
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBlue-emitting acridine-tagged silver(I)-bis-N-heterocyclic carbene†
Ganesan Prabusankar *a,
Nirmala Muthukumarana,
Moulali Vaddamanua,
Gembali Rajua,
Kavitha Velappanb,
Arruri Sathyanarayanac,
Yamane Masayac,
Shohei Sugiyamac,
Kyohei Hisanoc and
Osamu Tsutsumic
*a,
Nirmala Muthukumarana,
Moulali Vaddamanua,
Gembali Rajua,
Kavitha Velappanb,
Arruri Sathyanarayanac,
Yamane Masayac,
Shohei Sugiyamac,
Kyohei Hisanoc and
Osamu Tsutsumic
aDepartment of Chemistry, Indian Institute of Technology Hyderabad, India-502285. E-mail: prabu@iith.ac.in; Fax: +91 40 2301 6032; Tel: +91 40 2301 6089
bDepartment of Chemistry, GITAM University, Hyderabad, India-502329
cDepartment of Applied Chemistry, Ritsumeikan University, Kusatsu 525-8577, Japan
First published on 6th March 2019
Abstract
Herein, the photophysical properties of an acridine derivative of a bis-N-heterocyclic carbene silver complex were investigated. The HOMO and LUMO energy differences between 9-[(N-methyl imidazol-2-ylidene)]acridine and 4,5-bis[(N-methyl-imidazol-2-ylidene)methyl]acridine were theoretically compared. Based on the calculation, the 4,5-bis N-heterocyclic carbene-tethered acridine type of ligand was found to be a potential source for tuning the fluorescent nature of the resultant metal derivatives. Thus, a 4,5-bis N-heterocyclic carbene (NHC)-tethered acridine silver(I) salt was synthesized, and its photophysical properties were investigated. The 4,5-bis[(N-isopropylimidazol-2-ylidene)methyl]acridine silver(I) hexafluorophosphate complex was obtained from the reaction between [4,5-bis{(N-isopropylimidazolium)methyl}acridine] hexafluorophosphate and Ag2O in very good yield; this molecule was characterized by elemental analysis and FTIR, multinuclear (1H and 13C) NMR, UV-Vis, and fluorescence spectroscopic techniques. The molecular structure has been confirmed by single-crystal X-ray diffraction analysis, which has revealed that the complex is a homoleptic mononuclear silver(I) cationic solid. The charge of the Ag(I)–NHC cation is balanced by the hexafluorophosphate anion. The cationic moieties are closely packed in the chair and inverted chair forms where silver(I) possesses a quasi-linear geometry. Moreover, the silver complex provided blue emission from all the three excitations with good fluorescence quantum yield. The fluorescence lifetime of the silver(I) complex has been determined using the time-correlated single photon counting technique. Interestingly, the fluorescence decay pattern and the fluorescence lifetimes of the silver complex are largely different from those of the parent ligand acridine imidazolium salt. Moreover, the theoretical predictions have been found to be in good agreement with the experimental results.
Introduction
Silver metal complexes containing chromophore-tagged N-heterocyclic carbene (NHC) have attracted significant attention in medicinal chemistry and materials chemistry.1–11 Moreover, the silver–NHC complexes have been lately accredited as the building blocks of an effective antimicrobial agent to display phosphorescence emission.12–20 Over the past few decades, numerous luminescent silver(I)–NHC complexes have been described.21,22 Thus, it is clear that the N-substituents at the NHC counter ion and the coordination environment of the silver(I) center can significantly contribute to the photophysical properties of the silver(I)–NHC complexes.23 However, due to their light-sensitive nature, the structural and photophysical studies of the silver(I)–NHCs are challenging.17,21b,21gAmong several chromophoric sources, acridine has been recognized as one of the best chromophoric sources in organic luminous molecules and is known to have good biological properties.1 However, only a limited number of NHC metal complexes of acridine have been reported to date. The combination of NHCs with acridine in a Ag(I) complex is anticipated to result in interesting photoelectronic properties. Gimeno et al. have reported the first acridine chromophore-tagged NHC–silver complexes [(L)AgCl]2 (I and II) and [{(L)2Ag}BF4] (III and IV), where L = {N-methyl(imidazol-2-ylidene)}acridine or {N-benzyl(imidazol-2-ylidene)}acridine, with blue-green emission properties in 2012 (Chart 1).24 Subsequently, I was used as a precursor to isolate [(L)Au(S)], where S = mercaptopyridine or 2,3,4,6-tetra-O-acetyl-1-thio-β-D-glucopyranose with luminescence, cytotoxic and biodistribution properties.25 Later, {Ag[1-acridinyl-3-methylimidazoly-diene]2(PF6)} (V), {Ag[1-acridinyl-3-butylimidazoly-diene]2(PF6)CH3CN} (VI), {Ag[1-acridinyl-3-ethylimidazolydiene](PF6)}n (VII), and {Ag[1-acridinyl-3-benzylimidazoly-diene](PF6)}n (VIII) were isolated with fluorescence quenching effects (Chart 1).26 Interestingly, VI, VII, and VIII demonstrated anti-bacterial activity against Acinetobacter baumannii and P. aeruginosa; thus, the known NHC acridine derivatives were derived by derivatization of only the 9th position of acridine. In particular, acridine-tagged bis-NHC or the related chemistry was not explored until 2017.
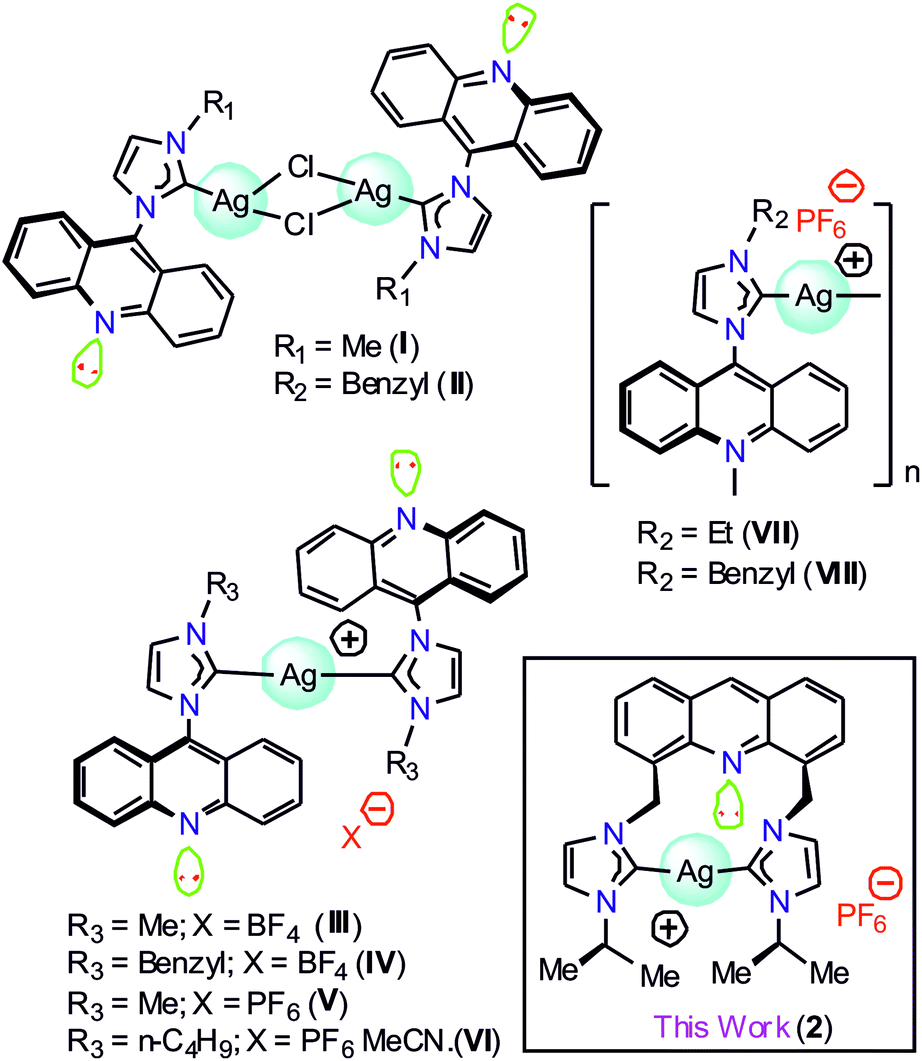 | ||
| Chart 1 The structurally characterized acridine-tagged NHC–silver derivatives. | ||
The first bis-NHC-acridine-supported metal derivative was reported in 2017.27 4,5-Bis[(N-isopropylimidazol-2-ylidene)methyl]acridine palladium dibromide was obtained from the reaction between the pincer type [4,5-bis{(N-isopropylimidazolium)methyl}acridine] dibromide and Pd(OAc)2 and utilized for the semi-hydrogenation of internal alkynes. 4,5-Bis[(N-isopropylimidazol-2-ylidene)methyl]acridine palladium dibromide was found to be highly active yet selective for the semi-hydrogenation of alkynes under mild conditions due to the presence of a non-coordinating acridine spacer. Notably, the photoluminescence role of the acridine chromophore attached with the bis-NHC metal derivative is not known. Thus, in this study, we report the structural and photophysical properties of 4,5-bis[(N-aryl-imidazol-2-ylidene)methyl]acridine silver(I) hexafluorophosphate (2). The photophysical properties of 2 were further investigated through theoretical analysis.
