DOI:
10.1039/C9RA00306A
(Paper)
RSC Adv., 2019,
9, 10897-10904
TUG1 knockdown enhances adriamycin cytotoxicity by inhibiting glycolysis in adriamycin-resistant acute myeloid leukemia HL60/ADR cells
Received
13th January 2019
, Accepted 29th March 2019
First published on 8th April 2019
Abstract
Taurine-upregulated gene 1 (TUG1) has been reported as an oncogenic long non-coding RNA (lncRNA) in acute myeloid leukemia (AML). Nevertheless, the roles and molecular mechanism of TUG1 in drug resistance of AML cells are still unclear. Glycolysis level was evaluated by detecting glucose consumption and lactate production. qRT-PCR and Western blot were performed to detect TUG1, hexokinase2 (HK2) and pyruvate kinase isoenzyme M2 (PKM2) expressions. Adriamycin (ADR) cytotoxicity and apoptosis were assessed by MTT assay and flow cytometry, respectively. The changes of the protein kinase B (Akt) pathway were determined by Western blot analysis of phosphorylated-Akt (p-Akt) (ser473) and Akt. Our results showed that glycolysis was increased in HL60/ADR cells, as evidenced by the elevated glucose consumption and lactate production, as well as the increased HK2 and PKM2 expressions at mRNA and protein levels. TUG1 was up-regulated in HL60/ADR cells and TUG knockdown inhibited glycolysis. TUG1 knockdown enhanced ADR-induced cytotoxicity and apoptosis in HL60/ADR cells. TUG1 knockdown inhibited the Akt pathway and activation of the Akt pathway by 740Y-P attenuated the effects of TUG1 knockdown on ADR-induced cytotoxicity and apoptosis, as well as glycolysis in HL60/ADR cells. Taken together, TUG1 knockdown enhances adriamycin cytotoxicity in HL60/ADR cells via inhibiting the glycolysis by inactivating the Akt pathway.
1. Introduction
Acute myeloid leukemia (AML) is a clinically and biologically heterogeneous hematopoietic malignant disorder characterized by the uncontrolled proliferation and accumulation of immature myeloid cells in the bone marrow and peripheral blood.1 AML is the most common form of leukemia in adults, comprising up to 12% of all leukemia in adults, with the lowest survival rate in all leukemia cases.2 Nowadays, anthracycline-based chemotherapy is the mainstay of the treatment approach for AML and adriamycin (ADR) is a commonly used anthracycline drug in the treatment of AML.3 Despite therapeutic advancement in treating AML during the last decades, the majority of patients diagnosed with AML experience relapse and eventually succumb to this disease after standard induction chemotherapy, and resistance to chemotherapy still represents a principle cause of relapse and treatment failure in AML.4 Thus, deciphering the molecular mechanism underlying the drug resistance of AML cells is necessary for exploring novel effective candidates for AML chemotherapy.
It is well known that cancer cells frequently exhibit abnormally high rates of aerobic glycolysis known as the Warburg effect, characterized by the increased glucose uptake and lactate production in an oxygen-independent manner.5 The up-regulated aerobic glycolysis is a well-recognized hallmark in human tumors, especially in drug resistant cancer cells.6 Many related enzymes of aerobic glycolysis such as hexokinase2 (HK2), and pyruvate kinase isoenzyme M2 (PKM2) have been reported to be linked to the development of drug resistance in tumor cells.7
Long non-coding RNAs (lncRNAs) are a class of evolutionarily conserved non-coding RNA molecules comprising more than 200 nucleotides in length which not translate protein.8 It is well documented that lncRNAs are implicated in various biological processes by regulating gene expression at epigenetic, transcriptional, posttranscriptional and translational levels.9 LncRNAs are frequently aberrantly expressed in almost all human tumors and play important roles in the initiation and progression of cancers.10 To date, increasing studies have reported the close association between the dysregulated lncRNAs and drug resistance in a wide range of tumors.11 LncRNA taurine-upregulated gene 1 (TUG1), a 7.1 kb lncRNA located at chromosome 22q12, was originally identified as a transcript which is up-regulated in response to taurine in the retinal cells of newborn mice.12 Recent studies have reported that TUG1 generally serves an oncogenic role in the development of several tumors including AML.13 However, no studies are available for the roles and molecular mechanism of TUG1 in the drug resistance of AML cells.
Phosphoinositide 3-kinase (PI3K) is constituted by two subunits, the p85 subunit (PI3K p85) and the p110 subunit (PI3K p110). Protein kinase B (Akt), generally regarded as one main PI3K effector, has been reported to play crucial roles in cell viability, apoptosis, and glycolysis.14–16 As a well known PI3K agonist, 740Y-P peptide can bind specifically to PI3K p85 subunit and activate p85, thus phosphorylate Akt on Ser 473.17,18 In our study, we demonstrated for the first time that TUG1 expression level and glycolysis were increased in ADR-resistant AML cells (HL60/ADR). Furthermore, we demonstrated that TUG1 knockdown enhanced ADR cytotoxicity by inhibiting glycolysis via inactivating the protein kinase B (Akt) pathway, shedding light on the roles and molecular mechanism of TUG1 in ADR resistance of AML cells.
2. Materials and methods
2.1. Cell culture and transfection
Human AML HL60 and HL60/ADR cells were purchased from the Chinese Academy of Sciences (Shanghai, China). Cells were incubated with RPMI 1640 medium (Hyclone, Logan, UT, USA) supplemented with 10% heat-inactivated fetal bovine serum (FBS, Gibco, Grand Island, NY, USA), 2 mM L-glutamine (Sigma-Aldrich, St. Louis, MO, USA), and 1% penicillin/streptomycin (Sigma-Aldrich) at 37 °C in a humidified air atmosphere of 95% air/5% CO2. To maintain the multidrug resistance phenotype, 1 μg mL−1 of ADR (Sigma-Aldrich) was added to the culture medium of HL60/ADR cells. Small interfering RNAs (siRNAs) targeting TUG1 (si-TUG1-1 and si-TUG1-2) and siRNA control (si-con) were purchased from GenePharma Co., Ltd. (Shanghai, China). For analysis of the function of TUG1 in AML cells, HL60 and HL60/ADR cells were seeded into 6-well plates and transfected with si-TUG1-1, si-TUG1-2, or si-con using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. On account of the higher knockdown efficiency of si-TUG1-2, we selected si-TUG1-2 (si-TUG1) for the following experiments (see Section 3.2 for more details).
2.2. Detection of glucose consumption and lactate production
HL60 and HL60/ADR cells were seeded into 24-well plates at a density of 2 × 104 cells per well and transfected with or without si-TUG1 or si-con, or combined with exposure to 15 μM 740Y-P (Tocris Bioscience, Shanghai, China) for 48 h. The culture medium was then collected and glucose concentration and lactate production were determined using a glucose assay kit (Biovision, Mountain View, CA, USA) and a Lactate Assay kit (Biovision), respectively.
2.3. Cell viability assay
The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was performed to evaluate cell viability. Briefly, HL60 and HL60/ADR cells (1 × 104 cells per well) were seeded in 96-well plates and treated with appointed concentrations of ADR or 2 mM glycolysis inhibitor 2-deoxy-D-glucose (2-DG; Sigma-Aldrich) for 48 h. In addition, HL60/ADR cells transfected with si-TUG1 or si-con were administrated with 5 μM ADR or in combination with 15 μM 740Y-P for 48 h. Subsequently, 20 μL of MTT dye (5 mg mL−1, Sigma-Aldrich) was added into each well, followed by incubation for another 4 h at 37 °C. After incubation, the culture medium was removed and 150 μL dimethyl sulfoxide (DMSO) was added to solubilize the purple formazan crystals with gentle shaking. The absorbance value was recorded at 570 nm using the ELx800 Absorbance Microplate Reader (Bio-rad, Hercules, CA, USA).
