
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe physicochemical investigation of hydrothermally reduced textile waste and application within carbon-based electrodes†
Edward P. Randviir *a,
Omar Kanoua,
Christopher M. Liauwb,
Gary J. Millerc,
Hayley G. Andrewsc and
Graham C. Smithd
*a,
Omar Kanoua,
Christopher M. Liauwb,
Gary J. Millerc,
Hayley G. Andrewsc and
Graham C. Smithd
aSchool of Science and the Environment, Faculty of Science and Engineering, Manchester Metropolitan University, Chester Street, Manchester M1 5GD, Lancs, UK. E-mail: E.Randviir@mmu.ac.uk; Fax: +44(0)1612476831; Tel: +44(0)1612471188
bSchool of Healthcare Science, Faculty of Science and Engineering, Manchester Metropolitan University, Chester Street, Manchester M1 5GD, Lancs, UK
cTechnical Services, Faculty of Science and Engineering, Manchester Metropolitan University, Chester Street, Manchester M1 5GD, Lancs, UK
dDepartment of Natural Sciences, Faculty of Science & Engineering, University of Chester, Thornton Science Park, Chester CH2 4NU, UK
First published on 10th April 2019
Abstract
Textile waste is on the rise due to the expanding global population and the fast fashion market. Large volumes of textile waste are increasing the need for new methods for recycling mixed fabric materials. This paper employs a hydrothermal conversion route for a polyester/cotton mix in phosphoric acid to generate carbon materials (hydrochars) for electrochemical applications. A combination of characterization techniques revealed the reaction products were largely comprised of two major components. The first is a granular material with a surface C![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :O ratio of 2:1 interspersed with phosphorous and titanium proved using energy dispersive X-ray spectroscopy, and the other is a crystalline material with a surface C:O ratio of 3:2 containing no phosphorous or titanium. The latter material was found via X-ray diffraction and differential scanning calorimetry to be terephthalic acid. Electrochemical experiments conducted using the hydrochar as a carbon paste electrode demonstrates an increase in current response compared to carbon reference materials. The improved current responses, intrinsically related to the surface area of the material, could be beneficial for electrochemical sensor applications, meaning that this route holds promise for the development of a cheap recycled carbon material, using straightforward methods and simple laboratory reagents.
:O ratio of 2:1 interspersed with phosphorous and titanium proved using energy dispersive X-ray spectroscopy, and the other is a crystalline material with a surface C:O ratio of 3:2 containing no phosphorous or titanium. The latter material was found via X-ray diffraction and differential scanning calorimetry to be terephthalic acid. Electrochemical experiments conducted using the hydrochar as a carbon paste electrode demonstrates an increase in current response compared to carbon reference materials. The improved current responses, intrinsically related to the surface area of the material, could be beneficial for electrochemical sensor applications, meaning that this route holds promise for the development of a cheap recycled carbon material, using straightforward methods and simple laboratory reagents.
1. Introduction
Second to oil, the fashion industry is thought to be the largest polluter in the world, with the industry's environmental impacts being made worse by the so-called “fast fashion” market. Textiles based on synthetic polymers such as polyethylene terephthalate (PET) are petroleum intensive products, which are presenting a challenge for innovators to find solutions on how to create the same materials from non-virgin sources. According to the Department for Environment, Food and Rural Affairs (DEFRA), the UK produces 27.3 million tonnes of household waste per year, of which 819000 tonnes (3%) is textile waste.‡ Of this textile waste, 120000 tonnes (2% of total dry recyclables) is recycled under current waste management systems in the UK,§ leaving 700000 tonnes that is either landfilled or incinerated within the existing waste management system. For the purposes of societal change towards a circular economic model, it is of paramount importance that materials are first reused, and then second recycled into new products, as a means of prevention of environmental harm, air pollution, and excessive landfill burden. This is especially pertinent since most fabrics are not constructed from one material: in fact most are constructed from mixed materials, for example PET and cotton (cellulose). In 2016, 100 million tonnes of fibres were produced for fabrics, with around 70% of these being synthetic fibres (e.g. PET), and 30% being natural fibres (e.g. cotton).¶
In order for textiles to become more sustainable, there is a need for a whole systems change in the way society designs, consumes, and disposes of fabrics. This paper focusses upon the disposal (recycling) aspect of this paradigm. Currently researched routes for recycling of PET waste include methods such as glycolysis or hydrolysis of the polymer fibres, which breaks the ester linkages in the polymer chain, returning it to monomers. These monomers can either be fed back into the polymer production process as a starting product (though quality is compromised due to catalyst solubility issues1), or more commonly incorporated into another product with a different application, thus replacing a virgin material within another market, but not necessarily the textiles market. Glycolysed products have been reported to be utilised in coatings applications2–4 with varying levels of success, while pyrolysis is also another conversion route to reduce the fibres into carbon-based materials. However, further methods are required to address the sheer volume of waste polymer materials that are available for recycling. Such a carbon-rich source may be utilised for applications in energy storage, in a similar way to academic research conducted on recycling of biomass fibres.5 Indeed, there are numerous reports of hydrothermal conversion of biomass fibres into hydrochar for use in energy storage applications, particularly where the hydrochars are chemically activated with potassium hydroxide.6,7
Academic literature focussing on the hydrothermal synthesis of carbon from PET is limited, since most hydrothermal conversion routes are performed on biomasses such as cotton. Work by Yang et al. investigates the adsorption ability of mesoporous carbon produced through glycolysis of amorphous PET at atmospheric pressure, with subsequent calcination of the product.8 The mesoporous carbon exhibited a large surface area (1251 m2 g−1), making it an ideal material for adsorption of pollutants such as phenol. Another contribution from Guo et al. focussed upon creating a wearable technology, where graphene and MnO2 were applied to a PET fabric and its electrochemical capabilities evaluated.9 The result was a flexible fabric electrode with a capacitance of 332 F g−1, which exhibited no demonstrable changes upon bending the material, an essential requirement for materials targeted at the wearable technology market. In fact, there are many research papers devoted to functionalized PET fabric,10–12 but these have not dealt with the issue of end-of-life fabrics, which is the focus of this paper. In this study the hydrothermal reaction products of a 65:35 PET:cotton textile will be comprehensively characterised, and the electrochemical characteristics of the hydrochar produced investigated. A near cost-neutral approach for the manufacture of carbon electrodes using waste textiles as a carbon source is proposed. The main cost of these electrodes is the energy required to complete the reaction, opening up a possible recycling route for end-of-life textiles into electrochemical sensor applications. The reaction was carried out at low temperature and high pressure; energy costs are minimised by keeping the reaction temperature below the melting point of PET.
2. Experimental
2.1 Materials
All chemicals were of the highest grade commercially available and obtained from Sigma-Aldrich (UK). All solutions were prepared using deionised water of resistivity no less than 18.2 MΩ cm. All electroactive materials were dissolved in pH 7.4 Phosphate Buffer Solution (PBS) with 0.1 M KCl as a supporting electrolyte. The electrochemical performance of the hydrochars were monitored using 1 mM hexamine-ruthenium(III) chloride as an outer-sphere redox probe, which is a standard approach in the field for examining the electrochemical performance of materials within proof-of-concept studies.13–16 Benchmarking electrodes were constructed from Super P® carbon, obtained from Alfa Aesar (UK). Super P® is a 99% carbon material with a very low bulk density of 0.16 g cm−3, a high specific surface area of 62.0 m2 g−1, and an absorption stiffness value of 32.0 mL (5 g)−1. The fabric used for the hydrothermal reaction was cut from a clean lab coat that was at the end of its life. The material was a 65:35 PET:cotton blended yarn with a total carbon content of 53.22% and total nitrogen content of 0.2872% by mass (as determined by an elemental analyser). The lab coat textile was white in colour; subsequent investigations indicated presence of titanium dioxide pigment.
2.2 Hydrothermal reaction
The PET/cotton textile was cut into small (<1 cm2) pieces. The reaction was conducted by adding 3 g of a 65:35 PET:cotton textile to a Teflon-lined stainless steel autoclave as described in Fig. 1.
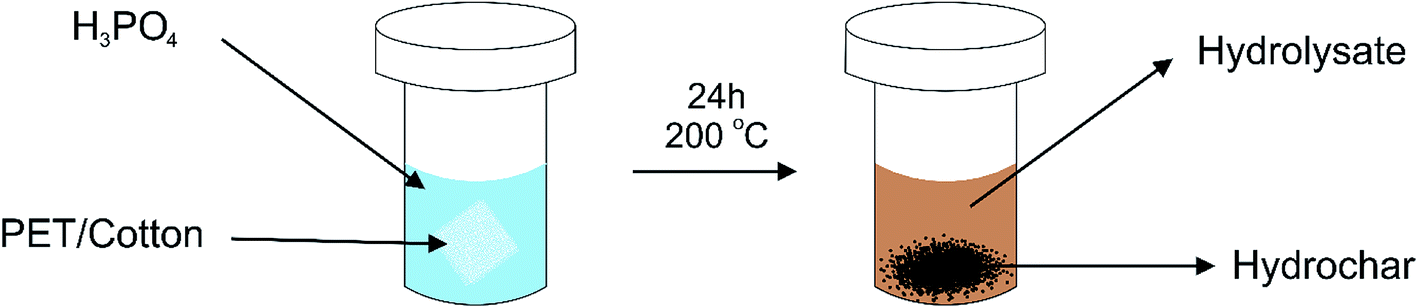 | ||
| Fig. 1 Schematic diagram of the hydrothermal reaction process. The textile material (3 g) is inserted into the Teflon®-lined stainless steel autoclave with a known concentration of H3PO4 (100 mL). The reaction vessel was placed in the oven at 200 °C for 24 h. | ||
The fabric was suspended in 100 mL of a known concentration of H3PO4 to promote the conversion of the fibres into a carbon material. The material was fully immersed in the acid prior to sealing the vessel to ensure the reaction was completely homogeneous and ran to completion. The reaction was conducted at 200 °C for 24 hours, which is 60 °C below the melting point of PET and significantly below pyrolysis temperatures. Previous reports have indicated that separation of PET and cotton fibres can occur under hydrothermal conditions, but the reactions must be performed in 1.5% HCl and below 160 °C to prevent the PET from reacting further.17 Thus, performing the reaction at a temperature higher than 160 °C allows PET conversion, yielding hydrochars containing an elevated carbon content, as measured by an elemental analyser, the results of which are given in ESI Table 1† (see later). After the reaction, it was noted that the liquor turned amber in colour, due to the formation of a range of chromophoric species that may include graphite oxide and various cellulose and PET degradation products. Formaldehyde was also likely to have been produced as indicated by its characteristic smell.