Results and discussion
Although the structural and photophysical properties of 9-[(N-alkyl or aryl imidazol-2-ylidene)]acridines are well known,24–26 the structural and photophysical properties of 4,5-bis[(N-alkyl or aryl-imidazol-2-ylidene)methyl]acridine are still unclear. Thus, DFT calculations were carried out at the B3LYP level to understand the HOMO and LUMO energy gap between 9-[(N-methyl imidazol-2-ylidene)]acridine (IX) and 4,5-bis[(N-methyl-imidazol-2-ylidene)methyl]acridine (X). A comparison of the HOMO and LUMO of IX and X was carried out (see ESI†). About 0.48 eV increase in the HOMO and LUMO energy gap was observed from IX to X. Moreover, the HOMO orbitals are largely located on the NHC in X, whereas the HOMO orbitals are typically located on acridine in IX. However, in IX and X, the LUMO is predominately located on the acridine moiety. Therefore, we speculate that the emission property of the silver–NHCs derived from 4,5-bis[(N-alkyl-imidazol-2-ylidene)methyl]acridine-type ligand should be much different from that of the silver–NHCs of 9-[(N-alkyl imidazol-2-ylidene)]acridines.The [4,5-bis{(N-isopropylimidazolium)methyl}acridine] hexafluoro-phosphate ligand (1) was synthesized in good yield as previously described.28 The reaction of 1 with Ag2O was carried out in the absence of light in an acetonitrile medium at 45 °C that led to the formation of a stable silver–NHC complex 2 (Scheme 1); 2 was obtained as an off-white, light-sensitive solid in excellent yield. Moreover, 2 was readily soluble in polar organic solvents, such as acetonitrile, acetone, DMSO, and DMF, sparingly soluble in dichloromethane and insoluble in non-polar organic solvents. In addition, 2 was fully characterized by elemental analysis and FTIR, 1H NMR and 13C NMR spectroscopy.
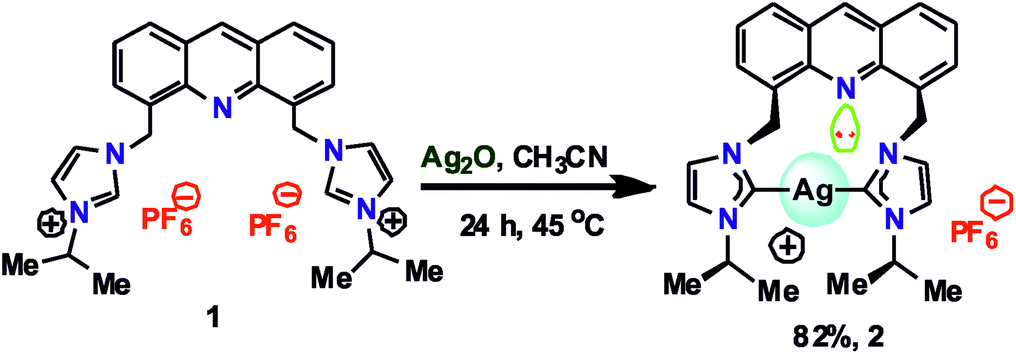 | ||
| Scheme 1 Synthesis of 2. | ||
As expected, the C2-proton (NCHN) resonance of the imidazolium salt was notably absent in the 1H NMR spectrum of 2.29 This observation confirmed the successful formation of the desired silver complex through the deprotonation of the azolium salt. In the 1H NMR spectrum, the aromatic protons appeared in the region of 8.19–7.64 ppm, and the singlet at 9.32 ppm belonged to the acridine proton. In the 13C NMR spectrum, the carbene carbon attached with silver(I) appeared at 179 ppm, which confirmed that the complex was intact in methanol.
The solid-state structure of 2 was further confirmed by the single-crystal X-ray diffraction technique (Fig. 1). The solid-state structure of 2 is reported in Fig. 1 along with the selected bond distances and angles. 2 is a mononuclear ionic solid. The cationic charge of Ag(I) is balanced by the hexafluorophosphate anion. The Ag(I) center is coordinated by two NHC moieties in a quasi-linear mode with the C(1)–Ag(1)–C(4) angle of 177.80(19)o. The C–Ag–C angle found in 2 falls within the range of the C–Ag–C angle found in III (173.62(16)o),24 V (180.00(1)o) and VI (180.00°).26 The Ag–C bond distances found in 2 are comparable to those of I (2.091(3) Å), III (2.096(4) Å)24 and known silver(I)-bis-carbene complexes.30
