DOI:
10.1039/C9RA00185A
(Paper)
RSC Adv., 2019,
9, 4806-4811
The identification of active N species in N-doped carbon carriers that improve the activity of Fe electrocatalysts towards the oxygen evolution reaction†
Received
9th January 2019
, Accepted 1st February 2019
First published on 6th February 2019
Abstract
Nitrogen-doped carbon nanomaterials have become some of the most effective carriers for transition metal-based electrocatalysts towards the oxygen evolution reaction. However, the specific active nitrogen species in nitrogen-doped carriers remains unclear up to now. To identify the active nitrogen species, herein, we prepare nitrogen-doped carbon nanospheres containing different types of nitrogen species and a small amount of Fe atoms. Electrochemical tests demonstrate that the Fe/nitrogen-doped carbon nanospheres with more graphitic nitrogen exhibit much higher activity for the oxygen evolution reaction than those with more pyridinic nitrogens and pyrrolic nitrogens in alkaline media, revealing that the graphitic nitrogen is the active species that greatly improves the activity of Fe catalysts. Density functional theory calculations further reveal that the graphitic nitrogen enhances the activity and stability of Fe-based catalysts mainly through increasing the adsorption energy, charge and spin densities of the Fe atoms loaded around it. These findings provide a brand-new perspective for rationally designing more effective transition metal-based electrocatalysts for the oxygen evolution reaction through controlling the active graphitic nitrogen distribution in carbon carriers.
1. Introduction
Transition metal-based electrocatalysts towards the oxygen evolution reaction (OER) in the fields of rechargeable fuel cells, metal–air batteries and water electrolysis have attracted much attention over the past decade due to their noble metal-like catalytic behaviors in acidic, neutral or alkaline media.1 Despite great progress, the high metal consumption, sophisticated operation and the poor durability are still challenges to be overcome. Currently, transition metal-based OER electrocatalysts with facile synthesis procedures, low metal loading, highly efficient electrocatalytic activity and robust stability are urgently needed. More recently, much advanced research has demonstrated that N-doped carbon nanomaterials could effectively improve the OER performance of transition metal-based electrocatalysts when they are employed as their carriers.2–11 As for the improvement mechanism, the M-pyridinic Nx (M represents Fe, Co, Ni, etc.) in some catalysts was regarded as the same active site for both OER and oxygen reduction reaction (ORR).3,9,10 Nevertheless, this undiscriminating active site seems quite perplexing because the OER and ORR are two completely opposite reactions. In fact, the M-pyridinic Nx generally exhibits more moderate OER activity relative to its robust catalytic behaviors in the ORR.9 Meanwhile, even though M-pyridinic Nx really serves as the OER active site just as it does in the ORR, this also does not mean no existence of other OER active structures because there existed quite a few graphitic N and pyrrolic N together with pyridinic N in the reports where the M-pyridinic Nx was considered to be the OER active site.9,11 So, up to now, the definite active N species which influences the OER activity of transition metal catalysts remains murky and needs to be investigated in depth. More specifically, among the three potential active N species of graphitic N, pyridinic N and pyrrolic N in N-doped carbon carriers, which one is really responsible for the OER performance improvement of transition metal catalysts is yet to be explored systematically from both experiments and theoretic calculations, which have greatly hindered the further targeted design of more effective transition metal-based electrocatalysts for the OER.
In general, for a given N-doped carbon carrier, different types of N species often exist simultaneously and are hard to be separated, innately making it quite difficult to definitively identify the real active N structure. Hence, synthesizing N-containing carbon carriers with specific N species through controlling synthesis conditions would be an effective strategy for the accurate identification of active N species. Meanwhile, Fe, as the cheapest and abundant transition metal, often shows remarkable activities in oxygen electrocatalysis reactions.9,12–16 To clearly identify the active N species, in this work, we selected elemental Fe as the active transition metal catalyst and the N-doped carbon nanospheres (N-CNSs) with specific N species as supports. Namely, experimentally, we loaded a small amount of Fe atoms onto a series of N-CNSs carriers with different N species distribution and evaluated their OER activities by electrochemical measurements in alkaline medium, respectively. Results indicated that the as-prepared Fe/N-CNSs with more graphitic N exhibited much higher activity for the OER than those with more pyridinic N and pyrrolic N in alkaline medium, and comparable activity and stability to the commercial noble metal IrO2 catalyst, revealing that the graphitic N in N-doped carriers is the active species which dramatically improves the OER activity and stability of Fe catalysts. These improvements could be attributed to the remarkable increase of the adsorption energy, charge and spin density of Fe atoms loaded on the graphitic N-doped carrier, as evidenced by density functional theory (DFT) calculations. As far as we are aware, no systematic study has been made on the definite effect of different types of N species upon the OER activity of transition metal catalysts.
2. Experimental
2.1 Preparation of materials
In a typical experiment, ammonia was input to remove the air in the quartz tube with a flow rate of 100 mL min−1 when the furnace was heated to 1500 °C. The furnace was cooled down to room temperature after 20 mL of aniline was thoroughly injected into the quartz tube at a flow rate of 4 mL h−1 with a stainless steel tube (diameter, 3 mm; length, 30 cm) as the transportation pipeline. The resultant sample was collected from the quartz tube and noted as Fe/N-CNSs. The controlling of N species distribution in N-CNSs was as follows: typically, two quartz boats with 0.5 g iron wire and 100 mg Fe/N-CNSs were put into a quartz tube in the furnace, respectively. The furnace was heated to 900 °C and kept for 1 h under a vacuum condition (vacuum degree: −1). The sample was collected and noted as Fe/N-CNSs1 after the furnace temperature was cooled down. In the same way, the Fe/N-CNSs2 and Fe/N-CNSs3 were prepared at 1300 °C for 1 h and 3 h, respectively. Meanwhile, to investigate the Fe distribution in prepared catalysts, parts of these catalysts were handled by concentrated hydrochloric acid under ultrasonic wave for 3 h, respectively.