2.3 Elemental analysis
Carbon and nitrogen analysis was performed on the hydrochars using a TruSpec™ CN LECO elemental analyser. The software was TruSpec™ version 2.7. For each measurement, a sample of mass 0.1–0.2 g was accurately weighed to 4 decimal places (exact masses reported in ESI Table 1†). The instrument was calibrated using an EDTA calibration standard supplied by LECO (41.09% C; 5.51% H; 9.56% N). The instrument creates a calibration curve from 10 repeats to ensure variation and drift is accounted for within the final results.2.4 Infrared spectroscopy
Attenuated total reflection-Fourier transform infrared spectroscopy (ATR-FTIR) analysis was performed on the hydrochar samples using a PerkinElmer Spectrum Two FT-IR Spectrometer fitted with a UATR single bounce ATR accessory with diamond (refractive index 2.40) internal reflection element (IRE) (45° angle of incidence) and LiTaO3 detector. Spectra (4000 to 500 cm−1) were made up of four co-added scans with three replicate spectra being obtained for each sample. The presented data is therefore an average of 12 accumulations in total. The background was run before each sample and spectra were collected at ambient temperature (21 °C). Spectra were converted to absorbance using external data processing software. It was assumed that the sample contact area with the IRE stayed the same for all the hydrochars. This was justified on the basis of the Scanning Electron Microscopy (SEM) images of the hydrochars (see ESI Fig. 4†) that indicate similar particle size distribution regardless of treatment conditions, hence reasonably constant contact area on the IRE. By virtue of the latter, it was possible to use absorbance values as a measure of amount of a particular species in the hydrochar. Functional group analysis of the hydrochars was performed using Attenuated Total Reflectance (ATR) spectroscopy supplied by PerkinElmer (Spectrum Two FT-IR spectrometer). The sample holder was cleaned with methanol in between each sample run and wiped with tissue paper.2.5 Raman spectroscopy
Raman analysis was performed using a Thermo Scientific DXR Raman Microscope. Samples were prepared by fixing the sample (fabric or hydrochar) to a piece of double sided sticky tape mounted upon a glass microscope slide. A 780 nm laser operating at 24 mW power was focussed onto the sample with an estimated spot size of 1.1 μm. Spectra were collected over 20 exposures each lasting for 2 s.2.6 X-ray photoelectron spectroscopy
X-ray Photoelectron Spectroscopy (XPS) was performed on the samples to gain further insight into the structures of the reference and newly synthesized materials. A 12 × 12 mm piece was cut from the cloth sample using clean scissors and mounted using a double-sided conducting adhesive pad. The powder samples were pressed onto a piece of doubled-sided conducting adhesive pad on an XPS sample stub against a clean agate surface to form a dense compressed layer. Samples were analysed by XPS using a Specs GmbH Phoibos 150 mm mean radius hemispherical sector analyser with 9-channeltron detection and a monochromatised Al source (Specs GmbH Focus 500, 1486.6 eV). Charge neutralisation was achieved using Specs GmbH FG20 low energy electron gun at ∼5 eV energy. Data were acquired using pass energies of 50 eV (survey scan) or 20 eV (scans over individual lines). The pressure during analysis was ∼7 × 10−10 torr. Under these conditions the typical resolution on the C 1s line from graphite is ∼0.7 eV FWHM. Data were analysed using CasaXPS v2.3.16.2.7 Scanning electron microscopy and energy-dispersive X-ray spectroscopy
SEM was conducted on the samples using a Carl Zeiss Supra 40VP microscope, with SmartSEM software. The samples were loaded onto aluminium pin stubs using adhesive carbon tabs. Excess sample material was removed by tapping the aluminium stub. An Angle Selective Backscatter (ASB) detector was used to image the sample. For backscattered electron imaging, the microscope was operated under variable pressure conditions with a chamber pressure of 0.3 mbar, an acceleration voltage of 20 kV and a working distance of 8.5 ± 0.3 mm. Energy-Dispersive X-ray (EDX) spectroscopy was conducted on the samples using an EDAX Inc. Apollo 40 SDD EDX spectrometer fitted onto the SEM, with Genesis software. EDX analysis was performed at an acceleration voltage of 20 kV and a working distance of approximately 15 mm. For EDX analysis, the microscope was operated under high vacuum conditions with a chamber pressure of 2 × 10−5 mbar.2.8 X-ray diffraction
Reflection geometry X-ray powder diffraction (XRD) data were collected using a PANalytical X'Pert diffractometer fitted with a PixCEL 1-D detector using a Cu anode (Kα1 λ = 1.5406 Å) with the generator set at 40 mA, 40 kV. Data were collected in the range 5–120° 2θ with a step size of 0.013° 2θ and a collection time of 118 s per step using automatic divergence and antiscatter slits with an observed length of 8.0 mm. Data were processed using HighScore Plus version 4.0.2.9 Differential scanning calorimetry
Differential Scanning Calorimetry (DSC) was performed using a Perkin-Elmer DSC-7 power compensation DSC. Samples were contained in aluminium pans and run in a nitrogen (30 mL min−1) atmosphere over the temperature range 20–500 °C. Data was normalized to account for the mass of sample used in each run.2.10 Electrochemical methods
All measurements were performed using a PalmSens EmStat in conjunction with PSTrace version 4.7. Experiments were performed using a three electrode cell consisting of a Carbon Paste Electrode (CPE) working electrode, a platinum auxiliary electrode, and a Saturated Calomel Electrode (SCE) reference (+0.248 V vs. SHE). All solutions were degassed using high purity nitrogen prior to electrochemical measurements to prevent interference from oxygen. Voltammetric measurements were conducted in the potential window of 0.3 to −0.7 V (unless otherwise stated). Data is interpreted through analysis of the current sensitivity to the applied scan rate as a function of acid concentration in the hydrochar synthesis step. A quasi-reversible Randles–Ševćik relationship is employed for this (see later). All datasets are normalized to the Super P® CPE. Electron transfer rate constants were calculated from the modified Nicholson method reported by Lavagnini et al.18 The normalized k0 data is calculated by dividing the effective k0 value by the electroactive surface area, which is also estimated using the Randles–Ševćik relationship. Experiments were performed at ambient temperature (21 °C).2.11 Electrodes
The hydrochars were tested for their electrochemical properties using a CPE approach; the general approach is presented in Fig. 2.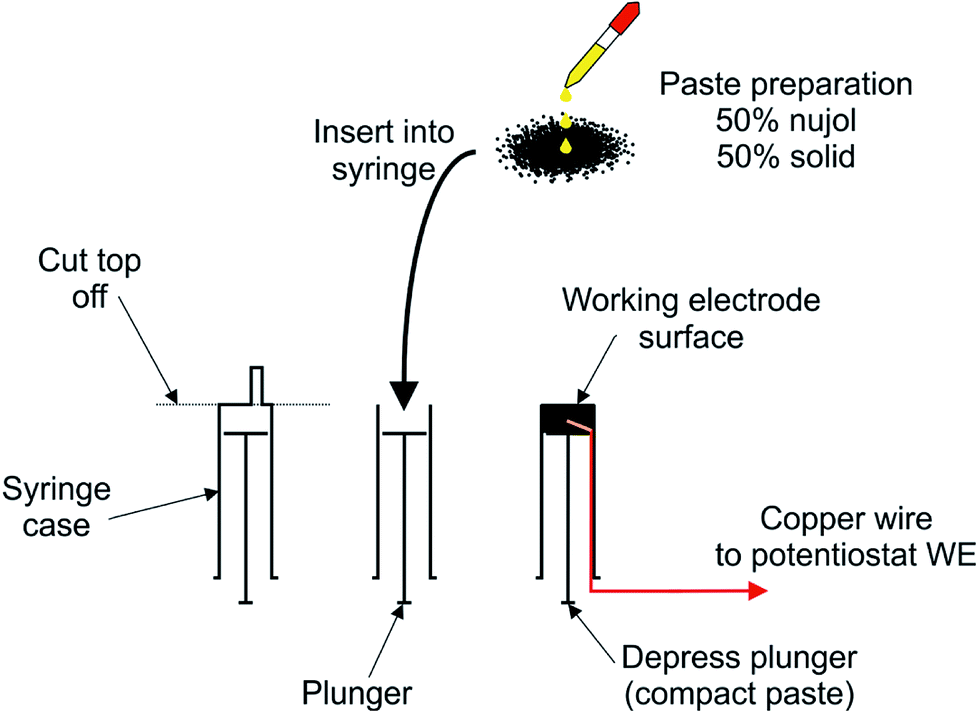 | ||
| Fig. 2 Preparation of the CPEs. The top of a syringe was cut using a scalpel, and a copper wire inserted such that the wire was exposed on the plunger surface. The paste material (made from SuperP® carbon and nujol) was inserted into the open end, defining the working electrode. The surface was levelled by depressing the plunger while the exposed carbon surface was pressed up against a sheet of white paper. | ||
The CPEs were built from a 5 mL syringe by cutting the end off the syringe with a scalpel and inserting a paste composed of 50% wt carbon based solids and 50% mineral oil (Nujol). The solid was a 50:50 mix of the hydrochar and Super P® carbon (denoted as conductive carbon henceforth). Previous (unpublished) work by the corresponding author revealed that electron transfer rates of CPEs mixed with plastic-derived hydrochars are unaffected up to 50% loading. The CPE is completed by connecting the paste to the working electrode port of the potentiostat using a length of copper wire. The electrode diameter was 10 mm and was polished in between each scan on a sheet of white paper until a flat homogeneous surface was obtained. The resulting CPE was used as the working electrode as described in work by Figueiredo-Filho et al.15
