
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, characterization and chemical degradation of poly(ester-triazole)s derived from D-galactose†
M. Verónica Rivasab,
Gabriela Petroselliab,
Rosa Erra-Balsells ab,
Oscar Varelaab and
Adriana A. Kolender*ab
ab,
Oscar Varelaab and
Adriana A. Kolender*ab
aUniversidad de Buenos Aires, Facultad Ciencias Exactas y Naturales, Departamento de Química Orgánica, Ciudad Universitaria, Pab. 2, C1428EHA, Buenos Aires, Argentina
bConsejo Nacional de Investigaciones Científicas y Técnicas (CONICET)-UBA Centro de Investigación en Hidratos de Carbono (CIHIDECAR), Argentina. E-mail: adrianak@qo.fcen.uba.ar
First published on 28th March 2019
Abstract
α-Azide-ω-alkynyl ester monomers were designed and synthesized in order to obtain hydrolytically degradable polymers. The monomers were prepared from D-galactose, as a renewable resource. Environmentally benign azido–alkyne cycloaddition polymerizations were conducted to afford poly(ester-triazole)s, with complete atom economy. Although polymer formation prevailed under optimized polymerization conditions, variable proportions of cyclic oligomer byproducts were detected. The Cu-catalyzed click polymerization led regioselectively to 1,4-disubstituted triazole linkages, while the thermal, metal-free polymerization produced a random distribution of 1,4- and 1,5-disubstituted triazoles in the polymer backbone. The poly(ester-triazole)s exhibited high molecular weights (Mw in the range 35–85 kDa). They were soluble in organic solvents but highly insoluble in water, thus removal of the Cu(I) catalyst was simplified. The polymers were stable up to 300 °C, and had Tg values in the range 90–100 °C. The materials were hydrolysed under either basic or strong acid conditions, and the degradation products have been characterized.
Introduction
Copper assisted azide–alkyne cycloaddition (CuAAC)1 applied to click polymerization2 is a powerful tool for the synthesis of polymers, due to high reaction rates and yields, combined with smooth reaction conditions in several solvents and high tolerance to many functional groups. The CuAAC reaction satisfies all the criteria established in the click chemistry concept by Sharpless.3 The 1,4-disubstituted 1,2,3-trizole ring formed is known as a low toxic group that has been exploited for the generation of many medicinal scaffolds exhibiting anti-HIV, anticancer, and antibacterial activities.4 The uncatalyzed thermal cycloaddition of azides and alkynes yields a mixture of 1,4- and 1,5-disubstituted regioisomers.5Azide and alkyne functionalities can be easily introduced into organic molecules and remain considerably robust in the presence of other functional groups and common chemical reagents. In carbohydrate chemistry, a sugar can be easily modified with alkyne or azide functionalities in routine synthetic protocols.6
Polymer synthesis from renewable biomass resources,7 in particular from carbohydrates,8 is a promising and developing field. Carbohydrates are seen as a readily available feedstock with interesting chemical properties, due to their diverse functionality and stereochemistry. The hydrophilicity endowed by the free hydroxyl groups present in native carbohydrates could be modulated by the use of protecting groups. Carbohydrate-derived linear AB polytriazoles,9 A2B2 polytriazoles,10 AB poly(amide-triazole)s,11,12 and A2B2 poly(ester-triazole)s13,14 were mainly reported by the groups of Drockenmuller and Galbis. In a previous work, we have reported the synthesis and characterization of carbohydrate-derived poly(amide-triazole)s with the secondary hydroxyl groups protected as their methylidene acetal derivatives.15 These materials, obtained from a rigid monomer derived from D-gluconolactone, were very insoluble and exhibited unusually high Tg values. In the present work we have designed a more flexible monomer in order to improve the solubility in common organic solvents and to study the influence of a flexible monomer on the properties of the resulting polymer. The synthesis was planned in order to follow the principles of Green Chemistry,16 namely renewable starting materials, high yields, safe reaction conditions, energy efficiency, catalysis, atom economy, and degradation.