2.2 Electrode preparation and electrochemical experiments
The glassy carbon electrode (GCE, 5.0 mm in diameter) were pretreated before each electrochemical tests. And 0.229 mg cm−2 of each example was loaded onto the surface of bare GCE. Electrochemical experiments were carried out at room temperature in a three-electrode cell connected to an electrochemical analyzer (Pine Research Instrumentation, USA). All as-prepared samples/GCE and IrO2/GCE were used as the working electrodes, and a graphite carbon as counter electrode. All potentials were measured and reported vs. the potential of Ag/AgCl electrode, which was converted to reversible hydrogen electrode (RHE) using the equation: E(RHE) = E(Ag/AgCl) + 0.059 × pH + 0.198. The LSV measurements were performed in the nitrogen-saturated 0.1 M KOH (pH = 13) solution. OER current densities of the samples were estimated in the potential range of 0.0 to 1.0 V at a rotation speed of 1600 rpm and scan rate of 10 mV s−1 at room temperature. Tafel slope was obtained through the Tafel equation (η = b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) log|J| + a, where η is the over potential, b is the Tafel slope, and J is the current density).
log|J| + a, where η is the over potential, b is the Tafel slope, and J is the current density).
2.3 Characterizations
The morphologies of the samples were characterized by scanning electron microscopy (SEM, ZEISS Merlin), transmission electron microscopy (TEM), high resolution and scanning transmission electron microscopy (HRTEM/STEM, Talos F200S). X-ray-diffraction (XRD) was performed with Cu Kα radiation. The surface area was measured by the Brunauer–Emmett–Teller (BET, Autosorb IQ). X-ray photoelectron spectroscopic (XPS) measurements were carried out on a Thermo Scientific ESCALAB 250XI using Al Kα radiation, and the C1s peak at 284.8 eV was taken as internal standard. X-ray absorption near edge structure (XANES) analysis was studied at the 1W1B beamline of the Beijing Synchrotron Radiation Facility, China. The Fe K-edge XANES were characterized in a fluorescence mode, with the standard Fe foil as reference. Crystallinity and defects of the synthesized sample were recorded with a Brucker Raman Spectroscopy Senterra. Thermogravimetric analyses (TG) were performed on a Setaram Labays Evo under an air flow of 10 mL min−1 from 30 °C to 850 °C at a heating rate of 5 °C min−1.
3. Results and discussion
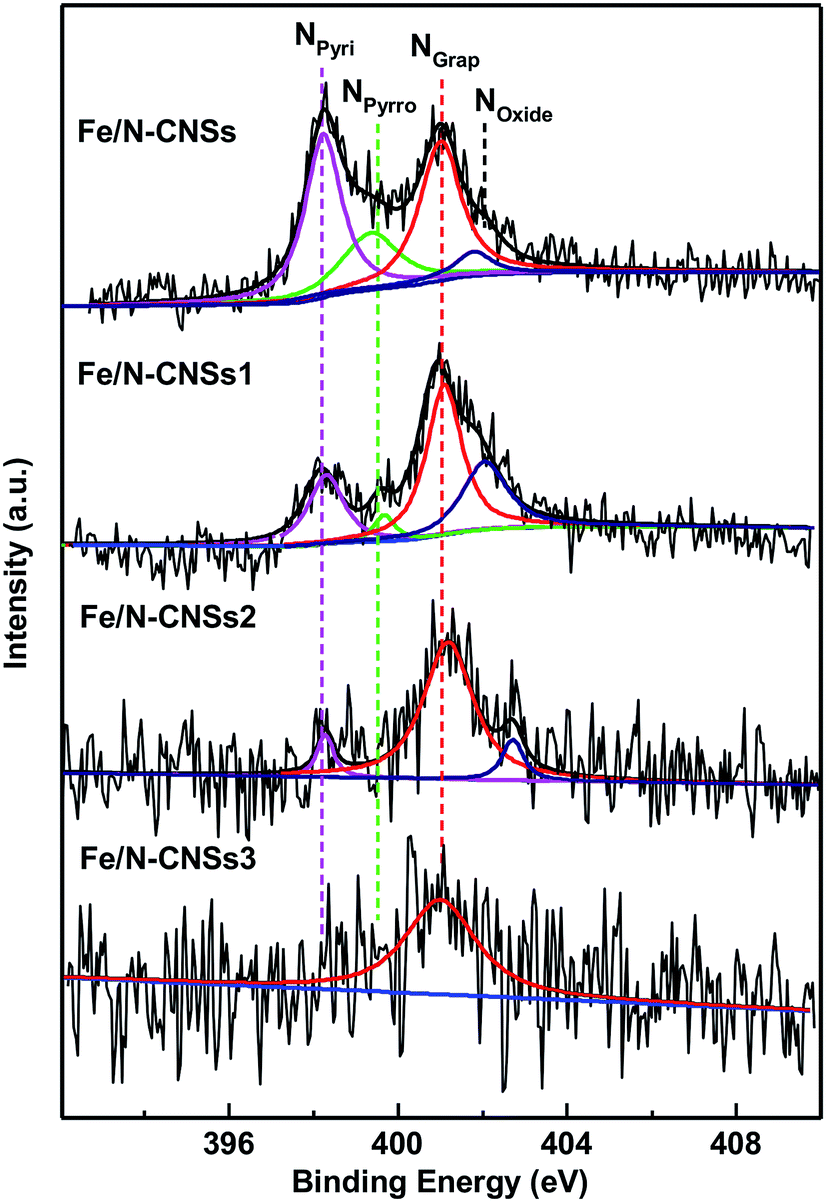
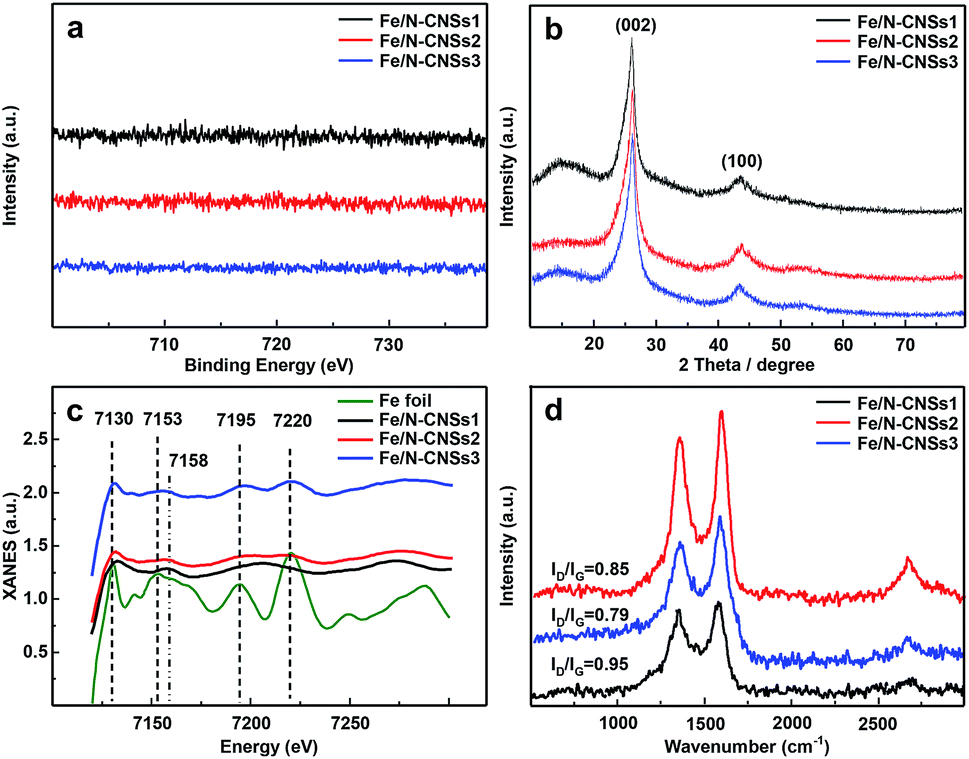
