DOI:
10.1039/C9RA00261H
(Paper)
RSC Adv., 2019,
9, 14024-14032
Infrared induced repeatable self-healing and removability of mechanically enhanced graphene–epoxy flexible materials†
Received
11th January 2019
, Accepted 24th March 2019
First published on 7th May 2019
Abstract
A repeatable self-healing epoxy composite mechanically enhanced by graphene nanosheets (GNS) was prepared from an epoxy monomer with Diels–Alder (DA) bonds, octanediol glycidyl ether (OGE) and polyether amine (D230). The GNS/epoxy composites, with a maximum tensile modulus of 14.52 ± 0.45 MPa and elongation at break more than 100%, could be healed several times under Infrared (IR) light with the healing efficiency as high as 90% through the molecule chain mobility and the rebonding of reversible DA bonds between furan and maleimide. Also, they displayed excellent recyclable ability by transforming into a soluble polymer, which offers a wide range of possibilities to produce epoxy flexible materials with healing and removable abilities.
1 Introduction
Epoxy polymeric materials have been used in a variety of fields, including the transport industry (cars, ships, and aircraft), civil engineering, electronics, etc., due to their high tensile strength, low shrinkage, excellent seal-ability, and good chemical and corrosion resistance.1–3 However, they are susceptible to failure after fracture or damage, which leads to a sharp decrease in their sustainability, safety, and lifetime.4,5 Thus, exploration of self-healing polymeric materials that can repair themselves after mechanical damage is in high demand.
For decades, scientists and engineers have paid great attention to developing self-healing polymeric materials to improve their integrity.6–11 In most cases, microcontainers, such as hollow fibers12,13 and microcapsules,14,15 which store healing agents, are embedded in the polymer matrix to form self-healing polymeric composites. When the mechanical damage (e.g., impact, compression, or abrasion) occurs, the healing agent would be released into the crack place due to the capillary effect, and then re-bond the crack. This kind of self-healing materials can heal themselves spontaneously and timely, but they can be healed only once at the same location because of exhaustion of the healing agents. To overcome this limitation, Wudl's group developed a reversible polymer that exhibited multiple cycles of crack mending with simple thermal treatment.16 In this system, a polymer with reversible bonds can be self-healed owing to the physical and/or chemical interactions at the broken interfaces. Inspired by this idea, a great deal of self-healing materials have been designed by embedding various reversible bonds such as Diels–Alder (DA) bond, disulfide bond (SS), and so on.2,17–20 Among them, DA reaction between furan and maleimide attracts great interests in self-healing due to the relatively high mechanical properties and healing efficiency.21–23 For example, Tian et al. has prepared a thermally remendable epoxy resin with the mechanical properties similar to those of commercial epoxy resin by reacting a reversible epoxy monomer containing both furan and epoxide groups with bismaleimide monomer and anhydride curing agent.21 Also, Kuang et al. prepared a reversible epoxy adhesive by curing a commercial epoxy oligomer with a diamine DA adduct cross-linker and found that the adhesive strength values about 3 MPa, which could be full recovered in the repeated fracture-thermal healing processes.22 As it were, thermally reversible self-healing materials have made great advances, some critical problems that hinder their practical applications still remain. For example, the relatively poor mechanical properties, the low flexibility and the single repair approach, which severely limits their possible applications in many cases, especially for the adhesives or sealers which need remotely or locally repair.
Graphene materials, due to their large π-conjugated system, have good compatibility with polymeric materials and been widely used as efficient fillers in fabrication of mechanical enhanced composite materials because of their ultrahigh mechanical strength.24–26 Furthermore, they also have some remarkable properties, including super chemical stability, outstanding thermal conductivity,27–29 good infrared (IR) absorbing capacity,30–32 which endows them with a strong response to IR light. These properties prompt us to believe that integrating graphene materials into epoxy materials might generate some novel self-healing epoxy composites, which not only have enhanced mechanical properties and appropriate flexibility but also can be healed via IR light repeatedly. To the best of our knowledge, not much study about repeatable self-healing epoxy flexible composites with enhanced mechanical properties has been reported so far.
Herein, we report a novel self-healing epoxy flexible material fabricated with graphene nanosheets (GNS) and epoxy polymer. Different from the conventional self-healing polymeric materials, in addition to its enhanced mechanical property, this GNS/epoxy self-healing material with high elongation (>100%) can be healed repeatedly with excellent healing efficiency higher than 90% and be removed via IR light.
2 Experimental
2.1 Materials
Furfurylglycidylether (FGE, 97 wt%) and 4,4′-methylenebis(N-phenylmaleimide) (DPMBMI, 95 wt%) were purchased from J&K Scientific Ltd. Graphene nanosheets (GNS), diethylenetriamine (DETA, A.R.), polyether amine (D230 A.R.) and other solvents were bought from Aladdin Reagent Ltd. Diglycidyl ether of bisphenyl A (DGEBA, Brand EP-4100HF, high-purity) and octanediol glycidyl ether (OGE, Brand ED-506, high-purity) were granted kindly by ADEKA Corporation of Japan. 2,2′-(Methylenebis(4,1-phenylene))bis(4-((oxiran-2-ylmethoxy)methyl)-3a,4,7,7a-tetrahydro-1H-4,7-epoxyisoin-dole-1,3(2H)-dione) (FDB) was synthesized with FGE and DPMBMI in our laboratory by a one-step method according to Min et al.2 All solvents and the above reagents were used as received without further purification.
