
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Retracted Article: Synthesis of deuterated isopentyl pyrophosphates for chemo-enzymatic labelling methods: GC-EI-MS based 1,2-hydride shift in epicedrol biosynthesis†
Madhukar S. Saidab,
Govinda R. Navaleab,
Jayant M. Gajbhiyeab and
Sandip S. Shinde *a
*a
aOrganic Chemistry Division, CSIR-National Chemical Laboratory (CSIR-NCL), Dr Homi Bhabha Road, Pune-411008, India. E-mail: ss.shinde@ncl.res.in
bAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad, 201 001, India
First published on 9th September 2019
Abstract
A sesquiterpene epicedrol cyclase mechanism was elucidated based on the gas chromatography coupled to electron impact mass spectrometry fragmentation data of deuterated (2H) epicedrol analogues. The chemo-enzymatic method was applied for the specific synthesis of 8-position labelled farnesyl pyrophosphate and epicedrol. EI-MS fragmentation ions compared with non-labelled and isotopic mass shift fragments suggest that the 2H of C6 migrates to the C7 position during the cyclization mechanism.
Terpenoids are one of the most valuable sub-classes of natural products, consisting of thousands of interesting structures with well functionalized scaffolds.1 Numerous terpene cyclase enzymes exist, isolated from bacteria, fungi and plants, but few have been investigated and characterized.2 The universal precursor isopentenyl pyrophosphate (IPP) plays a key role in extending linear alkylpyrophosphates, such as geranylpyrophosphates (GPP), farnesylpyrophosphates (FPP) and geranylgeranyl pyrophosphates (GGPP), which when finally triggered by terpene cyclase lead to the production of diverse hydrocarbon skeletons.3 This terpene cyclase catalysed mechanism involves specific rearrangements such as multiple cascade cyclisation, hydride shifts, and stereoselective reactions via cationic intermediates to achieve unique cyclic hydrocarbon skeletons.4
The biosynthesis mechanisms of enzymes have been investigated by feeding isotope-labelled precursors to terpene cyclase. These isotopes include deuterium, (2H), carbon (13C) and tritium (3H), and have been commonly used for the characterization of some enzymes, including patchouli alcohol synthase, cadinene synthase, and geosmin synthase etc.5 The conventional method to trace isotopically-labelled products derived from enzyme catalysts is nuclear magnetic resonance (NMR) spectroscopy, which generally requires milligram amounts of product for complete analysis, thus to synthesis a sufficient amount of product requires high quantities of labelled precursors and enzymes. Recently, the mechanisms of complex cyclic terpenes such as epicubebol,6 iso-dauc-8-en-11-ol,6 and pristinol synthase,7 diterpene synthase, phomopsene8 spiro-albatene9 and 18-hydroxydo labella-3,7-diene10 have been studied by gas chromatography coupled to electron impact mass spectrometry (GC/EI-MS). The key advantage of GC-MS analysis is that it requires less product compared to NMR spectroscopy, and is also a highly sensitivity, rapid method of analysis.11
The chemical synthesis of specific position labelled linear prenyl pyrophosphate is time consuming, laborious, and requires a multistep reaction process.12 To overcome these problems, a chemo-enzymatic strategy can be considered as an alternative protocol for the rapid synthesis of a specific precursor.13
A bioactive sesquiterpene epicedrol (EC, GenBank: AF157059) has been isolated from Artemisia annua L.14 Epicedrol cyclase (ECS) converts linear FPP into the unique tricyclic complex alcohol epicedrol. Recently, we studied the GC-EI-MS fragmentation of epicedrol derived from enzyme assay in H218O, which revealed that the source of the oxygen atom in EC was derived from water not from the pyrophosphate of FPP.15 In continuation of epicedrol biosynthesis study, herein a chemo-enzymatic strategy is applied to synthesise (2H)-labelled-EC and investigate the cyclisation mechanism of ECS by EI-MS fragmentation ions compared with non-labelled species and isotopic mass shift fragments (Fig. 1). In this strategy, the chemically prepared synthetic four (D)-IPPs 2–5 were used for the elongation to (2H)-FPP with dimethylallyl pyrophosphate (DMAPP) and GPP through FPP synthase from Santalum album L. and GPP synthase from Catharanthus roseus. The subsequent cyclisation by ECS yielded specific 8-position labelled EC analogues, which were analysed by GC-MS.