2.4. Flow cytometry analysis of apoptosis
HL60/ADR cells transfected with si-TUG1 or si-con were exposed to 5 μM ADR or combined with 15 μM 740Y-P for 48 h. After treatment, HL60/ADR cells were collected by centrifugation and apoptosis was assessed using Annexin V-fluorescein isothiocyanate (FITC) apoptosis detection kit (KeyGen, Nanjing, China). The apoptotic cells were examined by a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA, USA).
2.5. Quantitative real-time PCR (qRT-PCR)
HL60 and HL60/ADR cells were transfected with or without si-TUG1-1, si-TUG1-2, or si-con, or combined with treatment with 15 μM 740Y-P for 48 h and total RNA was then extracted from treated HL60 and HL60/ADR cells using TRIzol reagent (Invitrogen). Approximately 1 μg of total RNA was immediately reverse-transcribed into first-strand cDNA using a Reverse Transcription Kit (Takara, Dalian, China). For the detection of TUG1, HK2, and PKM2 mRNA expressions, qPCR was conducted using SYBR® Green Real-Time PCR Master Mixes (Thermo Fisher Scientific, Waltham, MA, USA) on the StepOnePlus qPCR system (Thermo Fisher Scientific). GAPDH was used as a quantitative normalization and the relative gene expression was calculated using the 2−ΔΔCt method. The primer sequences are as below: TUG1, forward 5′-TAG CAG TTC CCC AAT CCT TG-3′, reverse 5′-CAC AAA TTC CCA TCA TTC CC-3′; HK2 mRNA, forward 5′-TGT GCG TAA TGG CAA GCG GAG G-3′, reverse 5′-CCA CGG CAA CCA CAT CCA GGT C-3′; PMK2 mRNA, forward 5′-GAG TAC CAT GCG GAG ACC AT-3′, reverse 5′-GCG TTA TCC AGC GTG ATT TT-3′; GADPH mRNA, forward 5′-GAG TCA ACG GAT TTG GTC GT-3′, reverse 5′-TTG ATT TTG GAG GGA TCT CG-3′.
2.6. Western blot analysis
HL60 and HL60/ADR cells were treated with or without si-TUG1 or si-con, or combined with 5 μM ADR or 15 μM 740Y-P for 48 h. After treatment, HL60 and HL60/ADR cells were collected and lysed in Radioimmunoprecipitation Assay Lysis Buffer (Beyotime, Haimen, China) containing a protease inhibitor cocktail (Roche, Indianapolis, IN, USA). Proteins were harvested and protein concentration was quantified with a BCA protein assay kit (Beyotime). Protein samples (20 μg per lane) were subjected to 10% SDS-PAGE and electrotransferred onto the polyvinylidene difluoride membrane (PVDF, Millipore, Billerica, MA, USA). The membranes were blocked with 5% non-fat milk in TBS containing 1% Tween-20 (TBST) for 2 h and probed with primary antibodies including anti-HK2 (Cell Signaling Technology, Danvers, MA, USA), anti-PKM2 (Cell Signaling Technology), anti-phosphorylated Akt (p-Akt) (Ser473) (Cell Signaling Technology), anti-Akt (Cell Signaling Technology) and anti-β-actin (Cell Signaling Technology) overnight at 4 °C, followed by incubation with horseradish peroxidase-conjugated secondary antibodies (Cell Signaling Technology) for 1 h at room temperature. After being washed with TBST, the immunoreactive proteins were visualized using enhanced chemiluminescence reagent (Amersham Biosciences, Piscataway, NJ, USA).
2.7. Statistical analysis
All experimental results are expressed as mean ± standard deviation (SD) from three independent experiments. The statistical analysis was performed by the GraphPad Prism 6 (GraphPad Software, Inc., La Jolla, CA, USA) with Student's t-test and one-way analysis of variance (ANOVA). P-values < 0.05 were regarded as statistically significant.
3. Results
3.1. Glycolysis was increased in HL60/ADR cells
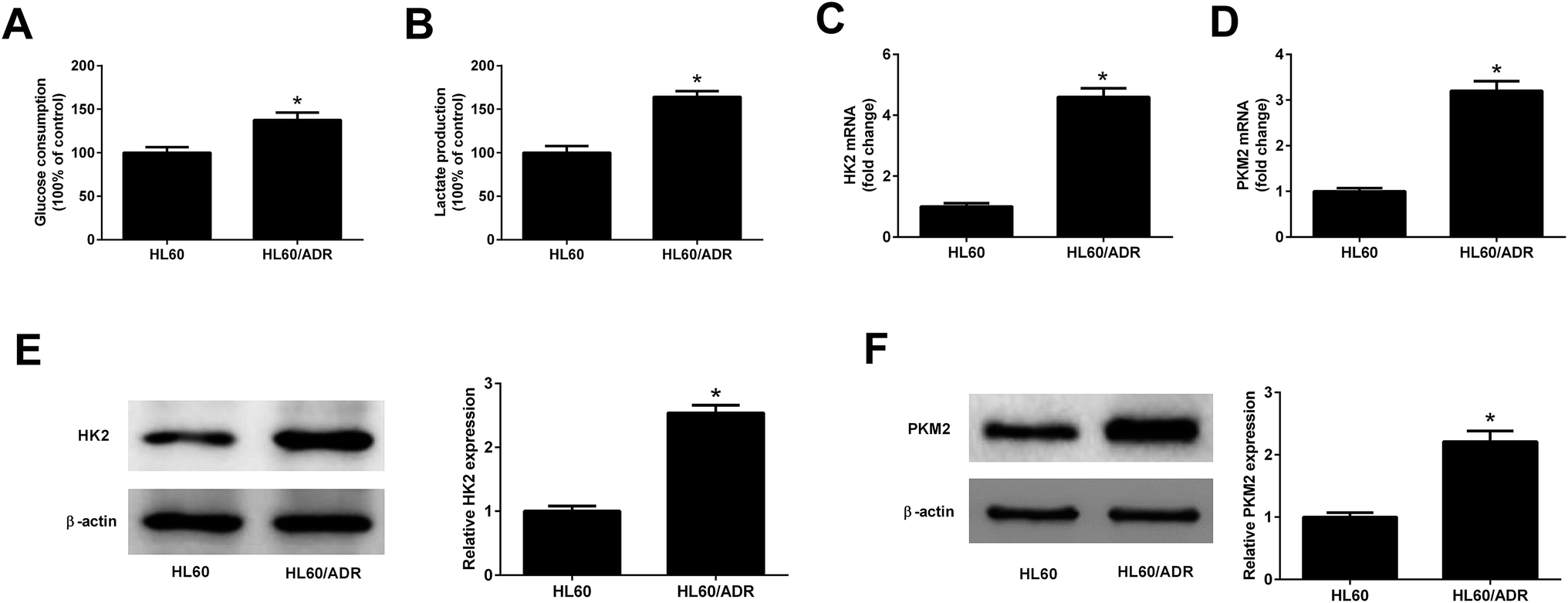
To determine the glycolysis level in ADR-resistant AML cells, glucose consumption and lactate production in HL60/ADR and HL60 cells were measured. As displayed in Fig. 1A and B, the glucose consumption and lactate production in HL60/ADR cells were increased compared to those in HL60 cells. HK2 and PKM2 are identified as two key enzymes in regulating aerobic glycolysis. qRT-PCR and Western blot analysis results demonstrated that the mRNA and protein levels of HK2 (Fig. 1C and E) and PKM2 (Fig. 1D and F) were increased in HL60/ADR cells with respect to those in HL60 cells. These results suggested that glycolysis was increased in HL60/ADR cells.