3. Results and discussion
This investigation aims to hydrothermally synthesize a conductive carbon material using a PET/cotton blend for incorporation into an electrochemical device. In order to fully understand the nature of the hydrochars synthesized within this work, a full physicochemical characterisation is presented using methods such as elemental analysis (LECO), IR, Raman, XPS XRD, SEM and EDX. The first section of the results will reveal the findings from the physicochemical characterization to build a picture of the chemical nature of the materials. The second part will investigate the surface interactions of the material with the outer-sphere redox probe, hexamine-ruthenium(III) chloride. The working hypothesis for this section is that an increase in acid concentration will produce a material with a higher C/O ratio, and thus exhibit increased conductive properties.3.1 Elemental analysis
Elemental analysis was performed on the obtained PET/cotton mix and all subsequent hydrochars. The results of the elemental analysis are presented in ESI Table 1,† and a plot of the carbon content versus reaction concentration is presented in ESI Fig. 1.† The carbon content of the PET/cotton mix was initially around 53% (ESI Fig. 1†), and increases to at least 55.5% after completion of the reaction. Interestingly, the data suggests that the concentration of acid promotes a higher carbon yield initially, as demonstrated by the increase in carbon content to 56.8% at 0.2 M, after which a decrease is observed when the acid concentration elevates further. Previous studies have shown that the concentration of H3PO4 has an effect on the activation energy of cellulose degradation, intermediate concentrations (ca. 3%) were found to reduce the activation energy for degradation relative to lower and higher concentrations, i.e. 2% and 5%.19 Indeed, the results presented within this section support such observations to some degree. There is also a decrease in nitrogen content by over 50% according to ESI Table 1.† The elemental analysis demonstrates oxygen removal during the reaction, which is to be expected within a hydrolysis-type reaction. It also potentially demonstrates that, though oxygen functionalities are targeted initially at low concentrations, carbon species are subsequently targeted at higher acid concentrations.3.2 Chemical bond analysis
As detailed in the Experimental section, the nature of chemical bonds were identified using and range of characterization techniques, including IR spectroscopy. Fig. 3 depicts the IR absorbance as a function of acid concentration of some selected peaks on the IR spectra (raw data presented in ESI Fig. 2†).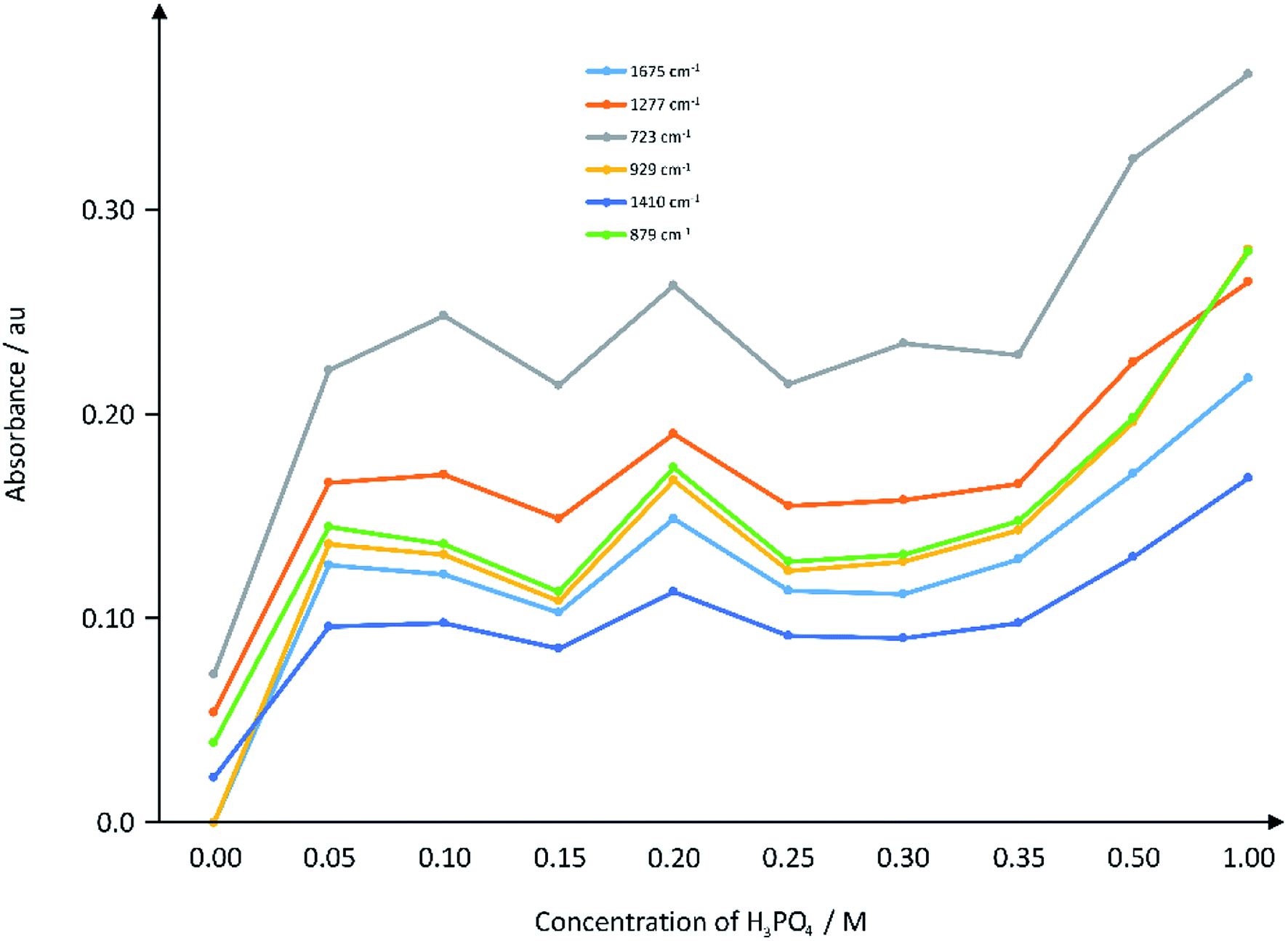 | ||
| Fig. 3 Plot of IR absorbance of selected wavenumbers against the concentration of acid in the hydrothermal reaction. The IR spectra were obtained from the ATR instrument as described in Section 2.4. | ||
The untreated PET/cotton textile was found to exhibit major absorption peaks at 1713 cm−1, 1241 cm−1, 1096 cm−1 and 723 cm−1. The peak at 1713 cm−1 is attributed to the ester carbonyl stretch in PET,20 whilst the peak at 1241 cm−1 is attributed to the phenolic C–O bond of the PET.21 The absorption at 1096 cm−1 is the C–O bending of the ester group in PET.22 There is also a strong absorption at 723 cm−1 attributed to C–H bonds of the ethylene glycol derived unit in the PET repeat unit.23 The aromatic C–H stretching bands in pure PET textile are very weak and are therefore non-detectable in the PET/cotton textile investigated here. The principle IR absorption bands of the cotton cellulose are at 3400 cm−1, 3360 cm−1 and 3267 cm−1 (hydrogen bonded OH stretching within the cellulose polymer crystalline regions) and C–O ether stretching in the hexose ring, centred at 1025 cm−1.
After the varying intensities of hydrothermal treatment, four completely new IR peaks were recorded at ca. 3100 cm−1, ∼1675 cm−1, ∼1410 cm−1 and ∼929 cm−1 compared to the PET/cotton mix on its own (Fig. 3 and ESI Fig. 2†). Aromatic C–H stretching bands (3100 cm−1) appear even at the lowest phosphoric acid concentration and remain at similar relative intensity at all acid concentrations; their formation is particularly significant as they indicate degradation and aromatisation of the polymers to structures that may have graphitic characteristics.24 The ester carbonyl band of the PET (1713 cm−1) reduces in intensity and becomes confounded with a new broad carbonyl band centred at 1675 cm−1 indicating that the hydrothermal reaction route favoured acid attack of the ester groups on the PET. This is a common degradation route for PET in non-accelerated conditions, thus the hydrothermal route appears to follow the same degradation route as previous reports.25 The absorption at ∼1675 cm−1 indicates a change to a ketone and/or carboxylic acid (the latter is supported by the presence of an –OH functionality at ∼1410 cm−1) that is conjugated with a phenyl ring or C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond, i.e. an α–β unsaturated carbonyl.21 The appearance of the aromatic C–H stretching bands may also be related to formation of terephthalic acid. Comparison with a reference spectrum for this acid indicates strong similarity to the hydrochar spectra that includes the unique shape of the OH and C–H stretching band envelope, the decrease in carbonyl stretching frequency to 1675 cm−1 and the doublet at ca. 1130 cm−1. The absorption at ∼929 cm−1 is thought to be a phosphorous-containing species, such as a P–O–P,26 derived from the phosphoric acid. The absorption at 723 cm−1 increases significantly after the reaction, which may indicate a contribution from aliphatic phosphines, while the increase in absorption at 1277 cm−1 may be an aliphatic phosphoryl contribution.27 The absorption at 1410 cm−1 is indicative of a P–O–CH2–CH3 functionality,26 which could easily form from the de-esterification of PET. EDX data (see later) supports the inclusion of phosphorous-containing compounds such as these.
C double bond, i.e. an α–β unsaturated carbonyl.21 The appearance of the aromatic C–H stretching bands may also be related to formation of terephthalic acid. Comparison with a reference spectrum for this acid indicates strong similarity to the hydrochar spectra that includes the unique shape of the OH and C–H stretching band envelope, the decrease in carbonyl stretching frequency to 1675 cm−1 and the doublet at ca. 1130 cm−1. The absorption at ∼929 cm−1 is thought to be a phosphorous-containing species, such as a P–O–P,26 derived from the phosphoric acid. The absorption at 723 cm−1 increases significantly after the reaction, which may indicate a contribution from aliphatic phosphines, while the increase in absorption at 1277 cm−1 may be an aliphatic phosphoryl contribution.27 The absorption at 1410 cm−1 is indicative of a P–O–CH2–CH3 functionality,26 which could easily form from the de-esterification of PET. EDX data (see later) supports the inclusion of phosphorous-containing compounds such as these.
Further analysis of the chemical bonds was conducted using Raman spectroscopy. During the Raman analysis, it was identified that the “crystalline” regions were providing a Raman fingerprint, while the “granular” regions were not (see SEM section for regional definitions). The Raman spectra obtained from the crystalline (see ESI Fig. 3†) were weak, however they bore resemblance to that of terephthalic acid,28 with characteristic Raman shifts appearing at 1628 cm−1, 1287 cm−1, 1178 cm−1, 1124 cm−1, 831 cm−1, 351 cm−1 and 264 cm−1.