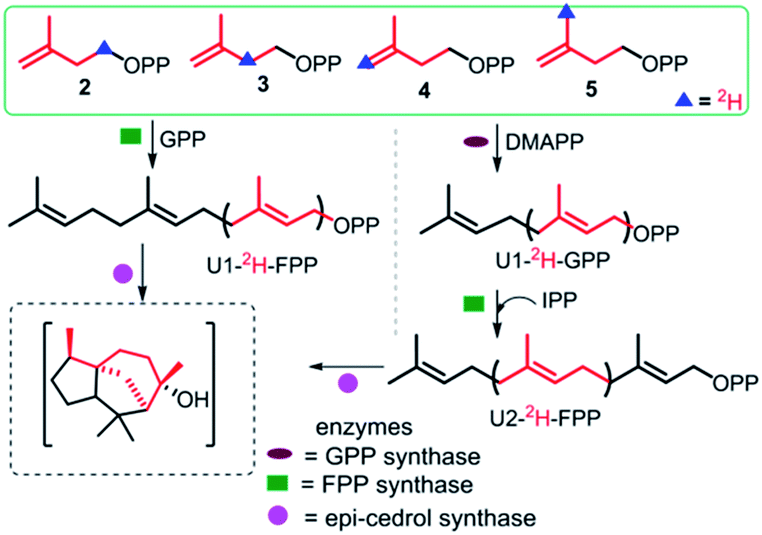 | ||
| Fig. 1 Strategy to specifically synthesise deuterium-labelled epicedrol using a chemo-enzymatic method. | ||
To explore the structure-based mechanistic pathway of epicedrol biosynthesis, initially all four (2H)-IPPs 2–5 were synthesized from the starting material methyl acetoacetate (1). The syntheses of labelled (1-2H2)-IPP (2) and (2-2H2)-IPP (3) were achieved by following the general synthesis steps shown in Scheme 1. Initially, the ketone protecting group of 1 reacts with ethylene glycol in the presence of PTSA (p-toluenesulfonic acid) under azeotropic conditions to afford the desired ester 6.
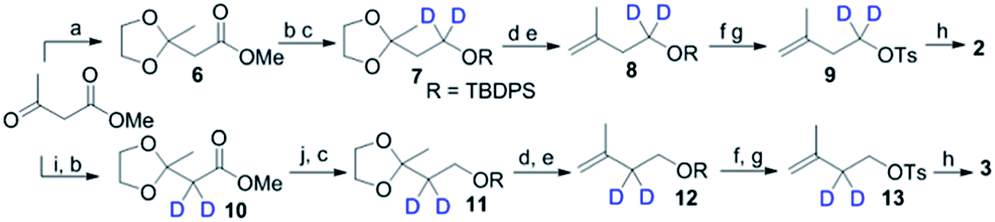 | ||
Scheme 1 Synthesis of 2 and 3. (a) Ethylene glycol, PTSA, toluene, reflux; (b) Na, MeOD, hexane 15 min; (c) TBDPSCl, TEA, DCM, 0 °C, 6 h; (d) PPTS, H2O/acetone 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, RT, 12 h; (e) PPh3CH3Br, n-BuLi, THF, 0 °C, 3 h; (f) TBAF, THF, RT, 2 h; (g) TsCl, DMAP, DCM, 0 °C, 10 h; (h) (Bu4N)3PO7H, ACN, RT; (i) D2O, RT, 12 h; (j) LiAlH4, THF, 2 h, 0 °C. :1, RT, 12 h; (e) PPh3CH3Br, n-BuLi, THF, 0 °C, 3 h; (f) TBAF, THF, RT, 2 h; (g) TsCl, DMAP, DCM, 0 °C, 10 h; (h) (Bu4N)3PO7H, ACN, RT; (i) D2O, RT, 12 h; (j) LiAlH4, THF, 2 h, 0 °C. | ||
Further, the ester group is reduced with 2H followed by the use of Bouveault–Blanc conditions16 to obtain the corresponding alcohol, which when treated with TBDPSCl (tert-butyl-diphenylchlorosilane) gives protected-OH 7. The selective deprotection of the ketone of 7 is achieved in the presence of PPTS (pyridinium p-toluenesulfonate) in aqueous acetone. Furthermore, the free ketone is subjected to carbon Wittig olefination using methyl triphenyl phosphonium bromide, to afford 8 in quantitative yield. The TBDPS group of 8 is deprotected in the presence of TBAF (tetra-n-butylammonium fluoride), then subsequent tosylation gives 9. The pyrophosphorylation of precursor 9 can be successfully performed according to the procedure by Davisson et al.,17 to achieve (1-2H2)-2. Proton exchange with a deuterium atom using D2O can then be carried out on the active methylene group of 1, followed by ketone protection to give 10. The reduction of 10 with LiAlH4, and subsequent protection with TBDPSCl affords 11. Further, 11–13 can be achieved by following the same protocol used for the synthesis of 7–9, where pyrophosphorylation of 13 and ion exchange give C2-deuterated 3.