Polyesters like polycaprolactone, polylactide, and polyhydroxybutyrate constitute important biodegradable materials.17 Thus, we were interested in combining the degradability of the ester linkage with carbohydrate monomers that could be subjected to CuAAC polymerizations, to give linear poly(ester-triazole)s. In addition, chiral AB carbohydrate monomers were designed in order to obtain stereoregular polymers. The monosaccharide of choice was the readily available and inexpensive D-galactose, which was oxidized to sodium D-galactonate or D-galactono-1,4-lactone (1) by an improved procedure developed in our laboratory.18 From lactone 1, the α-azide-ω-alkynyl ester monomer was prepared, with the secondary hydroxyl groups protected as labile isopropylidene acetals. The thermal properties and the chemical degradation of the resulting polymers were studied. It was expected that the change from amide to ester functionality, and methylidene to isopropylidene protective group would result in more degradable materials.
Experimental
Materials and methods
D-Galactose was purchased from B.D.H. Chemicals. Analytical thin layer chromatography (TLC) was performed on Silica Gel 60 F254 (Merck) aluminum-supported plates (layer thickness 0.2 mm). Visualization of the spots was effected by exposure to UV light, by charring with a solution of 5% (v/v) sulfuric acid in EtOH, containing 0.5% p-anisaldehyde or with cerium molybdate solution. Column chromatography was performed with Silica Gel 60 (230–400 mesh, E. Merck). Microwave reactions were carried out in an Anton Paar Monowave 300 reactor. Optical rotations were measured with a Perkin–Elmer 343 digital polarimeter at 25 °C and [α] is expressed in deg cm3 g−1 dm−1, concentrations (c) are given in g cm−3. Nuclear magnetic resonance (NMR) spectra were recorded with a Bruker AMX 500 instrument (1H: 500 MHz; 13C: 125.7 MHz) with residual solvent as the internal standard. The assignments were assisted by 2D COSY and HSQC techniques. IR spectra (neat sample for ATR spectra) were recorded with a Thermo Scientific Nicolet IS50 FTIR spectrometer. High resolution mass spectrometry (HRMS-ESI) was performed in a Bruker microTOF-Q II instrument.Gel permeation chromatography (GCP) was carried out at 30 °C with N,N-dimethylformamide (DMF), pure or containing 0.05% LiBr and 100 mM ethylenediamine as eluent, on a Waters 600 instrument equipped with a Styragel HR 4E (Waters) column.
Thermogravimetric analysis (TGA) was performed in a Shimadzu TGA-51 instrument; samples of about 2 mg were heated at a rate of 10 °C min−1, from 20 to 700 °C. Differential scanning calorimetry (DSC) was conducted with a DSC Q20 TA instrument. Samples of about 2 mg were heated from −10 to 250 °C at a rate of 10 °C min−1 (isothermic 5 min), then cooled at 10 °C min−1 to −10 °C (isothermic 5 min), then a second heating cycle at 10 °C min−1 to 250 °C (isothermic 5 min).
D-Galactono-1,4-lactone (1) and methyl 6-bromo-6-deoxy-2,3:4,5-di-O-isopropylidene-D-galactonate (2) were obtained as previously described.18
UV-MALDI-TOF analysis of the polymeric samples was performed using an Ultraflex II TOF/TOF mas spectrometer (Bruker Daltonics) equipped with a high-performance solid-state laser (λ = 355 nm). Mass spectra were acquired in linear positive and negative ion modes.
Monomer synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 hexane–EtOAC) was isolated compound 3 (1.250 g, 93%) as a colourless syrup.:H2O 3:1 (13 mL), KOH (1.3 g) was added, and it was stirred a room temperature for 3 h. After concentration under reduced pressure, water (20 mL) was added, and the aqueous mixture was extracted with EtOAc (3 × 15 mL). The aqueous phase was acidified to pH 2 with 1 M aqueous HCl and extracted with EtOAc (3 × 15 mL). The combined organic extracts were dried over anh MgSO4 and concentrated to give compound 6 (2.67 g, 93%) as a white solid.:1 hexane–EtOAC) to afford compound 5 (0.395 g, 91%) as a white solid.