XPS was also performed on the hydrochars, with the results presented in Table 1.
| PET/cotton | 0.05 M | 0.1 M | 0.2 M | 0.3 M | 0.5 M | 1.0 M | |
|---|---|---|---|---|---|---|---|
| O 1s | 26.01 | 23.34 | 24.31 | 25.12 | 24.39 | 25.02 | 28.81 |
| C 1s | 71.23 | 75.84 | 74.82 | 74.04 | 74.39 | 73.16 | 67.17 |
| P 2p | 0.25 | 0.49 | 0.85 | 1.21 | 1.82 | 4.02 | |
| S 2p | 0.27 | 0.37 | |||||
| N 1s | 0.87 | 0.57 | |||||
| Ca 2p | 0.5 | ||||||
| Si 2p | 1.11 |
The PET/cotton sample showed the highest proportion of oxygen to carbon (Fig. 4A), along with low levels of S, N, Ca and Si, which are thought to be impurities. Inspection of the survey scan from this sample showed that the S was present in an oxidised form, probably sulphate (SO42−), the N was present in an electronically neutral form (possibly amine or similar), the Ca was present in its highest stable oxidation state (i.e. Ca2+), and the Si was most likely in the form of silicone. The hydrochar samples all showed strong O 1s and C 1s signals in approximately the same ratio (Fig. 4B). In one case, (0.05 M) nitrogen was detected and in one case (0.1 M) sulfur was detected. All hydrochar samples showed the presence of P, in an oxide-like environment most likely phosphate (PO43−) or similar; this is clearly derived from the H3PO4 reactant. The only significant trend that could be observed in these composition data was the gradually increasing level of P across the series from 0.05 M to 1.0 M. The C 1s spectrum shown in Fig. 4C is a de-convoluted spectrum for the PET/cotton blend sample. The spectrum shows three relatively broad but clearly resolved components. The most intense component at 285.0 eV is due to carbon in C–C bonds, i.e. the polymer backbone of the PET. The second most intense component, in this case at 286.7 eV, is due to carbon singly bonded to oxygen. This may be attributed to hydroxyl C–OH or ester C–O–C type bonds in the PET and cotton components of the material. The third component at 288.7 eV is attributed to carbon double bonded to oxygen in ester bonds (i.e. –C(O)O–) and therefore must originate from the PET component of the PET/cotton blend. Overall, the C 1s spectrum shown in Fig. 4C is consistent with a PET/cotton cloth surface with the ratio between the PET and cotton components broadly in agreement with the known composition of the original cloth. The C 1s spectra from all hydrochar samples were very similar. The spectrum from the 0.5 M sample is shown in Fig. 4D. Compared to the PET/cotton reference C 1s spectrum, the hydrochar samples showed a much reduced oxygen-bonded intensity, with the single bonded and C(O)O components being closer in relative intensity. In addition, a weak component at higher binding energy of approximately 291.5 eV was detected in all cases. The spectrum is consistent with that expected from a pure PET reference29 with the 291.5 eV component being due to the π–π* shake-up of the PET aromatic ring. This suggests that the phosphoric acid has preferentially reacted with the cellulosic components of the PET/cotton blend, while the PET itself has not changed its chemical bonds quite as significantly.
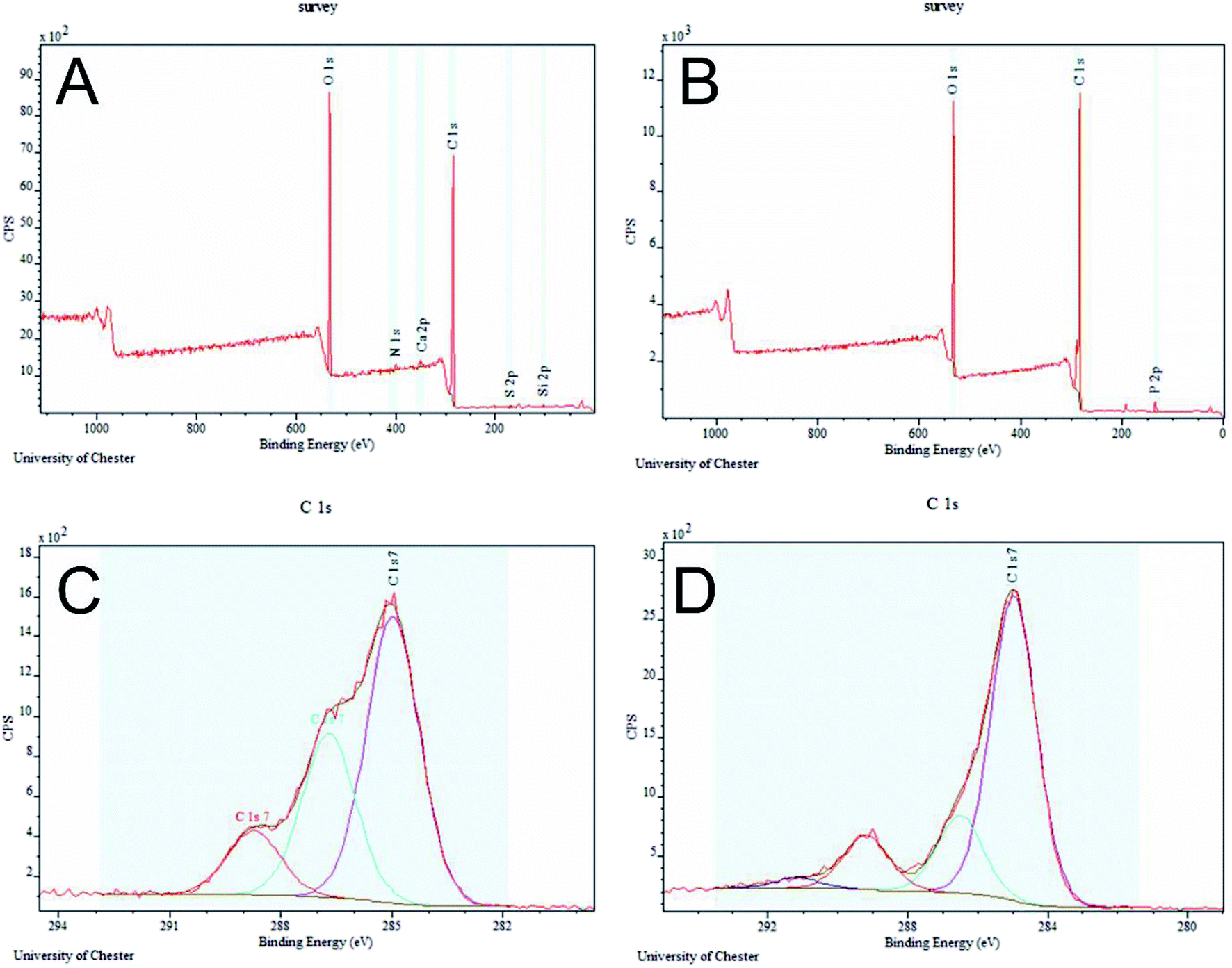 | ||
| Fig. 4 XPS spectra of (A) PET/cotton sample, (B) representative hydrochar sample from the 0.5 M reaction, (C) de-convoluted spectrum for the PET/cotton sample, and (D) de-convoluted spectrum for the 0.5 M hydrochar. | ||
In summary, the IR data indicates clear evidence of significant changes to the PET/cotton structure, the most significant is formation of aromatic structures, including terephthalic acid from depolymerisation (hydrolysis) of the PET. Formation of carbon rich, possibly graphitic species, featuring oxygen-based functional groups is also inferred, principally via degradation of the cellulose. These changes, together with the formation of phosphorus containing species, are likely to contribute significantly to the electrochemical activity (conductivity) of the hydrochars. Raman spectroscopy indicates that terephthalic acid may be formed in the reaction, which is also supported by the C 1s XPS spectrum for hydrochar samples, which show carbon species in highly oxygenated environments and an overall C 1s structure typical of a PET-like material with little or no cotton/cellulose present at the surface.
3.3 Hydrochar morphology
The morphology and surface texture of the products was investigated through the use of SEM. All backscattered electron images are depicted in ESI Fig. 4,† while the focus of this discussion is centred around Fig. 5. | ||
| Fig. 5 SEM images of (A) PET/cotton fibres and hydrochars synthesized from the hydrothermal reaction of PET/cotton fibres with (B) 0.15 M, (C) 0.25 M, and (D) 1.0 M H3PO4. The scale bar represents 100 μm. | ||
As can be seen in Fig. 5A, the PET/cotton fibre image shows an interwoven fibre structure with fibre diameters only a few microns thick. However, reaction under hydrothermal conditions yields significantly different surface morphologies. As can be seen in Fig. 5B, which was synthesized from 0.15 M H3PO4, there are two types of structure that have appeared in the material. First, there is a granular material with a particle size of no more than 10 μm, and a more crystalline material with dimensions in excess of 100 μm. There is no further information in the backscattered electron images to predict the origin of the crystalline material, though Fig. 5C and D, taken at higher concentrations of H3PO4, infer that the coverage of granular material changes as a function of concentration. ESI Fig. 4† demonstrates that the large crystalline component was present even at 0.05 M and remained largely unchanged over the full range of phosphoric acid concentrations investigated. There were subtle changes in the amount and character of the granular component; at 0.05 M H3PO4, there appears to be a reduced proportion of the latter relative that observed at 0.1 to 0.3 M. At 0.35 to 1.0 M the amount of granular component appeared to have reduced and was less defined as individual particles. EDX analyses conducted on the material are presented in ESI Fig. 5,† where it is apparent that the carbon content increases after the reaction is complete, while the oxygen content decreases as a function of acid concentration. Note this is in agreement with the working hypothesis. Interestingly, the oxygen content shows a steady decline with acid concentration, whereas the carbon content shows some variation with respect to acid concentration, in a similar way to the elemental analysis results presented in ESI Fig. 5.† Furthermore, EDX results indicate that phosphorous loading in the material increases as a function of acid concentration, indicating the production of an organophosphorus compound in the reaction. Further EDX analysis focusing on the individual granular and crystalline components are presented in Fig. 6, where it is revealed that the phosphorous is incorporated within the granular fraction of the material, whilst the crystalline material contains substantially less phosphorous.
