, DDPB (Tg – 151 and Td5 – 458 °C), DBDPA (Tg – 184 and Td5 – 490 °C) and CDDPI (Tg – 197 and Td5 – 495 °C), CDDPI exhibit high glass transition temperature (Tg) and thermal decomposition temperature (Td5) (Fig. 3). The higher thermal stability of CDDPI is because of the stronger rigidity of Cz than TPA which will be in favor of OLED stability.66–69 The improved Tg was probably due to the intermolecular interaction which can induce more condensed molecular packing and also implying that these compounds could form morphologically stable films under Joule heating.70–73 The thermal morphological stability of DDPB, DBDPA and CDDPI thin film was examined by atomic force microscopy (AFM) measurement at room temperature and also at 90 °C for 12 h. The root-mean-square roughness (RMS) of DDPB (0.29 nm), DBDPA (0.21 nm) and CDDPI (0.34 nm) show that there is no substantial changes before and after annealing (90 °C, 12 h) which also supports the suitability of these emissive materials for fabrication of OLEDs74–76 (Fig. 3). The carrier injection at interface between different layers in OLEDs is essential for high performance devices, thus, it is important for the emissive layer to have shallow HOMO energy (EHOMO) for improving the hole injection.77 The electronic energies (HOMO and LUMO: Fig. 1) of the non-doped blue emitters such as DDPB, DBDPA and CDDPI have been examined from redox potentials measured from potential vs. current plot (Fig. 3). From the oxidative onset potential, the EHOMO of −5.25 (DDPB), −5.14 eV (DBDPA) and −5.10 eV (CDDPI) (EHOMO = Eox + 4.8 eV) can be calculated and ELUMO −2.39 (DDPB), −2.56 eV (DBDPA) and −2.59 eV (CDDPI) have been deduced from ELUMO = EHOMO − 1239/λonset.78 The fully twisting molecular configuration of DDPB is likely to be the reason for higher energy gap of 2.86 eV (−2.58 eV – DBDPA; −2.51 eV – CDDPI). A single bond difference in the geometry of DDPB from DBDPA and CDDPI could change their photophysical properties and frontier energies. The space charge separation is found in these molecules which would be benefit for the injection of carrier from electrode.79 The partial overlap of HOMO/LUMO on phenanthrimidazole ring supports the charge transfer from π-linked binaphthyl to phenanthrimidazole moiety and the long π-linkers are beneficial to enhance the quantum yield. The DDPB, DBDPA and CDDPI ground state energy and their electron density delocalization at twist angle of 50° and 180° over HOMO and LUMO are shown in Fig. 4. Based on DFT energy, ϕ (50°) conformation of DDPB, DBDPA and CDDPI is more stable than ϕ (180°) conformation; in HOMO, the electron density is located on TPA and Cz and in LOMO the electron density is localized on imidazole moiety i.e., the HOMO and LUMO orbitals are separated from each other, therefore, CT transition of  is possible. However, at 50° (low energy twisting conformation) the electron density is partially localized on the frontier orbitals of DDPB, DBDPA and CDDPI. Therefore at 50°,
is possible. However, at 50° (low energy twisting conformation) the electron density is partially localized on the frontier orbitals of DDPB, DBDPA and CDDPI. Therefore at 50°,  transition is not pure CT but it is intercrossed between CT and LE transitions.59 The HOMO and LUMO of DDPB, DBDPA and CDDPI display adequate separation features and the differences are quite small which benefits the hole- and electron-transport properties (bipolar properties) and reduces the singlet–triplet splitting (ΔEST).80 Hence, the HOMO and LUMO moieties individually undertake the electron and hole transport functions. The calculated electron/hole transfer integrals of CDDPI (0.23/0.31 eV), DDPB (0.24/0.39 eV) and DBDPA (0.26/0.48 eV) reveal that these materials are bipolar materials. Moreover, these compounds displays both reduction and oxidation behaviour, revealing that these compounds possess good electron and hole transport abilities, hence, the synthesized materials are bipolar transport materials.81
transition is not pure CT but it is intercrossed between CT and LE transitions.59 The HOMO and LUMO of DDPB, DBDPA and CDDPI display adequate separation features and the differences are quite small which benefits the hole- and electron-transport properties (bipolar properties) and reduces the singlet–triplet splitting (ΔEST).80 Hence, the HOMO and LUMO moieties individually undertake the electron and hole transport functions. The calculated electron/hole transfer integrals of CDDPI (0.23/0.31 eV), DDPB (0.24/0.39 eV) and DBDPA (0.26/0.48 eV) reveal that these materials are bipolar materials. Moreover, these compounds displays both reduction and oxidation behaviour, revealing that these compounds possess good electron and hole transport abilities, hence, the synthesized materials are bipolar transport materials.81
Table 1 Photophysical and thermal properties and device performances of DDPB, DBDPA and CDDPI
| Parameters |
DDPB |
DBDPA |
CDDPI |
| Normalized absorption (λab) and emission (λem) spectra of DDPB, DBDPA and CDDPI in CH2Cl2 (10−5 M)/film. Tg/Td5 – glass transition temperature/thermal decomposition temperature at a weight percentage of 95%. ϕ (soln/film) – PL quantum yield was calculated in dichloromethane/solid state quantum yield has been measured on the quartz plate using an integrating sphere. EHOMO/LUMO – (EHOMO = Eox + 4.8 eV)/ELUMO = EHOMO − 1239/λonset. Eg – energy gap (EHOMO–ELUMO). ηex – external quantum efficiency; maximum internal quantum efficiency. ηIQE = ηex/ηout, ηout light out coupling efficiency (−20%); excitation utilization efficiency. ηs = ηIQE/ηPL. |
| Photophysical and thermal |
Properties |
| aλab (nm) (sol/film) |
253, 371/254, 373 |
254, 379/258, 380 |
249, 365/252, 369 |
| aλem (nm) (sol/film) |
435/449 |
442/461 |
429/446 |
| bTm/Tg/Td5 (°C) |
358/151/458 |
396/184/490 |
412/197/495 |
| cϕ (sol/film) |
0.68/0.61 |
0.75/0.74 |
0.82/0.73 |
| τ (ns) |
5.9 |
6.1 |
5.1 |
| dEHOMO/ELUMO (eV) |
−5.25/−2.39 |
−5.14/−2.56 |
−5.10/−2.59 |
| kr × 108 (s−1) |
1.1 |
1.2 |
1.6 |
| knr × 108 (s−1) |
0.5 |
0.4 |
0.3 |
|
| eEg (eV) |
−2.86 |
−2.58 |
−2.51 |
| Device efficiency |
| gηIQE (%) |
10.05 |
14.25 |
15.05 |
| hηs (%) |
16.48 |
19.26 |
26.62 |
| Von (V) |
4.3 |
3.7 |
3.0 |
| L (cd m−2) |
2010 |
3015 |
3992 |
| fηex (%) |
2.01 |
2.85 |
3.01 |
| ηc (cd A−1) |
1.61 |
2.01 |
2.56 |
| ηp (lm W−1) |
1.43 |
1.92 |
2.12 |
| EL (nm) |
447 |
459 |
444 |
| CIE (x, y) |
(0.16, 0.07) |
(0.15, 0.12) |
(0.15, 0.11) |
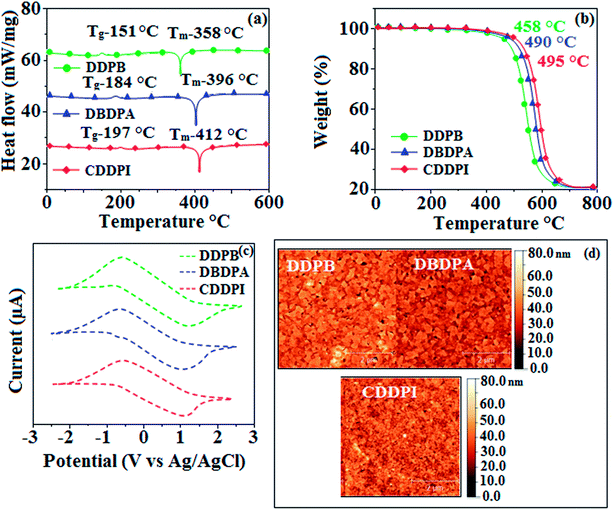 |
| | Fig. 3 (a) DSC graph; (b) TGA graph (c) cyclic voltammogram and (d) AFM images of DDPB, DBDPA and CDDPI. | |
 |
| | Fig. 4 (a) Frontier molecular orbitals of DDPB, DBDPA and CDDPI at 50° and 180° twist angles; (b) NTOs squint of DDPB, DBDPA and CDDPI. | |
3.3. Photophysical properties and HLCT character
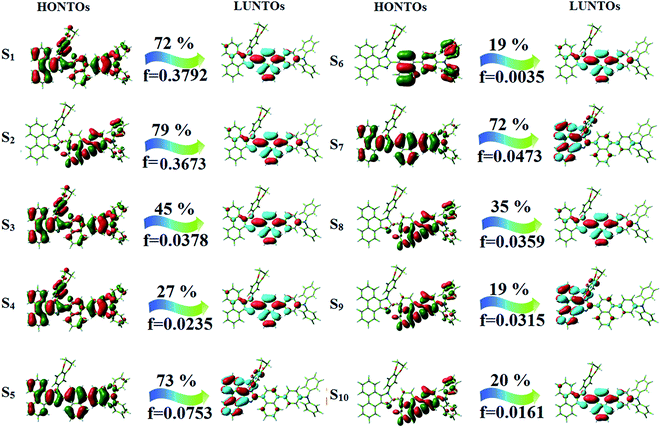
The photophysical properties of DDPB, DBDPA and CDDPI were investigated in solution and solid by absorption (λabs) and emission (λemi) studies (Fig. 5, Table 1).82 The strong absorption around 250 nm is due to π–π* transition originates from phenyl to imidazole ring. In addition, the absorption around 370 nm is attributed to the intramolecular charge transfer from donor to acceptor unit.83 The D–π–A derivatives (DDPB, DBDPA and CDDPI) exhibit higher blue shift with higher molar absorptivity when compared to their parent compounds and this might be due to the presence of strong and weak electron donor triphenylamine and a carbazole moiety which is expected to enhance the efficiencies.49 The extinction coefficient of D–π–A molecules is higher due to increase of conjugation length.84 The DDPB, DBDPA and CDDPI show very strong λobs (absorption) at 371 nm (εmax = 26954 cm−1 M−1), 379 nm (εmax = 26385 cm−1 M−1) and 365 nm (εmax = 27397 cm−1 M−1) on comparison with parent compounds BDBD and BDBPI due to intramolecular CT transition from donor to acceptor (Fig. 5). The film state of blue emissive materials show absorption at 373 nm (DDPB), 380 nm (DBDPA) and 369 nm (CDDPI) and the small shift shows the existence of weak π–π* intermolecular stacking.85 The parent compounds of DDPB and DBDPA/CDDPI exhibit emission at 382 and 401 nm with vibronic nature. The DDPB and DBDPA show red shifted emission at 435 and 442/446 nm, respectively without vibronic nature (Fig. 5). The observed solvatochromic red shift with increase of solvent polarity 45 nm (DDPB), 32 nm (DBDPA) and 24 nm (CDDPI) confirmed that the low-lying excited CT state of DDPB, DBDPA and CDDPI possesses large dipole moment (Fig. 6, S9 and S10†)51,86 and the red shifted emission (Tables S1–S3†) could be attributed to the twisted conformation which enable for the easier charge transfer from donor to accept or via naphthyl linker. The intramolecular charge transfer is further confirmed by molecular electrostatic potential (MEP) (Fig. 1). Compared with solution, the small red shift in their corresponding film reveal that suppressed π–π* stacking exist in solid state.87 The emission of DDPB, DBDPA and CDDPI is observed at 449, 461 and 446 nm in solid and the full-width at half-maximum (FWHM) is around 30 nm: the red-shift progress cannot be due to aggregation in its solid state and might be from the change of excited state configuration.9 The phenanthroimidazole derivatives DDPB, DBDPA and CDDPI show blue emission at 435, 442 and 429 nm, respectively in CH2Cl2 (Fig. 5). The PL spectra gradually widened and their peaks show red shift with increase of solvent polarity which indicates that their excited state have strong CT character when compared to ground state and further stabilized by polar solvents.87–89 The calculated singlet energy/triplet energy (ES/ET) of DDPB (2.52/2.29 eV), DBDPA (2.60/2.25 eV) and CDDPI (2.58/2.23 eV) shows that they have high triplet energy to sensitize phosphorescent dopants with ET below 2.3 eV. Compared with DBDPA, CDDPI exhibit higher blue shift in absorption and emission attributed to poor electron donor ability of Cz relative to TPA. The increased LE composition with decrease of CT in S1 emissive HLCT state is likely to be the reason for this blue shift. The FWHM in the absorption spectrum of CDDPI (24 nm) is narrowed compared to that of DBDPA (32 nm) and CDDPI (30 nm). This observation informed that decreased CT component of CDDPI in S1 state which is in good agreement with NTO description for S0 → S1 transition. The emission peak of DBDPA and CDDPI gives blue-shift relative to their parent compounds which is in controversy to the general observation i.e., extension of π-conjugation leads to red shifted emission.89 In addition to that there is an overlap between UV and PL spectra of both DBDPA and CDDPI because of enhanced LE character in DBDPA and CDDPI than their respective parent compounds. The CDDPI exhibits solvatochromic red shift (24 nm) which is smaller than DBDPA (32 nm) (Fig. S11, Tables S3 and S2†). Similarly, a small absorption shift about 28 nm and 20 nm has been observed for CDDPI and DBDPA, respectively (Fig. S11, Tables S3 and S2†). Solvatochromic shifts confirmed that low-lying S1 excited state of CDDPI and DBDPA must possess CT character.90,91 The % of CT character in S1 state of CDDPI is lower than DBDPA whereas % of LE character of CDDPI is higher than DBDPA (Table S4†). In S0–S1 and S0–S2 transitions, the HLCT was composed with CT state and LE state and exhibit larger oscillator strength [fS0–S1 = 0.5191 (DDPB): 0.6814 (CDDPI): 0.3792 (DBDPA) and fS0–S2 = 0.3023 (DDPB): 0.6712 (CDDPI): 0.3673 (DBDPA)] compared with other S0–Sn (n = 3, 4…) transitions as a result of major LE character in the HLCT state (Table 2; Fig. 7 – DBDPA; Fig. S12† – DDPB; Fig. S13† – CDDPI) which is necessary for higher efficiency OLEDs.92 In DBDPA and CDDPI, the hole and particle of S0–S1 and S0–S2 transitions of NTOs contained two transition configurations with close contributions; one squint towards the LE state and the other was like HLCT state (Fig. 4). These results implied that DBDPA and CDDPI exhibits a better mixed LE and CT state and the LE state dominated the fluorescence and intersection angle appeared between two directions in the phenanthrimidazole ring, namely one along C2-substituted direction and another along N1-substituted direction. Most of the CT component was nearly perpendicular to that of LE in the same mixed transition configuration which was a special excited state of the D–π–A structure. This novel mixed state led to low oscillator strength in DBDPA, (fS0–S1 – 0.3792) than that of CDDPI (fS0–S1 – 0.6814) and the transition energy barrier was very little from S1 to S4 (0.04 eV). Such slight differences usually cause stronger orbital coupling leading to free exciton transformation between the four excited states at room temperature and the emission species change or the number increases with changes in the external environment such as thermal activation. This internal conversion between different excited states would cause non-radiative decay ratio to increase photoluminescence efficiency with larger oscillator strength which is confirmed by the optical measurements.