Scheme 2 shows the synthesis of (4-2H2)-IPP 4 and (5-C2H3)-IPP 5. Building block 14 is produced by sequential reduction of ester 6, then TBDPS protection, and deketalation under similar reaction conditions to those used in Scheme 1. The Witting reaction of 14 using (2H3)-methyltriphenylphosphonium iodide affords compound 15. Further transformation to 16 involves the same protocols of TBDPS deprotection and tosylation of 9 (Scheme 1). Then, pyrophosphorylation of 16 gives C4-labelled 4. In the synthesis of IPP 5, compound 14 is subjected to a haloform reaction in the presence of Br2 in NaOH to afford 17 as a carboxylic acid.18 Acid 17 can be converted into an amide in the presence of CD3I and N,O-dimethyl hydroxylamine hydrochloride, then Grignard reaction with CD3MgI at −10 °C leads to the formation of product 18. Further synthesis of 20 is achieved via a similar reaction pathway as that of tosylated 9. Pyrophosphorylation of 20 gives C5-labelled 5.
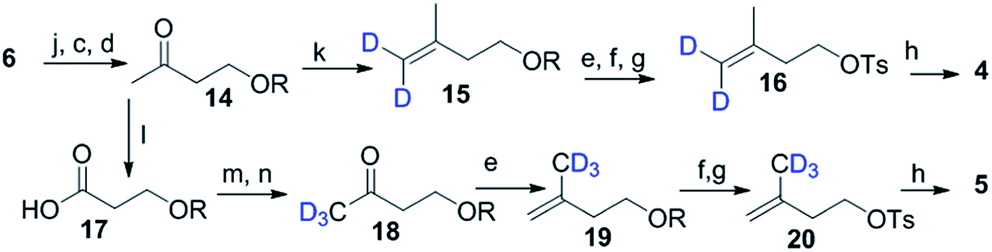 | ||
| Scheme 2 Synthesis of 4 and 5. (k) PPh3CD3Br (98% atom of D), n-BuLi THF, 0 °C, 3 h; (l) Br2, aq. NaOH, dioxane, 5 h; (m) CDI, CH3NHOCH3Cl, DCM 12 h; (n) CD3MgI (98% atom of D), THF, −78 °C, 3 h. | ||
Feeding enzyme assay of (2H)-IPPs 2–5 with GPP synthase and FPP in the presence of DMAPP and GPP afforded labelled FPPs. The unit-1 (U1) in the FPP labelling was achieved by FPP synthase assay between GPP and 2–5, to give (1-2H2), (2-2H), (4-2H2) and (15-C2H3)-FPP. Subsequent cyclisation by ECS yielded products to be analysed by GC/EI-MS. The obtained fragmentation ion data were compared with non-labeled epicedrol, as shown in Fig. 2.