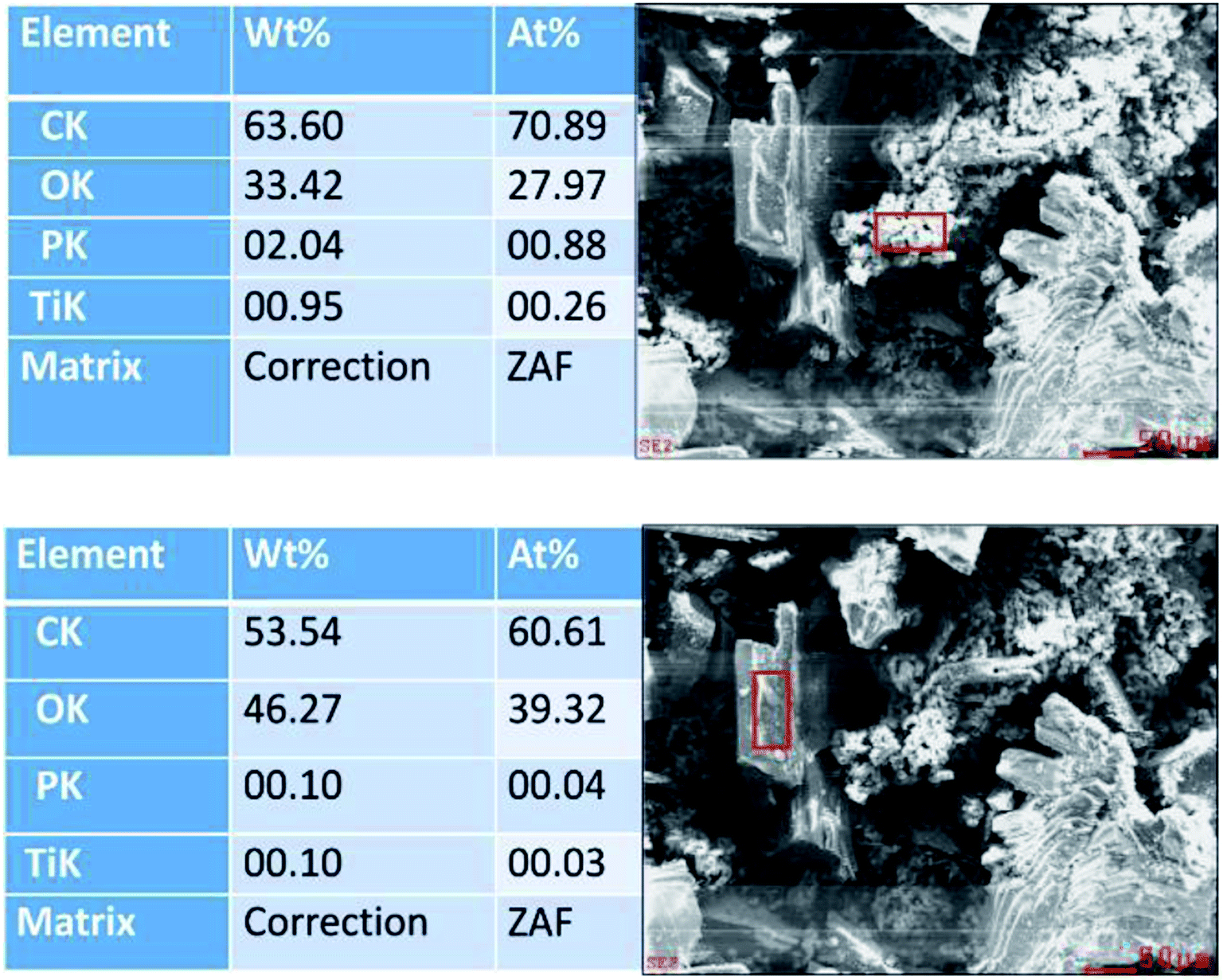 | ||
| Fig. 6 De-convoluted EDX-determined elemental distributions for the (top) granular and (bottom) crystalline regions of the hydrochar material. The reaction concentration is 0.25 M. | ||
It is significant that the granular component has a C:O atomic ratio of over 2:1, whilst the crystalline component has a C:O atomic ratio of 3:2, suggesting that these components originated from the PET and cotton cellulose fibres, indicating differing degradation mechanisms for the two polymers. Furthermore, titanium was found in the granular component of the material, but not in the crystalline component. This could be due to titanium dioxide pigment (likely to be rutile white), most probably added to the PET component as it is melt spun. Cellulose is readily swollen and degraded by H3PO4; at sufficiently high concentrations, the extent of swelling can be so high that the crystalline regions become solvated by the acid and the polymer becomes amorphous,30 if the H3PO4 concentration is reduced below 30%, the cellulose crystal structures can reform.31 It is likely from this understanding that the granular regions are the product resulting from treatment of the cellulose with phosphoric acid, the carbonisation is due to dehydration of the cellulose hydroxyl groups leaving chromophoric conjugated unsaturated structures, some of which are likely to have graphitic character.
3.4 XRD analysis of the hydrochars
The structures of the PET/cotton textile and the hydrochars were studied using XRD. The diffraction pattern for the PET/cotton mix is provided at the top of Fig. 7, where it can be seen that the material exhibits wide diffraction peaks (2θ) at 22.5° and 25.4°, where the former is due to cellulosic structures in the cotton ((200) reflection) and the latter is due to PET ((100) reflection).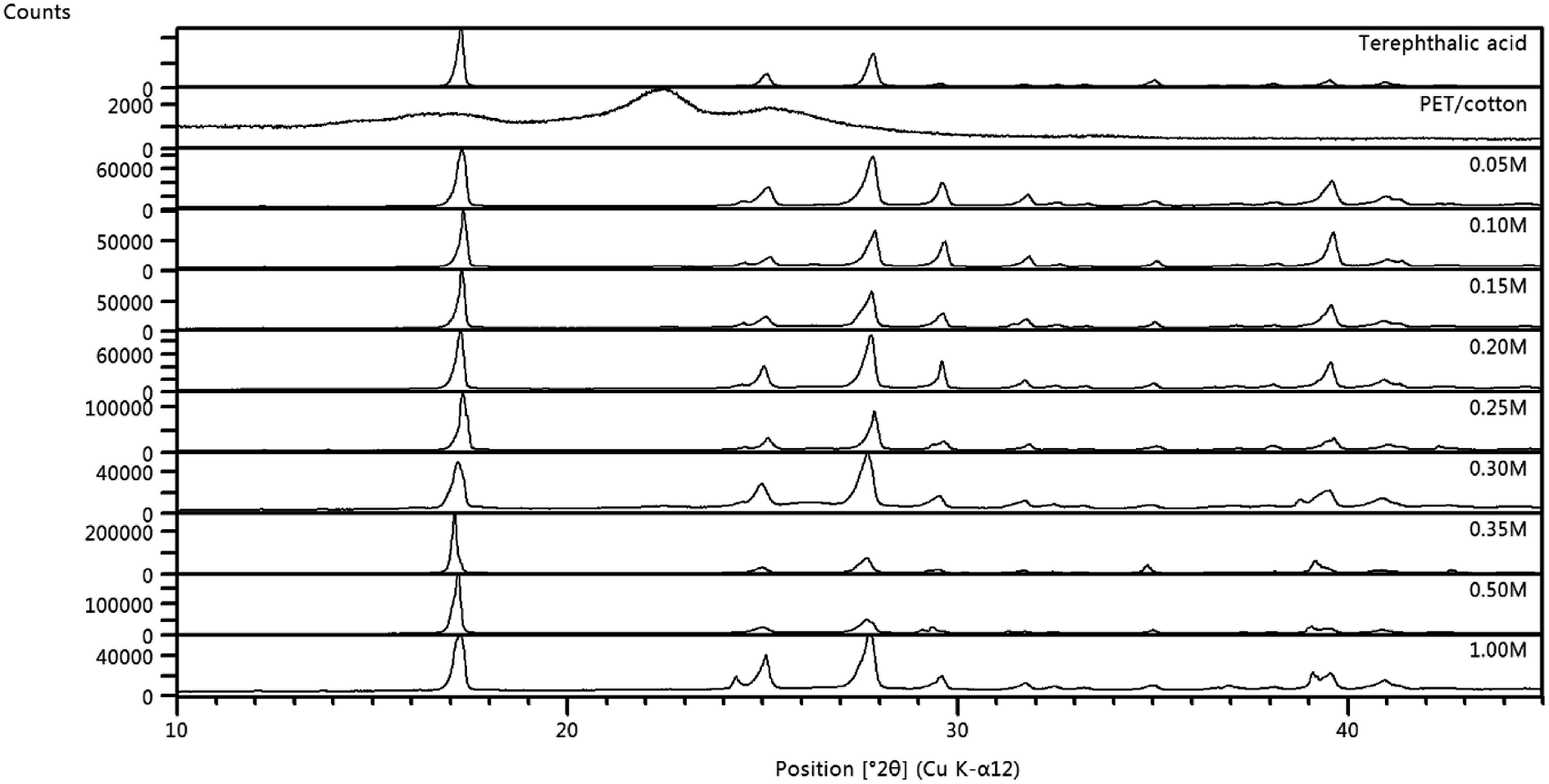 | ||
| Fig. 7 Diffraction patterns for all hydrochars, overlaid with the original PET/cotton fabric and terephthalic acid. Note the scale changes between materials. | ||
This is in agreement with previous literature reports on the diffraction patterns of PET/cotton blends.32 The shoulder (ca. 15°) on the broad peak at ∼17° may be the (1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0) reflection of cellulose, whilst the broad peak itself is the confounded (110) and (010) reflections of cellulose33 and PET, respectively. These appear at 16.3° and 18.0° in the respective pure textiles. Examining the hydrochar diffractograms in Fig. 7, it is evident from the counts scale that completely new materials with considerably higher crystalline content and structural uniformity have been formed. The former is apparent from the increase in the counts scale (103 for the textile and 105 for the hydrochars) and the latter is manifested as much sharper (narrower) diffraction peaks. The 2:1 C:O ratio identified by EDX is consistent with a benzodicarboxylic acid, possibly terephthalic acid.34–36 XRD data for a commercially available terephthalic acid sample was collected, and was shown to be very similar to that for the hydrochars, in terms of both the positions and relative intensities of the major diffraction peaks. The commercial terephthalic acid standard has an XRD pattern consistent with the known triclinic form I of the acid. The hydrochar patterns showed additional diffraction peaks, indicating the presence of additional minor phases. Combining the XRD, IR, XPS, and Raman data, it can be concluded that terephthalic acid has been formed in the reaction alongside a range of organophosphorus compounds possibly featuring chromophoric conjugated structures, some of which may have graphitic character.
0) reflection of cellulose, whilst the broad peak itself is the confounded (110) and (010) reflections of cellulose33 and PET, respectively. These appear at 16.3° and 18.0° in the respective pure textiles. Examining the hydrochar diffractograms in Fig. 7, it is evident from the counts scale that completely new materials with considerably higher crystalline content and structural uniformity have been formed. The former is apparent from the increase in the counts scale (103 for the textile and 105 for the hydrochars) and the latter is manifested as much sharper (narrower) diffraction peaks. The 2:1 C:O ratio identified by EDX is consistent with a benzodicarboxylic acid, possibly terephthalic acid.34–36 XRD data for a commercially available terephthalic acid sample was collected, and was shown to be very similar to that for the hydrochars, in terms of both the positions and relative intensities of the major diffraction peaks. The commercial terephthalic acid standard has an XRD pattern consistent with the known triclinic form I of the acid. The hydrochar patterns showed additional diffraction peaks, indicating the presence of additional minor phases. Combining the XRD, IR, XPS, and Raman data, it can be concluded that terephthalic acid has been formed in the reaction alongside a range of organophosphorus compounds possibly featuring chromophoric conjugated structures, some of which may have graphitic character.