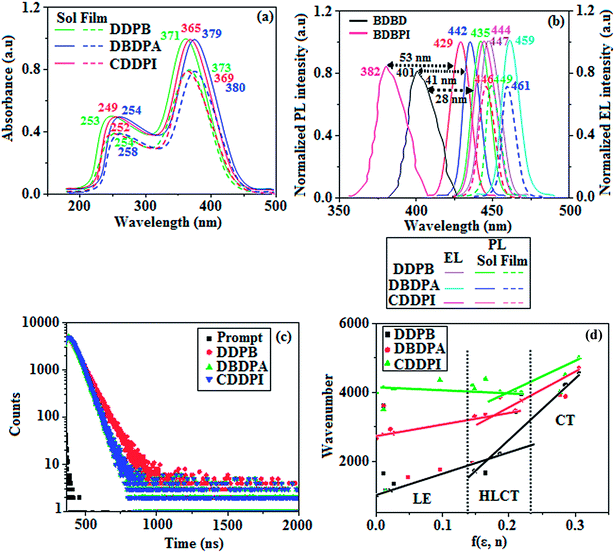 |
| | Fig. 5 (a) Normalized optical absorption spectra of DDPB, DBDPA and CDDPI; (b) normalized emission spectra BDBD and BDBPI in CH2Cl2 (10−5 M) along with EL spectra of DDPB, DBDPA and CDDPI and (c) life time decay curve of DDPB, DBDPA and CDDPI (d) Lippert–Mataga plot of DDPB, DBDPA and CDDPI. | |
 |
| | Fig. 6 Hole and particle distribution of DDPB [S1–S5 states:  -green increasing electron density and -green increasing electron density and  -blue decreasing electron density [density = transition = nIOp(6/8 = 3)]]. -blue decreasing electron density [density = transition = nIOp(6/8 = 3)]]. | |
Table 2 Calculated energies (E) and oscillator strength (f) of S1–S10 transitions from NTO of DDPB, DBDPA and CDDPI
| Transitions |
DDPB |
DBDPA |
CDDPI |
| E |
f |
NTO |
E |
f |
NTO |
E |
f |
NTO |
| S0 → S1 |
1.32 |
0.5191 |
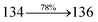 |
2.75 |
0.3792 |
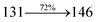 |
1.85 |
0.6814 |
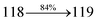 |
| S0 → S2 |
2.02 |
0.3023 |
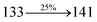 |
3.05 |
0.3673 |
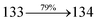 |
1.94 |
0.6712 |
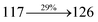 |
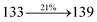 |
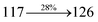 |
| S0 → S3 |
2.40 |
0.2983 |
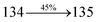 |
3.44 |
0.0378 |
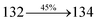 |
2.15 |
0.0485 |
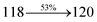 |
| S0 → S4 |
2.58 |
0.2428 |
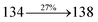 |
3.61 |
0.0235 |
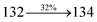 |
2.36 |
0.0620 |
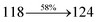 |
| S0 → S5 |
2.79 |
0.0292 |
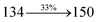 |
3.70 |
0.0753 |
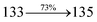 |
2.86 |
0.0414 |
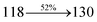 |
| S0 → S6 |
2.94 |
0.0678 |
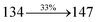 |
3.73 |
0.0035 |
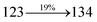 |
2.96 |
0.0188 |
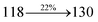 |
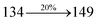 |
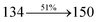 |
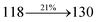 1 1 |
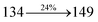 |
| S0 → S7 |
3.22 |
0.0489 |
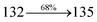 |
3.77 |
0.0473 |
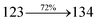 |
3.15 |
0.0553 |
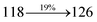 |
| S0 → S8 |
3.52 |
0.1293 |
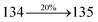 |
3.80 |
0.0359 |
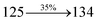 |
3.25 |
0.3998 |
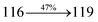 |
| S0 → S9 |
3.57 |
0.0142 |
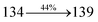 |
3.83 |
0.0315 |
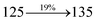 |
3.45 |
0.1324 |
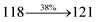 |
| S0 → S10 |
3.61 |
0.0271 |
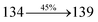 |
3.86 |
0.0161 |
 |
3.50 |
0.0119 |
 |
 |
| | Fig. 7 Highest occupied and lowest unoccupied natural transition orbitals of DBDPA. | |
The new born blue emitters show high quantum yield (solution/film) of DDPB (0.68/0.61), DBDPA (0.75/0.74) and CDDPI (0.82/0.73) and the high fluorescence yield is essential for efficient blue OLEDs (Table 1). The improved quantum yield is attributed to decreased proportion of non-radiative transition because of molecular interactions.78 Incorporation of binaphthyl into bulky phenanthrimidazole ring enhanced the intermolecular steric hindrance forced the molecule to form a more twisted structure when packing in solid which results in less aggregation and lower quantum yield in their solid state. The lower yield of DDPB is due to increased excited state intra molecular vibration results from the fully twisted DDPB whereas the high rigid geometry of DBDPA and CDDPI effectively reduced the radiative exciton which results high yield. The kr/knr (radiative transition rate and non-radiative transition rate) have been calculated from lifetime (τ) and quantum yield (ϕ) (Table 1). To analyze the relative contribution of radiative and non-radiative relaxation processes in the excited state deactivation, the radiative (kr) and non-radiative (knr) decay constants were calculated: kr = τ/Φ: 1.1 s−1 (DDPB); 1.2 s−1(DBDPA) and 1.6 s−1 (CDDPI); knr = τ/(1 − Φ): 0.5 s−1 (DDPB); 0.4 s−1 (DBDPA) and 0.3 s−1 (CDDPI) (Fig. 5). The CDDPI shows larger radiative rate constant (kr) and smaller non-radiative rate constant (knr) than those of DDPB and DBDPA.
3.4. Solvatochromism for HLCT character
The solvatochromic effect using Lippert–Mataga plot has been displayed in Fig. 5 (Tables S1–S3†). When solvent polarity increased the blue emitters exhibit a larger red shift which supports the charge transfer (CT) in these molecules.92 From Lippert–Mataga plot, the ground state dipole moment (μg) can be calculated: hc(![[small upsilon, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e131.gif) abs − flu) = hc(hcνacabs − hcνacflu) + 2(μe − μg)2/ao3[(ε − 1/2ε + 1) − 1/2(n2 − 1/2n2 + 1)] [μg and μe – ground state and excited state dipolemoment, abs and νacabs – solvent-equilibrated absorption maxima (λabs) and extrapolated to gas phase, flu and νacflu – solvent equilibrated fluorescence maxima (λemi) and extrapolated to gas-phase, respectively, ao – onsager cavity and ε and n solvent dielectric constant and refractive index, respectively]. The non-linear correlation of Stokes shift vs. solvent polarity function reveal that there is transformation of fitted line between ethyl ether and methylene chloride: non-linear correlation supports the presence of both locally excited state (LE) and charge transfer excited state (CT). The ground state dipole (μg) of blue emitting materials, DDPB, DBDPA and CDDPI could be estimated from density functional theory (DFT) calculation as, 3.12, 5.02 and 7.1 D, respectively which is attributed by local exciton (LE) transition and μe in high polar solvents is 21.9, 23.4 and 23.9 D, respectively.92 The large μe in high polar medium is in close with μe of charge-transfer molecule, 4-(N,N-dimethylamino)benzonitrile (23.0 D).93 All these results show that CT dominates in more polar medium and LE dominates in low polar solvent and there is mixed contribution of LE and CT in medium polar solvents. The high oscillator strength of S1 state of CDDPI results in higher PL efficiency (ηPL). Molecular modification from TPA to Cz causes an increasing % LE in S1 emissive state and enhanced ηPL of CDDPI. The overlap density between hole and particle depend upon the configuration of donor–acceptor architecture and the magnitude of overlap intensity tuned the % LE and % CT in S1 state (Fig. 6 – DDPB, Fig. S9† – DBDPA, Fig. S10† – CDDPI). The two different excited states of DDPB, DBDPA and CDDPI confirmed the two independent slopes of non-linear fitted line,91 i.e., LE and CT intercrossed excited state: in high polarity solvents CT state dominates, in low polar solvents LE state dominates and in moderate polar solvents intercrossed excited state of LE and CT exist. The intercrossed coupling of LE with CT states generates new hybridized local and charge transfer state (HLCT). The HLCT state in moderate polar solvent was confirmed by mono exponential time (Fig. 5) which supports the D–π–A molecular design.75,94 The LE and CT states of emissive materials DDPB, DBDPA and CDDPI show non-uniform properties in different polar solvents because of different excited-state dipole moments. As the polarity increases, CT state is stabilized due to strong interaction of the solvent field with CT excited state (large dipole moment) and LE remains unchanged,95 however, in low-polarity solvents, the LE being stabilized (low-lying excited state). The λemi of DBDPA and CDDPI film is close with that in ether which confirmed HLCT state formation.91
abs − flu) = hc(hcνacabs − hcνacflu) + 2(μe − μg)2/ao3[(ε − 1/2ε + 1) − 1/2(n2 − 1/2n2 + 1)] [μg and μe – ground state and excited state dipolemoment, abs and νacabs – solvent-equilibrated absorption maxima (λabs) and extrapolated to gas phase, flu and νacflu – solvent equilibrated fluorescence maxima (λemi) and extrapolated to gas-phase, respectively, ao – onsager cavity and ε and n solvent dielectric constant and refractive index, respectively]. The non-linear correlation of Stokes shift vs. solvent polarity function reveal that there is transformation of fitted line between ethyl ether and methylene chloride: non-linear correlation supports the presence of both locally excited state (LE) and charge transfer excited state (CT). The ground state dipole (μg) of blue emitting materials, DDPB, DBDPA and CDDPI could be estimated from density functional theory (DFT) calculation as, 3.12, 5.02 and 7.1 D, respectively which is attributed by local exciton (LE) transition and μe in high polar solvents is 21.9, 23.4 and 23.9 D, respectively.92 The large μe in high polar medium is in close with μe of charge-transfer molecule, 4-(N,N-dimethylamino)benzonitrile (23.0 D).93 All these results show that CT dominates in more polar medium and LE dominates in low polar solvent and there is mixed contribution of LE and CT in medium polar solvents. The high oscillator strength of S1 state of CDDPI results in higher PL efficiency (ηPL). Molecular modification from TPA to Cz causes an increasing % LE in S1 emissive state and enhanced ηPL of CDDPI. The overlap density between hole and particle depend upon the configuration of donor–acceptor architecture and the magnitude of overlap intensity tuned the % LE and % CT in S1 state (Fig. 6 – DDPB, Fig. S9† – DBDPA, Fig. S10† – CDDPI). The two different excited states of DDPB, DBDPA and CDDPI confirmed the two independent slopes of non-linear fitted line,91 i.e., LE and CT intercrossed excited state: in high polarity solvents CT state dominates, in low polar solvents LE state dominates and in moderate polar solvents intercrossed excited state of LE and CT exist. The intercrossed coupling of LE with CT states generates new hybridized local and charge transfer state (HLCT). The HLCT state in moderate polar solvent was confirmed by mono exponential time (Fig. 5) which supports the D–π–A molecular design.75,94 The LE and CT states of emissive materials DDPB, DBDPA and CDDPI show non-uniform properties in different polar solvents because of different excited-state dipole moments. As the polarity increases, CT state is stabilized due to strong interaction of the solvent field with CT excited state (large dipole moment) and LE remains unchanged,95 however, in low-polarity solvents, the LE being stabilized (low-lying excited state). The λemi of DBDPA and CDDPI film is close with that in ether which confirmed HLCT state formation.91
The excited-state properties of DBDPA (Fig. 7), DDPB (Fig. S12†) and CDDPI (Fig. S13†) were analyzed using natural transition orbitals (NTOs).41 The electron density delocalization over hole and particles confirmed that in S0 → S1 transition, LE state dominates with minor contribution of CT state in HLCT and shows higher when compared with S0 → S2 or other S0 → S10 transitions [fS0→S1 = 0.5191 > fS0→S2 = 0.3023 (DDPB): fS0→S1 = 0.3792 > fS0→S2 = 0.3673 (DBDPA): fS0→S1 = 0.6814 > fS0→S2 = 0.6712 (CDDPI)]. This is further evidence for higher % LE in HLCT state and is highly need for efficient OLED performances. The lower oscillator strength of DDPB reveal that DDPB exhibit completely hybridized LE and CT states than DBDPA and CDDPI and the eigen value (>0.95) further supports the better mixed LE and CT excited state to about 78% of transition which is supported by Δr > 2 for S1–S10 of DDPB, DBDPA and CDDPI (Tables S1–S3†). The electron density distribution on hole and particle of DBDPA and CDDPI were entirely differing from DDPB. The excitation energies of LE and CT states of the title materials were used as a tool to confirm the formation of HLCT state56 (Scheme 2, Tables 3–5).
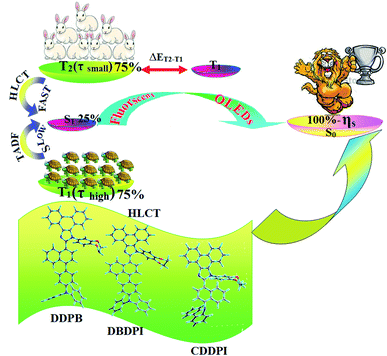 |
| | Scheme 2 Effect of TADF and HLCT on 100% exciton utilization efficiency (ηs). | |
Table 3 Excitation energy (E, eV), excitation co-efficient (ε), overlap integral (Δr, Å) for singlet and triplet states of DDPB
| State |
Singlet |
Triplet |
| E |
ε |
Δr |
E |
ε |
Δr |
| 1 |
1.3069 |
0.4716 |
2.2387 |
0.4016 |
0.4711 |
2.0157 |
| 2 |
2.0159 |
0.4033 |
2.6236 |
1.3068 |
0.4232 |
2.2177 |
| 3 |
2.4046 |
0.4522 |
4.8614 |
1.0737 |
0.4387 |
1.9531 |
| 4 |
1.7468 |
0.4260 |
4.9963 |
1.4491 |
0.4256 |
3.1194 |
| 5 |
2.7853 |
0.4327 |
3.4986 |
1.8658 |
0.4193 |
3.9187 |
| 6 |
2.9430 |
0.4250 |
3.0147 |
2.3521 |
0.3010 |
2.3141 |
| 7 |
1.7501 |
0.4195 |
1.4300 |
2.5457 |
0.4334 |
1.5606 |
| 8 |
3.5184 |
0.3508 |
4.5728 |
2.7755 |
0.3649 |
3.4988 |
| 9 |
3.5663 |
0.4113 |
4.2429 |
2.8379 |
0.2631 |
3.0845 |
| 10 |
3.6105 |
0.3381 |
4.4619 |
2.9442 |
0.1891 |
2.9672 |
Table 4 Excitation energy (E, eV), excitation co-efficient (ε), overlap integral (Δr, Å) for singlet and triplet states of DBDPA
| State |
Singlet |
Triplet |
| E |
ε |
Δr |
E |
ε |
Δr |
| 1 |
1.7539 |
0.4535 |
1.8665 |
1.0999 |
0.3407 |
1.2953 |
| 2 |
3.0502 |
0.4372 |
0.8836 |
1.7538 |
0.2743 |
3.5474 |
| 3 |
3.4377 |
0.3555 |
1.8897 |
2.6921 |
0.2809 |
2.7663 |
| 4 |
3.6106 |
0.3722 |
3.0340 |
2.7554 |
0.2250 |
3.1037 |
| 5 |
3.6978 |
0.3272 |
2.6956 |
2.8864 |
0.2590 |
2.5155 |
| 6 |
2.3895 |
0.3825 |
0.7581 |
2.0085 |
0.2620 |
3.2297 |
| 7 |
3.4778 |
0.3346 |
3.8811 |
2.2316 |
0.2572 |
1.8697 |
| 8 |
3.7976 |
0.2677 |
2.8953 |
2.2368 |
0.4446 |
1.8799 |
| 9 |
3.8287 |
0.2937 |
3.1646 |
2.4858 |
0.2958 |
3.0048 |
| 10 |
2.3105 |
0.3122 |
3.6380 |
2.5909 |
0.2057 |
1.4642 |
Table 5 Excitation energy (E, eV), excitation co-efficient (ε), overlap integral (Δr, Å) for singlet and triplet states of CDDPI
| State |
Singlet |
Triplet |
| E |
ε |
Δr |
E |
ε |
Δr |
| 1 |
1.4084 |
0.4348 |
2.0371 |
0.2898 |
0.4348 |
1.0780 |
| 2 |
1.9364 |
0.4457 |
5.6501 |
1.5683 |
0.4110 |
2.4346 |
| 3 |
1.4235 |
0.4520 |
4.9302 |
1.5863 |
0.9194 |
2.5722 |
| 4 |
2.3572 |
0.4627 |
2.0641 |
1.6340 |
0.6205 |
1.9457 |
| 5 |
2.8613 |
0.4260 |
1.9569 |
1.6904 |
0.6353 |
1.9177 |
| 6 |
2.9554 |
0.4091 |
3.9229 |
1.8919 |
0.7509 |
1.8641 |
| 7 |
3.1549 |
0.4237 |
2.4004 |
1.9710 |
0.6499 |
3.1570 |
| 8 |
3.2532 |
0.4152 |
3.9584 |
2.0172 |
0.6481 |
2.9625 |
| 9 |
3.4267 |
0.4419 |
4.9631 |
2.3568 |
0.8882 |
2.6737 |
| 10 |
3.4993 |
0.4282 |
2.6747 |
2.4278 |
0.6423 |
2.8688 |
In DDPB, CT state is stabilized than LE state and the energy gap is too small results effective hybridization whereas in DBDPA and CDDPI, the LE state is stabilized than CT state due to enhanced π-conjugation results weaker interstate coupling. The overlap between hole and particle of DDPB, DBDPA and CDDPI is displayed in Fig. 6, S9 and S10,† respectively. The more similar hole–electron wave function indicates the efficient hybridization between LE and CT states. The composition of HLCT in DDPB (Fig. 8), DBDPA (Fig. S14†) and CDDPI (Fig. S15†) can be analyzed by transition density matrix (TDM). The diagonal part reflects the LE component localized on main backbone while off-diagonal region represents CT component. Analysis of integral of electron, integral of hole and their overlap, integral of transition density and distance between hole and electron are displayed in Table S4† (DDPB), Table S5† (DBDPA) and Table S6† (CDDPI) and these excited state parameters reveal that the integral of hole and electron of DDPB is less than DBDPA and CDDPI whereas the electron density is close to the ideal value of zero which indicates the medium quality grid is enough for visualization. Computed electron–hole properties, distance between hole and electron, transition density, H and t indexes and RMSD of electron and hole of DDPB, DBDPA and CDDPI are displayed in Tables S7–S9,† respectively. Transition dipole moment calculated by integrating the same at uniform grid is almost same for both cases. The integral overlap of hole–electron distribution is a measure of spatial separation of hole and electron which is close to zero for DBDPA and CDDPI. Distance between centroid of hole and electron is a measure of CT length: the larger CT value indicates charge transfer takes place with longer length (Fig. 9, Table 6). The excited state characteristic of DDPB, DBDPA and CDDPI further supports that these materials capable of transporting electrons and holes in the fabricated devices. The coexisting LE/CT composition in DDPB, DBDPA and CDDPI harvested high ηPL and high ηs and enhanced OLEDs efficiencies. The new born blue emitters show high quantum yield (s/f) of DDPB (0.68/0.61), DBDPA (0.75/0.74) and CDDPI (0.82/0.73) and high fluorescence efficiencies are essential for efficient OLEDs (Table 1). The improved quantum yield is attributed to decreased proportion of the non-radiative transition because of molecular interactions, such as intermolecular interaction between cyano group and phenanthrimidazole plane. Incorporation of binaphthyl into bulky phenanthrimidazole enhanced the intermolecular steric hindrance forced the molecule to form a more twisted structure when packing in solid which results in less aggregation and lower quantum yield in solid state. It is a rare model for which the PL spectra are unchanged and the yields are increased by inserting bulky naphthyl group.