SEM, TEM, HRTEM and STEM images (Fig. 1a–d) of the representative sample (Fe/N-CNSs2) indicated that all as-prepared samples were comprised of well-defined coralline nanospheres, and no Fe nanoparticles were observed on their surface. The XPS spectra in Fig. 2 showed that the N species distributions in the four well-designed samples of Fe/N-CNSs, Fe/N-CNSs1, Fe/N-CNSs2 and Fe/N-CNSs3 were quite different (Fig. 2). There were pyridine N (ca. 398.2 eV), pyrrolic N (ca. 399.6 eV), graphitic N (ca. 401.0 eV) and N-oxide (ca. 402.1 eV) four N species in the Fe/N-CNSs and Fe/N-CNSs1, pyridine N (ca. 398.2 eV), graphitic N (ca. 401.1 eV) and N-oxide (ca. 402.6 eV) three N species in the Fe/N-CNSs2 and only graphitic N (ca. 401.0 eV) in the Fe/N-CNSs3. Table 1 displayed that the Fe/N-CNSs and Fe/N-CNSs1 contained much more pyridine N, pyrrolic N and N-oxide than the Fe/N-CNSs2 and Fe/N-CNSs3. However, the Fe/N-CNSs2 and Fe/N-CNSs3 possessed more graphitic N than the Fe/N-CNSs and Fe/N-CNSs1, suggesting that the graphitic-N in CNSs is more stable than the pyridine N, pyrrolic N and N oxide and the pyridine N or pyrrolic N could be partially converted into the graphitic N at high temperature. With respect to Fe, although elemental mapping (Fig. 1f) demonstrated the presence of Fe in the Fe/N-CNSs2, no Fe2p peaks were found from the XPS spectra of Fe/N-CNSs1, Fe/N-CNSs2 and Fe/N-CNSs3 (Fig. 3a). Meanwhile, from XRD data shown in Fig. 3b, no signals of elemental Fe and Fe-containing compounds were detected except the (002) and (100) plane peaks of graphite carbon at about 26° and 44°. These results indicated that there were only a small amount of Fe in these catalysts probably existing in the form of single atom or clusters with few Fe atoms.17,18 The Fe K-edge (XANES) analyses further showed the existence of little Fe. From Fig. 3c, it can be seen that the weak Fe signal peaks at 7130, 7195 and 7220 eV in the Fe/N-CNSs1, Fe/N-CNSs2 and Fe/N-CNSs3 were in good agreements with those of the standard Fe foil. The fine differences of the Fe signals at 7153 and 7158 eV from those of Fe foil indicated that the existence of a small amount of N species could influence the electron structure of Fe atom to some extent. TG analyses of these prepared samples before and after concentrated hydrochloric acid handling (Table 2 and Fig. S1†) further demonstrated that about 30–40% of Fe were anchored on the surface and in the mesoporous or macropore of carbon carriers and about 60–70% of Fe were encapsulated in carrier interior. These results indicated that the stainless steel transportation tube and iron wire at high temperature could be slightly vaporized and provide a smaller amount of Fe atoms to the N-doped carbon carriers. The relative ratio of ID/IG (0.95, 0.85 and 0.79) for the Fe/N-CNSs1, Fe/N-CNSs2 and Fe/N-CNSs3 in Raman spectra (Fig. 3d) revealed that the defects in the graphitic structures gradually decreased and the graphitization degree increased with the increase of heat temperature and the extension of heating time.