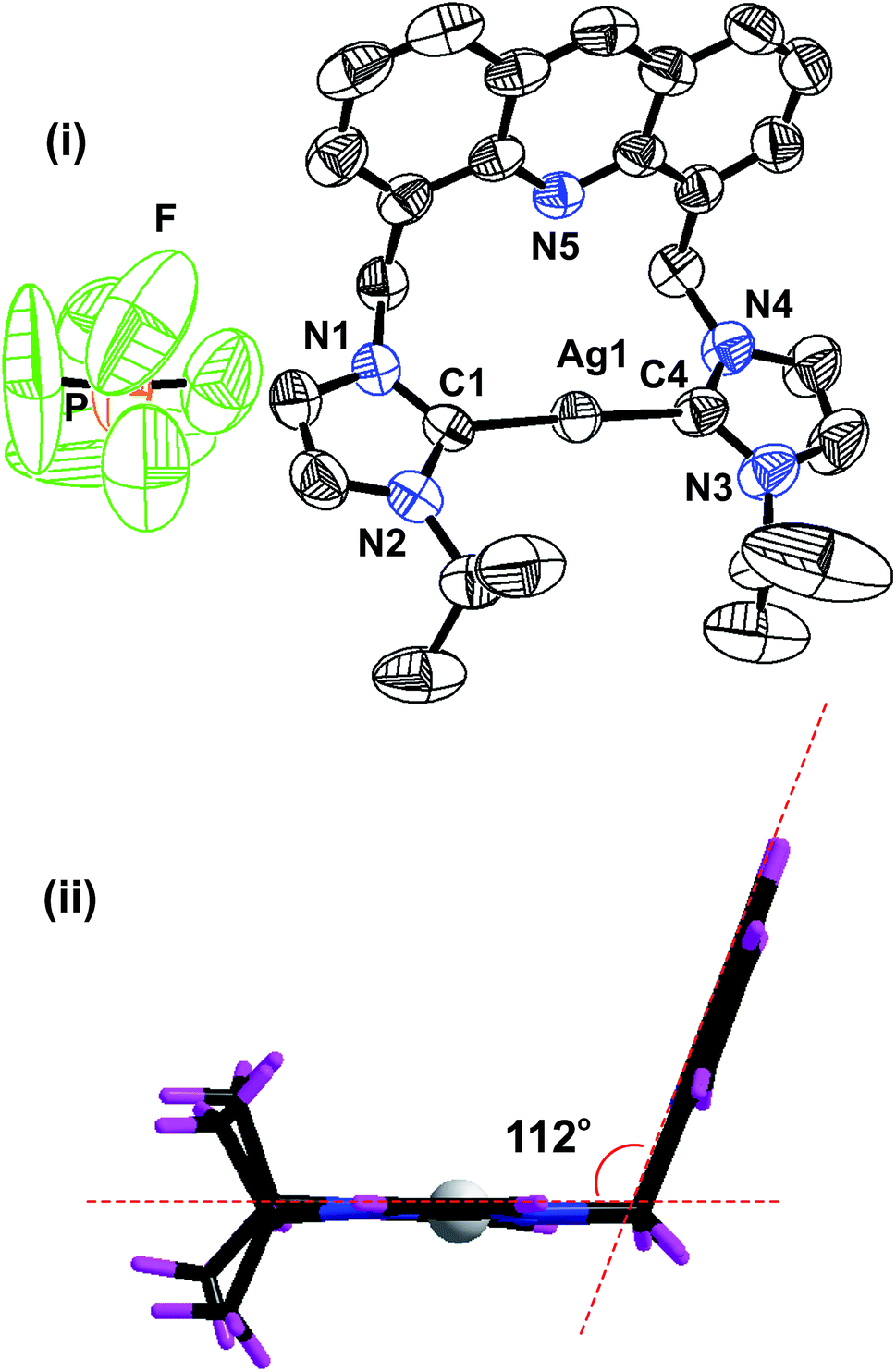 | ||
| Fig. 1 (i) The solid-state structure of 2; (ii) orientation of the imidazolium moieties and acridine unit in 2. The hexafluorophosphate anion has been omitted for clarity. Selected bond lengths (Å): Ag(1)–C(1), 2.069(5); Ag(1)–C(4), 2.074(5); N(1)–C(1), 1.341(6); C(1)–N(2), 1.350(6), and selected bond angles (°): C(1)–Ag(1)–C(4), 177.80(19); N(2)–C(1)–N(1), 105.0(4); N(3)–C(4)–N(4), 105.8(5). | ||
The distance between the acridine nitrogen and silver(I) is 3.251 Å. Thus, the position of acridine and the Ag–NHC plane are not suitable for the formation of a bond between the acridine nitrogen and the silver(I) center. However, as shown in the space filling model, the interaction between N(5) and Ag(I) cannot be ignored in solution (see ESI†). The acridine ring in 2 is nearly perpendicular to the NHC–Ag(I)–NHC plane. The two NHC ring systems and the silver(I) center are on the same plane. The molecular packing of 2 depicts that the cationic moieties are closely packed in the chair and inverted chair forms by stacking of the acridine moieties in a slightly sliding mode (see ESI†). The distance between two acridine moieties is 3.74 Å, and the parameters are not in the range for establishing a strong π–π interaction between two acridine moieties.
In addition, 2 shows inter and intra molecular P–F⋯H hydrogen bonding interactions between the phosphate anion and NHC–CH, ethyl CH and methyl CH. All the fluoride ions in the phosphate anions are involved in hydrogen bonding. Thus, the hydrogen-bonded packing of 2 results in a closely packed three-dimensional framework (see ESI†).
Photophysical properties of 1 and 2
The photophysical properties of 1 and 2 were investigated in methanol as a solvent using UV-Vis absorption spectrophotometry, fluorimetry, and time-correlated single photon counting techniques. The UV-Vis absorption spectra of 1 and 2 normalized at their longer wavelength absorption bands are shown in the Fig. 2. Both 1 and 2 exhibit a similar two-band absorption spectrum, with a strong band around 255 nm and a weak band around 356 nm. Based on the TD-DFT calculations,31–33 the 255 nm absorption can be assigned to the π → π* transition (vide infra). The longer wavelength absorption band for both 1 and 2 appeared at 356 nm similar to the absorption band of the acridine chromophore due to the π → π* transition. The molar extinction coefficients of 1 and 2 were calculated at 356 nm using the Beer–Lambert law and are shown in Fig. 3(i)–(iv).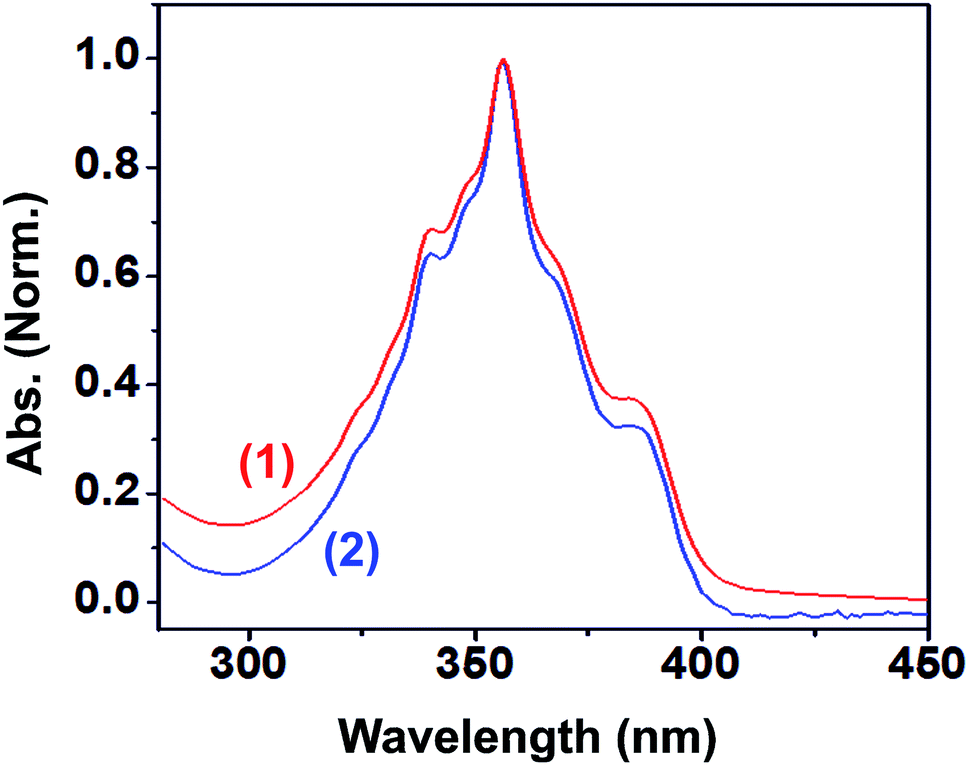 | ||
| Fig. 2 UV-Vis absorption spectra of 1 (blue) and 2 (red) normalized at their longer wavelength absorption bands, inset: longer wavelength absorption spectra of 1 (blue) and 2 (red). Solvent: methanol. | ||