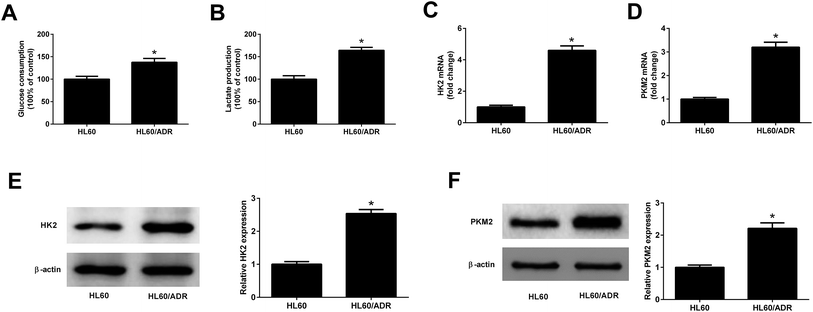 |
| | Fig. 1 Glycolysis was increased in HL60/ADR cells. (A and B) Glucose consumption and lactate production in HL60/ADR and HL60 cells. (C and D) qRT-PCR analysis of the mRNA expressions of HK2 and PKM2 in HL60/ADR and HL60 cells. (E and F) Western blot analysis of the protein levels of HK2 and PKM2 in HL60/ADR and HL60 cells. *P < 0.05. | |
3.2. TUG1 was up-regulated in HL60/ADR cells
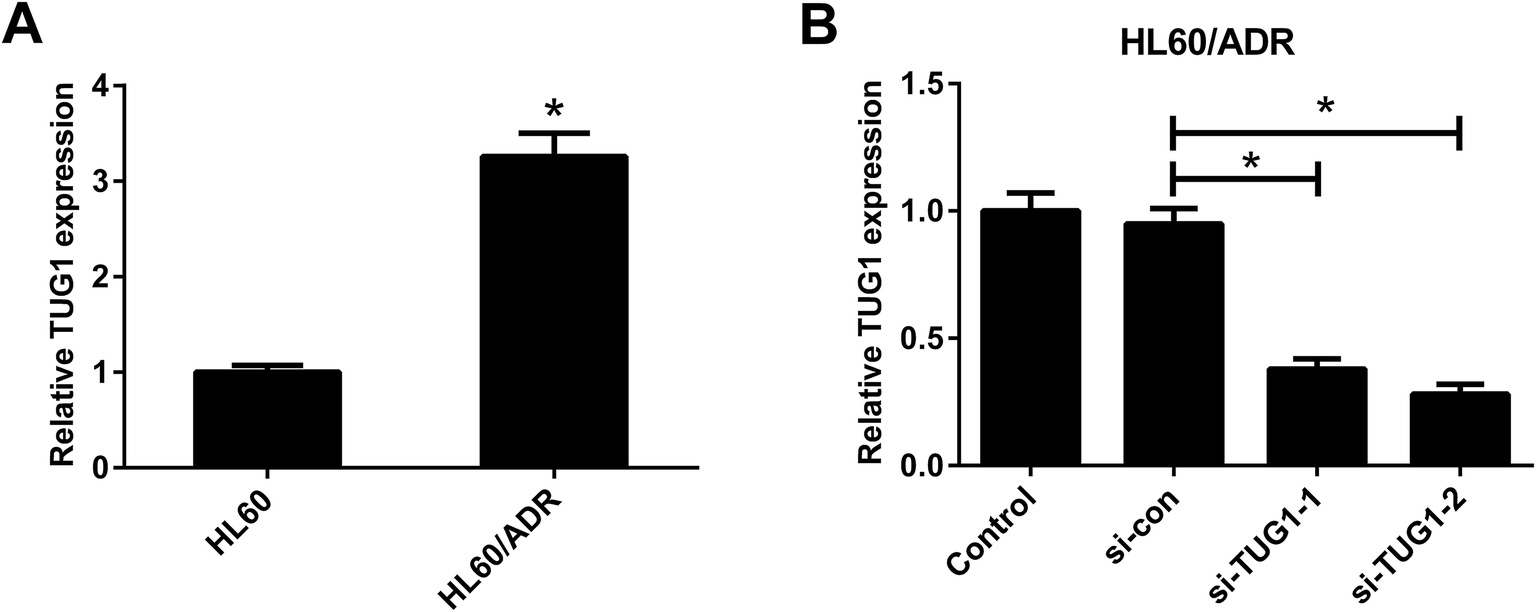
The expression profile of TUG1 in HL60/ADR and HL60 cells was detected by qRT-PCR. The results manifested that a higher expression of TUG1 was observed in HL60/ADR cells versus HL60 cells (Fig. 2A). To address the biological function of TUG1 in regulating ADR resistance of AML cells, HL60/ADR cells were transfected with si-TUG1-1, si-TUG1-2, or si-con. qRT-PCR results showed that TUG1 expression was reduced in HL60/ADR cells transfected with si-TUG1-1 or si-TUG1-2 in comparison with si-con-transfected group (Fig. 2B). Due to the higher knockdown efficiency of si-TUG1-2, we chose si-TUG1-2 (hereafter called si-TUG1) for subsequent experiments.
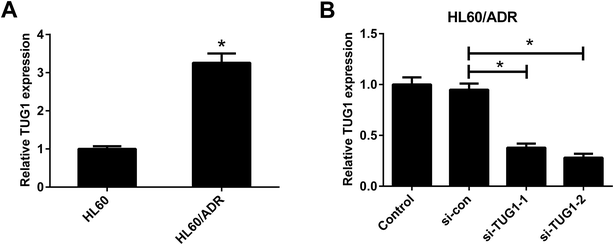 |
| | Fig. 2 TUG1 was highly expressed in HL60/ADR cells. (A) qRT-PCR analysis of TUG1 expression in HL60/ADR and HL60 cells. (B) qRT-PCR analysis of TUG1 expression in HL60/ADR cells transfected with si-TUG1-1, si-TUG1-2, or si-con. *P < 0.05. | |
3.3. TUG1 knockdown inhibited glycolysis in HL60/ADR cells
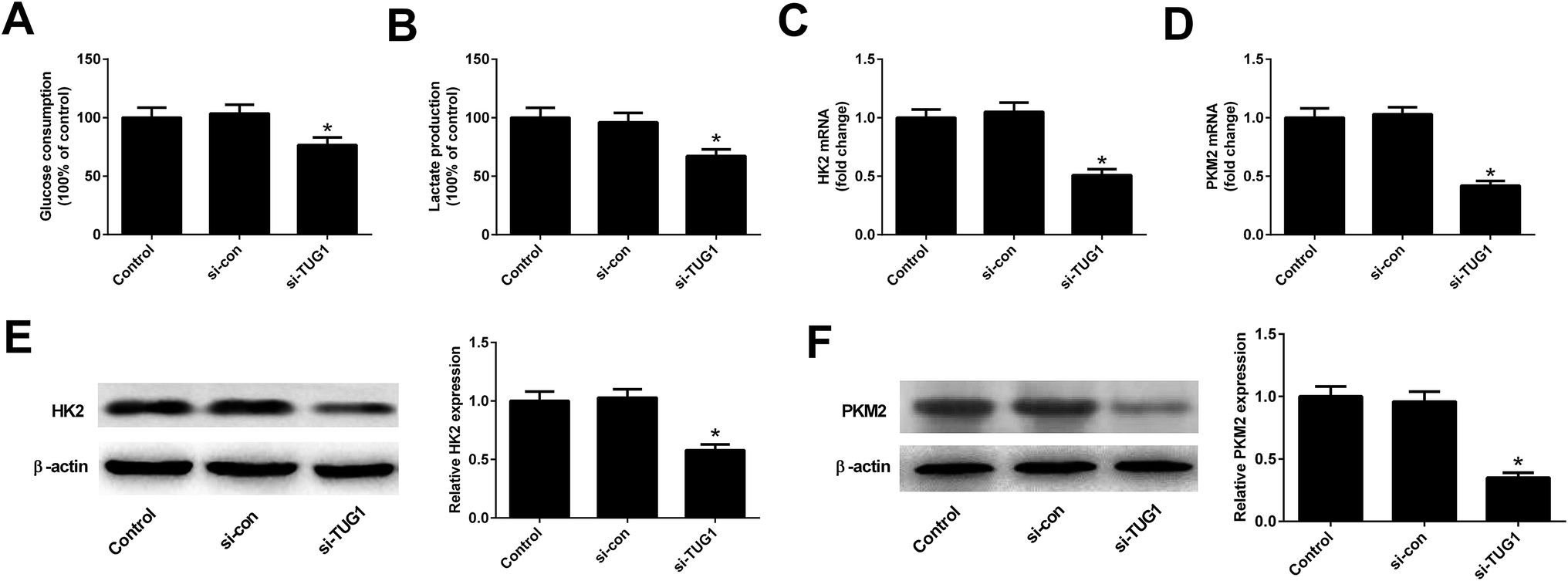
We analyzed the effects of TUG1 on glycolysis in HL60/ADR cells. The results displayed that TUG1 depletion led to a reduction of glucose consumption and lactate production in HL60/ADR cells when compared with si-con-transfected group (Fig. 3A and B). Meanwhile, the expressions of HK2 (Fig. 3C and E) and PKM2 (Fig. 3D and F) at mRNA and protein levels were decreased following transfection with si-TUG1 relative to si-con group in HL60/ADR cells. Collectively, we concluded that TUG1 knockdown inhibited glycolysis in HL60/ADR cells.