2.2 Preparation of epoxy polymers and GNS/epoxy composites with DA bonds
The thermally reversible GNS/epoxy composite samples were prepared by integrating GNS fillers into epoxy polymers with the weight contents of 0.1–0.7%. The preparation schematic diagram is as follows (Scheme 1). Typically, FDB monomer was mixed with OGE monomer firstly with a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5 and formed into a uniform epoxy mixture 1, then the GNS was filled into the above mixture 1 and mixed by SR-500 mixer (THINKY, Japan) at 1000 rpm for 10 min to form a uniform mixture 2. Next, the D230 amine curing agent was added into the mixture 2 with a stoichiometric ratio of 1:1 and mixed by SR-500 mixer at 1000 rpm for another 10 min. Finally, the mixture was degassed under vacuum of −0.09 MPa for 20 min. The well-mixed suspension was coated on glass slides or steel substrates using a micrometer controlled doctor blade and cured at 25 °C for 12 h and 50 °C for 48 h. Finally, the GNS/epoxy composites were obtained. The preparation process of epoxy polymers without GNS fillers were similar with GNS/epoxy composites except not adding GNS fillers into mixture 1.
:1.5 and formed into a uniform epoxy mixture 1, then the GNS was filled into the above mixture 1 and mixed by SR-500 mixer (THINKY, Japan) at 1000 rpm for 10 min to form a uniform mixture 2. Next, the D230 amine curing agent was added into the mixture 2 with a stoichiometric ratio of 1:1 and mixed by SR-500 mixer at 1000 rpm for another 10 min. Finally, the mixture was degassed under vacuum of −0.09 MPa for 20 min. The well-mixed suspension was coated on glass slides or steel substrates using a micrometer controlled doctor blade and cured at 25 °C for 12 h and 50 °C for 48 h. Finally, the GNS/epoxy composites were obtained. The preparation process of epoxy polymers without GNS fillers were similar with GNS/epoxy composites except not adding GNS fillers into mixture 1.
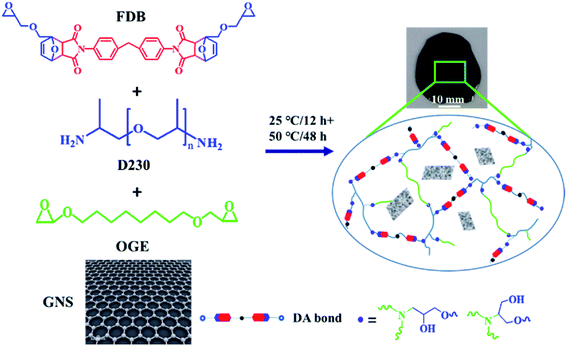 |
| | Scheme 1 The preparation schematic diagram of GNS/epoxy flexible composites embedded with DA bonds. | |
2.3 Measurement and characterization
The microstructure of GNS was characterized by X-ray diffraction (XRD, X'Pert PRO). Raman spectra of GNS were recorded using a HORIBA Scientific XploRA PLUS spectrometer with laser excitation source of 532 nm. The chemical composition of FDB/D230, OGE/D230, and FDB/OGE/D230 epoxy polymer were characterized by Fourier transform infrared spectrometer (FTIR, Bruker Vertex 70). The thermal reversible reaction of FDB/OGE/D230 was also monitored by in situ FTIR spectra (Bruker Vertex 70). The epoxy mixture was coated on the surface of a KBr tablet, and then the FTIR spectrum was collected after the tablet had been dried at curing conditions. The spectra were recorded in the range of 4000–500 cm−1. The glass transition temperature (Tg) was determined by differential scanning calorimetry (DSC, TA Q2000) at a temperature range of −50–200 °C with a heating rate of 10 °C min−1 in N2 atmosphere. The morphology of GNS and their dispersion in the epoxy matrix was characterized by transmission electron microscopy (TEM, Caiss). Before observation, the GNS/epoxy composite was cut by ultramicrotome (LEICA EM UC7 M80, Japan). The morphology of GNS and the fractured planes of epoxy polymer and GNS/epoxy composites were observed by field emission scanning electron microscopy (FESEM, FEI Helios Nanolab 600i). Prior to observation, the samples were sputter-coated with gold. The thermal conductivity of GNS/epoxy composites were determined by Thermal conductivity tester (NETZSCH LFA457). Thermogravimetric analysis (TGA) of epoxy polymers and GNS/epoxy composites was performed on a TA SDT Q600 TGA. The samples were heated from 25–600 °C at 10 °C min−1 under nitrogen atmosphere. The tensile modulus and elongation at break was measured by tensile testing machine (UTM-2360) in accordance with ASTM D638-2008 with a stretching rate of 5 mm min−1. The dynamic mechanical properties of epoxy polymers and GNS/epoxy composites were determined by Dynamic Mechanical Analyzer (DMA, TA Triton 2000) in the single cantilever beam mode at a temperature range of −50–180 °C with a heating rate of 5 °C min−1 and a vibration frequency of 1 Hz. The size of sample was 50 × 5 × 1 mm3 clamped by aluminum foil with a thickness of 0.1 mm. The self-healing property was measured by lap shear strength in accordance with ISO 9664-1995 with a glass substrate. The thickness of epoxy polymer and GNS/epoxy composites was about 300 μm controlled by glass beads. The bonded area was 25 mm × 12.5 mm. The healing efficiency (η) is defined as the ratio of maximum shear stress of healed materials (σhealed), to that of virgin ones (σvirgin):33
Five specimens were tested for each set of conditions and the mean values and their standard deviations were calculated. The removal property was evaluated by solubility experiment.