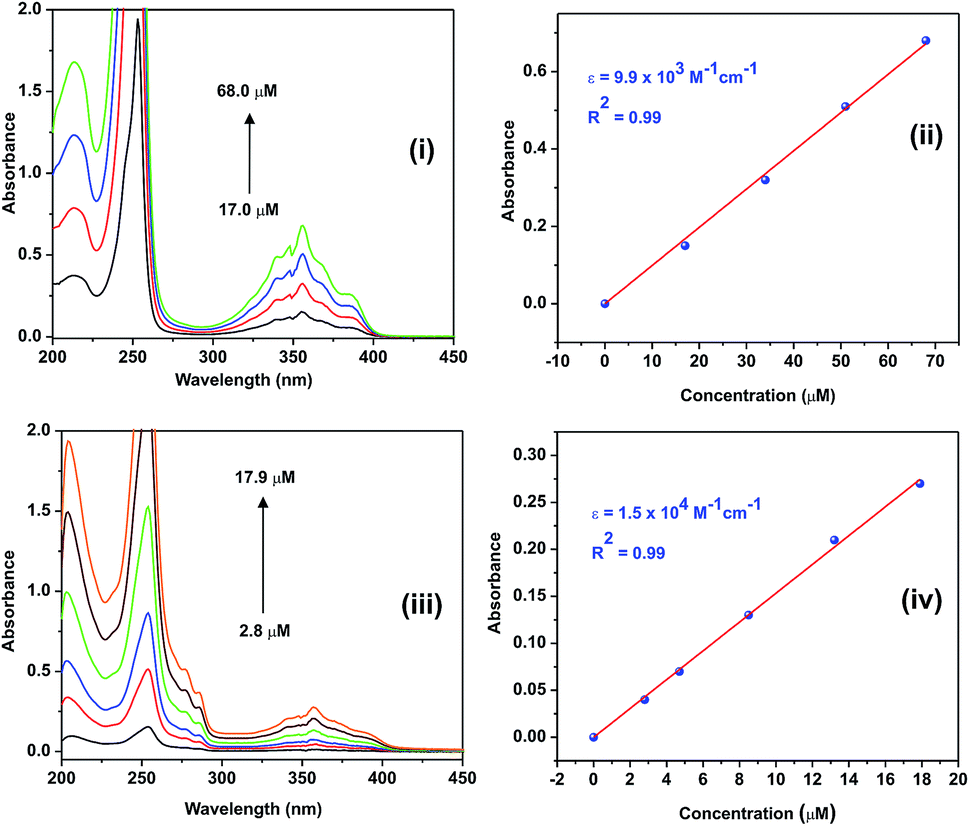 | ||
| Fig. 3 (i) The absorption spectra of 1 at various concentrations, (ii) the linear plot for the calculation of the molar extinction coefficient of 1, (iii) the absorption spectra of 2 at various concentrations, and (iv) the linear plot for the calculation of the molar extinction coefficient of 2. Solvent: methanol. | ||
When the absorbance of the analyte was plotted against its concentration, a straight line with an excellent correlation coefficient was obtained, as shown in Fig. 3(ii) and (iv). The molar extinction coefficients of 1 and 2 were graphically calculated from the slopes of Fig. 3(ii) and (iv) and found to be 9.9 × 103 and 1.5 × 104 M−1 cm−1, respectively. Note that the molar extinction coefficient of 2 is higher than that of 1 and comparable to the molar extinction coefficient of acridine.31,34 Moreover, the molar extinction coefficients have been mathematically calculated using the Beer–Lambert law (see ESI, Table S1†), and the values of 9.6 × 103 and 1.5 × 104 M−1 cm−1 are obtained for 1 and 2, respectively, which are in excellent agreement with the values obtained by the graphical method.
The methanolic solutions of 1 and 2 are colorless. However, they show blue luminescence under exposure to 365 nm UV radiation (see ESI†). Hence, the fluorescence spectra were obtained for 1 and 2 in methanol by exciting them at 356 nm. Both 1 and 2 display similar fluorescence spectra, as shown in the Fig. 4 (vide infra and see ESI†). A prominent peak is observed at 422 nm with a couple of shoulders appearing at 400 and 445 nm, which are in good agreement with the singlet-state emission spectrum of the neutral acridine molecule.35,36 Hence, the fluorescent state should be (π, π)* in nature. Notably, the solid-state luminescence spectra of 1 and 2 are not comparable (see ESI†). The solid-state emission bands are largely red shifted. This difference in the behavior of the luminescence properties can be due to the extended π–π interactions present in these molecules in the solid state that are not present in solution. The corresponding fluorescence quantum yields of 1 and 2 were calculated using a relative method according to the following equation:37
| ϕs = (Is/Ir) × (Ar/As) × (ns/nr)2 × ϕr |
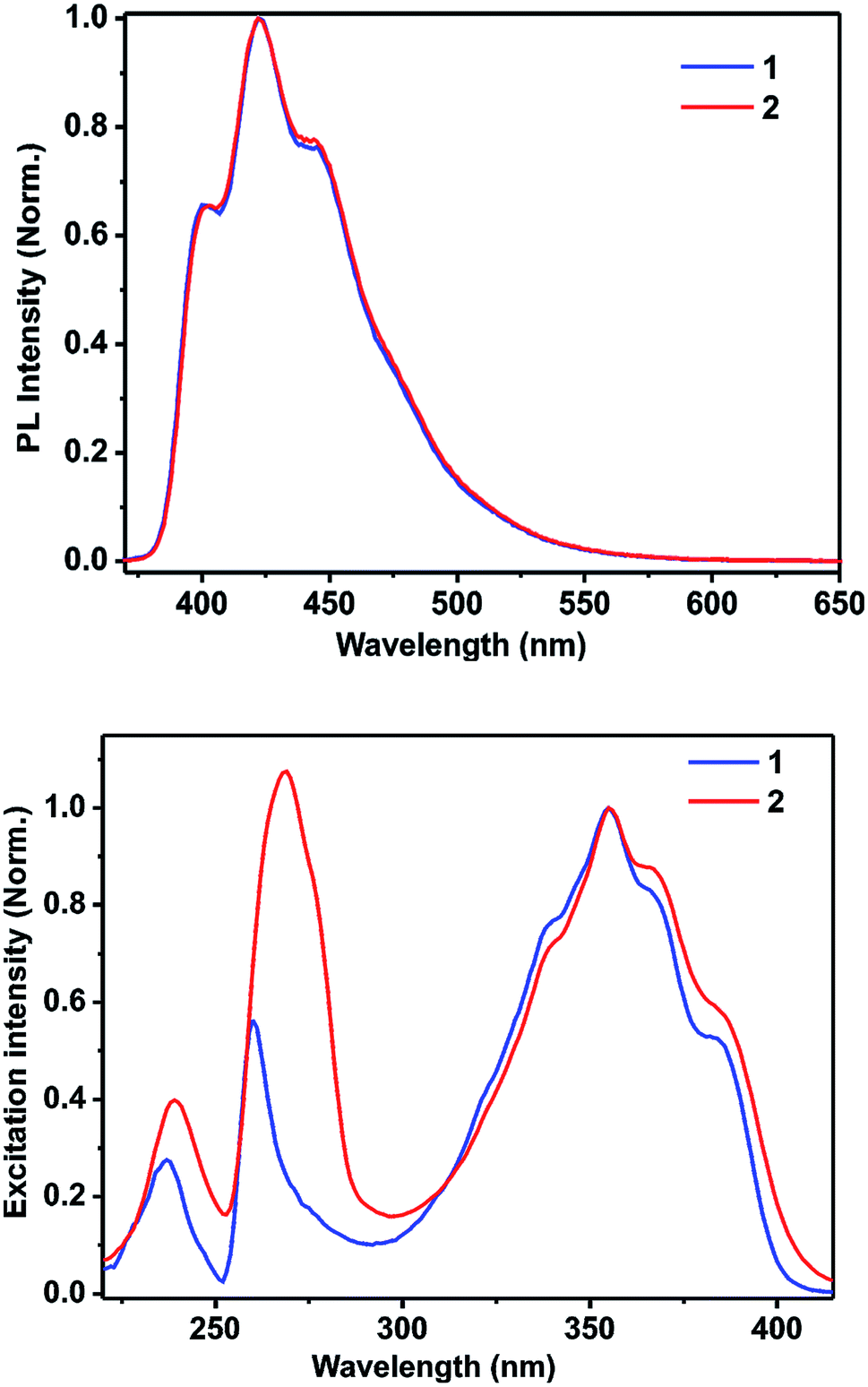 | ||
| Fig. 4 Top: the photoluminescence spectra of 1 and 2 in methanol (13.7 μM); bottom: the excitation spectra of 1 and 2 in methanol (13.7 μM). | ||
To explore the type of excitation responsible for the observed emission at 422 nm, excitation spectra were obtained for 1 and 2 by fixing the emission wavelength at 422 nm. The obtained excitation spectra were normalized at 356 nm and are shown in Fig. 4. The excitation spectra of 1 and 2 match well with their corresponding absorption spectra. It was found that all the three excitations contributed to the overall emission observed for 1 and 2.