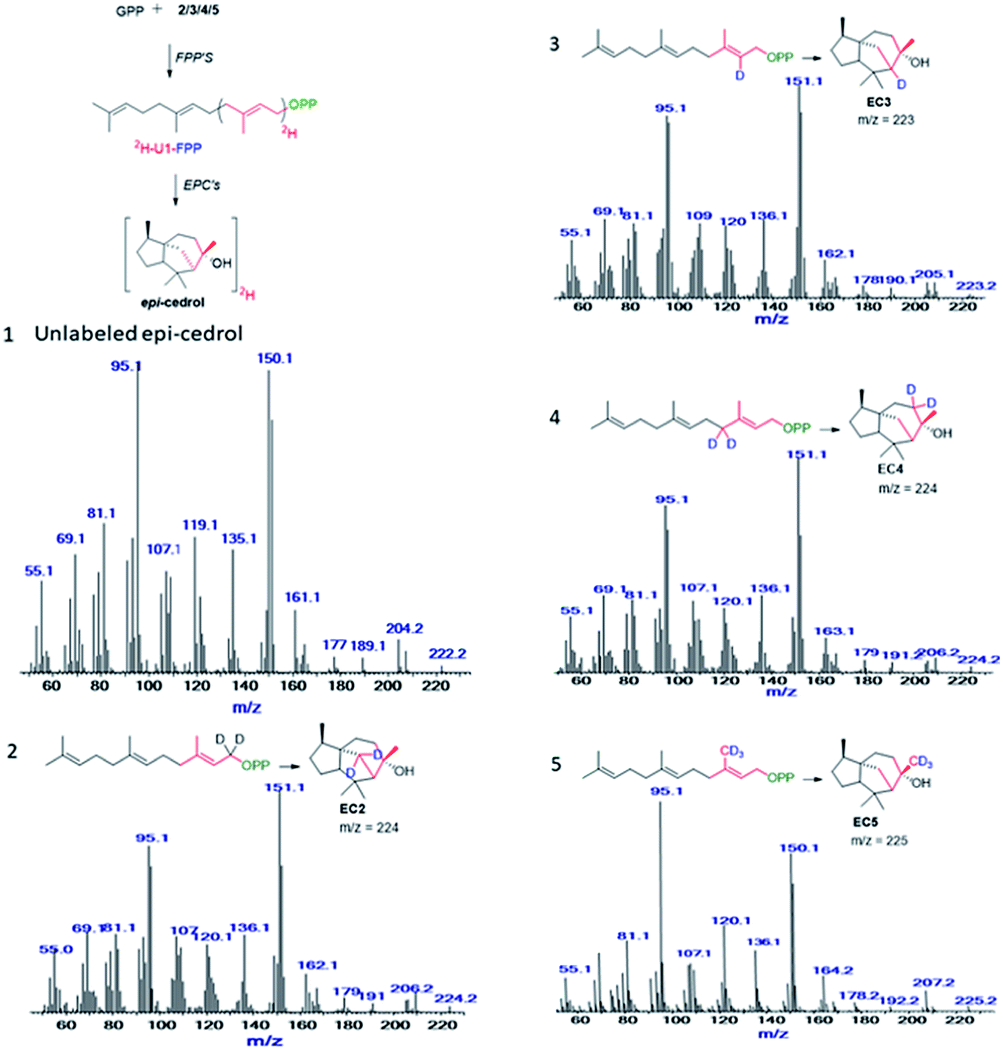 | ||
| Fig. 2 Mass spectra of epicedrol derived from (1), FPP, (2) C1–2H2 FPP, (3) C2–2H FPP, (4) C3–2H2 FPP, and (5) C15–2H3 FPP. | ||
The other four positions of the U2 of FPP were labeled as (5-2H2), (6-2H), (8-2H2) and (14-C2H3)-FPP by similar chemo-enzymatic methods using DMAPP, then subsequently exposed to FPP synthase, and ECS yielded deuterated EC analogues (as shown in ESI†). The products were analysed by GC/EI-MS and the comparative fragmentation patterns of unlabelled and (2H)-epicedrols were studied.
Their plausible structures were drawn after comparing the increased isotope mass shift fragments, as shown in Fig. 3.