3.5 Summary of physicochemical characterization
In summary of the physical characterization of the hydrochars, the evidence presented shows that the product of the hydrothermal reaction is a mixed material with a crystalline component constructed almost entirely from carbon and oxygen in a 3:2 ratio, whilst the granular aspects of the material show carbon and oxygen in a 2:1 ratio, with additional phosphorous and titanium components. IR spectroscopy of the hydrochars indicates that the dominant reaction is hydrolysis of the PET to terephthalic acid; any IR spectral information from graphitic structures formed is confounded with the strong aromatic C–H and hydrogen bonded OH stretching bands of the terephthalic acid. The IR spectra also allude to the formation of a range of P–O bonds, indicating that the acid reagent incorporates itself within the PET/cotton fabric during the reaction. Interestingly, the phosphorous shows a strong preference to incorporation into the granular material according to EDX analysis. The Raman spectroscopy experiments give a weak fingerprint for the crystalline areas of the material only, but within these areas the characteristic peaks for terephthalic acid are present. Comparison of the hydrochars to the XRD pattern of the commercial terephthalic acid show that most of the characteristic peaks are similar, providing strong evidence of terephthalic acid formation. The evidence seems to suggest that the H3PO4 preferentially reacts with the cellulosic material, which depolymerises to form a phosphorylated aromatic species, whilst proton-catalysed de-esterification of PET to form a phosphorylated alkyl chain and terephthalic acid is also likely. The two former species are likely to combine to form the granular material observed in the backscattered electron images, while the terephthalic acid crystallizes when dry to form the crystalline material. DSC was conducted on the hydrochars to evaluate the likelihood of terephthalic acid formation (see ESI Fig. 6†). Comparison of DSC data for pure terephthalic acid with that of the hydrochars reveals similar melting behaviour for both. Given the nature of the hydrochar production process and the mixed polymer feedstock, it is not surprising that the peak melting temperatures (Tmp) of the hydrochars are slightly depressed (Tmp 381–400 °C), relative to that of pure terephthalic acid (Tmp 422 °C). This is obviously due to possible inclusion of impurities such as cellulose degradation products and derivatives of the ethylene glycol monomer component of the PET. Characteristic sublimation of terephthalic acid was observed during DSC analysis of the hydrochars and the pure terephthalic acid.
3.6 Electrochemical properties
The electrochemical behaviour of the hydrochars were examined using CV as described in the Experimental section, the results of which are presented herein. The electrochemical data was examined in two ways. First, scan rate studies were employed for the range of hydrochars to examine the change in current behaviour with respect to the magnitude of the applied electric field, with the sensitivity of the peak current to the scan rate being used as an indicator of electron mobility. Second, the electron transfer rate constant, k0, was also extracted from the voltammetric data using a modified Nicholson method as reported by Lavagnini et al.18Fig. 8 depicts a typical cyclic voltammogram for a conductive carbon CPE, overlaid against electrodes constructed from 50:50 conductive carbon and hydrochar obtained from the chemical reaction of PET/cotton with H3PO4, as described previously.
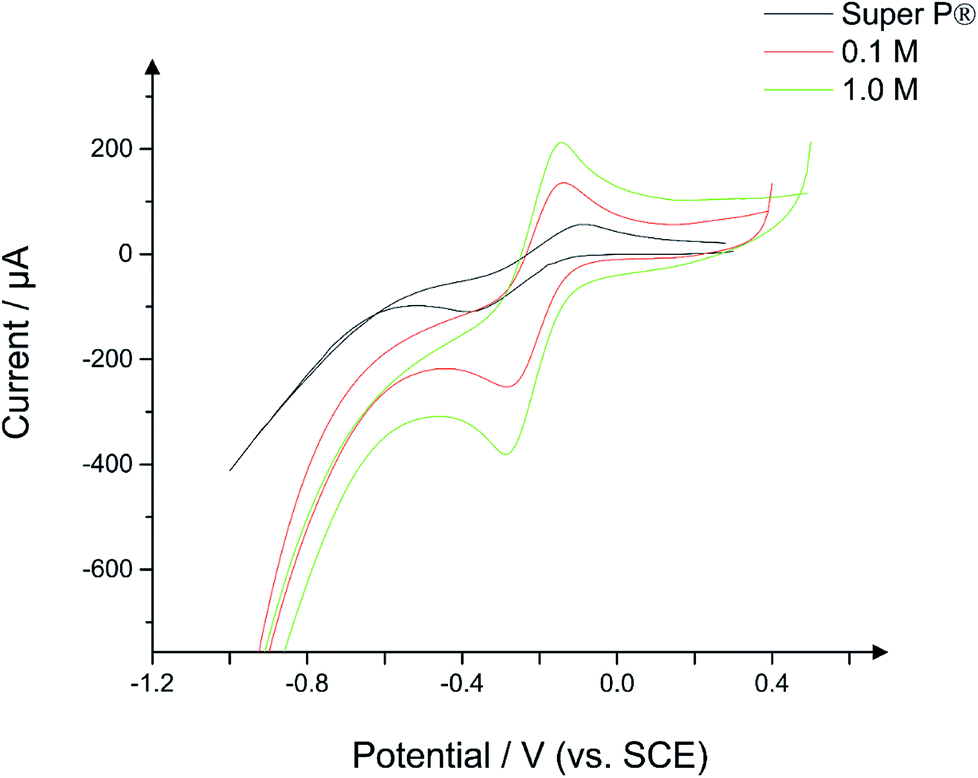 | ||
| Fig. 8 Cyclic voltammograms of CPEs constructed from 50:50 powder:nujol, where the powder is 100% conductive carbon in the case of the Super P® curve, while for the 0.1 M and 1.0 M curve the powder is constructed from 50% Super P® and 50% hydrochar. Scan rate: 100 mV s−1; redox probe: 1 mM hexamine-ruthenium (III) chloride (in pH 7.4 PBS and 0.1 M KCl). | ||
Note the CPEs are constructed from a 50:50 ratio of powder:nujol, where the powder contains 100% conductive carbon as a control, or 50% conductive carbon and 50% hydrochar for the new compounds. As can be seen in the case of the conductive carbon electrode, the voltammogram assumes a typical wave shape for hexamine-ruthenium(III) chloride, with an average (N = 3) peak current of 65 μA and a peak-to-peak separation of 167 mV. Given the highly conductive nature of conductive carbon as seen in previous literature,37–39 these results were unusual. Repeated testing of the CPEs over a range of scan rates indicated a %RSD of under 10%, which is similar to that of carbon SPEs,14 and as such the data can be relied upon as a benchmark. Turning towards the CPEs interspersed with hydrochar, it is apparent that there are significant changes in the current–potential profiles between the conductive carbon and the hydrochars. Fig. 8 demonstrates that the 0.1 M hydrochar gives an enhanced current response to hexamine-ruthenium(III) chloride, with an average (N = 3) peak oxidation current of 136 μA and a peak-to-peak separation of 100 mV, which is significantly reduced compared to the conductive carbon case. Increasing the acid concentration to 1.0 M resulted in an average (N = 3) peak oxidation current of 152 μA; an increase compared to both the conductive carbon and the 0.1 M hydrochar. A similar peak-to-peak separation was observed between the conductive carbon and the 1.0 M reaction (160 mV; N = 3). Scan rate studies were performed on the hydrochars, an example of which is presented in ESI Fig. 7.† As can be seen from ESI Fig. 7, using a hydrochar electrode synthesized from 0.3 M H3PO4, the peak oxidation current of 1 mM hexamine-ruthenium(III) chloride (in pH 7.4 PBS and 0.1 M KCl) linearly increased as a function of the square root of the scan rate (recorded current sensitivity up to 925 μA per unit of ν1/2). This indicates that diffusion is not limiting the current as described by the Randles–Ševćik equation for a quasi-reversible reaction, presented in eqn (1), where n is the number of electrons transferred, A is the electrode area, C is the concentration of electroactive species, D is the diffusion coefficient of the redox probe (9.1 × 10−6 cm2 s−1), and ν is the scan rate.40 Note the quasi–reversible equation has been chosen since the peak-to-peak separations deviate as the applied scan rate is changed. The sensitivity demonstrates the rate of change of current as a function of applied electric field, and in this case is used to demonstrate the change in behaviour of a range of hydrochars.
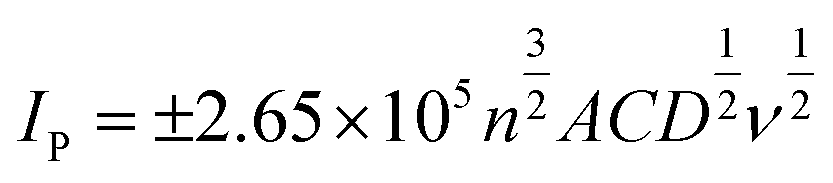 | (1) |
ESI Fig. 8† depicts the sensitivity values obtained for a range of hydrochars synthesized using different concentrations of H3PO4. The data was normalized by dividing each dataset by the lowest value in that dataset for ease of data presentation (the conductive carbon control electrode was the lowest value in each case). As can be seen in ESI Fig. 8,† a trend appears when the sensitivity values in the scan rate studies are plotted as a function of acid concentration. At low concentration (0.05 M), the sensitivity value (equating to 265 μA per unit of ν1/2) is relatively low with a small error margin. As the acid concentration increases, it becomes apparent that the sensitivity of the CPE increases significantly, with a maximum sensitivity obtained using a concentration of 0.25 M H3PO4 (507 μA per unit of ν1/2). Further increases in acid concentration result in a reduction in sensitivity compared to the mid-range concentrations as witnessed for the 0.5 M and 1.0 M cases. These results demonstrate that the hydrothermal reaction can be controlled to give an electrode with a high current response, where required (see later for comment on sensitivity error). While all the hydrochars appear to exhibit conductive properties, it was necessary to examine whether such conductivity could be observed using a hydrochar CPE without a loading of conductive carbon. For the case of 0.25 M, which was chosen because of the high current responses observed, a CPE was constructed using 66% hydrochar and 34% nujol. This ratio was chosen because the 50:50 hydrochar:nujol electrode was found to be more of a liquid than a paste, and therefore it was impossible to construct the electrode. Fig. 9 presents a scan rate study of the 100% hydrochar electrode, where a quasi-reversible response is apparent since the peak-to-peak separations increase significantly as the applied scan rate is increased. The voltammetric profile obtained in Fig. 9 is unequivocal evidence that the carbonized PET/cotton exhibits conductive properties, though it is also clear that the reaction must be improved/optimized in order to maximise the conductive characteristics of the material.