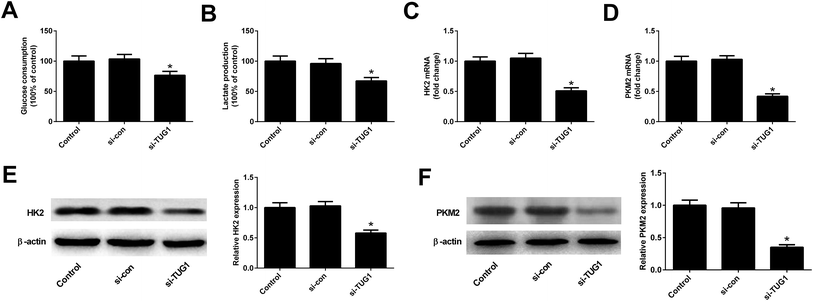 |
| | Fig. 3 TUG1 knockdown inhibited glycolysis in HL60/ADR cells. HL60/ADR cells were transfected with si-TUG1 or si-con for 48 h. (A and B) Glucose consumption and lactate production in HL60/ADR transfected with si-TUG1 or si-con. (C and D) qRT-PCR analysis of the mRNA expressions of HK2 and PKM2 in HL60/ADR cells introduced with si-TUG1 or si-con. (E and F) Western blot analysis of the protein levels of HK2 and PKM2 in si-TUG1- or si-con-treated HL60/ADR cells. *P < 0.05. | |
3.4. TUG1 knockdown enhanced ADR cytotoxicity in HL60/ADR cells
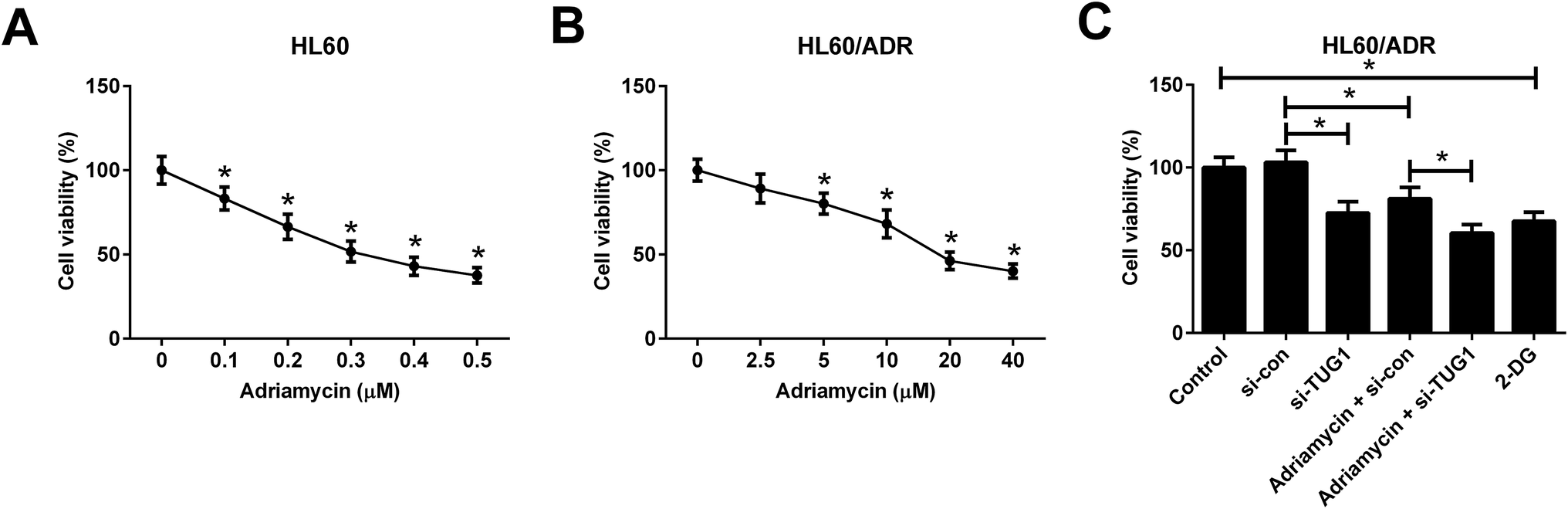
ADR cytotoxicity in HL60 and HL60/ADR cells was evaluated by MTT assay and the results showed that ADR suppressed cell viability in a dose-dependent manner in both HL60 and HL60/ADR cells but exhibited a higher cytotoxicity to HL60 cells (Fig. 4A and B), suggesting the ADR resistance of HL60/ADR cells. To clarify the effect of TUG1 on ADR cytotoxicity in HL60/ADR cells, 5 μM of ADR was selected for next analysis. MTT assay indicated that TUG1 knockdown suppressed cell viability and exacerbated ADR-induced cytotoxicity in HL60/ADR cells compared with si-con group (Fig. 4C). In addition, we found that treatment with 2-DG, a glycolysis inhibitor, also prominently repressed cell viability of HL60/ADR cells (Fig. 4C). According to the aforementioned results, it is reasonable to infer that TUG1 knockdown enhanced ADR cytotoxicity in HL60/ADR cells via inhibiting glycolysis.