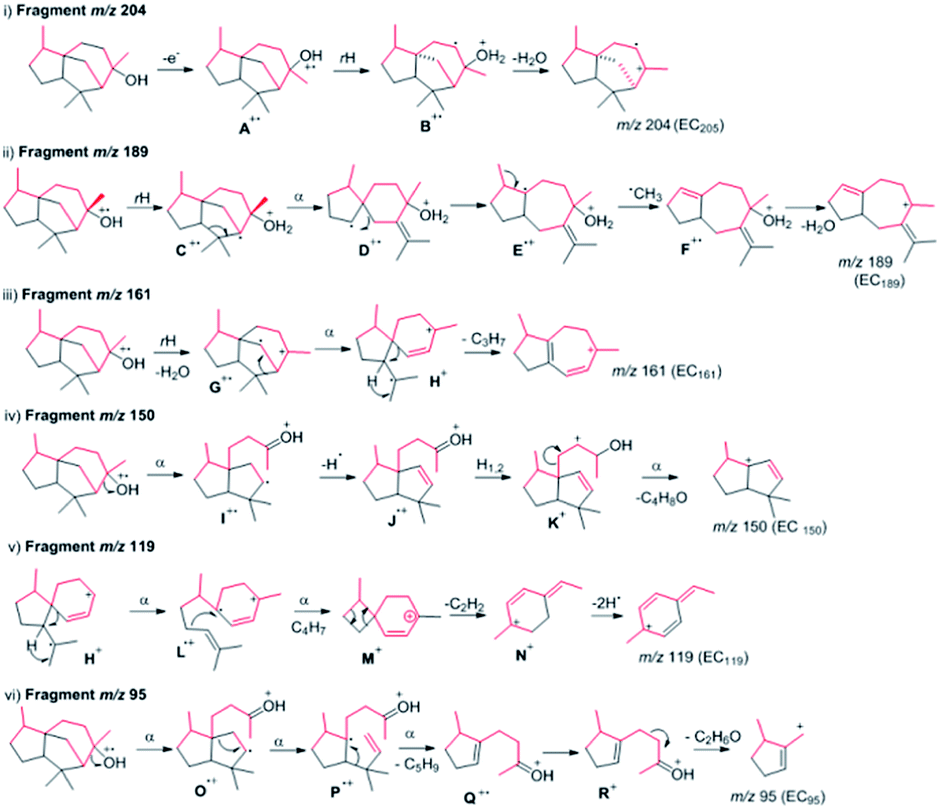 | ||
| Fig. 3 Possible fragmentation of epicedrol. The red lines indicate deuterium α-cleavage, rH: hydrogen rearrangement. | ||
The selected major fragments of epicedrol for analysis are the molecular ion [M]+ peak at m/z = 222, m/z = 95 (EC95), m/z = 204 (EC204), m/z = 161 (EC161), m/z = 150 (EC 150) and m/z 119 (EC119).
The origin of fragment EC204 reveals a position-specific mass shift in the formation of m/z = 204 by the elimination of water molecules with the loss of hydrogen. The fragment ion was observed for all 8 position-labelled ECs with an increased mass of +1, as observed for m/z = 205 in the experiment with (2-2H)-FPP. Mass shifts of +2 and +3 were observed at m/z = 206 and m/z = 207 in the spectra of (1-2H2), (4-2H2) and (15-C2H3)-FPP, as shown in Fig. 2. The electron impact ionization data of the ECs suggest the loss of an electron from the oxygen lone pairs to form B+˙, followed by hydrogen elimination with the loss of H2O to generate the fragment m/z = 204. This fragment ion structurally supports our previous H218O assay of epicedrol biosynthesis, where the tertiary cation intermediate was quenched with water.15
The structural fragment EC189 suggests that the generation of the fragment ion m/z = 189 can be traced through the methyl groups of FPP. Thus, in the fragmentation generated from the experiments on (14-C2H3)-FPP and (15-C2H3)-FPP, we compared the MS fragment ions to reveal that the labelled CD3 group disappears from (15-C2H3)-FPP at C15, but this is not observed in (14-C2H3)-FPP. This fragment ion indicates that the fragment originates from precursor m/z = 204 via the loss of a methyl radical.
The fragment m/z = 161 indicates the generation of the ion by the loss of a neutral iso-propyl group from fragment EC189. A plausible mechanism for this involves the sequential elimination of water then α-fragments from H+˙ followed by the loss of an isopropyl group. An alternative possibility for the formation of EC161 is via the cleavage of a C6–C10 bond to produce H+˙, which occurs during ring opening with the elimination of an isobutene unit, followed by an ethylene unit, with concomitant hydrogen rearrangement and multiple hydrogen eliminations to give N+ and the formation of ion m/z = 119. The mass fragment ion m/z = 150 clearly indicates its formation from epicedrol by the α-cleavage of the C2–C3 bond followed by inductive cleavage with the neutral loss of iso-butyraldehyde. This cation intermediate stabilizes via a tertiary carbocation and radical. After the initiation of A+˙ fragmentation, the formation of the structurally stable base ion fragment m/z = 95 is achieved after proton transfer to oxygen to yield P+˙ and subsequent hydrogen rearrangements via Q+ and R+ followed by inductive loss of acetone via a ring opening reaction. Furthermore, the B and C rings formed in epicedrol originate from isoprene U2 and U3-FPP.