:1 hexane–EtOAC) was isolated compound 3 (1.250 g, 93%) as a colourless syrup.:H2O 3:1 (13 mL), KOH (1.3 g) was added, and it was stirred a room temperature for 3 h. After concentration under reduced pressure, water (20 mL) was added, and the aqueous mixture was extracted with EtOAc (3 × 15 mL). The aqueous phase was acidified to pH 2 with 1 M aqueous HCl and extracted with EtOAc (3 × 15 mL). The combined organic extracts were dried over anh MgSO4 and concentrated to give compound 6 (2.67 g, 93%) as a white solid.:1 hexane–EtOAC) to afford compound 5 (0.395 g, 91%) as a white solid.Polymerization of compound 5
Several polymerization conditions were essayed as indicated in Table 1, the following general procedures were applied. The CuAAC polymerization in solution is described here, while the other polymerization procedures are reported in ESI.†| Entry | Conc. 5 (M) | Solvent | T (°C) | Time (h) | Catalyst (mol %) | Yield | Mna | Mwa | PDa | 1.4:1.5 ratio |
6:Cyclic compounds 8–10 ratio |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Determined by GPC.b MW irradiation.c 1,5-Disubstituted triazole units were not detected.d Conventional heating.e Cyclic compounds were not detected.f Not determined. | |||||||||||
| 1 | Bulk | — | 70b | 0.5 | 10 | 95 | 48700 |
67200 |
1.4 | —c | 10.7:1.0 |
| 2 | Bulk | — | 60–120b | 1 | — | 50 | 36300 |
78300 |
2.2 | 1.8:1.0 |
3.1:1.0 |
| 3 | Bulk | — | 70d | 16 | — | 86 | 34400 |
35200 |
1.02 | 1.7:1.0 |
2.7:1.0 |
| 4 | 0.25 | MeOH | 70b | 1 | 10 | 88 | 20300 |
30100 |
1.5 | — | —e |
| 5 | 0.3 | MeOH | 70b | 0.5 | 10 | 99 | 29900 |
34100 |
1.1 | — | 3.8:1.0 |
| 6 | 0.12 | MeOH | 25 | 16 | 10 | 99 | 13900 |
28900 |
2.1 | — | 5.7:1.0 |
| 7 | 0.3 | THF | 70d | 16 | — | 77 | 16600 |
20100 |
1.2 | 7.1:1.0 |
20.0:1.0 |
| 8 | 0.3 | THF | 70d | 16 | 10 | 72 | 30700 |
42500 |
1.4 | 41.0:1.0 |
— |
| 9 | 0.2 | THF | 70b | 2 | 10 | 91 | 49500 |
58200 |
1.2 | — | 2.5:1.0 |
| 10 | 0.2 | THF | 25 | 16 | 10 | 95 | 42500 |
52400 |
1.2 | — | 2.2:1.0 |
| 11 | 0.3 | DMF | 70d | 16 | — | 98 | 32400 |
54100 |
1.7 | — | 1.9:1.0 |
| 12 | 0.4 | DMF | 70b | 1 | 10 | 87 | 20500 |
21500 |
1.1 | — | 2.6:1.0 |
| 13 | 0.3 | DMF | 120b | 0.5 | 10 | 96 | 47100 |
55400 |
1.2 | — | 2.9:1.0 |
| 14 | 0.2 | DMF | 25 | 16 | 10 | 85 | 47300 |
69700 |
1.5 | — | 5.6:1.0 |
| 15 | 0.3 | DMF | 70b | 0.5 | 10 | 99 | 55700 |