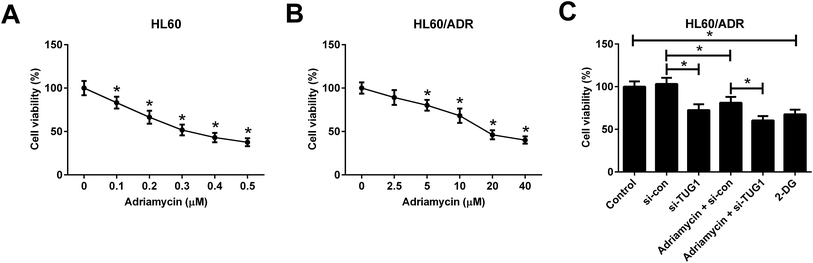 |
| | Fig. 4 TUG1 knockdown enhanced ADR cytotoxicity in HL60/ADR cells. (A) MTT assay was performed to detect cell viability in HL60 cells after exposure to the indicated concentrations of ADR (0, 0.1, 0.2, 0.3, 0.4, or 0.5 μM) for 48 h. *P < 0.05 vs. 0 μM ADR group. (B) ADR cytotoxicity was assessed by MTT assay in HL60/ADR cells treated with a series of concentrations of ADR (0, 2.5, 5, 10, 20, or 40 μM) for 48 h. *P < 0.05 vs. 0 μM ADR group. (C) Cell viability was detected by MTT assay in HL60/ADR cells after treatment with 2 mM 2-DG for 48 h, or transfected with si-TUG1 or si-con or along with treatment with 5 μM ADR for 48 h. *P < 0.05. | |
3.5. TUG1 knockdown enhanced ADR-induced apoptosis in HL60/ADR cells
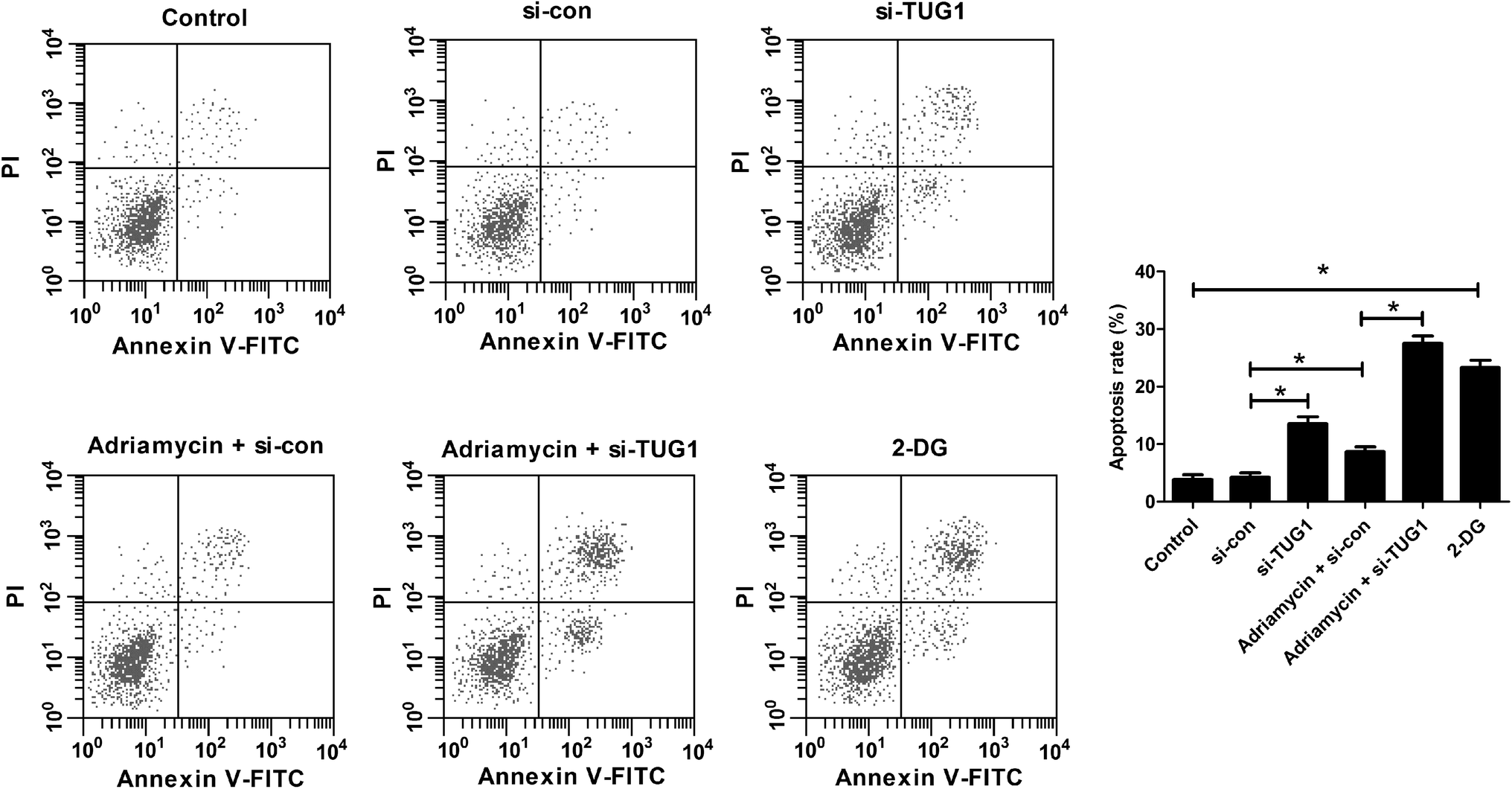
Furthermore, flow cytometry analysis demonstrated that silencing of TUG1 effectively promoted apoptosis in HL60/ADR cells (Fig. 5). Moreover, we found that ADR treatment induced apoptosis of HL60/ADR cells, while TUG1 depletion notably strengthened ADR-induced apoptosis in HL60/ADR cells (Fig. 5). Additionally, administration with 2-DG also increased the percentage of apoptotic cells in HL60/ADR cells with respect to control group (Fig. 5). These results suggested that TUG1 knockdown enhanced ADR-induced apoptosis in HL60/ADR cells via suppressing glycolysis.
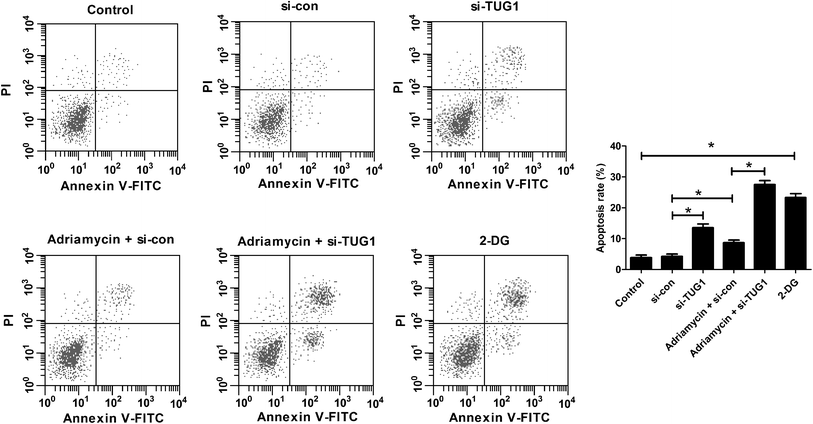 |
| | Fig. 5 TUG1 knockdown enhanced ADR-induced apoptosis in HL60/ADR cells. HL60/ADR cells were treated with 2 mM 2-DG for 48 h, or transfected with si-TUG1 or si-con or along with treatment with 5 μM ADR for 48 h, followed by evaluation of apoptosis by flow cytometry. *P < 0.05. | |
3.6. TUG1 knockdown inhibited the Akt pathway in HL60/ADR cells
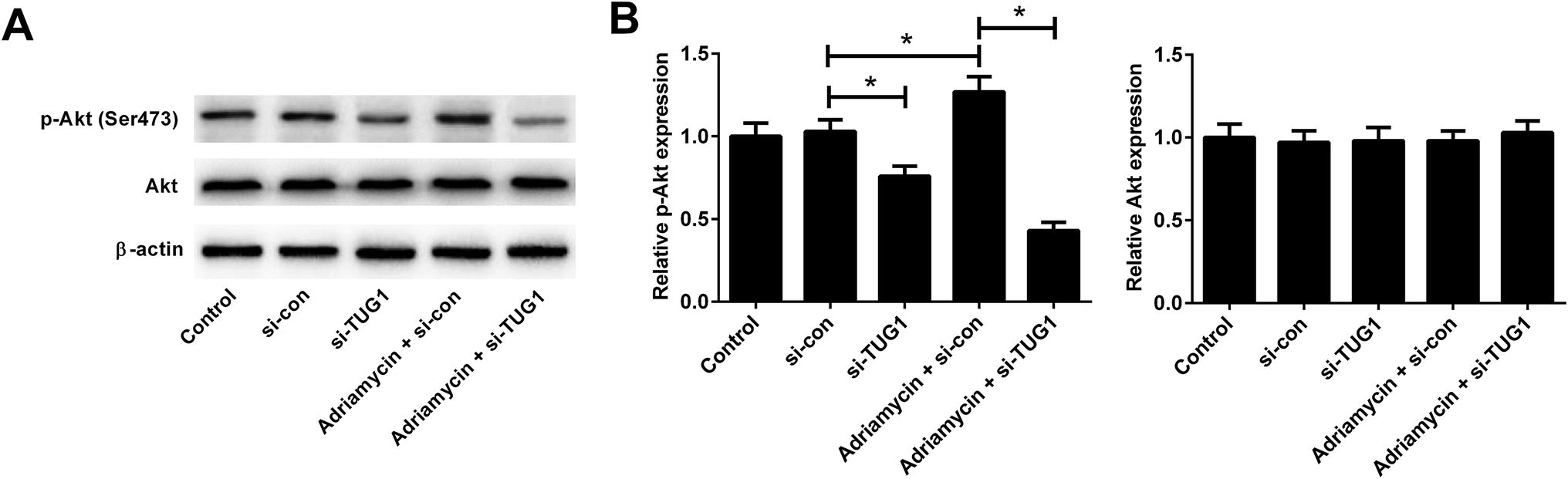
Recent studies have shown that the Akt pathway is frequently constitutively activated in AML and contributes to drug resistance.19 We therefore analyzed the effects of TUG1 on the Akt pathway in HL60/ADR cells by Western blot. The results implied that the phosphorylation of Akt was impeded following TUG1 knockdown in HL60/ADR cells (Fig. 6A and B). Moreover, TUG1 silencing also restrained ADR-induced increase of Akt phosphorylation in HL60/ADR cells (Fig. 6A and B). These findings demonstrated that TUG1 knockdown inhibited the Akt pathway in HL60/ADR cells.