Furthermore, to obtain a clear image about the contribution of various excitations to the overall emission, we obtained the 2D contour spectra for 1 and 2, which are shown in Fig. 5 (top and middle). Herein, note that both 1 and 2 excellently obey the mirror image rule, as shown in the ESI,† i.e. the S1 → S0 transition of the emission spectra is typically the mirror image of the corresponding S0 → S1 transition of their absorption spectra. Hence, it is revealed that the electronic excitation of 1 and 2 does not significantly change their nuclear geometry.39 The Commission Internationale de l'Eclairage (CIE) chromaticity diagram facilitates the quantitative evaluation of the photoluminescence color, as shown in Fig. 5 (bottom). Both 1 and 2 emit blue emission.
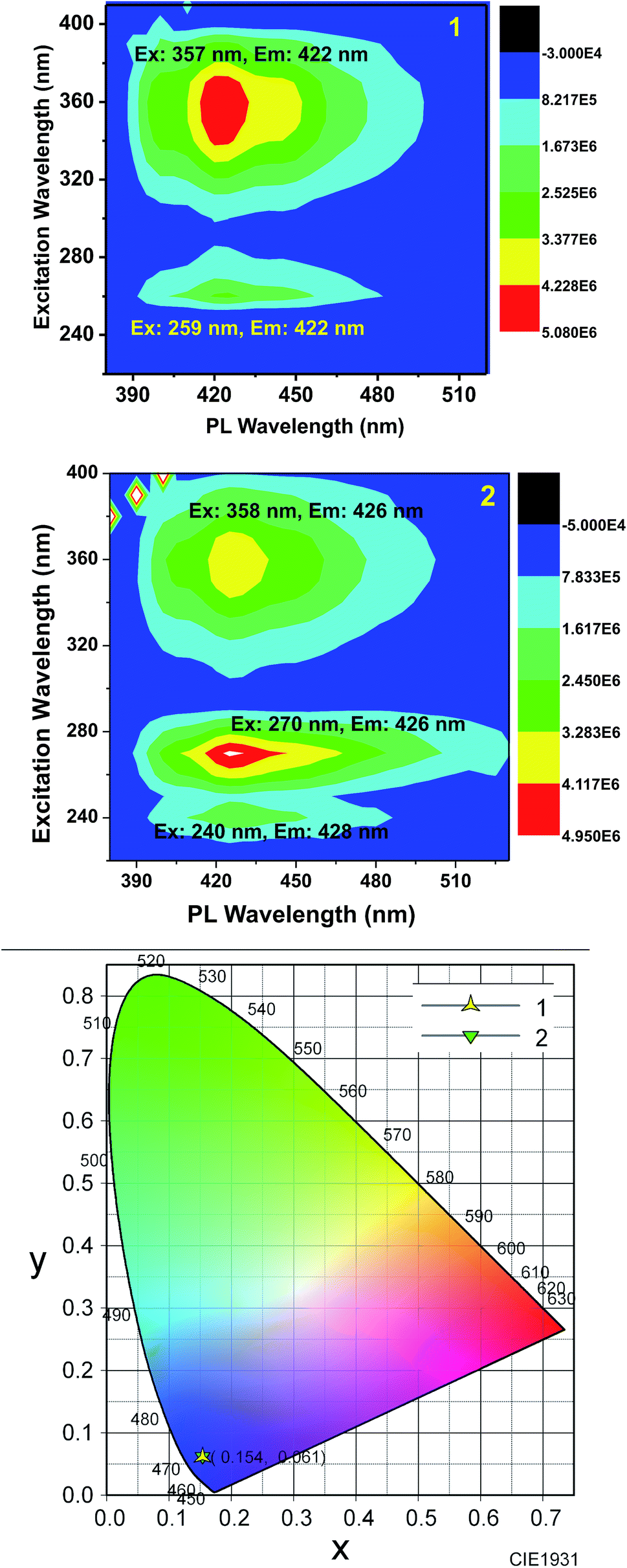 | ||
| Fig. 5 Top: the 2D contour spectrum of 1 in methanol (13.7 μM), Middle: the 2D contour spectrum of 2 in methanol (13.7 μM); and bottom: the CIE chromaticity diagram of 1 (star) and 2 (triangle) in the solution state. | ||
To explore the origin of the emission from 1 and 2, we measured their fluorescence lifetimes using the TCSPC technique. The ligand 1 is best-fitted to a bi-exponential function, whereas the complex 2 is fitted to a tri-exponential function (see ESI†).
The corresponding lifetimes of 1 are τ1 = 0.77 ± 0.02 ns (82.73%) and τ2 = 1.21 ± 0.04 ns (17.27%) with an average lifetime (τavg) of 0.85 ns. Similarly, the lifetimes of 2 are τ1 = 1.13 ± 0.01 ns (79.77%), τ2 = 0.32 ± 0.03 ns (12.40%), and τ3 = 6.85 ± 0.12 ns (7.83%), with a τavg of 1.46 ns (see ESI, Table S2†). Although the absorption spectra, emission spectra, and fluorescence quantum yields of 1 and 2 are similar to those of acridine, the fluorescence lifetimes of 1 and 2 are largely different from that of acridine.31,33,34 Moreover, the fluorescence decay pattern of 1 is quite different from that of 2. A new lifetime appears for 2, which supplements the complex formation between silver and 1. The parameters generated from the optical properties of 1 and 2 are summarized in Table 1.
| Optical data | 1 | 2 |
|---|---|---|
| λmax (abs) nm | 356 | 356 |
| εmax M−1 cm−1 | 9882 ± 140 | 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 333 ± 174 333 ± 174 |
| λmax (emi) nm | 422 | 422 |
| Stokes shift (cm−1) | 4393 | 4393 |
| ϕf (%) | 4 ± 0.02 | 2 ± 0.1 |
| τavg (ns) | 0.85 ± 0.09 | 1.46 ± 0.15 |
The DFT calculations were carried out at the B3LYP level of theory to gain further insights into the bonding situation in 2 (see ESI†). For silver, the Lanl2dz/def2svp/def2tzvp basis set was used, and for other elements, the 6-31G basis set was used. Herein, the important findings of our theoretical study have been described. The calculated geometrical parameters of 2 are in good agreement with the experimental data obtained from X-ray crystallography (see ESI, Table S3†).