3 Results and discussion
3.1 Characterization of graphene nanosheets
Fig. 1 gives the microstructure and chemical composition of GNS fillers. A narrow diffraction peak near 2θ = 26° is occurred for GNS (Fig. 1a), corresponding to the interval of 0.339 nm, indicates that the GNS has a good interlaminar crystal structure similar with natural graphite.30 Raman spectroscopy is a widely accepted tool to detect defects of carbon materials. As shown in Fig. 1b, two prominent peaks located at 1324 and 1566 cm−1 assigned to the D band and G band of GNS can be clearly observed.34 The relative intensity ratio of D to G band (ID/IG) is 8.05, suggesting that a large amount of graphitic carbon are present in GNS, which contributes to transporting heat.25 The GNS were dispersed uniformly in ethanol by ultrasonic, then dripped on aluminum foil or copper screen, and dried for SEM or TEM observation (Fig. 1c–e). It can be clearly seen that the GNS fillers are multilayered flakes (10–30 layers) with the diameter of about 2–10 μm.
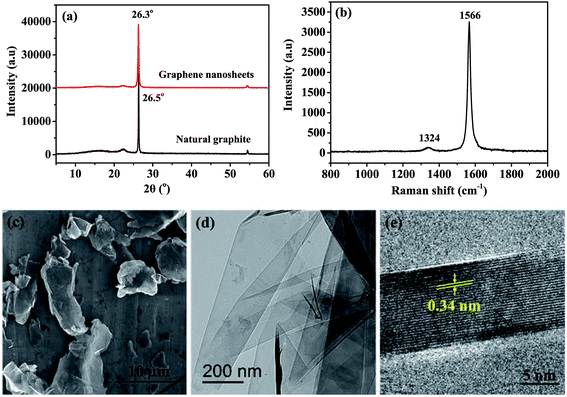 |
| | Fig. 1 XRD curves (a), Raman spectra (b), SEM image (c) and TEM graphs (d and e) of GNS fillers. | |
3.2 Preparation of epoxy polymers and GNS/epoxy composites
The FDB/D230, OGE/D230 and FDB/OGE/D230 epoxy polymers were prepared by curing the FDB or/and OGE monomers with D230 amine curing agent, and the FTIR spectra of these polymeric materials are shown in Fig. 2. The peaks attributed to the epoxide groups at 903 cm−1 (oxirane ring breathing) are disappeared entirely, indicating that epoxide groups have reacted with amine groups of D230 to form epoxy polymer networks. Accordingly, a characteristic peak of hydroxyl groups is emerged at 3465 cm−1 as a result of this crosslinking reaction. Besides, the DA bonds originated from the addition reaction of furan and maleimide groups are proved by the existence of the peaks at 1776 cm−1 in the cured sample (Fig. 2a and c), which provides some possibilities for self-healing and removability of epoxy polymers.
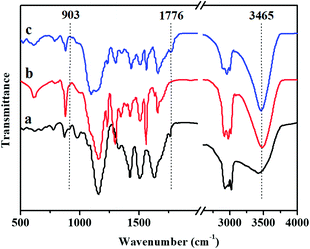 |
| | Fig. 2 FTIR spectra of (a) FDB/D230, (b) OGE/D230 and (c) FDB/OGE/D230 epoxy polymers. | |
In order to reveal how the DA bonds behave under thermal stimulus, in situ heating FTIR spectra and DSC of the cured FDB/OGE/D230 epoxy polymer were recorded (Fig. 3 and 4). As can be seen in Fig. 3, the peaks at 1776 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C in DA adduct) and 1191 cm−1 (C–C in DA adduct) decrease gradually with the increase of temperature, which indicates that DA adduct in the FDB/OGE/D230 epoxy polymer has been decomposed. Accordingly, the peaks at 1018 cm−1 (ring breathing and ether linkage in furan ring) and 1149 cm−1 (absorption of C–N–C in maleimide ring) increase until to 120 °C, and then decrease due to the self-polymerization of maleimide or furan.21 The thermally reversibility of FDB/OGE/D230 epoxy polymer was also evaluated by DSC curves (Fig. 4). The first heating trace shows a glass transition at ca. 47 °C and an endothermic peak at 118 °C as a result of the retro-DA reaction. Because there is not enough time for the recovered furan and maleimide moieties to be reconnected during the subsequent cooling and reheating processes, the consecutive cooling and second heating processes exhibited nearly flat curves.