85000 |
1.5 | — | 3.6:1.0 |
| 16 | 0.35 | DMF | 70d | 64 | 10 | 90 | 48700 |
57200 |
1.2 | — | 13.0:1.0 |
| 17 | 0.05 | DMF | 80d | 144 | 10 | 76 | n.d.f | n.d. | n.d. | — | 0.3:1.0 |
| 18 | 3.0 | DMF | 70d | 64 | 10 | 66 | 23000 |
42100 |
1.8 | — | — |
Hydrolysis of poly(ester-triazole) 6
Hydrolysis experiments were carried out on poly(ester-triazole) 6 (Table 1, entry 16). In a typical procedure, polymer 6 (0.010 g) was suspended in a buffer solution (1 mL) and vigorously stirred at room temperature or at 80 °C. After filtration, the solution was concentrated at reduced pressure. The residue was dissolved in D2O, while the material that remained insoluble after hydrolysis was dissolved in CDCl3. Both solutions were analyzed by NMR spectroscopy. Buffer solutions were: pH 2 (0.1 M H3PO4/NaCl buffer), pH 7.4 (0.1 M Na2HPO4/KH2PO4 buffer), and pH 10 (0.1 M glycine/NaCl). Alternatively, 0.5 M TFA (1 mL) was used and the mixture was stirred at 65 °C overnight.Results and discussion
Monomer synthesis
Poly(ester-triazole)s were synthetized using the α-azido-ω-alkyne ester 5 as monomer. Compound 5 (Scheme 1) was obtained from D-galactone-1,4-lactone (1). The brominated intermediate 2 was prepared from 1,18 which was treated with sodium azide to yield the 6-azido methyl ester 3. Alkaline hydrolysis of the ester gave the O-isopropylidene protected galactonic acid 4. Esterification of the acid with propargyl alcohol was achieved using 1-hydroxybenzotriazole (HOBt) and N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDCI) as condensing agents, in the presence of N,N-diisopropylethylamine (DIPEA). Monomer 5 was obtained in 68% overall yield from 1.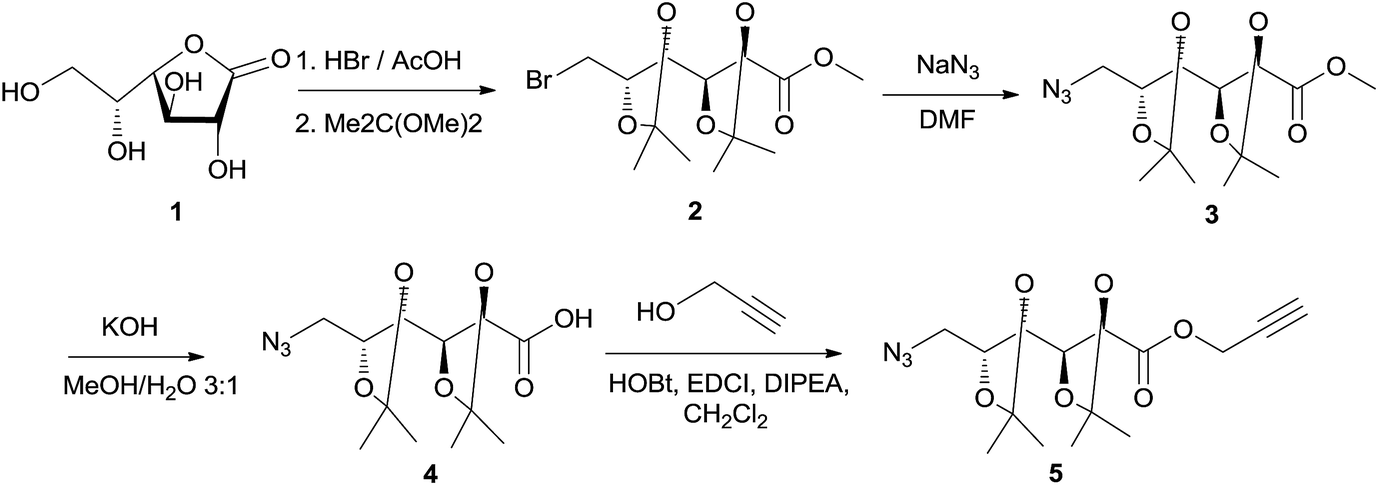 | ||
| Scheme 1 Synthesis of monomer 5 from D-galactono-1.4-lactone (1). | ||
The FTIR spectrum of 5 showed characteristic signals for alkyne and azide groups at 3243 and 2090 cm−1, respectively. The 1H NMR spectrum (Fig. 1(a)) exhibited characteristic resonances for H-1′a (4.80 ppm), H-1′b (4.78 ppm), and H-3′ (2.49 ppm), which confirmed the presence of the propargyl ester, together with signals from the sugar moiety. The 13C NMR spectrum of compound 5 showed the signals from the propargyl ester at 53.0, 77.0, 75.6, and 170.2 ppm (C-1′, C-2′, C-3′, and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, respectively).
O, respectively).