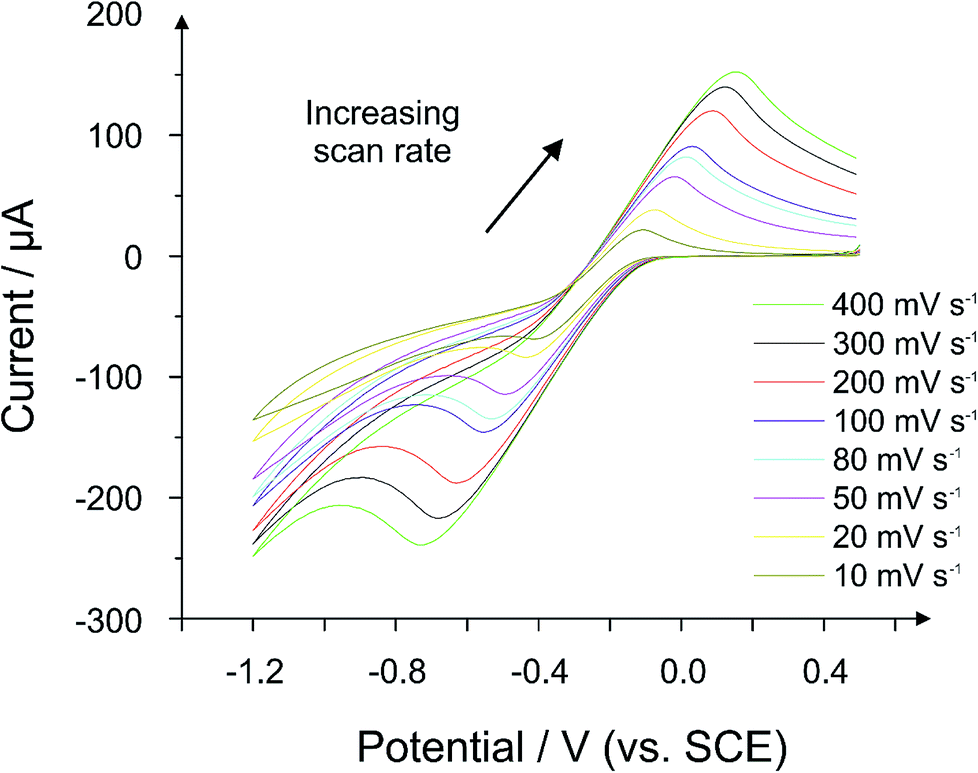 | ||
| Fig. 9 Voltammetric scan rate study using a 100% 0.25 M hydrochar electrode (66% hydrochar and 34% nujol). Probe: 1 mM hexamine-ruthenium(III) chloride (in pH 7.4 PBS and 0.1 M KCl). | ||
The increases in current response for all electrodes are assumed to indicate that the material exhibits conductivity features that are beneficial for electrochemical devices, though there are some limitations to this approach, not least evidenced by the error in sensitivity witnessed in ESI Fig. 8.† First, the CPE approach is known to exhibit some inconsistency in terms of the current outputs, especially when defining the electrode working area. This is circumvented in this work to a degree by using the same diameter syringes as a holder for the carbon paste, however minor deviations are apparent when slicing the tip from the syringe, meaning that there is an inherent error with the electrode design. Second, the mixing of the powder and nujol has to be very robust to ensure complete dispersion of the powder in the nujol. Even when the material appears to be completely mixed, there can be pockets of unmixed powder within the paste, leading to inconsistencies in the results. Thirdly, there is a disparity in the surface area and, therefore, the saturation of the powder/nujol mixture. Higher surface area materials, such as Super P®, uptake the nujol more efficiently than the hydrochars, creating more “powdery” paste. However, it was decided that the ratio must remain the same for comparative purposes. Note the differences in nujol uptake may be a factor in the changes in voltammetry observed. This could also be obviated in future work by mechanically grinding the hydrochars using a ball mill for a set period of time. This would ensure full breakup of the material, enhancing the surface area and improving the material's interaction with the nujol matrix. Finally, ensuring the surface of the CPE is homogeneous is difficult, but is achieved by polishing the electrode on a sheet of white paper. This process may give rise to errors in current response. Overall, the error for conductive carbon electrodes is demonstrated to be under 10% (%RSD), thus it is felt that this approach is satisfactory for comparison of materials performance.
Further electrochemical analysis was performed on the hydrochar electrodes through use of the modified Nicholson method for rate constant (k0) determination.18 Briefly, k0 is calculated from the magnitude of the peak-to-peak separation (X) through the use of eqn (2) and (3), where ψ is the so-called Nicholson parameter, F is the Faraday constant, R is the molar gas constant and T is the temperature.
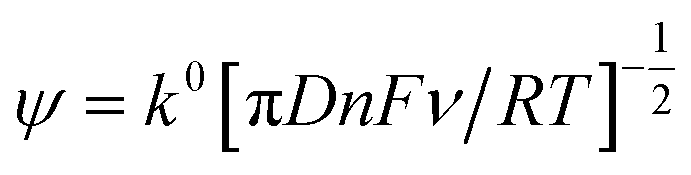 | (2) |
| ψ = (−0.6288 + 0.0021X)/(1 − 0.017X) | (3) |
The rate constants are presented graphically in Fig. 10, where it can be seen that the k0 value for the conductive carbon paste control electrode (denoted as concentration 0.0 in Fig. 10) exhibits a k0 value of 2.60 × 10−3 cm s−1.
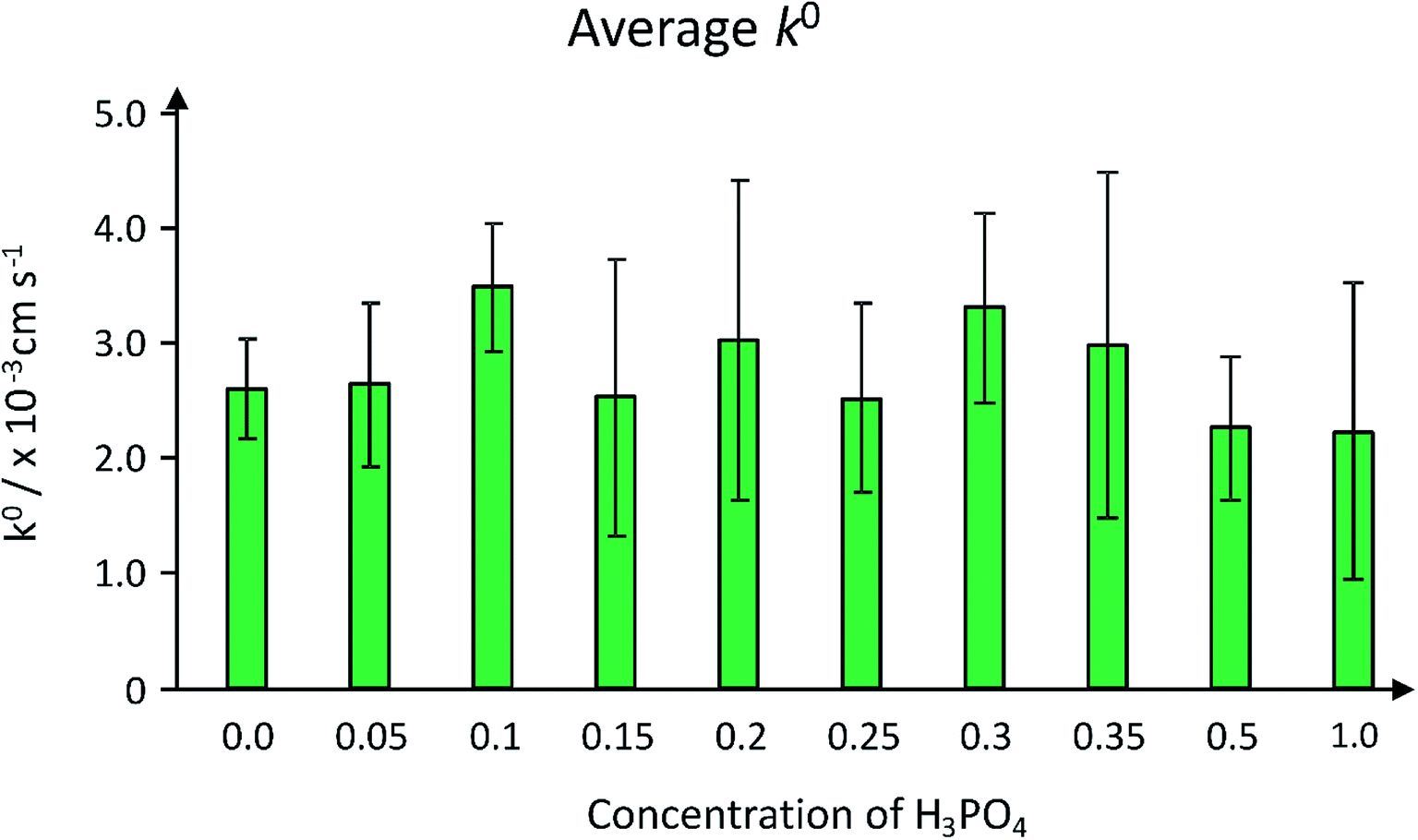 | ||
| Fig. 10 Graph of the electron transfer rate constant (k0) of the hydrochar carbon paste electrodes, as a function of acid concentration in the hydrothermal reaction. The error bars are standard deviations calculated from three separate experiments. | ||
Note this is an average k0 value calculated from three electrodes (%RSD is 17%; N = 3). A propensity for k0 to undergo an increase in electron transfer rate (Fig. 10) followed by a decrease as the concentration of acid in the reaction increases, with a maximum average k0 value arising from the 0.3 M reaction and the minimum arising from the 1.0 M reaction. This appears to be in line with the sensitivity trend observed in ESI Fig. 8.† The rate constants are comparable to several other carbon electrode materials: it is approximately one order of magnitude less than previously reported for an edge plane pyrolytic graphite electrode, and a similar order of magnitude to graphene-based nanomaterials.41–43
In summary of this section, the experiments have demonstrated that the hydrochar synthesized from textile waste can form a viable and potentially implementable carbon material for the detection of electroactive compounds. This is evidenced by the increase in current sensitivity towards the outer-sphere redox probe, hexamine-ruthenium(III) chloride compared to the conductive carbon CPE on its own. Further it is also shown that the materials may not be beneficial for electrochemical applications that require reductions in thermodynamic barriers for redox reactions (e.g. oxygen reduction). The approach is an inexpensive method of carbon material production, with the main cost of synthesis being the energy expenditure of the reaction (around £10), notwithstanding initial costs of the autoclave reactor. The approach uses simple, cheap, and readily available laboratory reagents in small quantities, highlighting an element of simplicity in the approach and overall cost-effective method for carbon electrode production, while also contributing towards increasing the recyclability of textile waste. This work serves as a proof-of-concept approach for this material, but further work is required to optimise the process to yield more consistent materials for use in electronic devices in the future. This will be achieved through screen-printing the carbon into a workable electrode solution, which will improve the repeatability and consistency of the material.
4. Conclusions
This paper has presented a robust investigation into the structural, morphological, and electrochemical properties of hydrochars synthesized by treating a PET/cotton textile with H3PO4 at a range of concentrations. Elemental analysis of the hydrochars found an increased carbon content in all hydrochars compared with the textile blend feedstock. EDX analysis indicated that the phosphorous content of the hydrochars increased as the concentration of H3PO4 increased. EDX analysis also revealed that the phosphorous content in the granular component of the material was much higher, leading to the deduction that latter component was a combination of a phosphorylated aromatic (potentially graphitic) species from the dehydration and depolymerisation of cellulose, and a phosphorylated alkyl chain from the de-esterification of PET. Backscattered electron imaging of the hydrochars indicated the formation of a crystalline material, suspected to be terephthalic acid, which was corroborated by XRD experiments. Investigation into the nature of chemical bonds in the hydrochars revealed that the hydrolysis of the ester linkages of PET was highly likely. There is also IR evidence for formation of alkylphosphorous groups. The grey/black colour of the hydrochars indicated formation of conjugated chromophoric structures that were most probably more plentiful in the granular component. The intensity of the IR absorptions of terephthalic acid masked any absorptions associated with such conjugated chromophoric structures. The C/O ratio of the material was also of interest, ranging from 3:2 for the crystalline material, to 2:1 for the granular material. XPS analysis revealed a reduction in overall oxygenation of the hydrochar compared to the original PET/cotton material, though some carbon bonds were present in highly oxygenated forms such as carboxylic acid groups. It is therefore highly likely that terephthalic acid has been at least partially synthesized in this approach.