 |
| | Fig. 1 The SEM, TEM, HRTEM and STEM images (a–d) of Fe/N-CNSs2. And the element mapping images of N and Fe (e and f). | |
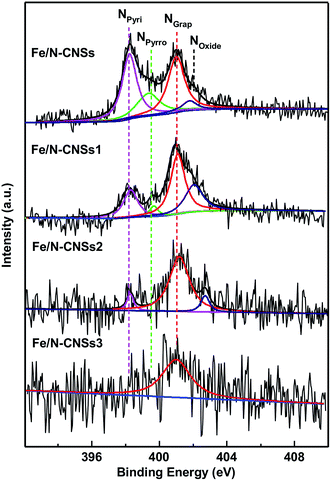 |
| | Fig. 2 The high-resolution N1s XPS spectra in the Fe/N-CNSs, Fe/N-CNSs1, Fe/N-CNSs2 and Fe/N-CNSs3. | |
Table 1 The elemental compositions of Fe/N-CNSs, Fe/N-CNSs1, Fe/N-CNSs2 and Fe/N-CNSs3 from XPS analyses
| Samples |
XPS (at%) |
| C |
O |
Npyri |
Npyrro |
Ngrap |
Noxide |
Gross N |
| Fe/N-CNSs |
89.55 |
7.08 |
1.28 |
0.70 |
1.22 |
0.17 |
3.37 |
| Fe/N-CNSs1 |
90.97 |
5.69 |
0.60 |
0.24 |
1.62 |
0.88 |
3.34 |
| Fe/N-CNSs2 |
94.42 |
2.85 |
0.23 |
— |
2.22 |
0.28 |
2.73 |
| Fe/N-CNSs3 |
93.83 |
4.19 |
— |
— |
1.98 |
— |
1.98 |
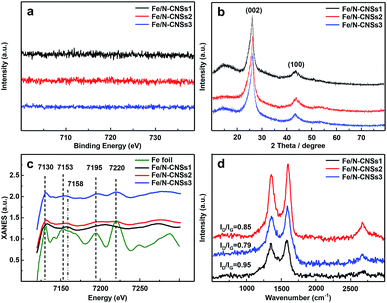 |
| | Fig. 3 The high-resolution Fe2p XPS spectra (a), XRD patterns (b), Fe-K edge XANES spectra (c) and the Raman spectra (d) of Fe/N-CNSs1, Fe/N-CNSs2 and Fe/N-CNSs3. | |
Table 2 The total Fe content, external Fe on the surface and in the mesopore and macropore dissolved by concentrated hydrochloric acid, and the interior Fe encapsulated in CNSs in the Fe/N-CNSs, Fe/N-CNSs1, Fe/N-CNSs2 and Fe/N-CNSs3 from TG analyses and the turnover frequency (TOF) of these four catalysts
| Samples |
TG (wt%) |
TOF |
| Fe-total |
Fe-external |
Fe-interior |
| Fe/N-CNSs |
2.31 |
0.93 |
1.38 |
0.014 |
| Fe/N-CNSs1 |
2.25 |
0.86 |
1.39 |
0.082 |
| Fe/N-CNSs2 |
2.05 |
0.63 |
1.42 |
0.165 |
| Fe/N-CNSs3 |
2.46 |
0.85 |
1.61 |
0.125 |
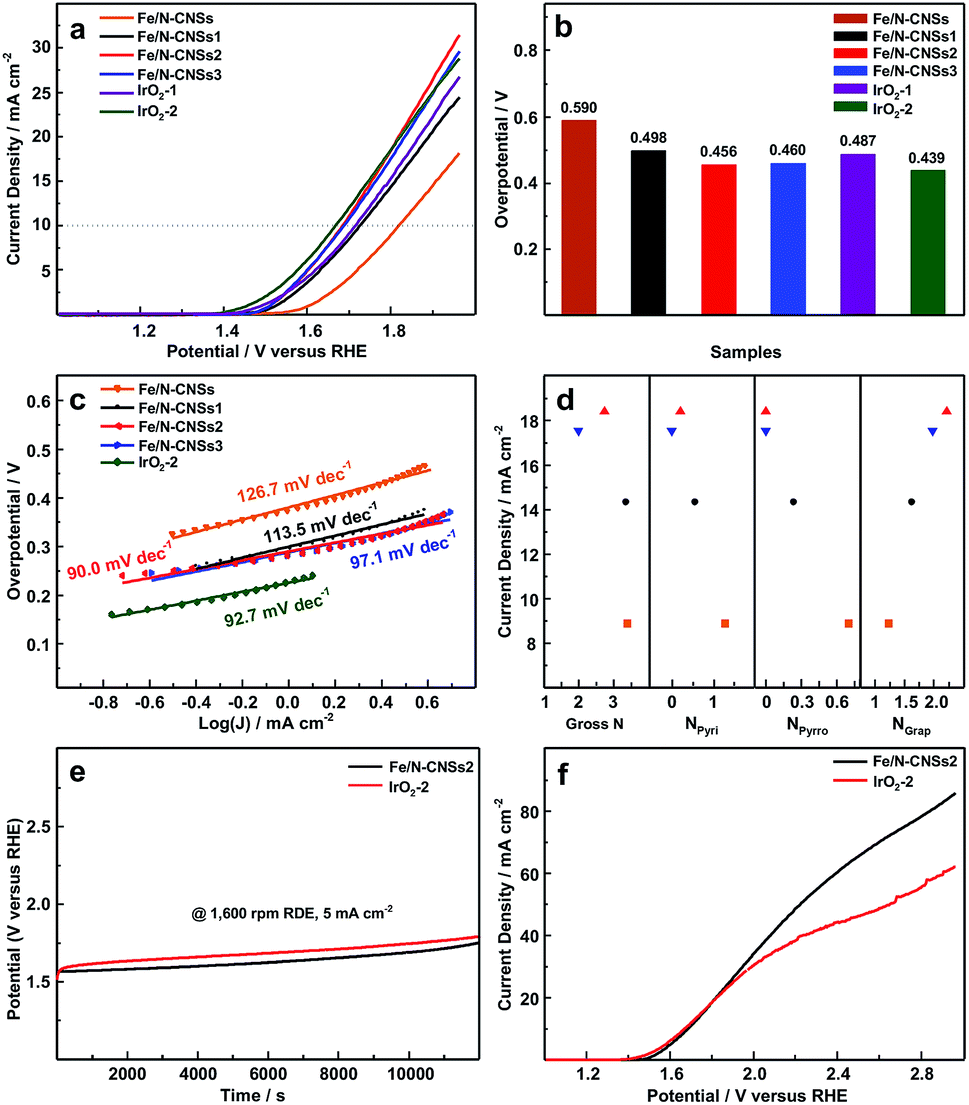
To investigate the OER electrocatalytic activities of the synthesized samples, the linear sweep voltammetry (LSV) measurements were carried out in N2-saturated 0.1 M KOH solution at a rotation speed of 1600 rpm. For a clear comparison, the commercial noble metal IrO2 catalysts (IrO2-1, 0.153 mg cm−2; IrO2-2, 0.229 mg cm−2) were subjected to the same test system. As displayed in Fig. 4a and b, the overpotentials (0.456 and 0.460 V) of Fe/N-CNSs2 and Fe/N-CNSs3 at the current density of 10 mA cm−2 were obviously lower than those (0.590, 0.498 and 0.487 V) of Fe/N-CNSs, Fe/N-CNSs1 and IrO2-1, and much close to that (0.439 V) of IrO2-2, indicating the outstanding OER electrocatalytic activities of Fe/N-CNSs2 and Fe/N-CNSs3. Meanwhile, in Fig. 4c, the Tafel slopes (90.0 and 97.1 mV dec−1) of Fe/N-CNSs2 and Fe/N-CNSs3 both were smaller than those (126.7, 113.5 and 92.7 mV dec−1) of Fe/N-CNSs, Fe/N-CNSs1 and IrO2, validating the higher OER kinetics of Fe/N-CNSs2 and Fe/N-CNSs3. The more excellent OER activities of Fe/N-CNSs2 and Fe/N-CNSs3 with more graphitic N than those of Fe/N-CNSs and Fe/N-CNSs1 with more pyridine N and pyrrolic N implied that the graphitic N is the real active N species which improves the Fe OER activity. To better understand the effect of N species on the OER activity of Fe catalyst, the dependences of the current densities of four catalysts on the N gross content, graphitic N, pyridine N and pyrrolic N species contents were further investigated at a selected potential of 1.8 V in the potential range from ca. 1.5 to 1.96 V, respectively. Fig. 4d showed that the current densities of these four catalysts increased only with the increase of graphitic N contents. The decrease or disappearance of gross N, pyridine N and pyrrolic N contents not only did not bring the activity decrease of Fe catalyst, but witnessed the enhancement of Fe OER activity, clearly indicating that the Fe OER activity is in positive correlation with the graphitic N contents and has no direct relationship with gross N, pyridine N and pyrrolic N (Table 1). To further ascertain the influence of these three N species on the OER activity, the intrinsic activities of these four Fe-based catalysts were investigated through turnover frequency (TOF), respectively. TOF values were calculated by the equation: TOF = I/4Fm, where I is current at the overpotential of 0.3 V, F is Faraday's constant (a value of 96485 C mol−1), and m is the number of moles of the active Fe anchoring on the surface and in the mesoporous and macropore on the working electrode.19 As seen from Table 2, all TOF values for Fe/N-CNSs, Fe/N-CNSs, Fe/N-CNSs2 and Fe/N-CNSs3 only gradual increased with the increase of the graphitic N content in these catalysts, confirming that the OER activity of Fe catalyst is directly related to the graphitic N in carbon carriers. These results indicated that among three potential active N species, the graphitic N is the active N species which remarkably improves the OER activity of transition metal Fe catalyst and even a small amount of transition metal atoms also could exhibit much high OER activity when they were atomically and uniformly loaded on graphitic N-doped carbon carriers.