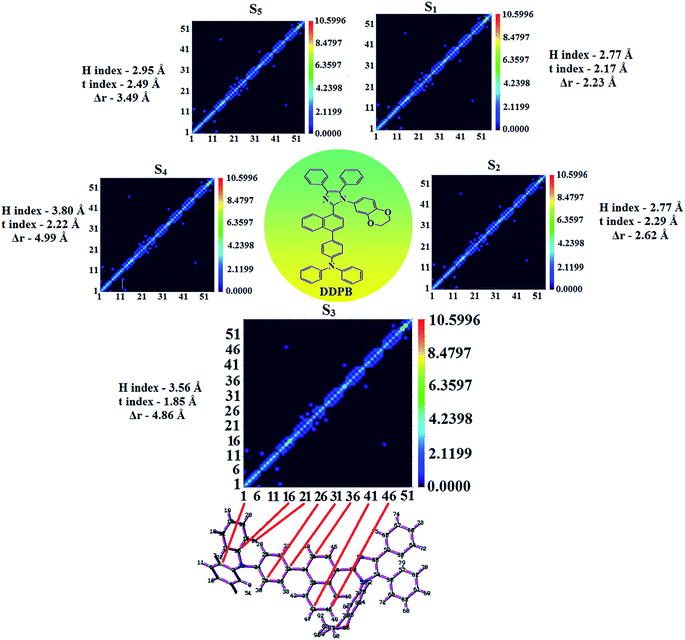 |
| | Fig. 8 Computed contour plots of transition density matrices (TDM) of DDPB [density = transition = n/IOp(6/8 = 3)]. | |
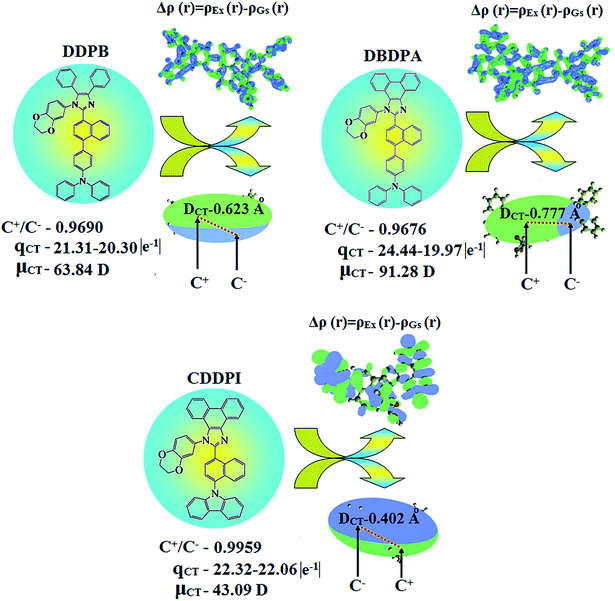 |
| | Fig. 9 Differences in total density for ground and excited states [Δρ(r) = ρEx(r) − ρGs(r); isosurface for DDPB, DBDPA, CDDPI, (0.0000006 a.u.) and for DDPB (0.15 a.u.)]; graphical representation of DCT and centroids of charges [C+(r)/C−(r); isosurface for DBDPA (0.29 a.u.) and for CDDPI (0.1 a.u.)]. | |
Table 6 Transferred charges (qCT), barycentres of electron density loss (R−)/gain (R+), distance between two barycenters (DCT), dipole moment of CT (μCT), RMSD of +ve/−ve parts, CT indices (H & t) and overlap integral of C+/C− of DDPB, DBDPA and CDDPI
| Blue emissive & host materials |
qCT |e−1| |
R+ (Å) |
R− (Å) |
DCT (Å) |
μCT (D) |
+ve RMSD |
−ve RMSD |
H/t indices (Å) |
Overlap integral (C+/C−) |
| x |
y |
z |
x |
y |
z |
| DDPB |
21.319–20.306 |
−0.03 |
−0.86 |
0.80 |
−0.12 |
−1.37 |
−0.25 |
0.623 |
63.84 |
11.87 |
12.53 |
6.45/6.19 |
0.9690 |
| DBDPA |
24.448–19.979 |
−2.35 |
1.05 |
−0.37 |
−2.93 |
−0.27 |
−0.13 |
0.777 |
91.28 |
11.05 |
12.93 |
6.32/5.75 |
0.9676 |
| CDDPI |
22.321–22.062 |
−1.89 |
0.99 |
0.28 |
−2.11 |
1.57 |
0.73 |
0.402 |
43.09 |
11.97 |
11.75 |
6.27/5.98 |
0.9959 |
3.5. Quasi-equivalent hybridization
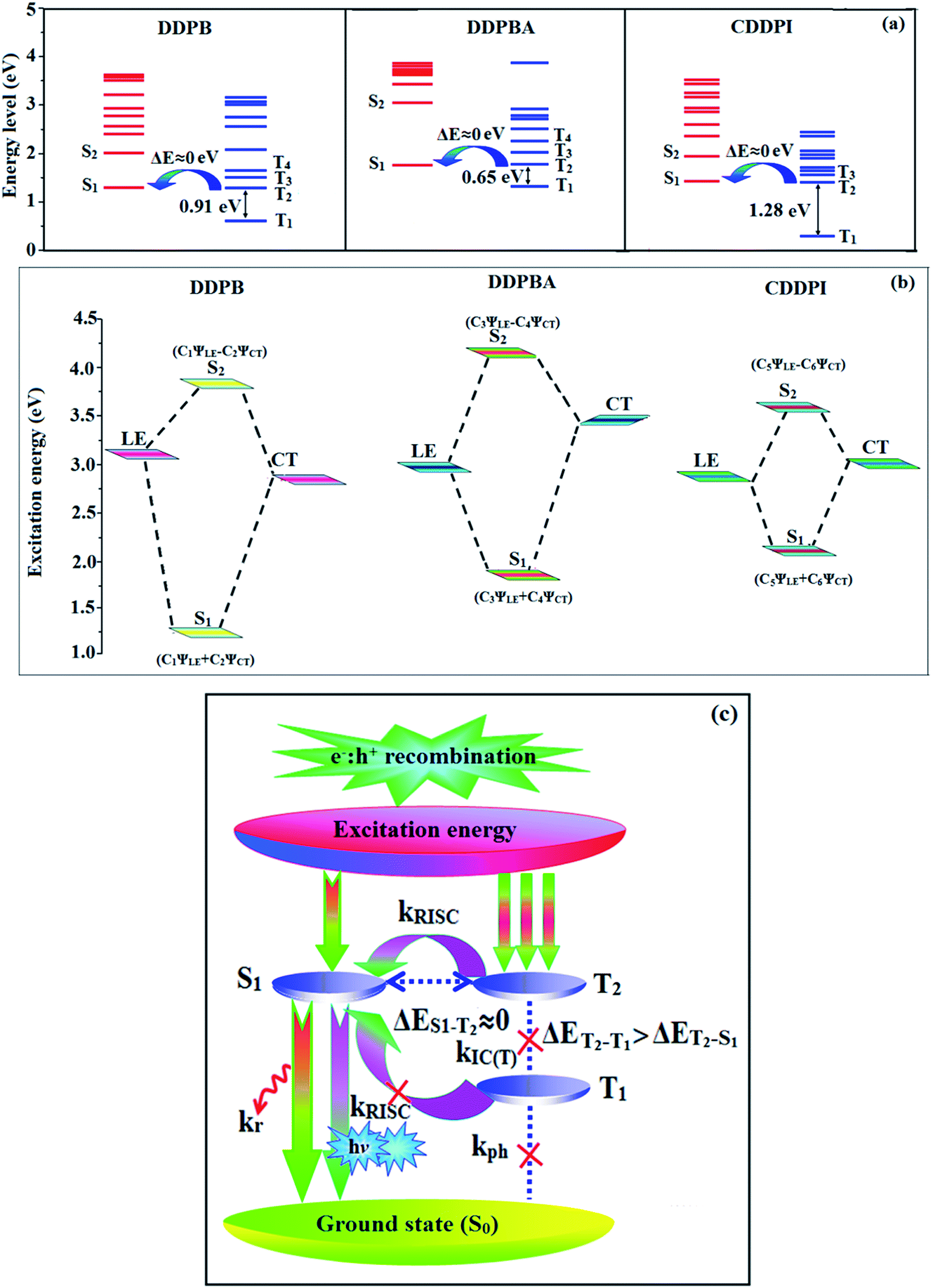
The HONTOs and LUNTOs of S1 and S2 excited states of DDPB, DBDPA and CDDPI exhibit a hybrid splitting state character that derives from interstate coupling of LE and CT levels to form HLCT (Tables 3–5). The hole contour on Cz or TPA moiety are in the opposite phase between S1 and S2 states whereas the particle on phenanthrimidazole moiety is same between S1 and S2 states for DDPB, DBDPA and CDDPI, respectively. This implied that the interstate hybridization coupling occurs through the positive and negative linear combination between LE and CT state wave function: ΨS1/S2 = cLEΨLE ± cCTΨCT. The percentage of CT level of CDDPI (62%) is less than that of DDPB (80%) and DBDPA (90%) as a result of weak donor ability of Cz than TPA results LE dominated S1 state in CDDPI (LE ∼ 45%), DDPB (LE ∼ 15%) and DBDPA (LE ∼ 10%) (Table S10†). As a result, CDDPI should exhibit higher photoluminescence efficiency (ηPL) and blue shifted emission relative to DDPB and DBDPA. The S1 and S2 excited states of DDPB, DBDPA and CDDPI are similar in energy, oscillator strength and HONTOs and LUNTOs distribution which indicate a quasi-equivalent hybridization between LE and CT states due to their almost iso-energetic initial states. In contrast, for nonequivalent hybridization the S1 and S2 excited states have quite significant difference in energy, oscillator strength and NTO image which is caused by non-equivalent hybridization between LE and CT initial states. Compared with non-equivalent hybridization, the quasi-equivalent hybridization is expected to achieve high ηPL and high ηS. The more balanced LE and CT components in HLCT state of DDPB, DBDPA and CDDPI enhanced the EL efficiency. The formation of HLCT state can be analyzed through the excitation energies of LE and CT states (Tables 3–5). A large energy gap between T1 and T2 for CDDPI (1.28 eV), DDPB (0.91 eV) and DBDPA (0.65 eV) arising from the same phenanthrimidazole acceptor and the energy gap between T1 and T2 of CDDPI is larger than DDPB and DBDPA (Fig. 10).96,97 A very small ΔEST ≈ 0 is observed between S1 and T3 states facilitating RISC (T2 → S1) process as a result of their HLCT state character. The increased LE component in S1 state of CDDPI enhanced the photoluminescence efficiency (ηPL), and high exciton utilization efficiency (ηS) and external quantum efficiency (ηex) have been harvested when compared with TPA emitters (Table 1). Excited state characters play a key role in PL and electroluminescence (EL) properties of OLEDs. Charge-transfer state is beneficial to enhance the singlet exciton utilization in fluorescent OLEDs by RISC due to small singlet and triplet energy splitting (ΔEST) in CT exciton. However, the dominant CT component in the emissive state reduces the PL efficiency in such materials. Here, the strategy is to carry out for fine excited state modulation to achieve combination of high PL efficiency using locally emissive (LE) component and high exciton utilizing CT component in one excited state. As a result, a quasi-equivalent hybridization of LE and CT components obtained in the emissive state upon addition of binaphthyl bridge in the newly synthesized material. Similar hole–electron wave function between S1 and S2 is observed in DDPB, DBDPA and CDDPI indicates a quasi-equivalent hybridization between LE and CT states as a result of their almost isoenergies of initial LE and CT states. Therefore, degree of hybridization between LE and CT states depends not only initial ELE–ECT energy gap but also their interstate coupling strength.98 Compared with non-equivalent hybridization, quasi-equivalent hybridization is expected to achieve the combination of high ηPL and high ηS to maximize EL efficiency of fluorescent OLED materials due to more balanced LE and CT components in HLCT state of DDPB, DBDPA and CDDPI. In DDPB, DBDPA and CDDPI, the LE state is stabilized than CT state and energy gap (ES2–ES1) is small when compared with their parent compounds results quasi hybridization. In the case of CDDPI, the energy gap (ES2–ES1) is reduced more when compared with DBDPA results effective hybridization and improves OLED efficiency. The qualitatively calculated percentage of LE and CT in S1–S10 and T1–T10 states are displayed in Table S10.† This also supports that HLCT state also contributes to hybridization apart from LE and CT states.