The geometrical changes in 2 are determined by comparing the optimized geometries of [4,5-bis{(N-methyl imidazol-2-ylidene)methyl}acridine] (in its singlet ground state) (Me-1) and [4,5-bis{(N-isopropyl imidazol-2-ylidene)methyl}acridine] (in its singlet ground state) (iPr-1) (see ESI†). Based on the bond parameters for Me-1, iPr-1, and 2, significant changes in the geometry were observed (Table 2). Although there are no studies available on a similar complex, our results can be compared with an earlier study reported by Frenking et al. on simple NHC ligands.40 In the complex 2, the geometrical changes undergone by the NHC ring when it is bound to Ag+ are significant. For instance, the NHC rings are in the same plane in 2, whereas in iPr-1, the NHC rings are twisted, as evident from the structure and the torsion angles. However, the changes involving the bond lengths and bond angles are very minimal. The N–Ccarbene–N angle in 2 is widened by about 3.0°, whereas the C–N bond length remains unchanged.
| Parameter | Me-1 | iPr-1 | 2 (Cal.) | 2 (Exp.) |
|---|---|---|---|---|
| Ccarbene–N | 1.3714 | 1.3698 | 1.3709 | 1.3354 |
| Ccarbene–N | 1.3729 | 1.3708 | 1.3720 | 1.3417 |
| Ccarbene–N | 1.3729 | 1.3708 | 1.3720 | 1.3421 |
| Ccarbene–N | 1.3714 | 1.3697 | 1.3732 | 1.3503 |
| N–Ccarbene–N | 101.53 | 101.73 | 104.43 | 105.02 |
| N–Ccarbene–N | 101.53 | 101.73 | 104.23 | 105.80 |
| Ccarbene–Nim–C–CAc | −94.69 | −95.87 | −89.41 | −91.84 (5) |
| Ccarbene–Nim–C–CAc | −94.71 | −96.02 | 90.66 | 95.18 (6) |
Moreover, the TDDFT, TDDFT-CPCM and TDDFT-SMD calculations were carried out to corroborate our experimental results (see ESI†). The computed HOMO–LUMO energy gap is in good agreement with the absorption spectrum of 2. A comparison of the HOMO–LUMO energy gap of 2 has been provided in the ESI.† The HOMO–LUMO energy gap for the free molecule is 0.13049 a.u., whereas in a solvent such as methanol, the energy difference is 0.13272 a.u. Note that in methanol, the energy of HOMO and LUMO has slightly increased. However, the energy difference between the HOMO and the LUMO is nearly the same (see ESI†). The calculations have predicted two absorptions for 2: a weak absorption at 391 nm (oscillator strength, f = 0.0773) due to the transition between the HOMO(π) and the LUMO(π*) and a strong absorption at 241 nm (oscillator strength, f = 0.9212) that can be attributed to the transition between the HOMO−3(π) and the LUMO(π*), the HOMO(π) and the LUMO+2(σ*), the HOMO−2(n/π) and the LUMO+2(σ*), or between the HOMO−1(n/π) and the LUMO+2(σ*) orbitals (see ESI†). The acridine moiety in 2 contributes to the absorbance at 391 nm, whereas the acridine and imidazole moieties are mainly involved in the 241 nm absorption. Moreover, a significant contribution (46.8%) of silver (HOMO) to acridine (LUMO) is observed at 241 nm, which may have merged with the predominant 241 nm transition (see ESI†). Therefore, it is clear that the contribution of various excitations to the overall emission must be considered as the excitation from the HOMO of acridine, silver and NHC to only the LUMO of the acridine moiety.
Experimental
Materials and methods
All starting materials, acridine, bromomethylmethylether, 1-isopropylimidazole, silver oxide (Ag2O) and potassium hexafluorophosphate (KPF6), were purchased from commercial suppliers and used as received. Solvents were of analytical grade and used without further purification except for acetonitrile, which was distilled over CaH2 and degassed by N2. All reactions involving the synthesis of the metal complex were carried out in oven- or flame-dried glassware with a magnetic stirrer under aerobic conditions. The silver reaction was conducted in the absence of light. [4,5-Bis{(N-isopropylimidazolium)methyl}acridine] hexafluorophosphate (1) was synthesized as reported.28 The FTIR measurement (neat) was carried out using the Bruker Alpha-P Fourier transform spectrometer. The microanalyses of carbon, hydrogen and nitrogen were carried out using the Euro EA – CHNSO Elemental Analyzer. The NMR spectra were obtained using the Bruker Ultrashield-400 spectrometer at 25 °C unless otherwise stated. Chemical shifts are provided relative to TMS and have been referenced to the solvent resonances as internal standards.The crystal structure of 2 was analysed using the Oxford Xcalibur 2 diffractometer. Single crystals of the complexes suitable for the single-crystal X-ray analysis were obtained from the reaction mixtures of the complexes at room temperature. A suitable crystal of 2 was selected and mounted on the SuperNova (Dual, Cu at zero, Eos) diffractometer. The crystal was kept at 298 K during data collection. Using Olex2,41 the structure was solved with the olex2.solve42 structure solution program using Charge Flipping and refined with the olex2.refine43 refinement package using the Gauss–Newton minimisation. The absorption corrections were performed on the basis of multi-scans. The non-hydrogen atoms were anisotropically refined. The hydrogen atoms were included in the refinement at the calculated positions riding on their carrier atoms. 2 provided a “B” level alert as low ‘MainMol’ Ueq as compared to the neighbors of C25. This may be considered as a false alarm for the terminal isopropyl methyl group.
The UV-Vis absorption spectra were obtained using a diode array single beam spectrophotometer, Agilent 8453. All the absorption spectra were baseline corrected prior to display. PL, excitation, and 3D contour spectra were obtained by the HORIBA JOBIN YVON Fluoromax 4P fluorimeter. The fluorescence decay measurements were carried out using the time-correlated single photon counting (TCSPC) technique with a 375 nm light-emitting diode (LED) as the excitation source. The TCSPC data analysis has been carried out by the software provided by IBH (DAS-6), which is based on the reconvolution technique using the iterative non-linear least-squares methods. The quality of the fit is normally identified by the reduced χ2, weighed residuals, and the autocorrelation function of the residuals. The relative fluorescence quantum yield (QY) was calculated by comparing the integrated PL intensities (excited at 355 nm) and the absorbance values of the samples at 355 nm with those of the reference quinine sulfate (purchased from Fluka). The known quantum yield of quinine sulfate is 0.543.
All the calculations were conducted using the Gaussian 16 suite of the program.44 The input files were generated using GaussView, Version 6.45 The initial geometry of the acridine-NHC–silver complex was obtained from the X-ray crystal structure data. The complex was optimized at the HF and B3LYP level of the theories using different basis sets having effective core potentials (ECP). For silver, the Lanl2DZ, def2svp and def2tzvp basis sets were used, and for other elements, the 6-31G basis set was used. The geometry obtained at the B3LYP level of theory is found to be in good agreement with the experimental data. We also carried out the time-dependant density functional theory (TDDFT) calculations to obtain further insight into the photophysical properties of the complex. TDDFT was performed for the free molecule and also in different solvents. The polarizable continuum model (PCM) and SMD (solvation model based on density – a universal solvation model) were used for the solvation studies. The vibrational frequency calculations were carried out to ensure that the structure was minima on the potential energy surface.