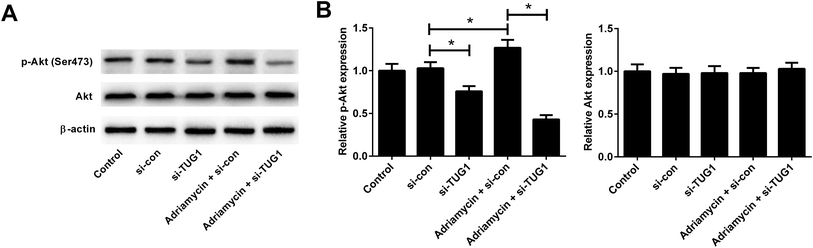 |
| | Fig. 6 TUG1 knockdown inhibited the Akt pathway in HL60/ADR cells. Western blot analysis was performed to detect the protein levels of p-Akt (Ser473) and Akt in HL60/ADR cells transfected with si-TUG1, si-con, or followed by treatment with 5 μM ADR for 48 h. (A) Representative blots are shown. (B) Densitometry was analyzed using the image J software and β-actin was used as an internal control. *P < 0.05. | |
3.7. Activation of Akt pathway attenuated the effect of TUG1 knockdown on ADR-induced cytotoxicity and apoptosis in HL60/ADR cells
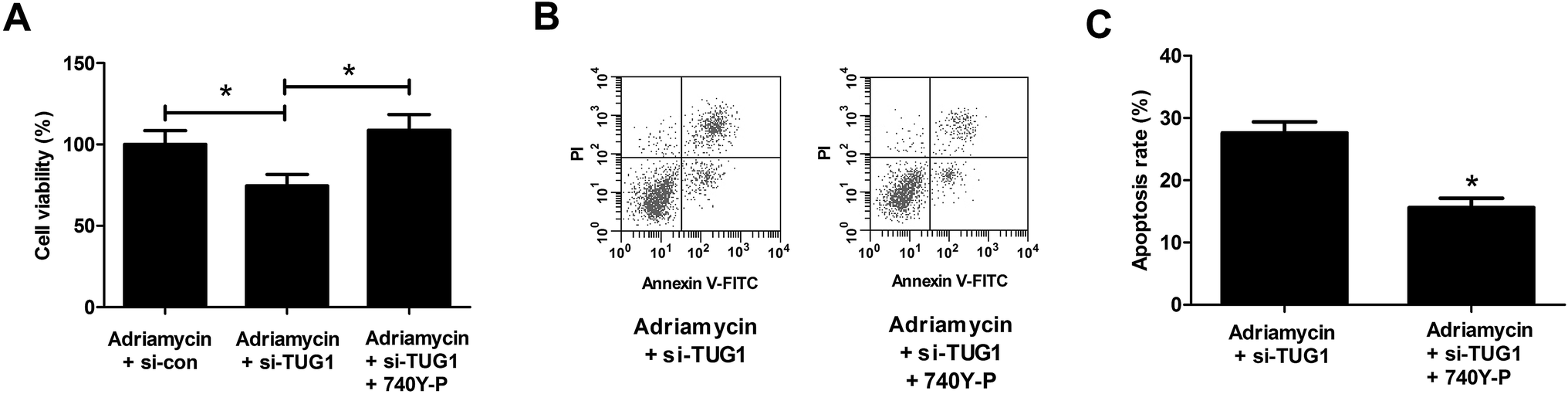
To further address whether the effects of TUG1 knockdown on cytotoxicity and apoptosis in HL60/ADR cells were mediated by the Akt pathway, HL60/ADR cells transfected with si-TUG1 or si-con were exposed to 5 μM ADR or combined with 15 μM 740Y-P, an agonist of the Akt pathway, for 48 h. MTT assay presented that 740Y-P treatment restored the promotive effect of TUG1 silencing on ADR-induced cytotoxicity in HL60/ADR cells (Fig. 7A). Flow cytometry analysis uncovered that 740Y-P treatment attenuated TUG1 knockdown-mediated reinforcement on ADR-induced apoptosis in HL60/ADR cells (Fig. 7B and C). These data indicated that activation of Akt pathway by 740Y-P attenuated the effects of TUG1 knockdown on ADR-induced cytotoxicity and apoptosis in HL60/ADR cells.
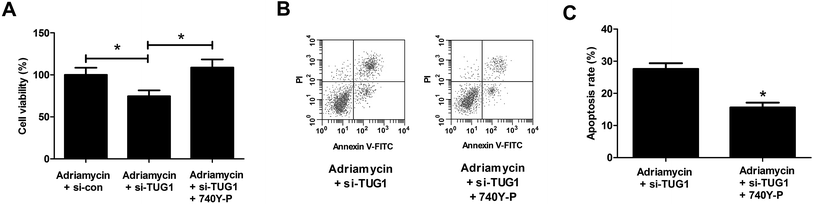 |
| | Fig. 7 Activation of Akt pathway attenuated the effects of TUG1 knockdown on ADR-induced cytotoxicity and apoptosis in HL60/ADR cells. HL60/ADR cells transfected with si-TUG1 or si-con were exposed to 5 μM ADR or combined with 15 μM 740Y-P for 48 h. (A) MTT assay was performed to examine ADR cytotoxicity in the treated HL60/ADR cells. (B) Flow cytometry was conducted to analyze apoptosis of the treated HL60/ADR cells. (C) The percentage of apoptotic HL60/ADR cells after treatment. *P < 0.05. | |
3.8. Activation of Akt pathway attenuated the effect of TUG1 knockdown on glycolysis in HL60/ADR cells
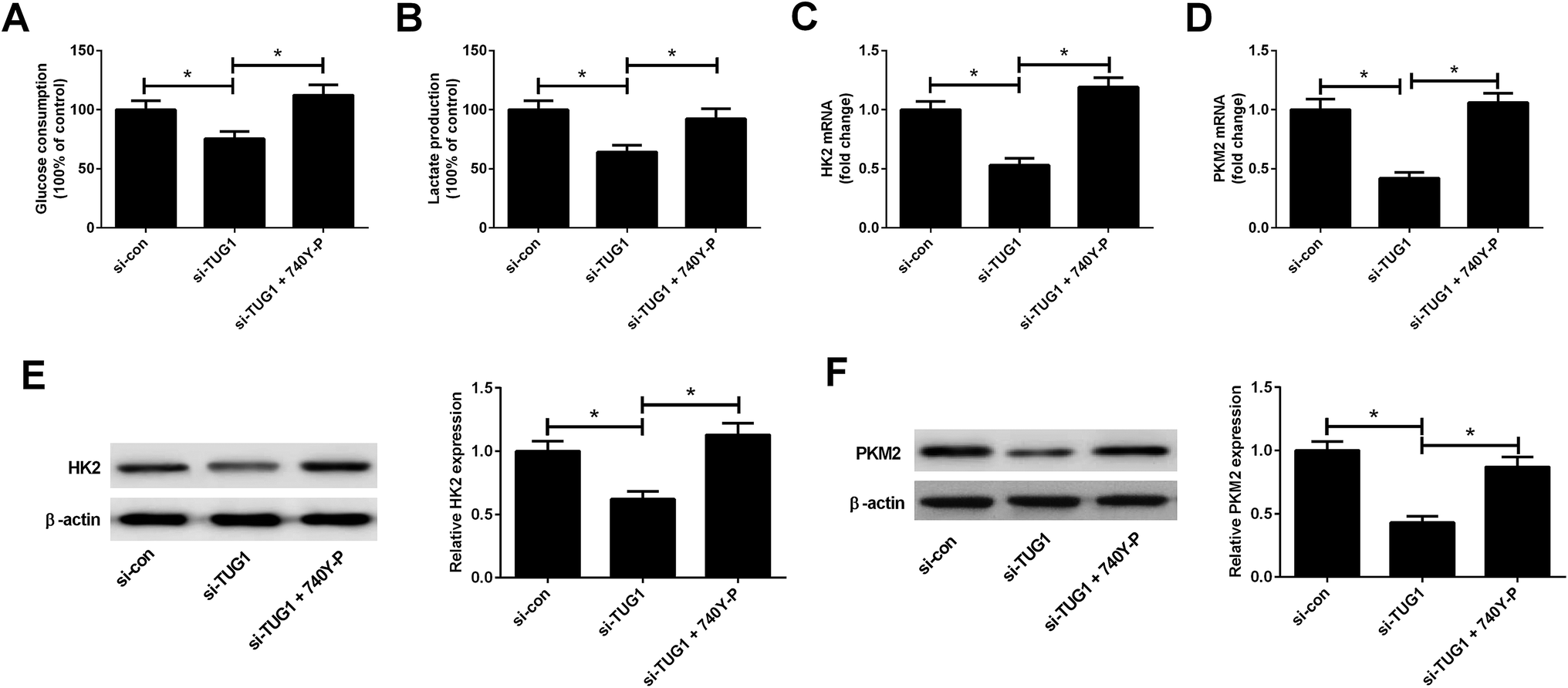
We explored whether the Akt pathway could affect the effect of TUG1 knockdown on glycolysis in HL60/ADR cells. As presented in Fig. 8A and B, 740Y-P treatment relieved the inhibitory effects of TUG1 knockdown on the glucose consumption and lactate production in HL60/ADR cells. Meanwhile, depletion of TUG1-mediated decreases of the mRNA and protein levels of HK2 (Fig. 8C and E) and PKM2 (Fig. 8D and F) in HL60/ADR cells were abolished following the addition of 740Y-P. Together, we concluded that activation of Akt pathway by 740Y-P attenuated the effect of TUG1 knockdown on glycolysis in HL60/ADR cells.
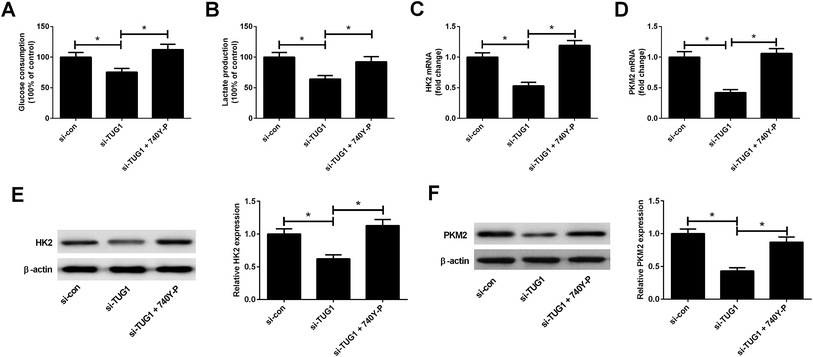 |
| | Fig. 8 Activation of Akt pathway attenuated the effect of TUG1 knockdown on glycolysis in HL60/ADR cells. HL60/ADR cells were transfected with si-TUG1 or si-con, or followed by exposure to 15 μM 740Y-P for 48 h. (A and B) Glucose consumption and lactate production in treated HL60/ADR cells. (C and D) The mRNA expressions of HK2 and PKM2 in the treated HL60/ADR cells were examined by qRT-PCR. (E and F) The protein levels of HK2 and PKM2 in the treated HL60/ADR cells were determined by Western blot. *P < 0.05. | |