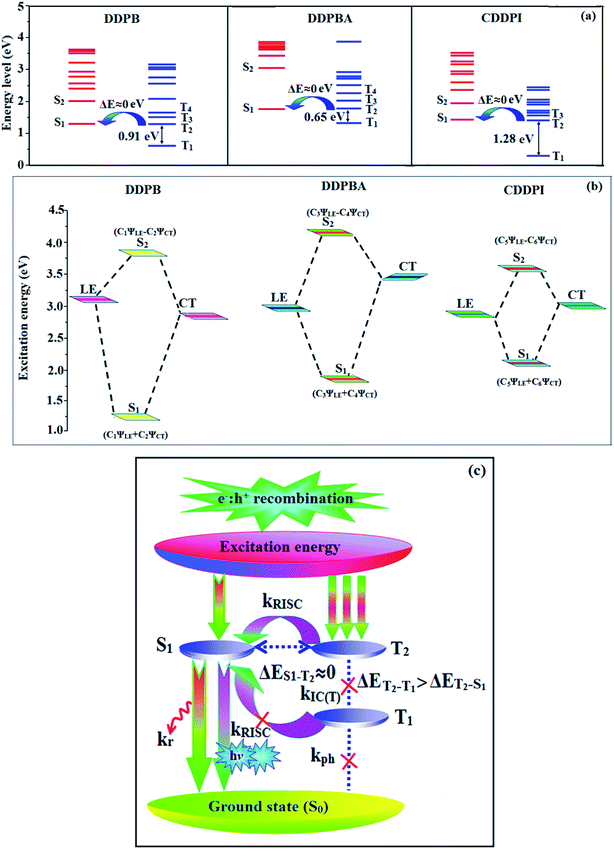 |
| | Fig. 10 (a) Schematic diagram of hybridization processes of LE and CT states of DDPB, DBDPA, CDDPI; (b) energy level of singlet (S) and triplet (T) states of DDPB, DBDPA, CDDPI; (c) scheme of exciton decay process after hole and electron recombination in OLEDs of D–π–A molecules. | |
3.6. Single carrier devices
To evaluate their carrier injection and transport properties, hole-only and electron-only devices have been fabricated: (a) ITO/HATCN (8 nm)/DDPB/DBDPA/CDDPI (50 nm)/HATCN (8 nm)/LiF (1 nm)/Al (100 nm) (hole-only device IV) and (b) ITO/TPBi (8 nm)/DDPB/DBDPA/CDDPI (50 nm)/TPBi (8 nm)/LiF (1 nm)/Al (100 nm) (electron-only device V). Fig. 11 shows the current density versus voltage characteristics of hole-only and electron-only devices. The electron current density of DDPB, DBDPA and CDDPI based device is higher than CBP-based device which reveal that these materials have effective electron injection and transport properties than CBP. The difference in current density between hole-only and electron-only devices based on DDPB, DBDPA and CDDPI is much smaller than that based on CBP at same voltage suggesting these materials are potential bipolar material capable of transporting electrons and holes in devices.99–101
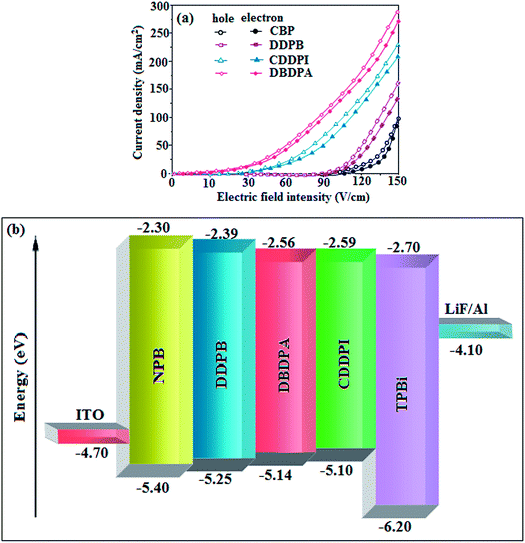 |
| | Fig. 11 (a) Hole-only and electron-only devices based on DDPB, DBDPA and CDDPI; (b) energy level diagram of non-doped devices. | |
3.7. Electroluminescent studies
The effective film forming properties of emissive materials are important for device efficiency. The better nanoscale morphology of annealed DBDPA thin film is attributed to low turn-on voltage. The only difference between DDPB and DBDPA and CDDPI is one single bond which affects the OLED performances. Energy-level diagram of the materials used for the fabrication of devices are shown in Fig. 11. The TADF material will show flat decay curve due to the time consuming TADF process for the exciton conversion from triplet to singlet however, the observed single-exponential sharp decay of DDPB, DBDPA and CDDPI shows that the radiative exciton are short-lived component without TADF contribution (Fig. 5). The lifetime measurement reveal that this intercrossed excited state in different polar solvents should be a hybridized local and charge transfer state (HLCT) instead of two species state through a simple addition of LE and CT. The mono exponential demonstrates that the intercrossed LE and CT in the moderate polar solvent formed as one hybridized HLCT state which supports the molecular design (Scheme 2). The exciton utilization efficiency (ηS) in DDPB, DBDPA and CDDPI are of neither TTA nor TADF mechanism.
The blue device with the configuration of ITO/NPB (60 nm)/DDPB/DBDPA/CDDPI (30 nm)/LiF (1 nm)/Al (100 nm) have been fabricated (Fig. 12). The electroluminescence (EL) spectra of the devices are similar to their PL spectra which shows both EL and PL originates from the same radiative decay of the singlet exciton. High device performances at low-turn on voltage are extracted from non-doped devices based on DDPB, DBDPA and CDDPI (Table 1). The CDDPI based device shows high efficiencies (ηc – 2.56 cd A−1; ηp – 2.12 lm W−1; ηex – 3.01%: Fig. 13) at 3.0 V with CIE (0.15, 0.11) and EL is 444 nm (FWHM-45 nm) than DBDPA (ηc – 2.01 cd A−1; ηp – 1.92 lm W−1; ηex – 2.85%) at 3.7 V with CIE (0.15, 0.13) and EL is 459 nm (FWHM-75 nm). The high ηex harvested from CDDPI and DBDPA based device is due to the co-emission from intercrossed excited state of LE and CT; the isoenergies of singlet (1CT) and triplet (3CT) make 3CT → 1CT transition as spin-allowed transition.102 The device with DDPB exhibit deep blue EL emission at 447 nm (narrow FWHM-37 nm) with CIE (0.16, 0.07) and the efficiencies are (ηc – 1.61 cd A−1; ηp – 1.43 lm W−1; ηex – 2.01%) at 4.3 V. The EL difference between DDPB and DBDPA is ∼12 nm whereas DBDPA exhibit blue emission with 75 nm normal FWHM. The EL and PL spectra of DDPB, DBDPA and CDDPI at 77 K and room temperature are shown in Fig. 5. The FWHM of PL spectrum of DDPB was gradually narrower (48 nm) from room temperature [77 K (40 nm)] to EL spectra (37 nm) whereas for DBDPA and CDDPI, the FWHM of EL spectra (75 nm – DBDPA and 69 nm – CDDPI) was larger than that of PL spectra at room temperature (65 nm – DBDPA and 60 nm – CDDPI) [77 K: (56 nm – DBDPA) and 50 nm – CDDPI]. On comparison with PL emission of DBDPA and CDDPI, the narrow EL spectrum is explained by weak microcavity effect and suppressed intramolecular vibration.103 The inevitable vibration splitting in the strongly rigid phenanthro[9,10-d]-imidazole structure of DBDPA and CDDPI may be enhanced in OLEDs to show large full peak width with red-shifted CIE. The external quantum efficiency of OLEDs can be calculated as follows: EQE = ηout × ηrc × ηγ × ΦPL,104 [ηout – light-out-coupling efficiency (20%), ηrc – product of the charge recombination efficiency (100%), ηγ – efficiency of radiative exciton production (25%) and ΦPL – photoluminescence quantum yield of the emitters]. The ηr calculated for DDPB (13–16%), DBDPA (19–21%) and CDDPI (16–21%) indicates γ is less than 100% due to very small unbalanced carrier transportation.105 This result could be attributed more balanced charge-transporting properties within the emissive layer achieved by better charge injection provided by hole transport layer. The ηIQE can be calculated from ηEQE/ηout as DDPB (10.1%), DBDPA (14.3%) and CDDPI (15.1%) and maximum ηs of DDPB (16.5%), DBDPA (19.3%) and CDDPI (26.6%) of EL devices can be estimated using the equation ηs = ηres × ηPL × ηout/ηEL, where ηout (≈1/2n2) is light out coupling efficiency (≈20%); ηrec is efficiency for electron hole recombination (100%). The enhanced ηs and ηIQE is probably due to the maintained CT component of D–π–A compounds. The device efficiencies are compared with already reported non-doped emitters efficiencies58,106–114 (Table 7) which shows that the newly synthesized non-doped devices based on DDPB, DBDPA and CDDPI are among the best in terms of efficiencies. These experimental results demonstrated that the additional triplet exciton have been utilized in the OLED applications for the HLCT character of DDPB, DBDPA and CDDPI as shown in Scheme 2 and showing the accuracy for our molecular design strategy. Devices with DDPB, DBDPA and CDDPI show maximum luminance (L) of 2010, 3015 and 3992 cd m−2, respectively. The EL brightness has a linear relationship with current density for these compounds indicating that the contribution from triplet–triplet annihilation was insignificant.115 The emission wavelength of DDPB, DBDPA and CDDPI in film is close to that in ethyl ether which supports the HLCT state formed in DDPB, DBDPA and CDDPI film.
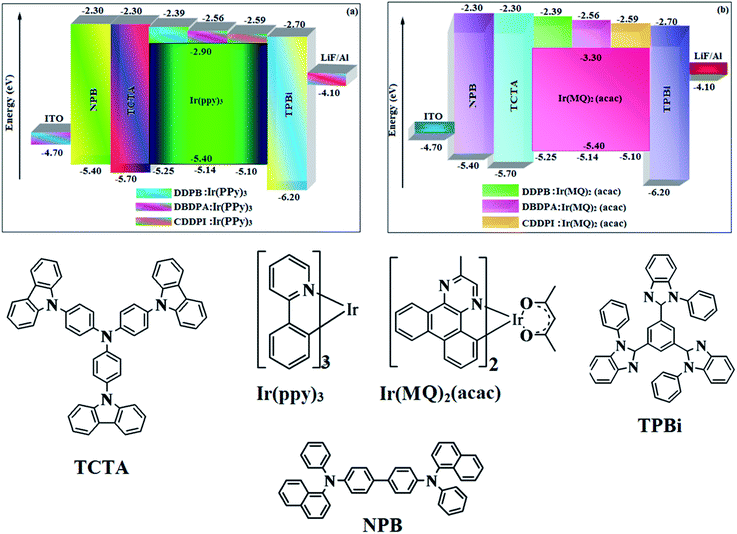 |
| | Fig. 12 Energy level diagram of green (a) and red (b) devices with molecular structures of functional materials used in devices. | |
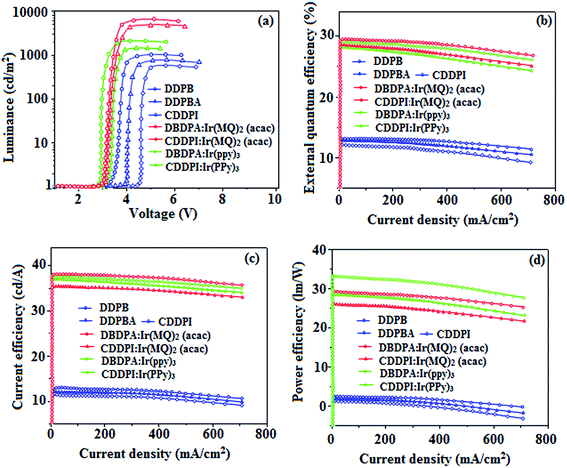 |
| | Fig. 13 Device performances of DDPB, DBDPA, CDDPI, DBDPA:Ir(ppy)3, CDDPI:Ir(PPy)3, DBDPA:Ir(MQ)2(acac) and CDDPI:Ir(MQ)2(acac): (a) luminance–voltage; (b) external quantum efficiency–current density; (c) current efficiency–current density and (d) power efficiency–current density. | |
Table 7 Summary of device efficiencies with reported non-doped emitters
| Emitter |
Von (V) |
L (cd m−2) |
EL (nm) |
ηc (cd A−1) |
ηp (lm W−1) |
CIE (x, y) |
Ref. |
| DDPB |
4.3 |
2010 |
447 |
1.61 |
1.43 |
(0.16, 0.07) |
This work |
| DBDPA |
3.7 |
3015 |
459 |
2.01 |
1.92 |
(0.15, 0.12) |
This work |
| CDDPI |
3.0 |
3992 |
444 |
2.56 |
2.12 |
(0.15, 0.11) |
This work |
| PPI |
3.8 |
3307 |
412 |
0.71 |
0.40 |
(0.161, 0.065) |
106 |
| mTPA-PPI |
3.2 |
4065 |
404 |
0.84 |
0.48 |
(0.161, 0.049) |
106 |
| L-BPPI (50 nm) |
8.5 |
70 |
440 |
0.01 |
— |
(0.16, 0.11) |
107 |
| L-BPPI (40 nm) |
6.5 |
295 |
440 |
0.13 |
— |
(0.16, 0.11) |
107 |
| L-BPPI (30 nm) |
5.0 |
420 |
440 |
0.40 |
— |
(0.16, 0.10) |
107 |
| L-BPPI (20 nm) |
4.5 |
391 |
440 |
0.68 |
— |
(0.16, 0.10) |
107 |
| Z-BPPI (50 nm) |
6.5 |
105 |
440 |
0.07 |
— |
(0.17, 0.12) |
107 |
| Z-BPPI (40 nm) |
5.0 |
502 |
440 |
0.34 |
— |
(0.16, 0.12) |
107 |
| Z-BPPI (30 nm) |
4.5 |
267 |
440 |
0.45 |
— |
(0.16, 0.12) |
107 |
| Z-BPPI (20 nm) |
5.0 |
100 |
440 |
0.31 |
— |
(0.16, 0.11) |
107 |
| TPA-PIM |
— |
4510 |
420 |
1.14 |
0.79 |
(0.161, 0.046) |
58b |
| MADN (BUBD) |
7.8 |
— |
440 |
2.1 |
— |
(0.15, 0.10) |
108 |
| CPPPI |
— |
3322 |
420 |
0.65 |
0.48 |
(0.165, 0.050) |
109 |
| PPICPPPI |
— |
4329 |
428 |
1.53 |
0.86 |
(0.166, 0.056) |
109 |
| PhBPI |
2.8 |
— |
450 |
1.87 |
1.85 |
— |
110 |
| Bilayer-TPBI |
3.2 |
— |
468 |
2.03 |
1.00 |
(0.15, 0.15) |
111 |
| TPA-BPI |
2.8 |
— |
448 |
1.83 |
1.58 |
(0.15, 0.09) |
112 |
| DPVBi |
7.5 |
— |
457 |
0.03 |
— |
(0.15, 0.13) |
113 |
| DPVICz |
4.2 |
— |
470 |
0.92 |
— |
(0.15, 0.22) |
113 |
| DPVTCz |
3.8 |
— |
470 |
1.94 |
— |
(0.14, 0.22) |
113 |
| 3,6-DPVTCz |
5.0 |
— |
449 |
0.11 |
— |
(0.15, 0.11) |
113 |
| PEDOt-PSS: 3 (100 nm) |
4.0 |
2800 |
460 |
0.61 |
0.14 |
(0.15, 0.14) |
114 |
| PEDOt-PSS: 3 (50 nm) |
3 |
10600 |
407 |
1.68 |
1.10 |
(0.16, 0.13) |
114 |
| PEDOt-PSS: 4 (40 nm) |
2.5 |
21200 |
392 |
1.90 |
1.55 |
(0.16, 0.14) |
114 |
The calculated triplet energy (ES/ET) of DDPB (2.29 eV), DBDPA (2.25 eV) and CDDPI (2.23 eV) shows that they have high triplet energy to sensitize phosphorescent dopants with ET below 2.3 eV. These DBDPA and CDDPI are also employed as host materials for green and red phosphorescent dopants. The fabricated green and red devices are having the configuration of ITO/NPB (40 nm)/TCTA (5 nm)/DBDPA (30 nm): 5 wt% Ir(ppy)3/CDDPI (30 nm): 5 wt% Ir(ppy)3/TPBI (50 nm)/LiF (1 nm)/Al (100 nm): ITO/NPB (40 nm)/TCTA (5 nm)/DBDPA (30 nm): 8 wt% Ir(MQ)2(acac)/CDDPI (30 nm): 8 wt% Ir(MQ)2(acac)/TPBI (50 nm)/LiF (1 nm)/Al (100 nm), respectively (Fig. 12), [Ir(ppy)3-fac-tris(2-phenylpyridine) iridium(III) and Ir(MQ)2(acac)-bis(2-methyldibenzo-[f,h]quinoxaline) acetylacetonate iridium(III) are used as emissive layers for green and red devices, respectively]. The device performances are shown in Fig. 13. The EL spectra are similar to PL spectra of the doped thin films (Fig. 5). The green device (432 nm) with CDDPI (30 nm): 5 wt% Ir(ppy)3 exhibits maximum luminance of 8812 cd m−2, maximum current and power efficiencies are of 27.5 cd A−1 and 33.0 lm W−1, respectively at 2.7 V with CIE (0.31, 0.60) (Table 4). The maximum external quantum efficiencies of the devices based on CDDPI:Ir(ppy)3 and DBDPA:Ir(ppy)3 [ηc – 27.5 cd A−1; ηp – 33.0 lm W−1; L – 8609 cd m−2; CIE (0.31, 0.60) with CIE (0.64, 0.34)] are 19.0 and 18.2%, respectively. Similar to green devices, red device (640 nm) with CDDPI:Ir(MQ)2(acac) exhibits maximum luminance of 39461 cd m−2 and excellent EL efficiencies (ηex – 19.2%; ηc – 27.9 cd A−1; ηp – 29.2 lm W−1 with CIE (0.64, 0.34) on compared with DBDPA:Ir(MQ)2(acac) based on device (L – 37621 cd m−2; ηex – 18.5%; ηc – 25.2 cd A−1; ηp – 25.8 lm W−1; CIE (0.64, 0.34) with EL 641 nm. The above experimental results demonstrate that CDDPI and DBDPA are universal host materials for green and red phosphorescent emitters (Table 8). The device performances reveal that CDDPI and DBDPA are universal host materials for green and red phosphorescent emitters.