The mechanism of hydride migration from C6 to C7 was studied by conversion of (6-2H)-FPP to its corresponding epicedrol. The position-specific mass shift analysis of m/z = 95 shows an increase in the mass by +1 and even a loss of the C6 portion in the case of the fragment m/z = 161 (see the ESI†). The extracted information from all the GC-EI-MS spectra reveals that an exceptional labelling fragmentation pattern was observed in the MS analysis of the C7-containing fragment. These results suggest that the hydrogen migrated in a 1,2-hydride shift manner to quench bisabolyl cation D.
Based on the comparative fragmentation analysis of the GC/EI-MS data of non-labeled and labelled epicedrol, we proposed a possible mechanism for epicedrol biosynthesis, as mapped out in Scheme 3. The isomerization of the OPP (O-pyrophosphate) group of FPP forms the stable isomer nerolidyl diphosphate intermediate B. The ring cyclisation of the C1 and C6 carbons generates intermediate C, then the axial hydride in the C6 position was transferred to C7 to generate bisabolyl carbocation D. The cyclisation driving force of the C10 double bond results in subsequent ring closure to form a new bond from C6–C10 and spiro-intermediate E followed by the formation of a third ring through the new bond between the C2 double bond and C11 carbocation via C-ring closure. Finally, carbocation G was quenched by an external water source to produce a stable epicedrol.
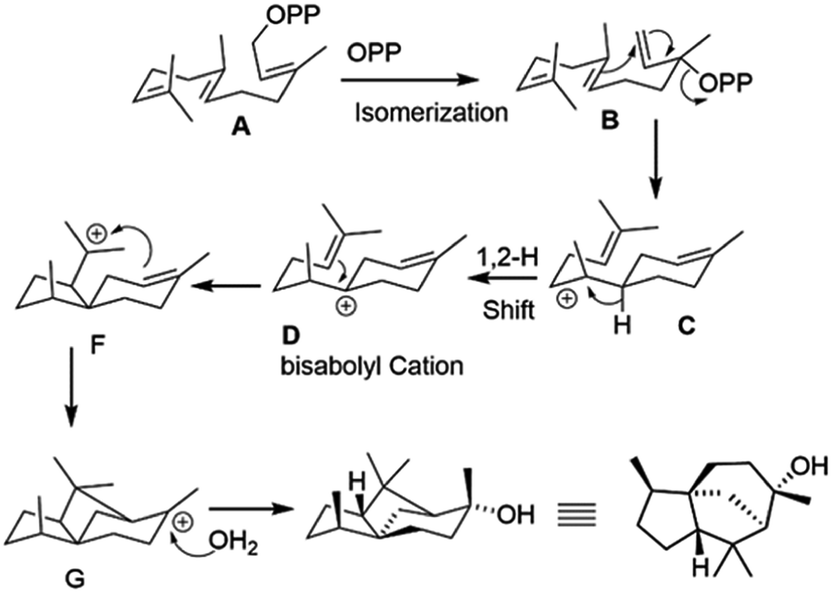 | ||
| Scheme 3 Proposed mechanism of epicedrol biosynthesis. | ||
In summary, four synthetic (2H)-IPP isomers 2–5 were prepared from the common starting material methyl acetoacetate and successfully used for (2H)-FPP synthesis through enzyme catalysts. The GC-EI-MS fragmentation based mechanism of epicedrol demonstrates that its biosynthesis proceeds via 1,2-hydride migration and cyclisation reactions. This strategy is highly sensitive and sufficient information on the proton/hydride rearrangement was obtained from the analysis of mass fragmented ions. Also, the rapid synthesis of any desired deuterium-labelled polyprenyl chain can be achieve using the four IPPs 2–5. Furthermore, we are finding an application for deuterated-IPP in a chemical and enzyme combination for valuable terpene synthesis.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) Dictionary of Natural Products, ed. J. Buckingham, Chapman & Hall, London, 1994, vol. 7 Search PubMed; (b) J. Buckingham, C. M. Cooper and R. Purchase, Natural Products Desk Reference, CRC Press, Taylor & Francis Group, Boca Raton, 2016, p. 235 Search PubMed.