Synthesis of 2
To a clear colorless solution of imidazolium salt 1 (0.36 g, 0.5 mmol) in acetonitrile (20 mL), silver(I) oxide (0.23 g, 1 mmol) was added followed by stirring at 45 °C for 24 h in the absence of light. The reaction mixture was then filtered through a bed of celite to remove the unreacted silver oxide and other by-products. The filtrate was concentrated to 50% of its volume. Subsequently, diethyl ether (50 mL) was added, and this resulted in the precipitation of an off-white amorphous solid. This solid was isolated by filtration, washed with fresh diethyl ether (3 × 3 mL) and air dried. The obtained complex was purified by fractional precipitation using an acetonitrile and diethyl ether mixture to obtain an off-white solid, 2. The suitable single crystals of 2 were obtained by slow diffusion of diethyl ether into a saturated acetonitrile solution of the complex at room temperature. Yield: 82% (based on 1). Anal. calcd for C27H29AgF6N5P (676.39): C, 47.43; H, 4.32; N, 10.35%. Found: C, 47.4; H, 4.4; N, 10.4%. FT-IR (cm−1, neat): 2023(w), 1703(m), 1614(m), 1552(w), 1396(w), 1327(w), 1271(w), 1141(s), 851(s), 746(m), 645(w), 560(w). 1H NMR (400 MHz, CDCl3/DMSO-d6) δ: 9.32 (s, 1H, Ar–H), 8.51–8.49 (d, J = 6.9 Hz, 1H, imi-H), 8.45–8.43 (d, J = 8.4 Hz, 1H, imi-H), 8.19 (s, 1H, Ar–H), 8.08 (s, 1H, Ar–H), 7.99–7.95 (t, 1H, Ar–H), 7.64 (s, 1H, Ar–H), 6.62 (s, 2H, CH2), 4.90–4.83 (m, 1H, CH), 1.82–1.81 (d, J = 6.9 Hz, 6H, CH3). 13C NMR (100 MHz, CDCl3/DMSO-d6) δ: 144.56 (Ar–C), 137.15 (Ar–C), 133.38 (Ar–C), 131.10 (Ar–C), 128.35 (Ar–C), 125.71 (imi-C), 124.67 (imi-C), 120.90 (Ar–C), 117.33 (Ar–C), 115.94 (Ar–C), 52.75 (–CH2), 47.38 (–CH), 22.58 (–CH3), C2-imidazole signal is absent. 1H NMR (400 MHz, CD3OD) δ: 9.01 (s, 1H, Ar–H), 8.12–8.10 (m, 4H, imi-H), 7.86 (s, 2H, Ar–H), 7.64–7.62 (d, J = 7.2 Hz, 2H, Ar–H), 7.62–7.60 (d, J = 7.2 Hz, 2H, Ar–H), 7.40–7.39 (d, J = 1.6 Hz, 2H, Ar–H), 6.29 (s, 4H, CH2), 4.63–4.52 (septet, 2H, CH), 1.47–1.46 (d, J = 6.8 Hz, 12H, CH3). 13C NMR (400 MHz, CD3OD) δ: 179.24 (Ag–C), 145.74 (Ar–C), 138.08 (Ar–C), 134.94 (Ar–C), 131.44 (Ar–C), 129.50 (Ar–C), 127.01 (imi-C), 125.42 (imi-C), 121.58 (Ar–C), 118.42 (Ar–C), 117.13 (Ar–C), 53.98 (–CH2), 47.09 (–CH), 22.86 (–CH3).Conclusion
In summary, an acridine moiety was incorporated into the Ag(I)–NHCs to obtain 2. We have theoretically compared the nature of the HOMO and LUMO energy differences between 9-[(N-methyl imidazol-2-ylidene)]acridine (IX) and 4,5-bis[(N-methyl-imidazol-2-ylidene)methyl]acridine (X). Bis-carbene X depicted notable deviations in the HOMO and LUMO energy gaps as compared to the mono-carbene IX. Thus, bis-imidazolium-tethered acridine ligand (1) was synthesized. Subsequently, we have synthesized a 4,5-bis NHC-tethered acridine silver(I) salt and reported its photophysical properties. The solid-state packing and space filling model of 2 show that the cationic moieties are closely packed without any π⋯π interaction between acridine ring systems. However, the intramolecular interaction between the acridine nitrogen and silver is close to the weak interaction zone. Moreover, the P–F⋯H type of inter- and intra-molecular hydrogen bonding interaction in 2 resulted in a closely packed three-dimensional framework. The homoleptic mononuclear silver(I) cationic solid 2 depicted blue emission from all the three excitations with good fluorescence quantum yield. The fluorescence lifetimes obtained using the time-correlated single photon counting technique have been demonstrated. Interestingly, the fluorescence decay pattern and the fluorescence lifetimes of the silver complex are largely different from those of the corresponding acridine imidazolium salt. Moreover, the spectral and structural features have been compared with the theoretical studies. The DFT calculations were carried out to understand the orbital contribution and bonding situation in 2. The calculated geometrical parameters of 2 are in good agreement with the experimental data obtained from X-ray crystallography. The calculated HOMO–LUMO energy gap for 2 is in good agreement with the absorption spectrum of 2. Exploration of the photophysical properties of other 4,5-bis NHC-tethered acridine metal derivatives is presently underway.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the JICA CKP ARC 2018-01 for providing the financial support. NM thanks DST-NPDF (PDF/2016/001834) for the DST-NPDF fellowship. VK thanks DST-WOS-A (WOS-A/CS-65/2016) for the fellowship. VM thanks UGC for the fellowship.Notes and references
- (a) R. Visbal and M. C. Gimeno, Chem. Soc. Rev., 2014, 43, 3551–3574 RSC; (b) T. V. Basova, A. Hassan and N. B. Morozova, Coord. Chem. Rev., 2019, 380, 58–82 CrossRef CAS; (c) S. Hameury, P. d. Frémont and P. Braunstein, Chem. Soc. Rev., 2017, 46, 632–733 RSC.
- N. Boysen, T. Hasselmann, S. Karle, D. Rogalla, D. Theirich, M. Winter, T. Riedl and A. Devi, Angew. Chem., Int. Ed., 2018, 57, 16224–16227 CrossRef CAS PubMed.
- L. Eloy, A.-S. Jarrousse, M.-L. Teyssot, A. Gautier, L. Morel, C. Jolivalt, T. Cresteil and S. Roland, ChemMedChem, 2012, 7, 805–814 CrossRef CAS PubMed.
- N. Sinha and F. E. Hahn, Acc. Chem. Res., 2017, 50, 2167–2184 CrossRef CAS PubMed.
- W. Liu and R. Gust, Chem. Soc. Rev., 2013, 42, 755–773 RSC.
- A. Citta, E. Schuh, F. Mohr, A. Folda, M. L. Massimino, A. Bindoli, A. Casini and M. P. Rigobello, Metallomics, 2013, 5, 1006–1015 RSC.
- B. Thati, A. Noble, B. S. Creaven, M. Walsh, M. McCann, K. Kavanagh, M. Devereux and D. A. Egan, Cancer Lett., 2007, 248, 321–331 CrossRef CAS PubMed.
- W. Liu and R. Gust, Coord. Chem. Rev., 2016, 329, 191–213 CrossRef CAS.
- S. Ray, R. Mohan, J. K. Singh, M. K. Samantaray, M. M. Shaikh, D. Panda and P. Ghosh, J. Am. Chem. Soc., 2007, 129, 15042–15053 CrossRef CAS PubMed.
- D. A. Medvetz, K. M. Hindi, M. J. Panzner, A. J. Ditto, Y. H. Yun and W. J. Youngs, Met.-Based Drugs, 2008, 2008, 384010 Search PubMed.
- L. Eloy, A. S. Jarrousse, M. L. Teyssot, A. Gautier, L. Morel, C. Jolivalt, T. Cresteil and S. Roland, ChemMedChem, 2012, 7, 805–814 CrossRef CAS PubMed.
- N. A. Johnson, M. R. Southerland and W. J. Youngs, Molecules, 2017, 22, 1263 CrossRef PubMed and references therein.
- D. Iacopetta, A. Mariconda, C. Saturnino, A. Caruso, G. Palma, J. Ceramella, N. Muià, M. Perri, M. S. Sinicropi, M. C. Caroleo and P. Longo, ChemMedChem, 2017, 12, 2054–2065 CrossRef CAS PubMed.
- J. C. Garrison and W. J. Youngs, Chem. Rev., 2005, 105, 3978–4008 CrossRef CAS PubMed.
- A. Kascatan-Nebioglu, M. J. Panzner, C. A. Tessier, C. L. Cannon and W. J. Youngs, Coord. Chem. Rev., 2007, 251, 884–895 CrossRef CAS.