Table 8 Electroluminescent efficiencies of green DBDPA:Ir(ppy)3, CDDPI:Ir(PPy)3 and red DBDPA:Ir(MQ)2(acac), CDDPI:Ir(MQ)2(acac)
| Emitters |
V1000 (V) |
L (cd m−2) |
ηex (%) |
ηc (cd A−1) |
ηp (lm W−1) |
CIE (x, y) |
EL (nm) |
| DBDPA:Ir(ppy)3 |
3.0 |
8609 |
18.2 |
27.0 |
28.6 |
0.31, 0.60 |
436 |
| CDDPI:Ir(PPy)3 |
2.7 |
8812 |
19.0 |
27.5 |
33.0 |
0.31, 0.60 |
432 |
| DBDPA:Ir(MQ)2(acac) |
3.2 |
37621 |
18.5 |
25.2 |
25.8 |
0.64, 0.34 |
641 |
| CDDPI:Ir(MQ)2(acac) |
3.0 |
39461 |
19.2 |
27.9 |
29.2 |
0.64, 0.34 |
640 |
4. Conclusion
In conclusion, the blue emissive, fully twisting DDPB based OLEDs exhibit quantum efficiency of 2.01% (FHWM of 37 nm) with CIE (0.16, 0.07). The λemi of DBDPA film is close with that in ether which confirmed HLCT state formation. The fully twisted DDPB with strong intramolecular vibration and weak microcavity effect produced narrow EL. The external quantum efficiency harvested from DBDPA based device is 2.85% with maximum current and power efficiency of 2.01 cd A−1 and 1.92 lm W−1, respectively. The enhanced ηs [DDPB – 16.5%, DBDPA – 19.3% and CDDPI – 26.7%] and ηIQE [DDPB – 10.1%, DBDPA – 14.3% and CDDPI – 15.1%] is probably due to the maintained CT component of D–π–A compounds. The blue emissive materials CDDPI and DBDPA used as a host to construct green and red phosphorescent OLEDs. The green device based on CDDPI (30 nm): 5 wt% Ir(ppy)3 exhibits maximum luminance of 8812 cd m−2, maximum current and power efficiencies are of 27.5 cd A−1 and 33.0 lm W−1, respectively at 2.7 V and red device based on CDDPI:Ir(MQ)2(acac) exhibits maximum luminance of 39661 cd m−2 and excellent EL efficiencies (ηex – 19.2%; ηc – 27.9 cd A−1; ηp – 29.2 lm W−1 with CIE (0.64, 0.34)) on compared with DBDPA:Ir(MQ)2(acac) based on device (L – 37621 cd m−2; ηex – 18.5%; ηc – 25.2 cd A−1; ηp – 25.8 lm W−1 with CIE (0.64, 0.34)). Fluorescent D–π–A emitters with HLCT state would be an effective practical strategy to develop low-cost and high-efficient blue organic electroluminescent materials.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
One of the author Dr J. Jayabharathi thank DST (Department of Science and Technology – EMR/2014/000094, F. No. SR/S1/1C-73/2010, F. No. SR/S1/1C-07/2007), DRDO (Defence Research and Development Organization – 213/MAT/10-11), CSIR (Council of Scientific and Industrial Research – No. 01/(2707)/13EMR-II), UGC (University Grant Commission – 36-21/2008, F. No. 30-71/2004(SR)) and DST-Nano Mission (SR/NM/NS-1001/2016) for financial support. Authors like to thank Mr P. Justin Jesuraj for scientific discussion.
References
-
(a) C. W. Tang and S. A. VanSlyke, Appl. Phys. Lett., 1987, 51, 913–915 CrossRef CAS;
(b) J. H. Burroughes, D. D. C. Bradley, A. R. Brown, R. N. Marks, K. Mackay, R. H. Friend, P. L. Born and A. B. Holmes, Nature, 1990, 347, 539–541 CrossRef CAS.
-
(a) K. Guo, J. Zhang, T. Xu, X. Gao and B. Wei, J. Disp. Technol., 2014, 10, 642–646 CAS;
(b) M. Liu, X. L. Li, D. C. Chen, Z. Z. Xie, X. Y. Cai, G. Z. Xie, K. K. Liu, J. X. Tang, S. J. Su and Y. Cao, Adv. Funct. Mater., 2015, 25, 5190–5198 CrossRef CAS.
-
(a) Y. Cao, I. D. Parker, G. Yu, C. Zhang and A. J. Heeger, Nature, 1999, 397, 414–417 CrossRef CAS PubMed;
(b) L. Yao, S. Zhang, R. Wang, W. Li, F. Shen, B. Yang and Y. Ma, Angew. Chem., Int. Ed., 2014, 53, 2119–2123 CrossRef CAS PubMed;
(c) Z. Gao, G. Cheng, F. Shen, S. Zhang, Y. Zhang, P. Lu and Y. Ma, Laser Photonics Rev., 2014, 8, L6–L10 CrossRef CAS.
-
(a) Z. Jiang, Z. Liu, C. Yang, C. Zhong, J. Qin, G. Yu and Y. Q. Liu, Adv. Funct. Mater., 2009, 19, 1227–1234 CrossRef;
(b) M. S. Gong, H. S. Lee and Y. M. Jeon, J. Mater. Chem., 2010, 20, 10735–10746 RSC.
-
(a) A. D. Gorse and M. Pesquer, J. Phys. Chem., 1995, 99, 4039–4049 CrossRef CAS;
(b) W. Y. Hung, G. C. Fang, Y. C. Chang, T. Y. Kuo, P. T. Chou, S. W. Lin and K. T. Wong, ACS Appl. Mater. Interfaces, 2013, 5, 6826–6831 CrossRef CAS PubMed.
-
(a) S. Chichibu, T. Azuhata, T. Sota and S. Nakamura, Appl. Phys. Lett., 1997, 70, 2822–2824 CrossRef CAS;
(b) T. Noda, H. Ogawa and Y. Shirota, Adv. Mater., 1999, 11, 283–285 CrossRef CAS.
-
(a) K. Kreger, M. Baete, C. Neuber, H. W. Schmidt and P. Strohriegl, Adv. Funct. Mater., 2007, 17, 3456–3461 CrossRef CAS;
(b) S. Tang, M. R. Liu, P. Lu, H. Xia, M. Li, Z. Q. Xie, F. Z. Shen, C. Gu, H. Wang, B. Yang and Y. G. Ma, Adv. Funct. Mater., 2007, 17, 2869–2877 CrossRef CAS;
(c) Y. Q. Mo, R. Y. Tian, W. Shi and Y. Cao, Chem. Commun., 2005, 39, 4925–4926 RSC.
-
(a) A. P. Kulkarni, C. J. Tonzola, A. Babel and S. A. Jenekhe, Chem. Mater., 2004, 16, 4556–4573 CrossRef CAS;
(b) P. Strohrieg and J. V. Grazulevicius, Adv. Mater., 2002, 14, 1439–1452 CrossRef.
- C. J. Kuo, T. Y. Li, C. C. Lien, C. H. Liu, F. I. Wu and M. J. Huang, J. Mater. Chem., 2009, 19, 1865–1871 RSC.
- K. C. Wu, P. J. Ku, C. S. Lin, H. T. Shih, F. I. Wu, M. J. Huang, J. J. Lin, I. C. Chen and C. H. Cheng, Adv. Funct. Mater., 2008, 18, 67–75 CrossRef CAS.
- P. I. Shih, C. Y. Chuang, C. H. Chien, E. W. G. Diau and C. F. Shu, Adv. Funct. Mater., 2007, 17, 3141–3146 CrossRef CAS.
- C. J. Tonzola, A. P. Kukarni, A. P. Gifford, W. Kaminsky and S. A. Jenekhe, Adv. Funct. Mater., 2007, 17, 863–874 CrossRef CAS.
-
(a) J. Shi and C. W. Tang, Appl. Phys. Lett., 2002, 80, 3201–3203 CrossRef CAS;
(b) Y. H. Kim, H. C. Jeong, S. H. Kim, K. Yang and S. K. Kwon, Adv. Funct. Mater., 2005, 15, 1799–1805 CrossRef CAS;
(c) Y. Y. Lyu, J. Kwak, O. Kwon, S. H. Lee, D. Kim, C. Lee and K. Char, Adv. Mater., 2008, 20, 2729 CrossRef PubMed.
- J. Jayabharathi, A. Prabhakaran, V. Thanikachalam and M. Sundharesan, RSC Adv., 2016, 6, 62208–62217 RSC.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- S. Zhang, L. Yao, Q. Peng, W. J. Li, Y. Pan, R. Xiao, Y. Gao, C. Gu, Z. Wang, P. Lu, F. Li, S. Su, B. Yang and Y. Ma, Adv. Funct. Mater., 2015, 25, 1755–1762 CrossRef CAS.
- Y. Tamai, H. Ohkita, H. Benten and S. Ito, Chem. Mater., 2014, 26, 2733–2742 CrossRef CAS.
- K. Okumoto, H. Kanno, Y. Hamada, H. Takahashi and K. Shibata, Appl. Phys. Lett., 2006, 89, 063504–063506 CrossRef.
- D. Y. Kondakov, T. D. Pawlik, T. K. Hatwar and J. P. Spindler, J. Appl. Phys., 2009, 106, 124510–124517 CrossRef.
- H. Fukagawa, T. Shimizu, N. Ohbe, S. Tokito, K. Tokumaru and H. Fujikake, Org. Electron., 2012, 13, 1197–1203 CrossRef CAS.
- A. Endo, M. Ogasawara, A. Takahashi, D. Yokoyama, Y. Kato and C. Adachi, Adv. Mater., 2009, 21, 4802–4806 CrossRef CAS PubMed.
- A. Endo, K. Sato, K. Yoshimura, T. Kai, A. Kawada, H. Miyazaki and C. Adachi, Appl. Phys. Lett., 2011, 98, 083302–083303 CrossRef.
- T. Nakagawa, S. Y. Ku, K. T. Wong and C. Adachi, Chem. Commun., 2012, 48, 9580–9582 RSC.
- S. Y. Lee, T. Yasuda, H. Nomura and C. Adachi, Appl. Phys. Lett., 2012, 101, 093306–093309 CrossRef.
- Q. Peng, W. Li, S. Zhang, P. Chen, F. Li and Y. Ma, Adv. Opt. Mater., 2013, 1, 362–366 CrossRef.
-
(a) M. Y. Lai, C. H. Chen, W. S. Huang, J. T. Lin, T. H. Ke, L. Y. Chen, M. H. Tsai and C. C. Wu, Angew. Chem., Int. Ed., 2008, 47, 581–595 CrossRef CAS PubMed;
(b) Y. Yuan, J. X. Chen, F. Lu, Q. X. Tong, Q. D. Yang, H. W. Mo, T. W. Ng, F. L. Wong, Z. Q. Guo, J. Ye, Z. Chen, X. H. Zhang and C. S. Lee, Chem. Mater., 2013, 25, 4957–4965 CrossRef CAS.
- H. Nakanotani, K. Masui, J. Nishide, T. Shibata and C. Adachi, Sci. Rep., 2013, 3, 2127–2135 CrossRef PubMed.
- S. Zhang, L. Yao, Q. Peng, W. Li, Y. Pan, R. Xiao, Y. Gao, C. Gu, Z. Wang, P. Lu, F. Li, S. Su, B. Yang and Y. Ma, Adv. Funct. Mater., 2015, 25, 1755–1762 CrossRef CAS.
- H. H. Chou and C. H. Cheng, Adv. Mater., 2010, 22, 2468–2471 CrossRef CAS PubMed.
-
(a) G. Qian and Z. Y. Wang, Can. J. Chem., 2010, 88, 192–201 CrossRef CAS;
(b) A. D. Gorse and M. Pesquer, J. Phys. Chem., 1995, 99, 4039–4049 CrossRef CAS.
- W. J. Li, D. D. Liu, F. Z. Shen, D. G. Ma, Z. M. Wang, T. Fei, B. Yang and Y. G. Ma, Adv. Funct. Mater., 2012, 22, 2797–2803 CrossRef CAS.
- W. J. Li, Y. Y. Pan, R. Xiao, Q. M. Peng, S. T. Zhang, D. G. Ma, F. Li, F. Z. Shen, Y. H. Wang, B. Yang and Y. G. Ma, Adv. Funct. Mater., 2014, 24, 1609–1614 CrossRef CAS.
- S. Tang, W. Li, F. Shen, D. Liu, B. Yang and Y. Ma, J. Mater. Chem. A, 2012, 22, 4401 RSC.
- J. R. Sheats, H. Antoniadis, M. Hueschen, W. Leonard, J. Miller, R. Moon, D. Roitman and A. Stocking, Science, 1996, 273, 884–888 CrossRef CAS PubMed.
- D. Chaudhuri, E. Sigmund, A. Meyer, L. Rcck, P. Klemm, S. Lautenschlager, A. Schmid, S. R. Yost, T. Van, S. Bange, S. Hcger and J. M. Lupton, Angew. Chem., 2013, 52, 13449–13452 CrossRef CAS PubMed.
-
(a) J. N. Moorthy, P. Natarajan, P. Venkatakrishnan, D. F. Huang and T. J. Chow, Org. Lett., 2007, 9, 5215–5218 CrossRef CAS PubMed;
(b) S. Tao, Z. Peng, X. Zhang, P. Wang, C. S. Lee and S. T. Lee, Adv. Funct. Mater., 2005, 15, 1716–1721 CrossRef CAS.