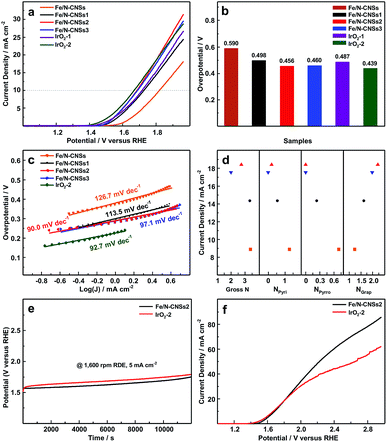 |
| | Fig. 4 The OER polarization curves of Fe/N-CNSs, Fe/N-CNSs1, Fe/N-CNSs2, Fe/N-CNSs3 and IrO2 in N2-saturated 0.1 M KOH solution at the rotation rate of 1600 rpm (a). Corresponding overpotentials at a chosen current density of 10 mA cm−2 (b) and Tafel plots (c) derived from (a). The dependences of the current densities of Fe/N-CNSs (■), Fe/N-CNSs1 (◆), Fe/N-CNSs2 (▲) and Fe/N-CNSs3 (▼) on the N gross content, graphitic N, pyridine N and pyrrolic N species contents (d). The chronopotentiometry responses of Fe/N-CNSs2 and IrO2 (e). And The OER polarization curves of Fe/N-CNSs2 and IrO2 in N2-saturated 0.1 M KOH solution and the potential range of ca. 1.0 to 2.97 V at the rotation rate of 1600 rpm (f). | |
Since the stability often plays a crucial role during the process of practical application, the stability of the Fe/N-CNSs2 with graphitic N and more prominent OER activity was estimated by the chronoamperometric response for 12000 s in the N2-saturated 0.1 M KOH solution together with that of commercial IrO2 catalyst. From Fig. 4e, it can be found that the Fe/N-CNSs2 underwent a positive potential shift of ca. 0.18 V while IrO2 went through a potential rise of ca. 0.24 V after 12000 s at a constant current density of 5 mA cm−2 and a rotation speed of 1600 rpm. Although the onset potential (1.52 V) of IrO2 at the constant current density of 5 mA cm−2 was lower than that (1.57 V) of Fe/N-CNSs2, it degenerated more sharply within 200 s in comparison with that of Fe/N-CNSs2. Meanwhile, since the OER catalysts often suffer from the detachment from the surface of carriers or working electrode, the stabilities of Fe/N-CNSs2 and IrO2 on the working electrode were also estimated comparatively by the LSV measurements in the potential range of ca. 1.0 to 2.97 V. Fig. 4f showed that the current densities of Fe/N-CNSs2 continued to proportionally increase with the increase of potential till ca. 2.3 V while those of IrO2 slowed at ca. 1.9 V, indicating that the Fe/N-CNSs2 is more stable than the commercial IrO2 catalyst and does not easily split off from the surface of carriers or working electrode during the practical application process. These results demonstrated that the Fe/N-CNSs2 possessed more excellent stability than the commercial noble metal IrO2 catalyst, showing a greatly promising practical application prospect in the fields of water splitting, regenerative fuel cells and rechargeable metal–air batteries.
Our previous work has demonstrated that it is the pyridine N and pyrrolic N rather than graphitic N in nitrogen-contain mesoporous carbon materials which could obviously improve the OER activity of carbon materials both in acidic and alkaline media.20 And the defects are also believed to serve as catalytic active sites for the OER directly.21,22 However, in this work, for the four N-containing Fe-based catalysts, namely, Fe/N-CNSs, Fe/N-CNSs1, Fe/N-CNSs2 and Fe/N-CNSs3, the Fe/N-CNSs and Fe/N-CNSs1 contained more pyridine N, pyrrolic N and defects, but exhibited the worse OER activity. The Fe/N-CNSs2 with more graphitic N and less defects, and the Fe/N-CNSs3 with only graphitic N species and least defects, both displayed much higher OER activity than the Fe/N-CNSs and Fe/N-CNSs1. These results indicated that the graphitic N itself probably could not ameliorate the OER activity of carbon materials considerably unlike the pyridine N and pyrrolic N, but it could improve the OER activity of Fe catalyst more drastically than the pyridine N and pyrrolic N. To further understand the difference of N species in the OER performance of these prepared Fe-based catalysts, the binding energy (Eb), charge distribution (CD) and spin density (SD) of Fe atom loaded on non-N or N-doped graphene with graphitic N, pyridine N or pyrrolic N were calculated by DFT implemented in Gaussian 09 program,23 respectively. The B3LYP density functional combined with the 6-31G(d) basis set were used for all calculations.24–26 The binding energy was calculated through the following expression: Eb = EFe/N-graphene − EN-graphene − EFe, where EFe/N-graphene, EN-graphene and EFe denote the total energies of the optimized Fe binding on N-graphene, the N-graphene and a single Fe atom. To reduce the computational cost, the configurations for Fe atom binding at the ortho-, meta-, and para-positions of the N atom are considered. From Fig. S2,† it is interesting to find that on the surface of graphene only doped with graphitic N, four stable Fe loading configurations (Fe/NgrapG1, Fe/NgrapG2, Fe/NgrapG3 and Fe/NgrapG4) were identified while only two stable configurations were found on the surface of graphene only with pyridine N or pyrrolic N (Fe/NpyriG1, Fe/NpyriG2 or Fe/NpyrroG1, Fe/NpyrroG2). Surprisingly, Table 3 further revealed that the spin densities of Fe atoms located at four different sites on graphitic N-doped graphene all strikingly increased from ca. 0 to +2.4 when comparing to those of Fe atom on the non-N graphene. Meanwhile, the charge densities of Fe atoms in the Fe/NgrapG2, Fe/NgrapG3 and Fe/NgrapG4 also increased by ca. 0.03 except that in the Fe/NgrapG1, indicating that the graphitic N could change the charge and spin densities of Fe atom notably. In contrast, both spin and charge densities of Fe atoms in the Fe/NpyriG1, Fe/NpyriG2, Fe/NpyrroG1 and Fe/NpyrroG2 almost unchanged in comparison with those of Fe atom on the non-N graphene, meaning that the doping of pyridine N and pyrrolic N could not arouse the remarkable redistribution of spin and charge density of Fe atoms loaded around them. Furthermore, more intriguingly, the binding energies (3.2, 3.1, 3.2 and 3.0 eV) of Fe atoms at the four different stable sites on graphitic N-doped graphene all were more than twice that (ca. 1.4 eV) of Fe atom on the non-N graphene or graphene doped with pyridine N or pyrrolic N. Since the positive atomic spin and charge densities often determine the catalytic capability of the materials through controlling the adsorption bond strengths and positional selectivity of adsorbate radicals,27–29 and the larger binding energy often means higher stability for metal-based catalysts,30,31 it can be concluded that as the active species, unlike the pyridine N and pyrrolic N, the graphitic N in carbon networks could effectively improve the OER activity and stability of Fe catalysts mainly through increasing the charge and spin density and binding energy of Fe atoms.
Table 3 The binding energy of Fe atom, the charge and spin densities of Fe and N atoms from DFT calculations
| |
Eb (eV) |
Fe CD |
Fe SD |
N CD |
N SD |
| Fe/NgrapG1 |
3.2 |
0.24 |
2.12 |
−0.82 |
−0.10 |
| Fe/NgrapG2 |
3.1 |
0.70 |
2.49 |
−0.83 |
−0.07 |
| Fe/NgrapG3 |
3.2 |
0.69 |
2.47 |
−0.89 |
−0.08 |
| Fe/NgrapG4 |
3.0 |
0.71 |
2.56 |
−0.85 |
−0.06 |
| Fe/NpyriG1 |
1.4 |
0.66 |
0.00 |
−0.51 |
−0.00 |
| Fe/NpyriG2 |
1.4 |
0.66 |
0.00 |
−0.52 |
−0.00 |
| Fe/NpyrroG1 |
1.1 |
0.65 |
0.00 |
−0.55 |
−0.00 |
| Fe/NpyrroG2 |
1.4 |
0.66 |
0.00 |
−0.57 |
−0.00 |
| Fe/non-NG |
1.4 |
0.66 |
0.00 |
— |
— |
4. Conclusions
In summary, we have systematically investigated the influence of three potential active N species in N-doped carbon nanospheres on the OER activity and stability of Fe-based catalysts through electrochemical experiments and DFT theoretical calculations. The elemental mapping and Fe K-edge XANES analyses testified that stainless steel and iron wire could serve as Fe precursor and provide smaller amount of Fe atoms at high temperature. Electrochemical tests demonstrated that Fe atoms loaded on the surface of graphitic N-doped carbon nanospheres exhibited much higher OER activity than those on pyridine N or pyrrolic N-doped ones, and comparable activity and stability to the commercial noble metal IrO2 catalyst. Moreover, the theoretical calculations further revealed that the graphitic N in graphene carrier could dramatically increase the charge and spin density and binding energy of Fe atoms unlike the pyridine N and pyrrolic N, directly leading to the OER activity and stability enhancement of Fe-based catalysts. These in-depth insights into the influence of different N species on Fe-based catalysts would provide a bran-new perspective for the rational design of more effective transition metal-based OER electrocatalysts in the future.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We acknowledge the financial support from the Fundamental Research Funds for the Central Universities of China (No. 2017XKQY064) and National Supercomputing Center in Shenzhen for providing the computational resources and Gaussian 09 suite of programs (Revision D.01).
Notes and references
- V. Vij, S. Sultan, A. M. Harzandi, A. Meena, J. N. Tiwari, W.-G. Lee, T. Yoon and K. S. Kim, ACS Catal., 2017, 7, 7196–7225 CrossRef CAS.
- Y. Xu, W. Tu, B. Zhang, S. Yin, Y. Huang, M. Kraft and R. Xu, Adv. Mater., 2017, 29, 1605957–1605965 CrossRef PubMed.
- I. S. Amiinu, X. Liu, Z. Pu, W. Li, Q. Li, J. Zhang, H. Tang, H. Zhang and S. Mu, Adv. Funct. Mater., 2018, 28, 1704638–1704646 CrossRef.