- M. S. Sinicropi, D. Iacopetta, C. Rosano, R. Randino, A. Caruso, C. Saturnino, N. Muià, J. Ceramella, F. Puoci, M. Rodriquez, P. Longo and M. R. Plutino, J. Enzyme Inhib. Med. Chem., 2018, 33, 434–444 CrossRef CAS PubMed.
- S. Kankala, N. Thota, F. Bjoerkling, M. K. Taylor, R. Vadde and R. Balusu, Silver carbene complexes: an emerging class of anticancer agents, Drug Dev. Res., 2018, 1–12 Search PubMed.
- J. C. Y. Lin, R. T. W. Huang, C. S. Lee, A. Bhattacharyya, W. S. Hwang and I. J. B. Lin, Chem. Rev., 2009, 109, 3561–3598 CrossRef CAS PubMed.
- G. Achar, C. R. Shahini, S. A. Patil and S. Budagumpi, J. Organomet. Chem., 2017, 833, 28–42 CrossRef CAS.
- P. L. Arnold, Heteroat. Chem., 2002, 13, 534–539 CrossRef CAS.
- (a) T. Lu, J.-Y. Wang, L.-X. Shi, Z.-N. Chen, X.-T. Chen and Z.-L. Xue, Dalton Trans., 2018, 47, 6742–6753 RSC; (b) V. J. Catalano, L. B. Munro, C. E. Strasser and A. F. Samin, Inorg. Chem., 2011, 50, 8465–8476 CrossRef CAS PubMed; (c) M. Kriechbaum, M. List, R. J. F. Berger, M. Patzschke and U. Monkowius, Chem.–Eur. J., 2012, 18, 5506–5509 CrossRef CAS PubMed; (d) V. J. Catalano and M. A. Malwitz, Inorg. Chem., 2003, 42, 5483–5485 CrossRef CAS PubMed; (e) V. J. Catalano, M. A. Malwitz and A. O. Etogo, Inorg. Chem., 2004, 43, 5714–5724 CrossRef CAS PubMed; (f) V. J. Catalano and A. L. Moore, Inorg. Chem., 2005, 44, 6558–6566 CrossRef CAS PubMed; (g) V. J. Catalano and A. O. Etogo, Inorg. Chem., 2007, 46, 5608–5615 CrossRef CAS PubMed; (h) D. Rios, M. M. Olmstead and A. L. Balch, Inorg. Chem., 2009, 48, 5279–5287 CrossRef CAS; (i) S. D. Adhikary, L. Jhulki, S. Seth, A. Kundu, V. Bertolasi, P. Mitra, A. Mahapatra and J. Dinda, Inorg. Chim. Acta, 2012, 384, 239–246 CrossRef.
- (a) C. Xia, K. Yang, W. Sun, X. Lu and J. Xie, J. Coord. Chem., 2017, 70, 615–625 CrossRef CAS; (b) S. D. Adhikary, S. K. Seth, M. R. Senapati and J. Dinda, J. Mol. Struct., 2013, 1042, 123–128 CrossRef; (c) Y. Zhou, X. Zhang, W. Chen and H. Qiu, J. Organomet. Chem., 2008, 693, 205–215 CrossRef CAS; (d) J. Dinda, S. D. Adhikary, S. K. Seth and A. Mahapatra, New J. Chem., 2013, 37, 431–438 RSC; (e) B. Liu, W. Chen and S. Jin, Organometallics, 2007, 26, 3660–3667 CrossRef CAS; (f) Q.-X. Liu, F.-B. Xu, Q.-S. Li, X.-S. Zeng, X.-B. Leng, Y. L. Chou and Z.-Z. Zhang, Organometallics, 2003, 22, 309–314 CrossRef CAS; (g) Y. Zhou and W. Chen, Organometallics, 2007, 26, 2742–2746 CrossRef CAS; (h) M. Kriechbaum, J. Hölbling, H.-G. Stammler, M. List, R. J. F. Berger and U. Monkowius, Organometallics, 2013, 32, 2876–2884 CrossRef CAS.
- P. de Fremont, N. M. Scott, E. D. Stevens, T. Ramnial, O. C. Lightbody, C. L. B. Macdonald, J. A. C. Clyburne, C. D. Abernethy and S. P. Nolan, Organometallics, 2005, 24, 6301–6309 CrossRef CAS.
- M. C. Gimeno, A. Laguna and R. Visbal, Organometallics, 2012, 31, 7146–7157 CrossRef CAS.
- R. Visbal, V. Fernández-Moreira, I. Marzo, A. Laguna and M. C. Gimeno, Dalton Trans., 2016, 45, 15026–15033 RSC.
- Z. He, K. Huang, F. Xiong, S.-F. Zhang, J.-R. Xue, Y. Liang, L.-H. Jing and D.-B. Qin, J. Organomet. Chem., 2015, 797, 67–75 CrossRef CAS.
- G. Prabusankar, A. Sathyanarayana, G. Raju and C. Nagababu, Asian J. Org. Chem., 2017, 6, 1451–1459 CrossRef CAS.
- G. Raju, S. Vishwanath, A. Prasad, B. K. Patel and G. Prabusankar, J. Mol. Struct., 2016, 1107, 291–299 CrossRef CAS.
- G. Achar, P. Agarwal, K. N. Brinda, J. G. Małecki, R. S. Keri and S. Budagumpi, J. Organomet. Chem., 2018, 854, 64–75 CrossRef CAS.
- M. G. Gardiner and C. C. Ho, Coord. Chem. Rev., 2018, 375, 373–388 CrossRef CAS and references therein.
- A. Kellmann, J. Phys. Chem., 1977, 81, 1195–1198 CrossRef CAS.
- A. Elangovan, H. Chiu, S. Yang and T. Ho, Org. Biomol. Chem., 2004, 2, 3113–3118 RSC.
- A. J. Christy and M. Umadevi, Int. J. ChemTech Res., 2015, 8, 383–390 CAS.
- S. J. Ladner and R. S. Becker, J. Phys. Chem., 1963, 67, 2481–2486 CrossRef CAS.
- A. Gafni and L. Brand, Chem. Phys. Lett., 1978, 58, 346–350 CrossRef CAS.
- M. Goez, Cyclic and linear photoionizations of acridine derivatives and xanthone investigated by nanosecond laser flash photolysis, PhD thesis, Martin-Luther-University Halle-Wittenberg, 2005.
- K. Koichi, K. Kunihiko, K. Akihiko, U. Koji and K. Hiroshi, J. Phys. Chem., 1985, 89, 868–871 CrossRef.
- L. G. Samsonova, N. I. Selivanov, T. N. Kopylova, V. Ya. Artyukhov, G. V. Maier, V. G. Plotnikov, V. A. Sazhnikov, A. A. Khlebunov and M. V. Alfimov, High Energy Chem., 2009, 43, 105–115 CrossRef CAS.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, Singapore, 3rd edn, 2006 Search PubMed.
- C. Boehme and G. Frenking, Organometallics, 1998, 17, 5801–5809 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- L. J. Bourhis, O. V. Dolomanov, R. J. Gildea, J. A. K. Howard and H. Puschmann, Acta Crystallogr., 2015, A71, 59–75 CrossRef PubMed.
- L. J. Bourhis, O. V. Dolomanov, R. J. Gildea, J. A. K. Howard and H. Puschmann, Acta Crystallogr., 2015, A71, 59–75 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- R. Dennington, T. A. Keith and J. M. Millam, GaussView, Version 6, Semichem Inc., Shawnee Mission, KS, 2016 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: FT-IR, NMR, X-ray data, figures, and tables. 1H and 13C NMR spectra of the synthesized complex, solid state packing, photophysical data, computational data. CCDC 1879132. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra00281b |
| This journal is © The Royal Society of Chemistry 2019 |