C in DA adduct) and 1191 cm−1 (C–C in DA adduct) decrease gradually with the increase of temperature, which indicates that DA adduct in the FDB/OGE/D230 epoxy polymer has been decomposed. Accordingly, the peaks at 1018 cm−1 (ring breathing and ether linkage in furan ring) and 1149 cm−1 (absorption of C–N–C in maleimide ring) increase until to 120 °C, and then decrease due to the self-polymerization of maleimide or furan.21 The thermally reversibility of FDB/OGE/D230 epoxy polymer was also evaluated by DSC curves (Fig. 4). The first heating trace shows a glass transition at ca. 47 °C and an endothermic peak at 118 °C as a result of the retro-DA reaction. Because there is not enough time for the recovered furan and maleimide moieties to be reconnected during the subsequent cooling and reheating processes, the consecutive cooling and second heating processes exhibited nearly flat curves.
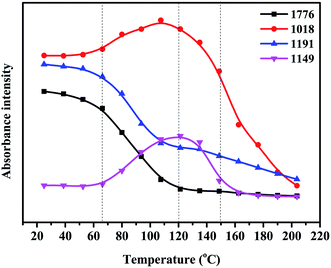 |
| | Fig. 3 The characteristic absorbance intensity of FDB/OGE/D230 epoxy polymer with reaction temperature. | |
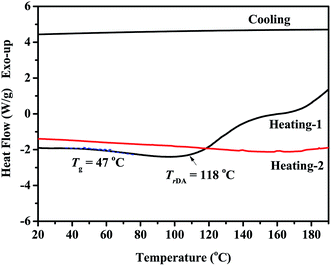 |
| | Fig. 4 DSC curves of FDB/OGE/D230 epoxy polymer with reaction temperature. | |
A series of GNS/epoxy composites were prepared by filling different contents of GNS into FDB/OGE/D230 epoxy polymer (the molar ratio of FDB and OGE was 1:1.5). Fig. 5 illustrates the DSC results of the cured epoxy polymer reacted at 25 °C for 12 h and 50 °C for 48 h. The Tgs of FDB/D230 and OGE/D230 epoxy polymers are about 94 °C and 17 °C (Fig. 5a and b) respectively, indicating that the former is rigid polymer and the latter is soft at room temperature. The polymer with more rigid chains is easier to fracture and produces some cracks in the matrix under stress.2 When some soft OGE monomers are participated in the crosslinked reaction of FDB and D230, the Tg of FDB/D230 epoxy polymer drops quickly, from 94 °C to 47 °C (the molar ratio of FDB and OGE was 1:1.5, Fig. 5c). The decreased Tg of epoxy polymer means the increase of the mobility of molecule chains, which is contributed for both sides of crack to contacting with each other and healing the cracks. After filling only 0.1 wt% GNS into the above epoxy polymer, the Tg increases about 5 °C (attained to 52 °C) due to the hindering effect of GNS fillers. In addition, a retro-DA reaction can be found at the temperature range of 110–130 °C for the epoxy polymers which contain DA bonds, demonstrating that the prepared GNS/FDB/OGE/D230 epoxy composites have the ability of self-healing and recyclability.2,21–23
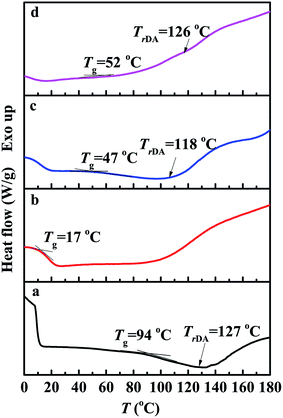 |
| | Fig. 5 DSC results of the cured epoxy polymers (N2 atmosphere, 10 °C min−1). (a) FDB/D230, (b) OGE/D230, (c) FDB/OGE/D230, (d) GNS/FDB/OGE/D230 epoxy polymers. | |
Fig. 6 shows the Tgs of GNS/epoxy composites with different loadings of GNS fillers. The Tgs of these epoxy materials increase with GNS content because GNS are rigid fillers. When filling into the epoxy matrix, they would hinder the mobility of molecule chains in the cross-linked networks and delay the glass transition of epoxy polymers. Therefore, too much GNS fillers are not conducive to the repair of epoxy composites.
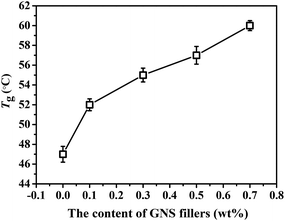 |
| | Fig. 6 Tgs of GNS/FDB/OGE/D230 epoxy composites with different GNS contents. | |
3.3 Mechanical properties of GNS/epoxy composites
Graphene materials are often used as fillers in fabricating mechanical enhanced composite materials because of their outstanding mechanical property. As shown in Fig. 7, there is an optimized weight fraction of GNS in the epoxy matrix and the tensile strengths of GNS/epoxy composites are overall enhanced partially due to the well-dispersed graphene sheets in the matrix (shown in Fig. S1, ESI†). The maximum tensile modulus of GNS/epoxy composite could be attained to 14.52 ± 0.45 MPa when the content of GNS is 0.5 wt%, which is lower than that of heat reversible polyurethane composites (about 36 MPa) enhanced by hydramine-functionalized graphene nanosheets (FGNS) due to strong interfacial interaction.32 In contrast, the conventional self-healing materials may showed compromised or poor mechanical strength (ESI, Table S1†).6,8,12 The dramatic improvement of the mechanical strength, e.g. about 60% increase of tensile strength from the pure epoxy polymer (FDB/OGE/D230) after incorporation of GNS into the network is attributed to the efficient load transfer between GNS fillers and the epoxy matrix.30,32 Besides, all of the GNS/epoxy composites exhibit higher elongation at break (exceeding to 100%) than traditional self-healing epoxy polymers.12–14 The enhanced mechanical property and the good flexibility partially highlight the potential of GNS/epoxy composites for practical applications, e.g. epoxy adhesives and sealers.