4. Discussion
Resistance to chemotherapeutic drugs constitutes one of the major obstacles in the clinical treatment of AML. Therefore, it is desperately needed to elucidate the molecular mechanism behind drug resistance and find novel therapeutic approaches for reversing drug resistance. Recently, the key roles of lncRNAs in drug resistance in multiple tumors have received increasing attention. Recent studies have manifested that TUG1 was highly expressed in several tumors, such as epithelial ovarian cancer,20 osteosarcoma,21 and AML,13 whereas its expression was generally lowly expressed in non-small cell lung carcinoma,22 suggesting the context-dependent roles in different types of tumors. Moreover, TUG1 has been demonstrated to be associated with drug resistance in several malignancies. For instance, TUG1 increased cisplatin sensitivity of triple negative breast cancer cells by regulating miR-197/nemo-like kinase (NLK) via inactivating the Wnt pathway.23 On the contrary, TUG1 promoted cell growth and chemoresistance of small cell lung cancer cells by regulating LIMK2b (a splice variant of LIM-kinase 2) expression via binding with enhancer of zeste homolog 2 (EZH2).24 It is previously reported that high expression of TUG1 associated with advanced disease and worse prognosis in AML patients, and induced AML cell proliferation and repressed cell apoptosis.13 However, the roles and molecular mechanism of TUG1 in regulating drug resistance in AML cells remain largely unknown. In our study, we firstly demonstrated that TUG1 was up-regulated in HL60/ADR cells compared with HL60 cells. Functional analysis revealed that TUG1 knockdown enhanced ADR-induced cytotoxicity and apoptosis in HL60/ADR cells.
Recently, several mechanisms for drug resistance have been proposed, among which the relevance of altered energy metabolism, especially aerobic glycolysis, to drug resistance has been the focus of current researches.25 Experimental data are accumulating suggesting that elevated aerobic glycolysis is observed in drug-resistant cancer cells and is identified as an essential contributor to the development of drug resistance in various cancer cells including AML.26,27 Therefore, increasing studies have proposed that inhibition of glycolysis may be an effective approach to reverse drug resistance in drug-resistant cancer cells.28,29 miR-125 overexpression increased sensitivity of 5-fluorouracil (5-FU)-resistant cells to 5-FU through inhibition of glycolysis by targeting HK2.30 Knockdown of lncRNA UCA1 suppressed the chemoresistance of pediatric AML through suppressing glycolysis.31 In our study, we found the elevated glucose consumption and lactate production in HL60/ADR cells, as well as the increased HK2 and PKM2 expressions at mRNA and protein levels in HL60/ADR cells, indicating that glycolysis was increased in HL60/ADR cells, consistently with the previous studies.26,27 Meanwhile, we demonstrated that TUG1 knockdown suppressed glycolysis in HL60/ADR cells. Inhibition of glycolysis by 2-DG repressed cell viability and induced apoptosis in HL60/ADR cells. Based on the above results, we inferred that knockdown of TUG1 increased sensitivity to ADR in HL60/ADR cells via inhibition of glycolysis.