- C. H. Chien, C. K. Chen, F. M. Hsu, C. F. Shu, P. T. Chou and C. H. Lai, Adv. Funct. Mater., 2009, 19, 560–566 CrossRef CAS.
- J. Ye, Z. Chen, M. K. Fung, C. Zheng, X. Ou and X. H. Zhang, Chem. Mater., 2013, 25, 2630–2637 CrossRef CAS.
- W. Qin, Z. Yang, Y. Jiang, J. W. Y. Lam, G. Liang, H. S. Kwok and B. Z. Tang, Chem. Mater., 2015, 27, 3892–3901 CrossRef CAS.
-
(a) B. Liu, J. W. Zhao, C. Y. Luo, F. Lu, S. L. Tao and Q. X. Tong, J. Mater. Chem. C, 2016, 4, 2003–2010 RSC;
(b) G. Li, J. W. Zhao, D. Zhang, Z. C. Shi, Z. L. Zhu, H. Q. Song, J. J. Zhu, S. L. Tao, F. Lu and Q. X. Tong, J. Mater. Chem. C, 2016, 4, 8787–8794 RSC.
- W. C. Chen, Y. Yuan, G. F. Wu, H. X. Wei, L. Tang, Q. X. Tong, F. L. Wong and C. S. Lee, Adv. Opt. Mater., 2014, 2, 626–631 CrossRef CAS.
- W. C. Chen, G. F. Wu, Y. Yuan, H. X. Wei, F. L. Wong, Q. X. Tong and C. S. Lee, RSC Adv., 2015, 5, 18067–18074 RSC.
- X. Ouyang, X. Li, L. Ai, D. Mi, Z. Ge and S. J. Su, ACS Appl. Mater. Interfaces, 2015, 7, 7869–7877 CrossRef CAS PubMed.
- K. Wang, S. Wang, J. Wei, Y. Miao, Y. Liu and Y. Wang, Org. Electron., 2014, 15, 3211–3220 CrossRef CAS.
- N. Su, F. Meng, J. Chen, Y. Wang, H. Tan, S. Su and W. Zhu, Dyes Pigm., 2016, 128, 68–74 CrossRef CAS.
- H. Liu, P. Chen, D. Hu, X. Tang, Y. Pan, H. Zhang, W. Zhang, X. Han, Q. Bai, P. Lu and Y. Ma, Chem.–Eur. J., 2014, 20, 2149–2153 CrossRef CAS PubMed.
- R. Kim, S. J. Yoo, E. K. Kim, H. S. Yu, S. C. Shin, S. K. Lee, S. K. Kwon and Y. H. Kim, Dyes Pigm., 2012, 95, 384–391 CrossRef CAS.
- I. Cho, S. H. Kim, J. H. Kim, S. Park and S. Y. Park, J. Mater. Chem., 2012, 22, 123–129 RSC.
- W. C. Chen, Y. Yuan, G. F. Wu, H. X. Wei, J. Ye, M. Chen, F. Lu, Q. X. Tong, F. L. Wong and C. S. Lee, Org. Electron., 2015, 17, 159–166 CrossRef CAS.
- X. Tang, L. Yao, H. Liu, F. Shen, S. Zhang, Y. Zhang, H. Zhang, P. Lu and Y. Ma, J. Mater. Chem. C, 2014, 2, 5019–5027 RSC.
- S. Zhuang, R. Shangguan, H. Huang, G. Tua, L. Wang and X. Zhu, Dyes Pigm., 2014, 101, 93–102 CrossRef CAS.
- B. Wang, G. Y. Mu, H. J. Tan, Z. H. Lei, J. J. Jin and L. Wang, J. Mater. Chem. C, 2015, 3, 7709–7719 RSC.
- S. K. Kim, B. Yang, Y. I. Park, Y. Ma, J. Y. Lee, H. J. Kim and J. Park, Org. Electron., 2009, 10, 822–833 CrossRef CAS.
- J. Huang, N. Sun, Y. Dong, R. Tang, P. Lu, P. Cai, Q. Li, D. Ma, J. Qin and Z. Li, Adv. Funct. Mater., 2013, 23, 2329–2337 CrossRef CAS.
-
(a) K. Wang, F. C. Zhao, C. G. Wang, S. Y. Chen, D. Chen, H. Y. Zhang, Y. Liu, D. G. Ma and Y. Wang, Adv. Funct. Mater., 2013, 23, 2672–2680 CrossRef CAS;
(b) K. Wang, S. P. Wang, J. B. Wei, S. Y. Chen, D. Liu, Y. Liu and Y. Wang, J. Mater. Chem. C, 2014, 2, 6817–6826 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Revision A.02, Gaussian, Inc., Wallingford.,CT, 2009 Search PubMed.
- T. L. Bahers, C. Adamo and I. Ciofini, J. Chem. Theory Comput., 2011, 7, 2498–2506 CrossRef PubMed.
-
(a) Y. Tan, Z. Zhao, L. Shang, Y. Liu, C. Wei, J. Li, H. Wei, Z. Liu, Z. Bian and C. Huanga, J. Mater. Chem. C, 2017, 5, 11901–11909 RSC;
(b) W. Li, L. Yao, H. Liu, Z. Wang, S. Zhang, R. Xiao, H. Zhang, P. Lu, B. Yang and Y. Ma, J. Mater. Chem. C, 2014, 2, 4733–4736 RSC.
- W. Li, Y. Pan, L. Yao, H. Liu, S. Zhang, C. Wang, F. Shen, P. Lu, B. Yang and Y. Ma, Adv. Opt. Mater., 2014, 2, 892–901 CrossRef CAS.
- R. Kim, S. Lee, K. H. Kim, Y. J. Lee, S. K. Kwon, J. J. Kim and Y. H. Kim, Chem. Commun., 2013, 49, 4664–4666 RSC.
- H. Liu, L. Yao, L. Bao, X. Chen, Y. Gao, S. Zhang, W. Li, P. Lu, B. Yang and Y. Ma, Chem. Commun., 2016, 52, 7356–7359 RSC.
- W. Liu, C. J. Zheng, K. Wang, Z. Chen, D. Y. Chen, F. Li, X. M. Ou, Y. P. Dong and X. H. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 18930–18936 CrossRef CAS PubMed.
- Y. Tao, Q. Wang, C. Yang, C. Zhong, K. Zhang, J. Qin and D. Ma, Adv. Funct. Mater., 2010, 20, 304–311 CrossRef CAS.
- C. Fan, Y. H. Chen, Z. Q. Jiang, C. L. Yang, C. Zhong, J. G. Qin and D. G. Ma, J. Mater. Chem., 2010, 20, 3232–3237 RSC.
- Z. Huang, B. Wang, Q. Zhang, S. Xiang, X. Lv, L. Ma, B. Yang, Y. Gao and L. Wang, Dyes Pigm., 2017, 140, 328–336 CrossRef CAS.
- P. Y. Chou, H. H. Chou, Y. H. Chen, T. H. Su, C. Y. Liao, H. W. Lin, W. C. Lin, H. Y. Yen, I. C. Chena and C. H. Cheng, Chem. Commun., 2014, 50, 6869–6871 RSC.
- Z. Wang, X. Li, K. Xue, H. Li, X. Zhang, Y. Liu, Z. Yu, P. Lu and P. Chen, J. Mater. Chem. C, 2016, 4, 1886–1894 RSC.
- Z. Gao, Z. Wang, T. Shan, Y. Liu, F. Shen, Y. Pan, H. Zhang, X. He, P. Lu, B. Yang and Y. Ma, Org. Electron., 2014, 15, 2667–2676 CrossRef CAS.
- C. Wang, X. Li, Y. Pan, S. Zhang, L. Yao, Q. Bai, W. Li, P. Lu, B. Yang, S. Su and Y. Ma, ACS Appl. Mater. Interfaces, 2016, 8, 3041–3049 CrossRef CAS PubMed.
-
(a) Y. Zhang, S. L. Lai, Q. X. Tong, M. F. Lo, T. W. Ng, M. Y. Chan, Z. C. Wen, J. He, K. S. Jeff, X. L. Tang, W. M. Liu, C. C. Ko, P. F. Wang and C. S. Lee, Chem. Mater., 2012, 24, 61–70 CrossRef;
(b) Y. Zhang, S. L. Lai, Q. X. Tong, M. Y. Chan, T. W. Ng, Z. C. Wen, G. Q. Zhang, S. T. Lee, H. L. Kwong and C. S. Lee, J. Mater. Chem., 2011, 21, 8206–8214 RSC;
(c) Y. Zhang, J. H. Wang, J. Zheng and D. Li, Chem. Commun., 2015, 51, 6350–6353 RSC.
-
(a) H. Huang, Y. X. Wang, S. Q. Zhuang, X. Yang, L. Wang and C. L. Yang, J. Phys. Chem. C, 2012, 116, 19458–19466 CrossRef CAS;
(b) S. Zhuang, R. Shangguan, H. Huang, G. Tua, L. Wang and X. Zhu, Dyes Pigm., 2014, 101, 93–102 CrossRef CAS;
(c) B. Wang, G. Y. Mu, J. H. Tan, Z. H Lei, J. J. Jin and L. Wang, J. Mater. Chem. C, 2015, 3, 7709–7719 RSC.
-
(a) J. L. Wang, W. Y. Lin and W. L. Li, Biomaterials, 2013, 34, 7429–7436 CrossRef CAS PubMed;
(b) X. Ouyang, D. Chen, S. Zeng, X. Zhang, S. Su and Z. Ge, J. Mater. Chem., 2012, 22, 23005–23011 RSC;
(c) D. Kumar, K. R. J. Thomas, C. Lin and J. Jou, Chem.–Asian J, 2013, 8, 2111–2124 CrossRef CAS PubMed;
(d) R. Misra, T. Jadhav, B. Dhokale and S. M. Mobin, Chem. Commun., 2014, 50, 9076–9078 RSC.
-
(a) W. J. Li, D. D. Liu, F. Z. Shen, D. G. Ma, Z. M. Wang, T. Feng, Y. X. Xu, B. Yang and Y. G. Ma, Adv. Funct. Mater., 2012, 22, 2797–2803 CrossRef CAS;
(b) S. T. Zhang, W. J. Li, L. Yao, Y. Y. Pan, B. Yang and Y. G. Ma, Chem. Commun., 2013, 49, 11302–11304 RSC.
- Z. M. Wang, X. H. Song, Z. Gao, D. W. Yu, X. J. Zhang, P. Lu, F. Z. Shen and Y. G. Ma, RSC Adv., 2012, 2, 9635–9642 RSC.
- H. Zhuang, Q. Zhou, Q. Zhang, H. Li, N. Li, Q. Xua and J. Lu, J. Mater. Chem. C, 2015, 3, 416–422 RSC.
- A. Islam, D. Zhang, X. Ouyang, R. Yang, T. Lei, L. Hong, R. Peng, L. Duan and Z. Ge, J. Mater. Chem. C, 2017, 5, 6527–6536 RSC.
- C. Li, Z. Li, X. Yan, Y. Zhang, Z. Zhang and Y. Wang, J. Mater. Chem. C, 2017, 5, 1973–1980 RSC.
-
(a) Y. P. Li, F. Li, H. Y. Zhang, Z. Q. Xie, W. J. Xie, H. Xu, B. Li, F. Z. Shen, L. Ye, M. Hanif, D. G. Ma and Y. G. Ma, Chem. Commun., 2007, 231–233 RSC;
(b) Y. P. Li, F. Z. Shen, H. Wang, F. He, Z. Q. Xie, H. Y. Zhang, Z. M. Wang, L. L. Liu, F. Li, M. Hanif, L. Ye and Y. G. Ma, Chem. Mater., 2008, 20, 7312–7318 CrossRef CAS.
-
(a) L. Duan, J. Qiao, Y. D. Sun and Y. Qiu, Adv. Mater., 2011, 23, 1137–1144 CrossRef CAS PubMed;
(b) C. J. Zheng, J. Wang, J. Ye, M. F. Lo, X. K. Liu, M. K. Fung, X. H. Zhang and C. S. Lee, Adv. Mater., 2013, 25, 2205–2211 CrossRef CAS PubMed;
(c) D. D. Zhang, L. Duan, Y. L. Li, H. Y. Li, Z. Y. Bin, D. Q. Zhang, J. Qiao, G. D. Dong, L. D. Wang and Y. Qiu, Adv. Funct. Mater., 2014, 24, 3551–3561 CrossRef CAS.
-
(a) L. Duan, J. Qiao, Y. D. Sun and Y. Qiu, Adv. Mater., 2011, 23, 1137–1144 CrossRef CAS PubMed;
(b) D. D. Zhang, L. Duan, Y. L. Li, H. Y. Li, Z. Y. Bin, D. Q. Zhang, J. Qiao, G. D. Dong, L. D. Wang and Y. Qiu, Adv. Funct. Mater., 2014, 24, 3551–3561 CrossRef CAS.
- C. Li, S. Wang, W. Chen, J. Wei, G. Yang, K. Ye, Y. Li and Y. Wang, Chem. Commun., 2015, 51, 10632–10635 RSC.
-
(a) W. Z. Yuan, S. M. Chen, J. W. Y. Lam, C. M. Deng, P. Lu, H. M. H. Sung, I. D. Williams, H. S. Kwok, Y. M. Zhang and B. Z. Tang, Chem. Commun., 2011, 47, 11216–11218 RSC;
(b) X. J. Xu, S. Y. Chen, G. Yu, C. A. Di, H. You, D. G. Ma and Y. Q. Liu, Adv. Mater., 2007, 19, 1281–1285 CrossRef CAS.
- C. H. Chien, C. K. Chen, F. M. Hsu, C. F. Shu, P. T. Chou and C. H. Lai, Adv. Funct. Mater., 2009, 19, 2834–2843 CrossRef.
- R. L. Martin, J. Chem. Phys., 2003, 118, 4775–4777 CrossRef CAS.
- S. Tretiak and S. Mukamel, Chem. Rev., 2002, 102, 3171–3756 CrossRef CAS PubMed.
- Z. Ma, E. Wang, M. E. Jarvid, P. Henriksson, O. Inganas, F. Zhang and M. R. Andersson, J. Mater. Chem., 2012, 22, 2306–2314 RSC.
- V. Bulovic, A. Shoustikov, M. A. Baldo, E. Bose, V. G. Kozlov, M. E. Thomoson and S. R. Forrest, Chem. Phys. Lett., 1998, 287, 455–460 CrossRef CAS.
- X. Du, G. Li, J. Zhao, S. Tao, C. Zheng, H. Lin, Q. Tong and X. Zhang, Adv. Opt. Mater., 2017, 1700498–1700510 CrossRef.
- Z. R. Grabowski, K. Rotkiewicz and W. Rettig, Chem. Rev., 2003, 103, 3899–4032 CrossRef PubMed.
- C. Liu, Q. Fu, Y. Zou, C. Yang, D. Ma and J. Qin, Chem. Mater., 2014, 26, 3074–3083 CrossRef CAS.
- E. Lippert, W. Liider and H. Boos, in Advances in Molecular Spectroscopy, ed. A. Mangini, Pergamon Press, Oxford, 1962, p. 443 Search PubMed.
- Z. Wang, Y. Feng, S. Zhang, Y. Gao, Z. Gao, Y. Chen, X. Zhang, P. Lu, B. Yang, P. Chen and Y. M. S. Liu, Phys. Chem. Chem. Phys., 2014, 16, 20772–20779 RSC.
-
(a) S. P. Jagtap, S. Mukhopadhyay, V. Coropceanu, G. L. Brizius, J. Bré das and D. M. Collard, J. Am. Chem. Soc., 2012, 134, 7176–7185 CrossRef CAS PubMed;
(b) S. Shirai, S. Iwata, T. Tani and S. Inagaki, J. Phys. Chem. A, 2011, 115, 7687–7699 CrossRef CAS PubMed.
- Y. P. Yurenko, J. Novotný, V. Sklenářabc and R. Marek, Phys. Chem. Chem. Phys., 2014, 16, 2072–2084 RSC.