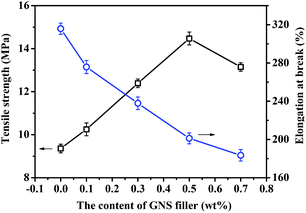 |
| | Fig. 7 The tensile strength and elongation at break of GNS/epoxy composites with different GNS contents. | |
The fracture morphology of epoxy polymer with or without 0.5 wt% GNS fillers were observed by SEM (Fig. 8). A typical brittle fracture morphology (river pattern) is observed for FDB/D230 epoxy polymer, which is caused by the immobility of rigid chains and the weak binding force of DA bonds (Fig. 8a). In contrast, an obvious ductile fracture characteristics are presented for FDB/OGE/D230 due to the flexibility of OGE monomer (Fig. 8b). After integrating 0.5 wt% GNS into FDB/OGE/D230 epoxy matrix, the dimple fracture is presented (Fig. 8c and d). This is because when the epoxy composites are subjected to external stress, the uniformly-dispersed GNS fillers could be acted as the stress concentration center and absorb a large amount of strain energy transmitted from the epoxy matrix, which would improve the mechanical properties of epoxy composites.24,32
 |
| | Fig. 8 The fracture morphology of (a) FDB/D230 epoxy polymer, (b) FDB/OGE/D230 epoxy polymer and (c and d) GNS/FDB/OGE/D230 epoxy composites filled with 0.5 wt% GNS. | |
Fig. 9 gives the dynamic mechanical properties of FDB/D230, FDB/OGE/D230 epoxy polymers and GNS/FDB/OGE/D230 epoxy composites filled with 0.5 wt% GNS. The storage modulus of all of the epoxy polymers decrease with the increase of temperatures. There are two maximum values of loss tangent occurred and correspondingly two storage modulus platforms presented. The first loss tangent represents the glass transition of the epoxy polymer and the second represents the thermal reversible decomposition of DA bonds in the epoxy polymer. For the common epoxy polymers, when the temperature is lower than the glass transition temperature, the storage modulus is higher due to the frozen cross-linking network and molecular segment of the polymer. With the increase of temperature, the polymer enters into the glass transition region, and some chain segments with low molecular weight begin to move, thus the storage modulus begins to decrease. As the temperature continues to rise, the mobility of molecular segment steadily increases until all the molecular segments are completely free and the polymer enters into a high elastic state. For the epoxy polymer without reversible bond, the dynamic mechanical properties are in accordance with the above law. However, when the epoxy polymers contain reversible bonds (e.g. FDB/D230), the whole cross-linking network would be decomposed into low molecular weight segments due to the de-bonding of the furan–maleimide reversible covalent bonds in the polymer matrix, so a second loss tangent maximum value appears at about 120 °C (Fig. 9a). As a result of the plasticizing effect of small molecules, the storage modulus drops sharply and a second platform appears. After some soft monomers (e.g. OGE) participating in the crosslinked reaction of FDB and D230, the Tg of FDB/D230 epoxy polymer drops quickly, from 94 °C to 47 °C (the molar ratio of FDB and OGE is 1:1.5, Fig. 9b) due to the increase of moveable molecular segment. When some rigid GNS fillers were added into the FDB/OGE/D230 epoxy matrix, the Tg increases about 9 °C and the storage modulus is significantly improved (Fig. 9c). Since the activation energy required for the thermal decomposition of furan–maleimide reversible covalent bonds is same for three samples, the decomposition temperature appears all located at the range of 110–130 °C (117 °C for FDB/D230, 120 °C for FDB/OGE/D230 and 122 °C for GNS/FDB/OGE/D230). In contrast to the glass transition temperature measured by DMA and DSC, it is found that the Tg determined by DMA was 1–10 °C higher than that of DSC, which is mainly related to the test method of DMA and DSC.35 The high Tg and the wide peak of FDB/D230 may be caused by the superposition of glass transition and heat decomposition of DA bonds.