- (a) J. S. Dickschat, Nat. Prod. Rep., 2016, 33, 87 RSC; (b) C. Joseph, Annu. Rev. Plant Physiol. Plant Mol. Biol., 1995, 46, 521 CrossRef; (c) S. Bharat and S. Ram, 3 Biotech., 2015, 2, 129 Search PubMed.
- (a) D. J. Miller, J. Gao, D. G. Truhlar, N. J. Young, V. Gonzaleza and R. K. Allemann, Org. Biomol. Chem., 2008, 6, 2346 RSC; (b) C. A. Citron, P. Rabe, L. Barra, C. Nakano, T. Hoshino and J. S. Dickschat, Eur. J. Org. Chem., 2014, 34, 7648 Search PubMed; (c) R. S. Nett, J. S. Dickschat and R. J. Peters, Org. Lett., 2016, 18, 5974 CrossRef CAS PubMed; (d) V. Jo Davison, T. M. Zabriskie and C. D. poulter, Bioorg. Chem., 1986, 14, 46 CrossRef.
- (a) D. W. Christianson, Chem. Rev., 2006, 106, 3412 CrossRef CAS PubMed; (b) M. Baunach, J. Franke and C. Hertweck, Angew. Chem., Int. Ed., 2015, 54, 2604 CrossRef CAS PubMed; (c) D. W. Christianson, Chem. Rev., 2017, 117, 11570 CrossRef CAS PubMed; (d) E. Oldfield and F. Lin, Angew. Chem., Int. Ed., 2012, 51, 1124 CrossRef CAS PubMed.
- (a) J. Jiaoyang and D. E. Cane, J. Am. Chem. Soc., 2008, 130, 428 CrossRef PubMed; (b) N. G. Vattekkatte, A. T. Köllner, J. Degenhardt, J. Gershenzon and W. Boland, Chem. Commun., 2015, 51, 3797 RSC; (c) J. A. Faraldos, D. J. Miller, V. González, Z. Yoosuf-Aly, O. Cascón, A. Li and R. K. Allemann, J. Am. Chem. Soc., 2012, 134, 5900 CrossRef CAS PubMed; (d) A. A. Scholte and J. C. Vederas, Org. Biomol. Chem., 2006, 4, 730 RSC; (e) J. A. Faraldos, S. Wu, J. Chappell and R. M. Coates, J. Am. Chem. Soc., 2010, 132, 2998 CrossRef CAS PubMed; (f) T. J. Savage and R. Croteau, Arch. Biochem. Biophys., 1993, 305, 581 CrossRef CAS PubMed.
- P. Rabe and J. S. Dickschat, Beilstein J. Org. Chem., 2016, 12, 1380 CrossRef CAS PubMed.
- T. A. Klapschinski, P. Rabe and J. S. Dickschat, Angew. Chem., Int. Ed., 2016, 55, 1 CrossRef PubMed.
- S. S. Shinde, A. Minami, Z. Chen, T. Tokiwano, T. Toyomasu, N. Kato, T. Sassa and H. Oikawa, J. Antibiot., 2017, 70, 632 CrossRef CAS PubMed.
- (a) J. Rinkel, L. Lauterbach, P. Rabe and J. S. Dickschat, Angew. Chem., 2018, 57, 3268 Search PubMed; (b) J. S. Dickschat, J. Rinkel, P. Rabe, A. B. Kashkooli and H. J. Bouwmeester, Beilstein J. Org. Chem., 2017, 13, 1770 CrossRef CAS PubMed.