-
(a) M. S. Tsai, Y. C. Hsu, J. T. Lin, H. C. Chen and C. P. Hsu, J. Phys. Chem. C, 2007, 111, 18785–18793 CrossRef CAS;
(b) Y. N. Yan, W. L. Pan and H. C. Song, Dyes Pigm., 2010, 86, 249–258 CrossRef CAS;
(c) T. W. Kim, S. H. Oh, H. Choi, G. Wang, H. Hwang, D. Y. Kim and T. Lee, Appl. Phys. Lett., 2008, 92, 253308–253313 CrossRef.
- M. R. Zhu and C. L. Yang, Chem. Soc. Rev., 2013, 42, 4963–4976 RSC.
- H. H. Chou, Y. H. Chen, H. P. Hsu, W. H. Chang, Y. H. Chen and C. H. Cheng, Adv. Mater., 2012, 24, 5867–5871 CrossRef CAS PubMed.
- Q. Zhang, J. Li, K. Shizu, S. Huang, S. Hirata, H. Miyazaki and C. Adachi, J. Am. Chem. Soc., 2012, 134, 14706–14709 CrossRef CAS PubMed.
- Y. Liu, L. S. Cui, M. F. Xu, X. B. Shi, D. Y. Zhou, Z. K. Wang, Z. Q. Jiang and L. S. Liao, J. Mater. Chem. C, 2014, 2, 2488–2495 RSC.
- Y. H. Lou, M. F. Xu, L. Zhang, Z. K. Wang, S. Naka, H. Okada and L. S. Liao, Org. Electron., 2013, 14, 2698–2704 CrossRef CAS.
- Z. Wang, Y. Lou, S. Naka and H. Okada, Appl. Phys. Lett., 2011, 98, 063302–063304 CrossRef.
- J. Liu, Z. Zeng, X. Cao, G. Lu, L. H. Wang, Q. L. Fan, W. Huang and H. Zhang, Small, 2012, 8, 3517–3522 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
-
(a) S. Tokito, T. Tsutsui and Y. Taga, J. Appl. Phys., 1999, 86, 2407–2411 CrossRef CAS;
(b) S. H. Cho, J. R. Oh, H. K. Park, H. K. Kim, Y. H. Lee, J. G. Lee and Y. R. Do, Opt. Express, 2010, 18, 1099–1104 CrossRef CAS PubMed.
- A. O. Eseola, O. Adepitan, H. Gorls and W. Plass, New J. Chem., 2012, 36, 891–902 RSC.
- H. Liu, Q. Bai, L. Yao, H. Zhang, H. Xu, S. Zhang, W. Li, Y. Gao, J. Li, P. Lu, H. Wang, B. Yang and Y. Ma, Chem. Sci., 2015, 6, 3797–3804 RSC.
- Z. Wang, Y. Feng, H. Li, Z. Gao, X. Zhang, P. Lu, P. Chen, Y. Ma and S. Liu, Phys. Chem. Chem. Phys., 2014, 16, 10837–10843 RSC.
- Z. Gao, Y. Liu, Z. Wang, F. Shen, H. Liu, G. Sun, L. Yao, Y. Lv, P. Lu and Y. Ma, Chem.–Eur. J., 2013, 19, 2602–2605 CrossRef CAS PubMed.
- X. L. Li, X. Ouyang, D. Chen, X. Cai, M. Liu, Z. Ge, Y. Cao and S. J. Su, Nanotechnology, 2016, 27, 124001–124011 CrossRef PubMed.
- Y. Zhang, T. W. Ng, F. Lu, Q. X. Tong, S. L. Lai, M. Y. Chan, H. L. Kwong and C. S. Lee, Dyes Pigm., 2013, 98, 190–194 CrossRef CAS.
- A. Fisher, S. Chenais, S. Forget, M. C. Castex, D. Ades, A. Siove, C. Denis, P. Maisse and B. Geffroy, J. Phys. D: Appl. Phys., 2006, 39, 917–922 CrossRef.
- Y. Zhang, S. L. Lai, Q. X. Tong, M. F. Lo, T. W. Ng, M. Y. Chan, Z. C. Wen, J. He, K. S. Jeff, X. L. Tang, W. M. Liu, C. C. Ko, P. F. Wang and C. S. Lee, Chem. Mater., 2012, 24, 61–70 CrossRef.
- F. I. Wu, P. I. Shih, M. C. Yuan, A. K. Dixit, C. F. Shu, Z. M. Chung and E. W. G. Diau, J. Mater. Chem., 2005, 15, 4753–4760 RSC.
- Y. L. Liao, C. Y. Lin, K. T. Wong, T. H. Hou and W. Y. Hung, Org. Lett., 2007, 9, 4511–4514 CrossRef CAS PubMed.
- J. Yang, Q. Guo, J. Wang, Z. Ren, J. Chen, Q. Peng, D. Ma and Z. Li, Adv. Opt. Mater., 2018, 6, 1800342–1800348 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra00135b |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *,