- X. Zhao, P. Pachfule, S. Li, J. R. J. Simke, J. Schmidt and A. Thomas, Angew. Chem., Int. Ed., 2018, 57, 8921–8926 CrossRef CAS PubMed.
- Y. Hou, S. Cui, Z. Wen, X. Guo, X. Feng and J. Chen, Small, 2015, 11, 5940–5948 CrossRef CAS PubMed.
- G. Fu, Z. Cui, Y. Chen, Y. Li, Y. Tang and J. B. Goodenough, Adv. Energy Mater., 2017, 7, 1601172–1601179 CrossRef.
- J. Wang and F. Ciucci, Small, 2017, 13, 1604103–1604117 CrossRef PubMed.
- H. F. Wang, C. Tang and Q. Zhang, Adv. Funct. Mater., 2018, 1803329–1803350 CrossRef.
- L. Ma, S. Chen, Z. Pei, Y. Huang, G. Liang, F. Mo, Q. Yang, J. Su, Y. Gao, J. A. Zapien and C. Zhi, ACS Nano, 2018, 12, 1949–1958 CrossRef CAS PubMed.
- C. Su, H. Cheng, W. Li, Z. Liu, N. Li, Z. Hou, F. Bai, H. Zhang and T. Ma, Adv. Energy Mater., 2017, 7, 1602421–16024212 Search PubMed.
- K. Liu, H. Zhong, F. Meng, X. Zhang, J. Yan and Q. Jiang, Mater. Chem. Front., 2017, 1, 2155–2173 RSC.
- J. Li, J. Chen, H. Wang, Y. Ren, K. Liu, Y. Tang and M. Shao, Energy Storage Mater., 2017, 8, 49–58 CrossRef.
- J. Wang, H. Wu, D. Gao, S. Miao, G. Wang and X. Bao, Nano Energy, 2015, 13, 387–396 CrossRef CAS.
- J. L. Shui, N. K. Karan, M. Balasubramanian, S. Y. Li and D. J. Liu, J. Am. Chem. Soc., 2012, 134, 16654–16661 CrossRef CAS PubMed.
- J. Liu, X. J. Sun, P. Song, Y. W. Zhang, W. Xing and W. L. Xu, Adv. Mater., 2013, 25, 6879–6883 CrossRef CAS.
- Q. Li, R. G. Cao, J. Cho and G. Wu, Adv. Energy Mater., 2014, 4, 1301415–1301433 CrossRef.
- P. Yin, T. Yao, Y. Wu, L. Zheng, Y. Lin, W. Liu, H. Ju, J. Zhu, X. Hong, Z. Deng, G. Zhou, S. Wei and Y. Li, Angew. Chem., Int. Ed., 2016, 55, 10800–10805 CrossRef CAS PubMed.
- C. Zhao, X. Dai, T. Yao, W. Chen, X. Wang, J. Wang, J. Yang, S. Wei, Y. Wu and Y. Li, J. Am. Chem. Soc., 2017, 139, 8078–8081 CrossRef CAS PubMed.
- M. B. Stevens, L. J. Enman, A. S. Batchellor, M. R. Cosby, A. E. Vise, C. D. M. Trang and S. W. Boettcher, Chem. Mater., 2017, 29, 120–140 CrossRef CAS.
- M. Li, Z. W. Liu, F. Wang and J. J. Xuan, J. Energy Chem., 2017, 26, 422–427 CrossRef.
- Z. J. Liu, Z. H. Zhao, Y. Y. Wang, S. Dou, D. F. Yan, D. D. Liu, Z. H. Xia and S. Y. Wang, Adv. Mater., 2017, 29, 1606207–1606214 CrossRef PubMed.
- Z. W. Liu, F. Peng, H. J. Wang, H. Yu, W. X. Zheng and J. Yang, Angew. Chem., Int. Ed., 2011, 50, 3257–3261 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks and H. B. Schlegel, et al., Gaussian 09, Revision D.01 Inc., Wallingford, CT, 2009 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- W. J. Hehre, L. Radom, P. V. R. Schleyer and J. A. Pople, Ab initio molecular orbital theory, Wiley-Interscience, New York, 1986 Search PubMed.
- L. Zhang and Z. Xia, J. Phys. Chem. C, 2011, 115, 11170–11176 CrossRef CAS.
- S. Wang, L. Zhang, Z. Xia, A. Roy, D. W. Chang, J. B. Baek and L. Dai, Angew. Chem., 2012, 124, 4285–4288 CrossRef.
- L. Zhang, J. Niu, L. Dai and Z. Xia, Langmuir, 2012, 28, 7542–7550 CrossRef CAS PubMed.
- Z. W. Liu, Q. Q. Shi, R. F. Zhang, Q.-D. Wang, G. J. Kang and F. Peng, J. Power Sources, 2014, 268, 171–175 CrossRef CAS.
- Z. W. Liu, J. F. Qu, X. Fu, Q.-D. Wang, G. Y. Zhong and F. Peng, Mater. Lett., 2014, 142, 115–118 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra00185a |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Fei Han,
Guo-Jun Kang,
Ling Liu,
Jinlong Liu and
Quan-De Wang*
*,
Fei Han,
Guo-Jun Kang,
Ling Liu,
Jinlong Liu and
Quan-De Wang*