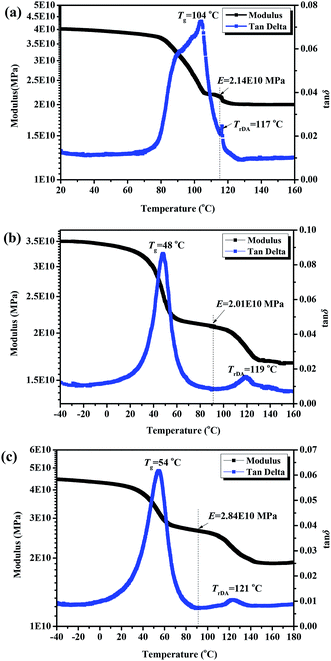 |
| | Fig. 9 Dynamic mechanical properties of (a) FDB/D230, (b) FDB/OGE/D230 epoxy polymers and (c) GNS/FDB/OGE/D230 epoxy composites filled with 0.5 wt% GNS. | |
3.4 Thermal conductivity of epoxy composites with different GNS contents
Fig. 10 gives the thermal conductivity of GNS/epoxy composites with different GNS loadings. The thermal conductivity of original FDB/OGE/D230 epoxy polymer is zero. When filling some GNS fillers into the epoxy polymer (0.1 wt%), the coefficient of heat conductivity immediately increases due to the high heat conductivity of GNS. With the increase of GNS, a complete heat conduction network would be formed in the epoxy matrix and the heat energy would be transferred through it, which improves the thermal conductivity of GNS/epoxy composites. The enhanced thermal conductivity provides the basis for the infrared repair of the epoxy composites.
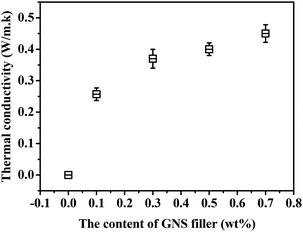 |
| | Fig. 10 The thermal conductivity of GNS/epoxy composites with different GNS contents. | |
3.5 Healing properties of GNS/epoxy composites under IR light
The self-healing performances of GNS/epoxy composites were investigated using IR light (Fig. 11). The fractured samples were positioned under a conventional IR lamp, and the power density delivered to the samples was ∼0.2 W cm−2.
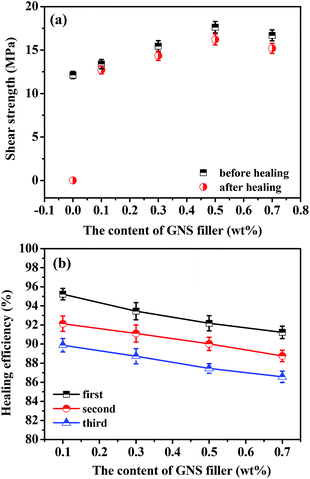 |
| | Fig. 11 The healing properties of GNS/FDB/OGE/D230 epoxy composites with different GNS contents under IR light (a) shear strength; (b) healing efficiency. | |
As shown in Fig. 11a, the first healing efficiencies of the GNS/epoxy samples with different GNS loadings are all as high as 90%. The representative shear strength for the virgin and the healed samples also indicated that the GNS/epoxy samples can be healed almost completely. The time to achieve such high healing efficiencies is all in the range of several minutes (∼20 min) for all the samples with different GNS loadings. On the contrary, the epoxy samples broke immediately upon loading and offered zero healing efficiency even after longer healing (∼30 min). The results indicate clearly that the addition of GNS remarkably improved the healing performances of epoxy polymer, and it is believed that two important intrinsic properties of graphene lead to these results. Firstly, graphene has good IR absorbing capacity,30–32 and the addition of GNS could make the epoxy composites show strong IR absorption. Secondly, the excellent thermal conductivity of graphene would make it efficiently transfer the Joule energy from the IR absorption into the epoxy matrix (shown in Fig. 11). Thus, in the healing process, GNS works as a nanoscale heater and transfer unit to generate the required energy and then transport the energy to the matrix efficiently.30,32 Therefore, once exposed to IR light, the GNS/epoxy samples were heated rapidly and homogeneously, and then the epoxy chains at the broken interface diffused, and recombined again to heal the fracture. Healing repeatability is also very important for the self-healing polymeric materials. Fig. 11b gives the self-healing repeatability of GNS/epoxy composites. All of the GNS/epoxy samples could be healed for more than 3 times under IR light and the healing efficiencies are higher than 85%, although some decrease in each healing cycle caused by the self-polymerization of furan and maleimide group.21 This result suggests that this self-healing polymeric material with the increased reliability and service life may have a great potential to be used in a wide variety of applications ranging from adhesives to sealers.