The hydrochars were used to construct CPEs to examine the electrochemical performance of the material. This work found that the hydrochars outperformed the reference conductive carbon CPE, as evidenced by an increase in peak oxidation current and decrease in peak-to-peak separation of the electrodes. Furthermore, the hydrochar prepared with 0.25 M H3PO4 was shown to conduct without the need for a conductive carbon matrix, opening up a future possibility of a 100% PET/cotton-derived electrode. It has been noted however, that the case of 100% loading reduces the current profile and results in a tendency towards irreversibility. Furthermore, normalized k0 values suggest that the electron transfer rate of the hydrochar materials actually decreases if the overall electroactive surface area is taken into account. Future work in this area will be targeted at improvement of the synthetic route for the hydrochars to optimise the conducting ability of the product by performing the reaction within an inert atmosphere to optimise carbon yield, and incorporation of the product into a more workable (and repeatable) electrode solution such as screen-printed electrodes. In conclusion, it has been shown that textile waste-derived hydrochars have potential applications as electroanalytical sensors due to increased current outputs relative to other conductive carbons used for such applications. This work, if optimised and scaled up, could therefore contribute towards the societal recycling challenge by reducing the volume of textile waste sent to landfill and incineration plants. The production of terephthalic acid via the method investigated should not be overlooked as this could facilitate circular economics in relation to production of virgin PET and other polymers and resins. Segregation of terephthalic acid from the original reaction products will be the focus of future works.
Conflicts of interest
The authors declare no conflicts of interest with this work.Acknowledgements
The authors acknowledge several colleagues who have provided support to this project: Professor Craig Banks (MMU) and his team for use of electrochemical instrumentation and reagents; Alex Garcia-Ferrari for his help and support in training Mr Kanou on the electrochemical equipment; and David McKendry for elemental analysis. The authors also acknowledge the Nuffield Foundation for facilitating the project for Mr Kanou to participate within.References
- F. Chen, G. Wang, W. Li and F. Yang, Ind. Eng. Chem. Res., 2013, 52, 565–571 CrossRef CAS.
- S. M. Cakić, I. S. Ristić, M. M. Cincović, N. Č. Nikolić, L. B. Nikolić and M. J. Cvetinov, Prog. Org. Coat., 2017, 105, 111–122 CrossRef.
- V. Jamdar, M. Kathalewar and A. Sabnis, J. Coat. Technol. Res., 2018, 15, 259–270 CrossRef CAS.
- V. Jamdar, M. Kathalewar, K. A. Dubey and A. Sabnis, Prog. Org. Coat., 2017, 107, 54–63 CrossRef CAS.
- C. Zequine, C. K. Ranaweera, Z. Wang, P. R. Dvornic, P. K. Kahol, S. Singh, P. Tripathi, O. N. Srivastava, S. Singh, B. K. Gupta, G. Gupta and R. K. Gupta, Sci. Rep., 2017, 7, 1174 CrossRef PubMed.
- A. Jain, R. Balasubramanian and M. P. Srinivasan, Chem. Eng. J., 2016, 283, 789–805 CrossRef CAS.
- A. Jain, C. Xu, S. Jayaraman, R. Balasubramanian, J. Y. Lee and M. P. Srinivasan, Microporous Mesoporous Mater., 2015, 218, 55–61 CrossRef CAS.
- J. Yang, Y. X. Jin, X. P. Yu and Q. F. Yue, J. Sol-Gel Sci. Technol., 2017, 83, 413–421 CrossRef CAS.
- M.-X. Guo, S.-W. Bian, F. Shao, S. Liu and Y.-H. Peng, Electrochim. Acta, 2016, 209, 486–497 CrossRef CAS.
- M. Ashraf, P. Champagne, C. Campagne, A. Perwuelz, F. Dumont and A. Leriche, J. Ind. Text., 2014, 45, 1440–1456 CrossRef.
- D. Kucuk, O. Balci and M. Tutak, International Journal of Clothing Science and Technology, 2016, 28, 340–367 CrossRef.
- M. Z. Khan, M. Ashraf, T. Hussain, A. Rehman, M. M. Malik, Z. A. Raza, Y. Nawab and Q. Zia, Fibers Polym., 2015, 16, 1092–1097 CrossRef CAS.
- D. A. C. Brownson, P. J. Kelly and C. E. Banks, RSC Adv., 2015, 5, 37281–37286 RSC.
- E. P. Randviir, D. A. C. Brownson, J. P. Metters, R. O. Kadara and C. E. Banks, Phys. Chem. Chem. Phys., 2014, 16, 4598–4611 RSC.
- L. C. S. Figueiredo-Filho, D. A. C. Brownson, M. Gómez-Mingot, J. Iniesta, O. Fatibello-Filho and C. E. Banks, Analyst, 2013, 138, 6354–6364 RSC.
- D. A. C. Brownson, L. J. Munro, D. K. Kampouris and C. E. Banks, RSC Adv., 2011, 1, 978–988 RSC.
- W. Hou, C. Ling, S. Shi, Z. Yan, M. Zhang, B. Zhang and J. Dai, Fibers Polym., 2018, 19, 742–750 CrossRef CAS.
- I. Lavagnini, R. Antiochia and F. Magno, Electroanalysis, 2004, 16, 505–506 CrossRef CAS.
- S. M. Shaik, C. Y. Koh, P. N. Sharratt and R. B. H. Tan, Thermochim. Acta, 2013, 566, 1–9 CrossRef CAS.
- E. Žagar and J. Grdadolnik, J. Mol. Struct., 2003, 658, 143–152 CrossRef.
- N. Arahman, A. Fahrina, S. Amalia, R. Sunarya and S. Mulyati, F1000Research, 2017, 6, 668 Search PubMed.
- P. González Morones, S. Fernández Tavizón, E. Hernández Hernández, C. Alberto Gallardo Vega and A. De León Santillán, RSC Adv., 2016, 6, 18413–18418 RSC.
- I. N. Strain, Q. Wu, A. M. Pourrahimi, M. S. Hedenqvist, R. T. Olsson and R. L. Andersson, J. Mater. Chem. A, 2015, 3, 1632–1640 RSC.
- B. Hu, K. Wang, L. Wu, S.-H. Yu, M. Antonietti and M.-M. Titirici, Adv. Mater., 2010, 22, 813–828 CrossRef CAS PubMed.
- R. A. Holser, Mater. Chem. Phys., 2011, 128, 10–11 CrossRef CAS.
- R. M. Silverstein and F. X. Webster, Spectrometric Identification of Organic Compounds, John Wiley & Sons, Inc., 6th edn, 1998 Search PubMed.
- L. Daasch and D. Smith, Anal. Chem., 1951, 23, 853–868 CrossRef CAS.
- C. A. S. Téllez, E. Hollauer, M. A. Mondragon and V. M. Castaño, Spectrochim. Acta, Part A, 2001, 57, 993–1007 CrossRef.
- G. Beamson and D. Briggs, High Resolution XPS of Organic Polymers: The Scienta ESCA300 Database, Wiley, New York, 1992 Search PubMed.
- S. Wei, V. Kumar and G. S. Banker, Int. J. Pharm., 1996, 142, 175–181 CrossRef CAS.
- V. V. Vinogradov, L. N. Mizerovskii and O. P. Akaev, Fibre Chem., 2002, 34, 167–171 CrossRef CAS.
- D. Mitcham, R. J. Berni and V. W. Tripp, Text. Res. J., 1977, 47, 386–388 CrossRef CAS.
- Y. Dong, Z. Bai, L. Zhang, R. Liu and T. Zhu, J. Appl. Polym. Sci., 2005, 99, 286–291 CrossRef.
- M. Bailey and C. J. Brown, Acta Crystallogr., 1967, 22, 387–391 CrossRef CAS.
- S. Gražulis, A. Daškevič, A. Merkys, D. Chateigner, L. Lutterotti, M. Quirós, N. R. Serebryanaya, P. Moeck, R. T. Downs and A. Le Bail, Nucleic Acids Res., 2012, 40, D420–D427 CrossRef PubMed.
- S. Gražulis, D. Chateigner, R. T. Downs, A. F. T. Yokochi, M. Quirós, L. Lutterotti, E. Manakova, J. Butkus, P. Moeck and A. Le Bail, J. Appl. Crystallogr., 2009, 42, 726–729 CrossRef PubMed.
- G. Zou, H. Hou, C. W. Foster, C. E. Banks, T. Guo, Y. Jiang, Y. Zhang and X. Ji, Adv. Sci., 2018, 5, 1800241 CrossRef PubMed.
- E. Martínez-Periñán, C. W. Foster, M. P. Down, Y. Zhang, X. Ji, E. Lorenzo, D. Kononovs, A. I. Saprykin, V. N. Yakovlev, G. A. Pozdnyakov and C. E. Banks, C, 2017, 3, 20 Search PubMed.
- S. J. Rowley-Neale, M. Ratova, L. T. N. Fugita, G. C. Smith, A. Gaffar, J. Kulczyk-Malecka, P. J. Kelly and C. E. Banks, ACS Omega, 2018, 3, 7235–7242 CrossRef CAS.
- D. A. C. Brownson and C. E. Banks, The Handbook of Graphene Electrochemistry, Springer, 2014 Search PubMed.
- E. P. Randviir, D. A. C. Brownson, M. Gomez-Mingot, D. K. Kampouris, J. Iniesta and C. E. Banks, Nanoscale, 2012, 4, 6470–6480 RSC.
- S. J. Rowley-Neale, E. P. Randviir, A. S. Abo Dena and C. E. Banks, Appl. Mater. Today, 2018, 10, 218–226 CrossRef.
- E. P. Randviir and C. E. Banks, Graphene-Based Electrochemical Sensors, 2018 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra00175a |
| ‡ DEFRA, statistics on waste managed by local authorities in England 2016/17, 2017, https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/664594/LACW_mgt_annual_Stats_Notice_Dec_2017.pdf, accessed October 2018. |
| § DEFRA, UK Statistics on Waste, 2018, https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/683051/UK_Statisticson_Waste_statistical_notice_Feb_2018_FINAL.pdf, accessed October 2018. |
| ¶ Textile Beat, what are your clothes made from?, 2018, https://textilebeat.com/wp-content/uploads/2018/04/Slow-Clothing-fibres-across-time-web.jpg, accessed September 2018. |
| This journal is © The Royal Society of Chemistry 2019 |