The Akt pathway is a well-known important signaling pathway involved in the regulation of many cellular functions, such as metabolism, survival, apoptosis and chemotherapy resistance.32 Recent studies show that the Akt pathway is hyper-activated in various human malignancies including AML.33 Constitutive activation of the Akt pathway may contribute to cancer progression and chemotherapy resistance in AML.14 Thus, targeting the Akt pathway might provide an effective strategy to reverse drug resistance in cancers.34 Herein, our study proved that TUG1 knockdown suppressed the Akt pathway in HL60/ADR cells. Activation of the Akt pathway attenuated the effects of TUG1 knockdown on ADR-induced cytotoxicity and apoptosis, as well as glycolysis in HL60/ADR cells, suggesting that TUG1 knockdown increased sensitivity to ADR in HL60/ADR cells via inhibiting the glycolysis by inactivating the Akt pathway.
5. Conclusion
In summary, we found that TUG1 was up-regulated in HL60/ADR cells and TUG1 knockdown increased ADR cytotoxicity in HL60/ADR cells via inhibiting the glycolysis by inactivating the Akt pathway, which provide a theoretical foundation for the development of novel AML therapeutic strategies. Therefore, TUG1 may be a potential therapeutic sensitizer for overcoming ADR resistance in ADR-resistant AML cells.
Conflicts of interest
The authors declare that they have no conflict of interest.
References
- J. Leptidis, S. Aloizos, P. Chlorokostas and S. Gourgiotis, Am. J. Emerg. Med., 2014, 32, 1294.e1291–1292 CrossRef PubMed.
- R. Siegel, J. Ma, Z. Zou and A. Jemal, Ca-Cancer J. Clin., 2014, 64, 9–29 CrossRef PubMed.
- P. A. von dem Borne, L. C. de Wreede, C. J. Halkes, W. A. Marijt, J. H. Falkenburg and H. Veelken, Leuk. Res., 2016, 46, 45–50 CrossRef CAS PubMed.
- M. S. Tallman, D. G. Gilliland and J. M. Rowe, Blood, 2005, 106, 1154–1163 CrossRef CAS PubMed.
- M. G. Vander Heiden, L. C. Cantley and C. B. Thompson, Science, 2009, 324, 1029–1033 CrossRef CAS PubMed.
- B. Bhattacharya, M. F. Mohd Omar and R. Soong, Br. J. Pharmacol., 2016, 173, 970–979 CrossRef CAS PubMed.
- Y. Zhao, E. B. Butler and M. Tan, Cell Death Dis., 2013, 4, e532 CrossRef CAS PubMed.
- M. Guttman and J. L. Rinn, Nature, 2012, 482, 339–346 CrossRef CAS PubMed.
- M. K. Iyer, Y. S. Niknafs, R. Malik, U. Singhal, A. Sahu, Y. Hosono, T. R. Barrette, J. R. Prensner, J. R. Evans, S. Zhao, A. Poliakov, X. Cao, S. M. Dhanasekaran, Y. M. Wu, D. R. Robinson, D. G. Beer, F. Y. Feng, H. K. Iyer and A. M. Chinnaiyan, Nat. Genet., 2015, 47, 199–208 CrossRef CAS PubMed.
- M. Haemmerle and T. Gutschner, Int. J. Mol. Sci., 2015, 16, 1395–1405 CrossRef CAS PubMed.
- D. Ayers and J. Vandesompele, Genes, 2017, 8, 95 CrossRef PubMed.
- T. L. Young, T. Matsuda and C. L. Cepko, Curr. Biol., 2005, 15, 501–512 CrossRef CAS PubMed.
- X. Wang, L. Zhang, F. Zhao, R. Xu, J. Jiang, C. Zhang, H. Liu and H. Huang, Ann. Hematol., 2018, 97, 1375–1389 CrossRef CAS PubMed.
- Z. Song, Y. Guo, M. Zhou and X. Zhang, Metabolism, 2014, 63, 1324–1333 CrossRef CAS PubMed.
- Y. Zhou, N. Lu, C. Qiao, T. Ni, Z. Li, B. Yu, Q. Guo and L. Wei, Mol. Carcinog., 2016, 55, 1317–1328 CrossRef CAS PubMed.
- Q. Lu, S. Yan, H. Sun, W. Wang, Y. Li, X. Yang, X. Jiang, Y. Che and Z. Xi, Cell Death Dis., 2015, 6, e2005 CrossRef CAS PubMed.
- D. Derossi, E. J. Williams, P. J. Green, D. J. Dunican and P. Doherty, Biochem. Biophys. Res. Commun., 1998, 251, 148–152 CrossRef CAS PubMed.
- E. J. Williams and P. Doherty, Mol. Cell. Neurosci., 1999, 13, 272–280 CrossRef CAS PubMed.
- S. Dinner and L. C. Platanias, J. Cell. Biochem., 2016, 117, 1745–1752 CrossRef CAS PubMed.
- T. H. Li, J. J. Zhang, S. X. Liu and Y. Chen, Medicine, 2018, 97, e0575 CrossRef CAS PubMed.
- C. Xie, B. Chen, B. Wu, J. Guo and Y. Cao, Biomed. Pharmacother., 2018, 97, 1645–1653 CrossRef CAS PubMed.
- E. B. Zhang, D. D. Yin, M. Sun, R. Kong, X. H. Liu, L. H. You, L. Han, R. Xia, K. M. Wang, J. S. Yang, W. De, Y. Q. Shu and Z. X. Wang, Cell Death Dis., 2014, 5, e1243 CrossRef CAS PubMed.
- T. Tang, Y. Cheng, Q. She, Y. Jiang, Y. Chen, W. Yang and Y. Li, Biomed. Pharmacother., 2018, 107, 338–346 CrossRef CAS PubMed.
- Y. Niu, F. Ma, W. Huang, S. Fang, M. Li, T. Wei and L. Guo, Mol. Cancer, 2017, 16, 5 CrossRef PubMed.
- B. Bhattacharya, S. H. H. Low, C. Soh, N. Kamal Mustapa, M. Beloueche-Babari, K. X. Koh, J. Loh and R. Soong, Br. J. Pharmacol., 2014, 171, 3255–3267 CrossRef CAS PubMed.
- K. Song, M. Li, X. Xu, L. I. Xuan, G. Huang and Q. Liu, Oncol. Lett., 2016, 12, 334 CrossRef CAS PubMed.
- M. Tamada, O. Nagano, S. Tateyama, M. Ohmura, T. Yae, T. Ishimoto, E. Sugihara, N. Onishi, T. Yamamoto and H. Yanagawa, Cancer Res., 2012, 72, 1438 CrossRef CAS PubMed.
- J. Kluza, M. Jendoubi, C. Ballot, A. Dammak, A. Jonneaux, T. Idziorek, S. Joha, V. Dauphin, M. Malet-Martino, S. Balayssac, P. Maboudou, G. Briand, P. Formstecher, B. Quesnel and P. Marchetti, PLoS One, 2011, 6, e21924 CrossRef CAS PubMed.
- E. Hulleman, K. M. Kazemier, A. Holleman, D. J. VanderWeele, C. M. Rudin, M. J. Broekhuis, W. E. Evans, R. Pieters and M. L. Den Boer, Blood, 2009, 113, 2014–2021 CrossRef CAS PubMed.
- J. X. Jiang, S. Gao, Y. Z. Pan, C. Yu and C. Y. Sun, Mol. Med. Rep., 2014, 10, 995–1002 CrossRef CAS PubMed.
- Y. Zhang, Y. Liu and X. Xu, J. Cell. Biochem., 2018, 119, 6296–6308 CrossRef CAS PubMed.
- T. Brotelle and J. O. Bay, Bull. Cancer, 2016, 103, 18–29 CrossRef PubMed.
- A. M. Martelli, P. L. Tazzari, C. Evangelisti, F. Chiarini, W. L. Blalock, A. M. Billi, L. Manzoli, J. A. McCubrey and L. Cocco, Curr. Med. Chem., 2007, 14, 2009–2023 CrossRef CAS PubMed.
- H. A. Burris, 3rd, Cancer Chemother. Pharmacol., 2013, 71, 829–842 CrossRef PubMed.
Footnote |
| † These authors contributed equally to this work and should be considered as co-first authors. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Chao Wang and
Ning Hu
*,
Chao Wang and
Ning Hu