Through the self-healing process of GNS/epoxy composites use the IR light stimuli, it should be always the case that the self-healing mechanism of GNS/epoxy composites may be as follows (Fig. 12): when the GNS/epoxy composites are damaged under external stress, the well dispersed graphene sheets would convert the energy absorbed from IR light into thermal energy and transfer it to the epoxy matrix efficiently. Then the temperature of the whole GNS/epoxy composites increased rapidly and the self-healing of the GNS/epoxy composites are accomplished by the wetting, diffusion or re-entanglement of the flexible chains at a temperature above the Tg of epoxy and recombinement of furan–maleimide reversible DA bonds.21,22,32 As discussed above, the excellent IR absorption and the efficient energy transfer of graphene are the cause of the light healing.30–32
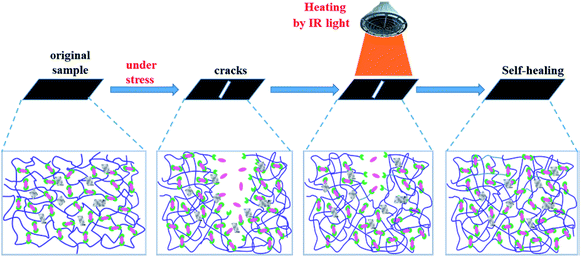 |
| | Fig. 12 The healing mechanism of GNS/FDB/OGE/D230 epoxy composites. | |
3.6 Removability of GNS/epoxy composites under IR light
The thermal properties of GNS/epoxy composites was studied by TGA (Fig. 13). From the thermogravimetric curves of FDB/D230, FDB/OGE/D230 and GNS/FDB/OGE/D230 epoxy polymers, the weight loss of these cross-linked epoxy polymers with DA units commences at about 130 °C and reduces to 88.8%, 91.1% and 96.5% by 260 °C, which implies that the DA units in the crosslink component of the epoxy polymer are broken and the crosslink density decreased rapidly. Due to the scissioning of the epoxy polymer structure by the rDA reaction, this material is free to move. When the temperatures are above 260 °C, the weight is reduced sharply, which means that the main chains of the epoxy polymer become unstable and started to decompose at that temperature. Compared with the TGA trace of the epoxy polymer with or without DA units, the scissioning temperature of all the thermal reversible epoxy polymer (FDB/D230, FDB/OGE/D230 and GNS/FDB/OGE/D230, about 260 °C) was lower than that of the thermal irreversible epoxy polymer (OGE/D230, 285 °C).
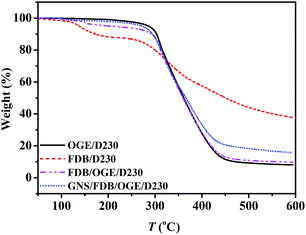 |
| | Fig. 13 TGA curves of epoxy polymers and GNS/epoxy composite. | |
The removability is also meaningful for the crosslinked epoxy materials and evaluated by solubility experiment (Fig. 14). The OGE/D230 epoxy polymer and GNS/FDB/OGE/D230 epoxy composite were immersed in dimethyl sulfoxide (DMSO) at 25 °C for 2 h (Fig. 14b and f), they were all insoluble. After heated under IR light for 20 min, most of the GNS/FDB/OGE/D230 epoxy composites became readily soluble (Fig. 14g and h) due to the rDA reaction of furan–maleimide bond while the OGE/D230 epoxy polymer was still insoluble (Fig. 14c and d), showing that the GNS/FDB/OGE/D230 epoxy composites can be removed from the metal substrates under IR light. Therefore, the GNS/FDB/OGE/D230 epoxy composites could be utilized as a new class of thermally reversible cross-linked polymers which are with enhanced mechanically properties and removability.
 |
| | Fig. 14 The OGE/D230 epoxy polymer (a–d) and GNS/FDB/OGE/D230 epoxy composites (e–h) in DMSO solvent before and after IR light for 20 min. | |
4 Conclusions
A repeatable self-healing GNS/epoxy flexible composites was prepared via a simple method. They exhibited improved mechanical properties and could be healed effectively by IR light. Furthermore, they also can be removed rapidly. These results indicate that the GNS/epoxy composites can act as a promising self-healing and removable material and find wide applications in transport industries, electronics, and so on.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research is support by the Natural Science Foundation of China (51573172, 21805252), Director's Funds of China Academy of Engineering Physics (SJZD201506) and Discipline Development Foundation of Science and Technology on Surface Physics and Chemistry Laboratory (XKFZ201710).
References
- D. Y. Wu, S. Meure and D. Solomon, Prog. Polym. Sci., 2008, 33, 479 CrossRef CAS.
- Y. Q. Min, S. Y. Huang, Y. X. Wang, Z. J. Zhang, B. Y. Du, X. H. Zhang and Z. Q. Fan, Macromolecules, 2015, 48, 316 CrossRef CAS.
- B. J. Blaiszik, S. L. B. Kramer, S. C. Olugebefola, J. S. Moore, N. R. Sottos and S. R. White, Annu. Rev. Mater. Res., 2010, 40, 179 CrossRef CAS.
- C. E. Diesendruck, N. R. Sottos, J. S. Moore and S. R. White, Angew. Chem., Int. Ed., 2015, 54, 2 CrossRef PubMed.
- A. Stoddart, Nat. Rev. Mater., 2016, 2, 16004 CrossRef.
- D. A. Davis, A. Hamilton, J. L. Yang, L. D. Cremar, D. Van Gough, S. L. Potisek, M. T. Ong, P. V. Braun, T. J. Martinez, S. R. White, J. S. Moore and N. R. Sottos, Nature, 2009, 459, 68 CrossRef CAS PubMed.
- N. R. Sottos, Nat. Chem., 2014, 6, 381 CrossRef CAS PubMed.
- L. M. Meng, Y. C. Yuan, M. Z. Rong and M. Q. Zhang, J. Mater. Chem., 2010, 20, 6030 RSC.