- (a) R. P. Adam, Identification of Essential Oil Component by Gas Chromatography/Mass Spectrometry, Allured Publishing Corporation: Carol Stream, IL, USA, 2009 Search PubMed; (b) Mass spectra, in NIST Chemistry Web book, ed. P. J. Linstrom and W. G. Mallard, National Institutes of Standard and Technology, Gaithersburg, USA, 2016 Search PubMed; (c) S. Fuchs, T. Beck, S. Burkardt, M. Sandvoss and A. Mosandl, J. Agric. Food Chem., 1999, 47, 3053 CrossRef CAS PubMed; (d) M. Hea, Z. Yanga, T. Yang, Y. Ye, J. Nie, Y. Hu and P. Yana, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2017, 158, 1052 Search PubMed; (e) S. Bartram, A. Jux, G. Gleixner and W. Boland, Phytochemistry, 2006, 67, 1661 CrossRef CAS PubMed; (f) D. J. Miller, F. Yu and R. K. Allemann, ChemBioChem, 2007, 8, 1819 CrossRef CAS PubMed; (g) W. Wardencik, M. Michulec and J. Curyło, J. Food Sci. Technol., 2004, 39, 703 CrossRef.
- (a) P. Rabe, L. Barra, J. Rinkel, R. Riclea, C. A. Citron, T. A. Klapschinski, A. Janusko and J. S. Dickschat, Angew. Chem., Int. Ed., 2015, 54, 13448 CrossRef CAS PubMed; (b) Y. Jin, D. C. Williams, M. Hezari, R. Croteau and R. M. Coates, J. Org. Chem., 2005, 70, 4667 CrossRef PubMed; (c) A. Meguro, Y. Motoyoshi, K. Teramoto, S. Ueda, Y. Totsuka, Y. Ando, T. Tomita, S. Kim, T. Kimura, M. Igarashi, R. Sawa, T. Shinada, M. Nishiyama and T. Kuzuyama, Angew. Chem., Int. Ed., 2015, 54, 19 CrossRef PubMed; (d) Y. Totsuka, S. Ueda, T. Kuzuyama and T. Shinada, Bull. Chem. Soc. Jpn., 2015, 88, 575 CrossRef CAS; (e) T. J. Zahn, M. Eilers, Z. Guo, M. B. Ksebati, M. Simon, J. D. Scholten, S. O. Smith and R. A. Gibbs, J. Am. Chem. Soc., 2000, 122, 7153 CrossRef CAS.
- (a) M. Kobayashi, M. Ito, T. Koyama and K. Ogura, J. Am. Chem. Soc., 1985, 107, 4588 CrossRef CAS; (b) M. Ito, M. Kobayashi, T. Koyama and K. Ogura, Biochemistry, 1987, 26, 4745 CrossRef CAS PubMed.
- (a) Y. Zhang, L. Han, S. Chen, J. Guan, F. Qu and Y. Zhao, Biomed. Pharmacother., 2016, 83, 641 CrossRef CAS PubMed; (b) L. K. Eneh, H. Saijo, A. K. Borg-Karlson, J. M. Lindh and G. K. Rajarao, Malar. J., 2016, 15, 478 CrossRef PubMed.
- (a) P. Mercke, J. Corck, R. Croteau and E. J. Brodelius, Arch. Biochem. Biophys., 1999, 399, 213 CrossRef PubMed; (b) L. Hua and S. P. T. Matsuda, Arch. Biochem. Biophys., 1999, 399, 208 CrossRef PubMed; (c) B. E. Jackson, E. A. Hart-Wells and S. P. T. Matsuda, Org. Lett., 2003, 5, 1629 CrossRef CAS PubMed.
- S. S. Shinde, G. R. Navale, M. S. Said and H. V. Thulasiram, Tetrahedron Lett., 2016, 57, 1161 CrossRef CAS.
- M. Han, X. Ma, S. Yao, Y. Ding, Z. Yan, A. Adijiang, Y. Wu, H. Li, Y. Zhang, P. Lei, Y. Ling and J. An, J. Org. Chem., 2017, 82, 1285 CrossRef CAS PubMed.
- (a) V. J. Davisson, A. B. Woodside, T. R. Neal, K. E. Stremler, M. Muehlbacher and D. C. Poulter, J. Org. Chem., 1986, 51, 4768 CrossRef CAS; (b) V. J. Davisson, A. B. Woodside and C. D. Poulter, Methods Enzymol., 1985, 110, 130 CAS.
- M. K. Brown and E. J. Corey, Org. Lett., 2010, 12, 172 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR, GCMS, and HRMS data of pure compound. See DOI: 10.1039/c9ra00163h |
| This journal is © The Royal Society of Chemistry 2019 |