Venugopal Thanikachalam
*,
Venugopal Thanikachalam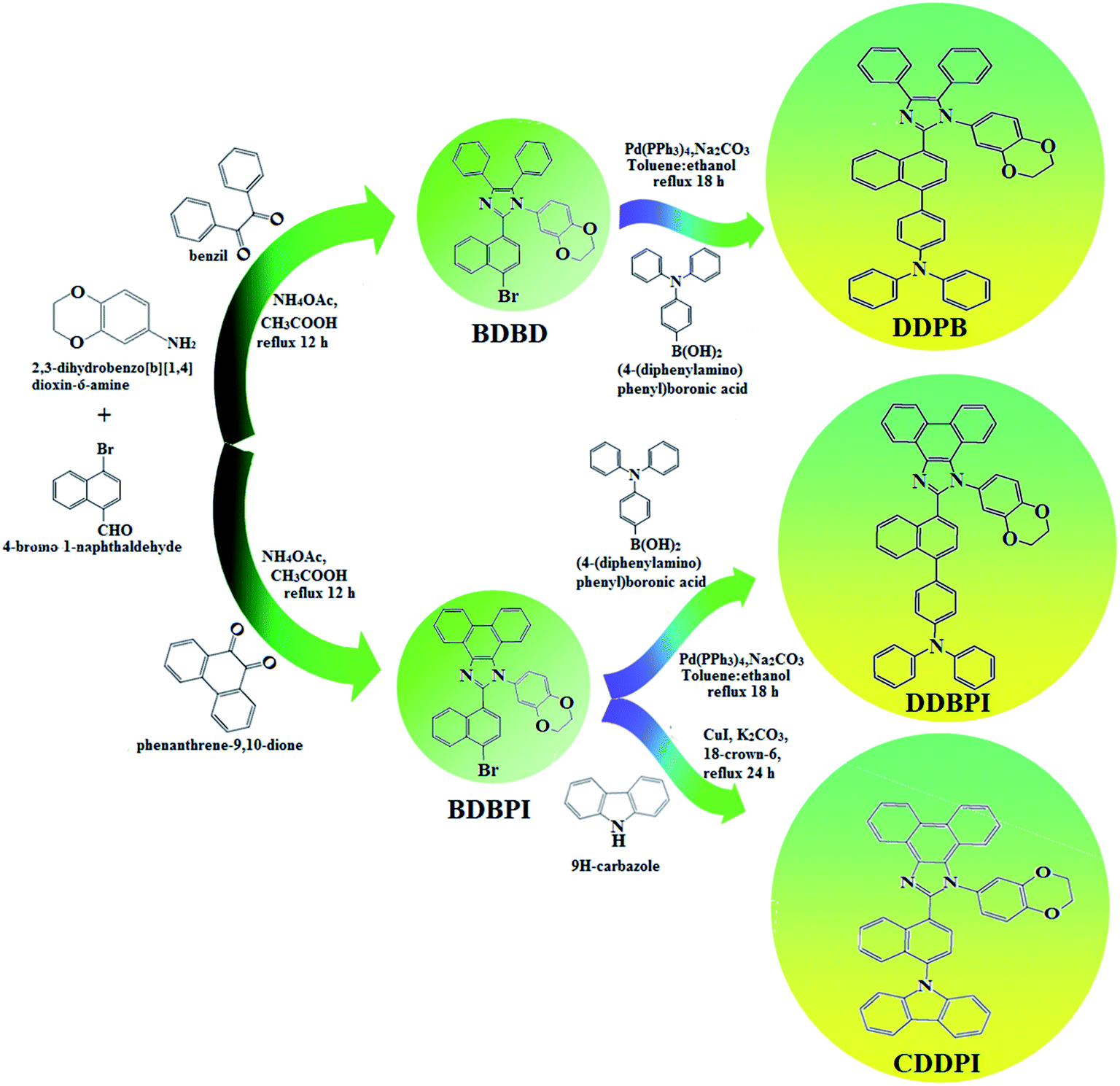

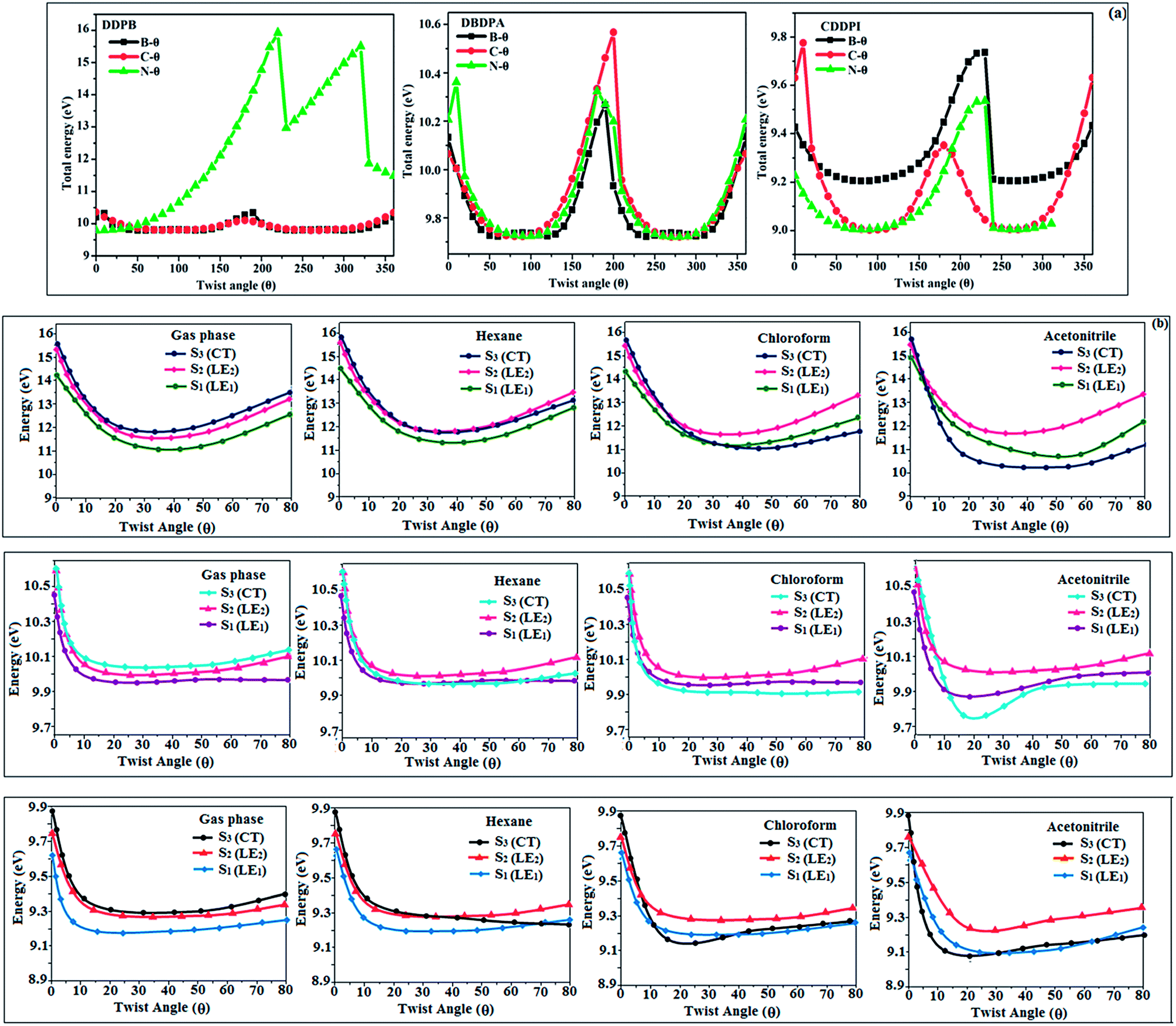
 is possible. However, at 50° (low energy twisting conformation) the electron density is partially localized on the frontier orbitals of DDPB, DBDPA and CDDPI. Therefore at 50°,
is possible. However, at 50° (low energy twisting conformation) the electron density is partially localized on the frontier orbitals of DDPB, DBDPA and CDDPI. Therefore at 50°,  transition is not pure CT but it is intercrossed between CT and LE transitions.59 The HOMO and LUMO of DDPB, DBDPA and CDDPI display adequate separation features and the differences are quite small which benefits the hole- and electron-transport properties (bipolar properties) and reduces the singlet–triplet splitting (ΔEST).80 Hence, the HOMO and LUMO moieties individually undertake the electron and hole transport functions. The calculated electron/hole transfer integrals of CDDPI (0.23/0.31 eV), DDPB (0.24/0.39 eV) and DBDPA (0.26/0.48 eV) reveal that these materials are bipolar materials. Moreover, these compounds displays both reduction and oxidation behaviour, revealing that these compounds possess good electron and hole transport abilities, hence, the synthesized materials are bipolar transport materials.81
transition is not pure CT but it is intercrossed between CT and LE transitions.59 The HOMO and LUMO of DDPB, DBDPA and CDDPI display adequate separation features and the differences are quite small which benefits the hole- and electron-transport properties (bipolar properties) and reduces the singlet–triplet splitting (ΔEST).80 Hence, the HOMO and LUMO moieties individually undertake the electron and hole transport functions. The calculated electron/hole transfer integrals of CDDPI (0.23/0.31 eV), DDPB (0.24/0.39 eV) and DBDPA (0.26/0.48 eV) reveal that these materials are bipolar materials. Moreover, these compounds displays both reduction and oxidation behaviour, revealing that these compounds possess good electron and hole transport abilities, hence, the synthesized materials are bipolar transport materials.81
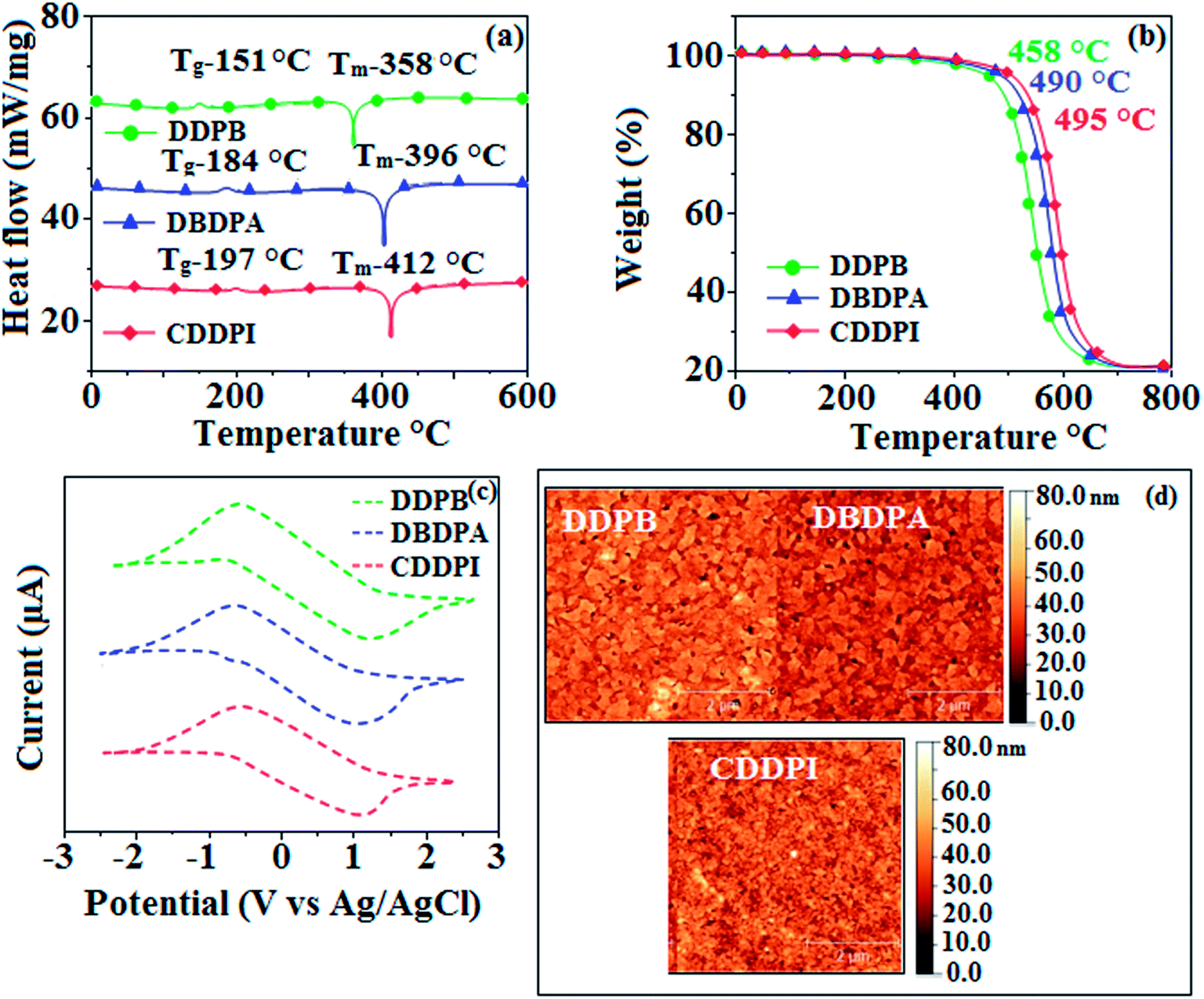
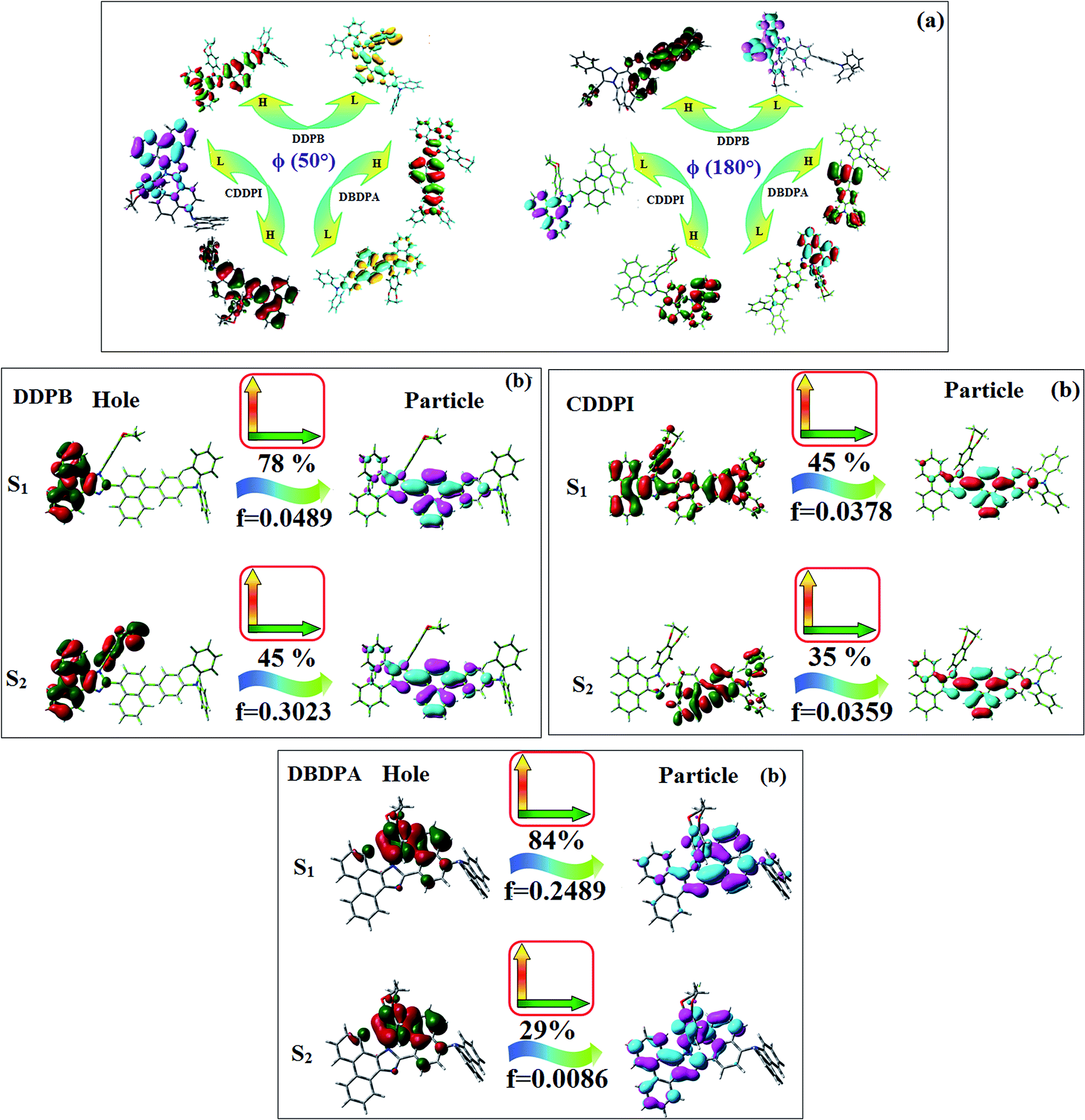
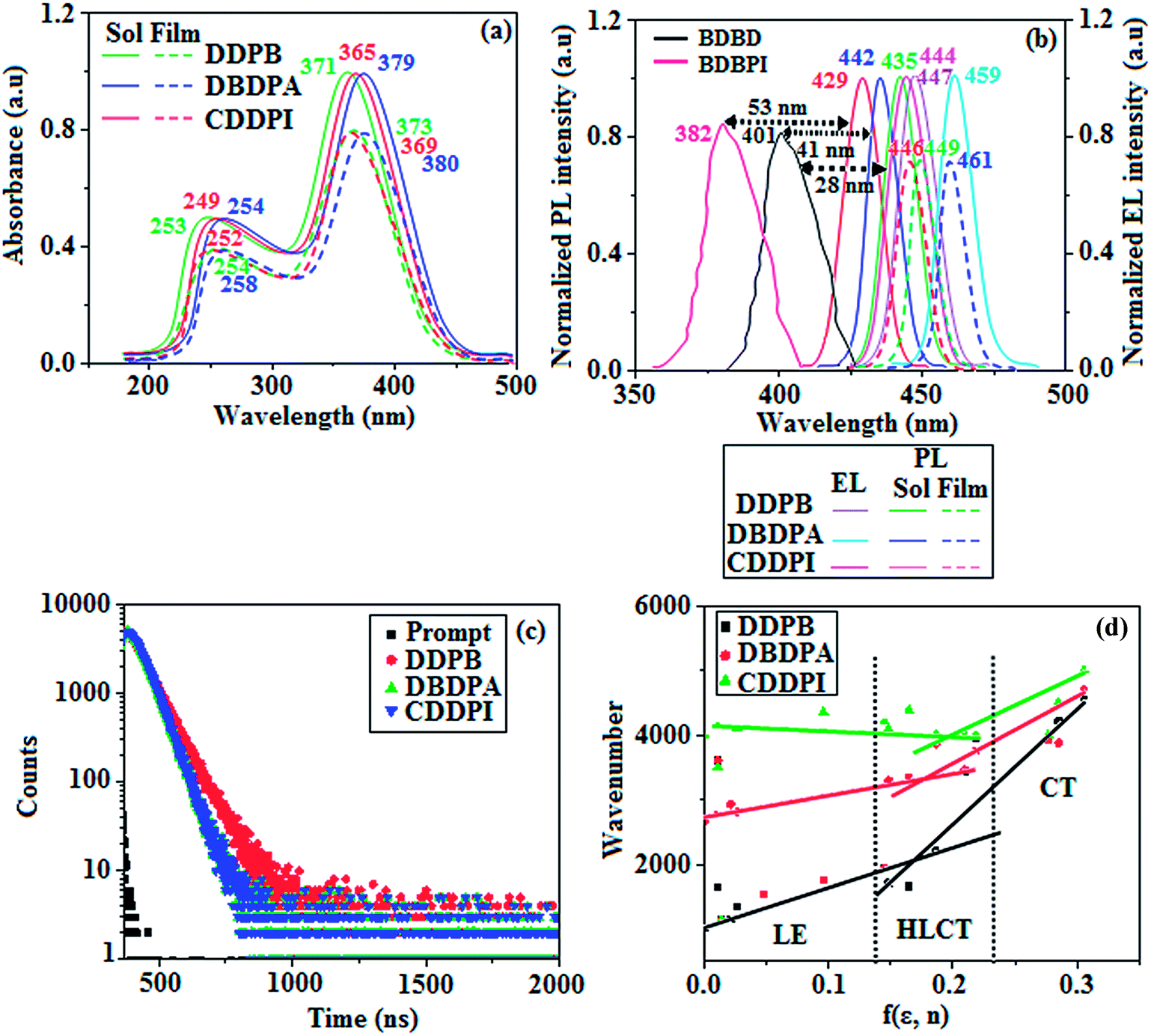
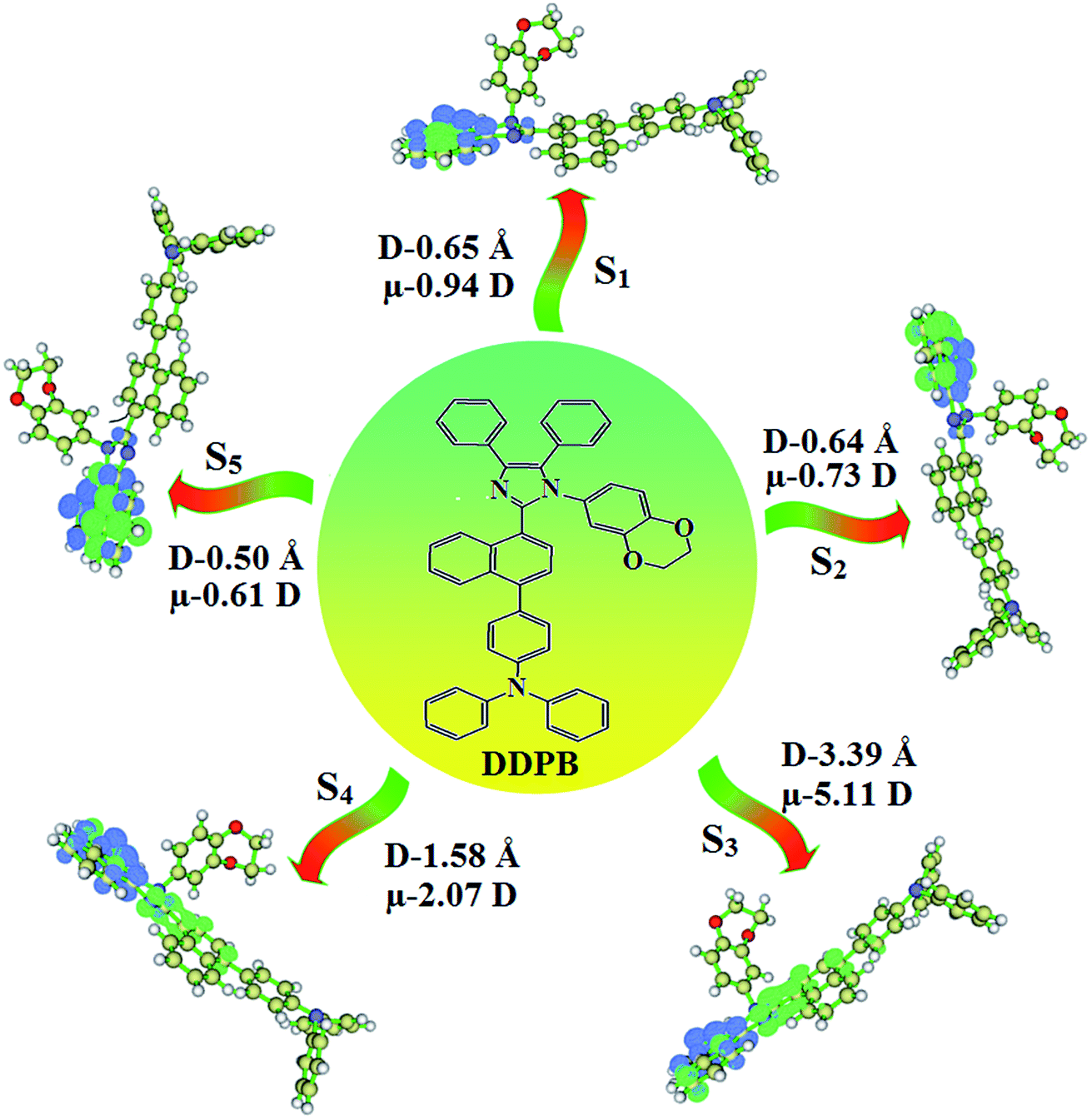
 -green increasing electron density and
-green increasing electron density and  -blue decreasing electron density [density = transition = nIOp(6/8 = 3)]].
-blue decreasing electron density [density = transition = nIOp(6/8 = 3)]].

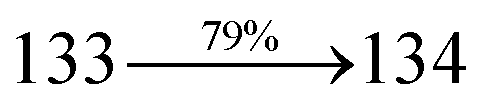
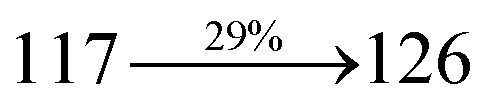
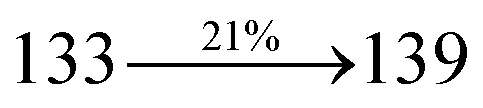
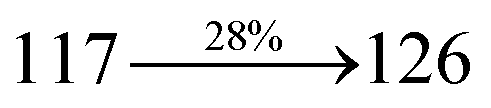
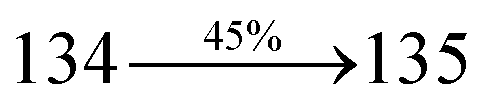
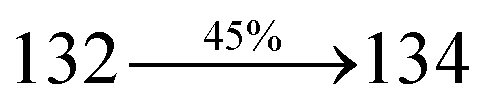
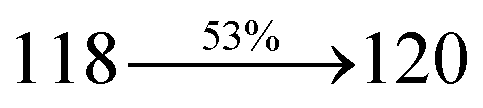
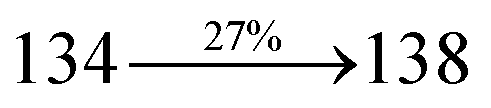

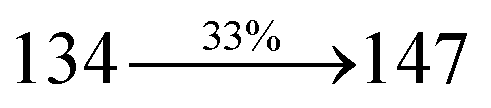
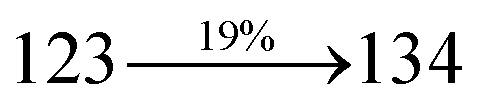
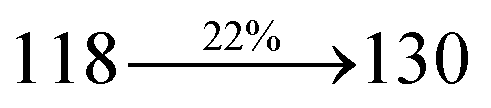
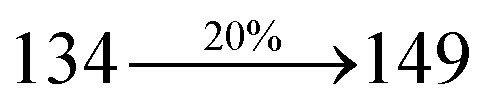
 1
1