- Y. Yang, X. Ding and M. W. Urban, Prog. Polym. Sci., 2015, 49–50, 34 CrossRef CAS.
- J. K. Christopher and N. B. Christopher, Chem. Soc. Rev., 2013, 42, 7161 RSC.
- X. Huang, Z. Huang, J. C. Lai, L. Li, G. C. Yang and C. H. Li, Compos. Sci. Technol., 2018, 167, 346 CrossRef CAS.
- J. W. C. Pang and I. P. Bond, Compos. Sci. Technol., 2005, 65, 1791 CrossRef CAS.
- Y. K. Guo, L. Chen, D. G. Xu, J. R. Zhong, G. Z. Yue, D. Astruc, M. B. Shuai and P. Zhao, RSC Adv., 2016, 6, 65067 RSC.
- H. H. Jin, C. L. Mangun, A. S. Griffin, J. S. Moore, N. R. Sottos and S. R. White, Adv. Mater., 2014, 26, 282 CrossRef CAS PubMed.
- I. L. Hia, E. S. Chan, S. P. Chai and P. Pasbakhsh, J. Mater. Chem. A, 2018, 6, 8470 RSC.
- X. X. Chen, M. A. Dam, K. Ono, A. Mal, H. B. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 2002, 295, 1698 CrossRef CAS PubMed.
- Y. L. Liu and T. W. Chuo, Polym. Chem., 2013, 4, 2194 RSC.
- C. Zeng, H. Seino, J. Ren, K. Hatanaka and N. Yoshie, Macromolecules, 2013, 46, 1794 CrossRef CAS.
- W. Gao, M. Bie, F. Liu, P. Chang and Y. Quan, ACS Appl. Mater. Interfaces, 2017, 9, 15798 CrossRef CAS PubMed.
- L. M. Johnson, E. Ledet, N. D. Huffman, S. L. Swarner, S. D. Shepherd, P. G. Durham and G. D. Rothrock, Polymer, 2015, 64, 84 CrossRef CAS.
- Q. Tian, Y. C. Yuan, M. Z. Rong and M. Q. Zhang, J. Mater. Chem., 2009, 19, 1289 RSC.
- X. Kuang, G. M. Liu, X. Dong, X. G. Liu, J. J. Xu and D. J. Wang, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 2094 CrossRef CAS.
- N. Bai, K. Saito and G. P. Simon, Polym. Chem., 2013, 4, 724 RSC.
- X. Huang, X. Y. Qi, F. Boey and H. Zhang, Chem. Soc. Rev., 2012, 41, 666 RSC.
- Y. T. Park, Y. Qian, C. Chan, T. Suh, M. G. Nejhad and C. W. Macosko, Adv. Funct. Mater., 2015, 25, 575 CrossRef CAS.
- Y. Zhang, Y. Li, P. Ming, Q. Zhang, T. Liu, L. Jiang and Q. Cheng, Adv. Mater., 2016, 28, 2834 CrossRef CAS PubMed.
- S. H. Song, K. H. Park, B. H. Kim, Y. W. Choi, G. H. Jun and D. J. Lee, Adv. Mater., 2013, 25, 732 CrossRef CAS PubMed.
- Y. X. Fu, Z. X. He, D. C. Mo and S. S. Lu, Int. J. Therm. Sci., 2014, 86, 276 CrossRef CAS.
- L. Peng, Z. Xu, Z. Liu, Y. Guo, P. Li and C. Gao, Adv. Mater., 2017, 29, 1700589 CrossRef PubMed.
- L. Huang, N. Yi, Y. Wu, Y. Zhang, Q. Zhang and Y. Huang, Adv. Mater., 2013, 25, 2224 CrossRef CAS PubMed.
- C. Cai, Y. Zhang, X. Zou, R. Zhang, X. Wang and Q. Wu, RSC Adv., 2017, 7, 46336 RSC.
- S. W. Wu, J. H. Li, G. P. Zhang, Y. M. Yao, G. Li, R. Sun and C. P. Wong, ACS Appl. Mater. Interfaces, 2017, 9, 3040 CrossRef CAS PubMed.
- E. Tsangouri, D. Aggelis and D. Van Hemelrijck, Prog. Polym. Sci., 2015, 49, 154 CrossRef.
- X. Tang, B. Yu, R. V. Hansen, X. Chen, X. Hu and J. Yang, Adv. Mater. Interfaces, 2015, 2, 1 Search PubMed.
- M. Abouhamzeh, J. Sinke, K. M. B. Jansen and R. Benedictus, Compos. Struct., 2015, 124, 19 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Comparison of the prepared self-healing epoxy polymers in this paper with other conventional epoxy polymer, TEM graph of GNS/FDB/OGE/D230 epoxy composite filled with 0.5 wt% GNS. See DOI: 10.1039/c9ra00261h |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Dongli Zoub,
Wanqiu Zhuc,
Xiaojiao Yanga,
Pengxiang Zhaod,
Changan Chen*a and
Maobing Shuai*b
a,
Dongli Zoub,
Wanqiu Zhuc,
Xiaojiao Yanga,
Pengxiang Zhaod,
Changan Chen*a and
Maobing Shuai*b