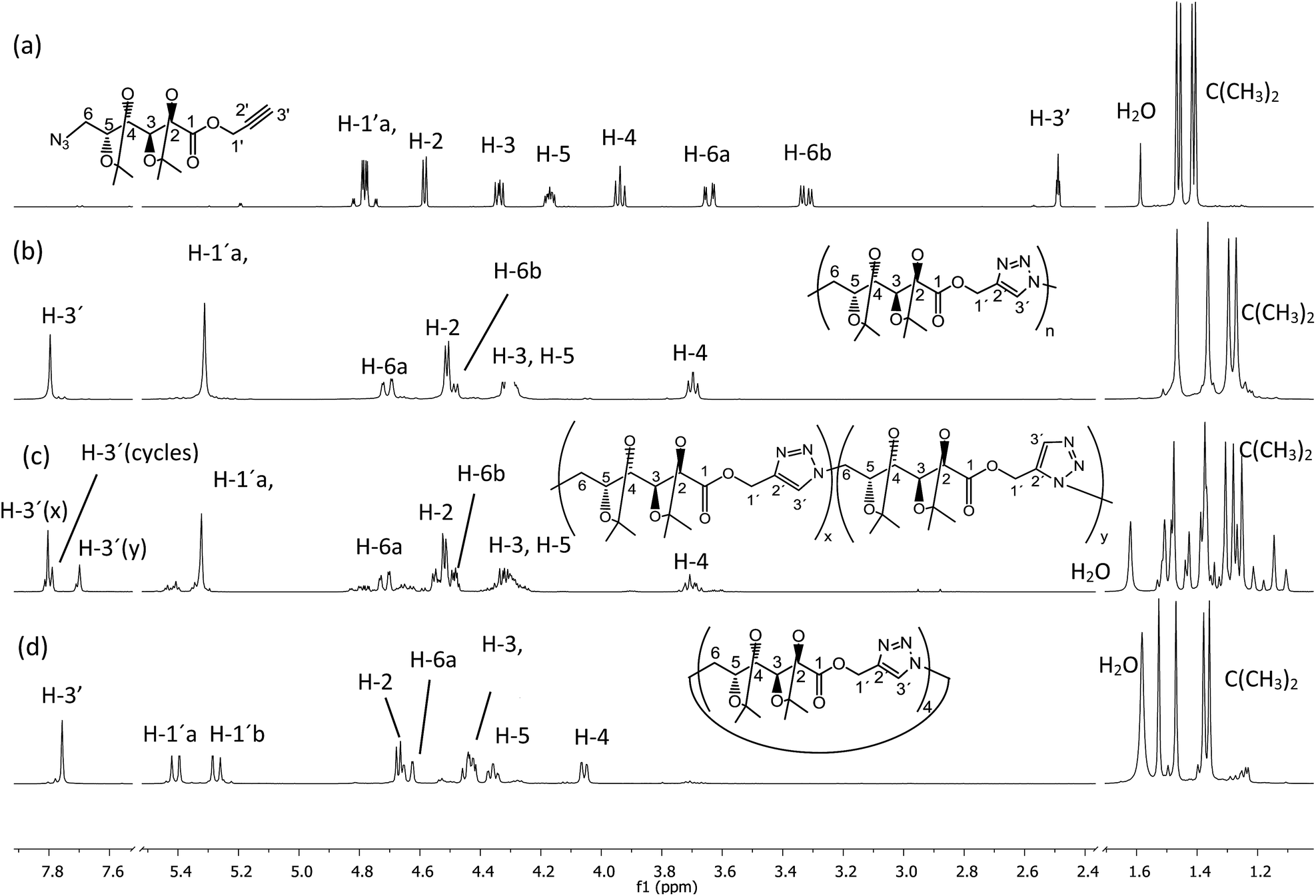 | ||
| Fig. 1 1H NMR (500 MHz, CDCl3) spectra of (a) monomer 5, (b) poly(ester-triazole) 6, (c) poly(ester-triazole) 7, and (d) cyclotetramer 10. | ||
Click polymerization
In order to optimize the polymerization conditions, the CuAAC reaction was carried out under varied monomer concentrations, heating conditions (conventional heating or microwave irradiation), reaction times and using solvents of different polarity (DMSO, DMF, THF, MeOH, or in bulk) as shown in Table 1. Thermal cycloaddition in the absence of copper was attempted as well. The products were analyzed by NMR and FTIR spectroscopies, and the molecular weight distributions were estimated by GPC.The CuAAC polymerization in bulk (Table 1, entry 1) gave mainly the expected poly(ester-triazole) 6 (Scheme 2). The FTIR spectrum suggested a high polymerization degree, since the absorption at 2089 cm−1 (from azide terminal group) was not observed. The CO band changed from 1758 cm−1 (monomer) to 1741 cm−1 in the poly(ester-triazole), thus absence of hydrogen bond was assumed for the carbonyl group.20
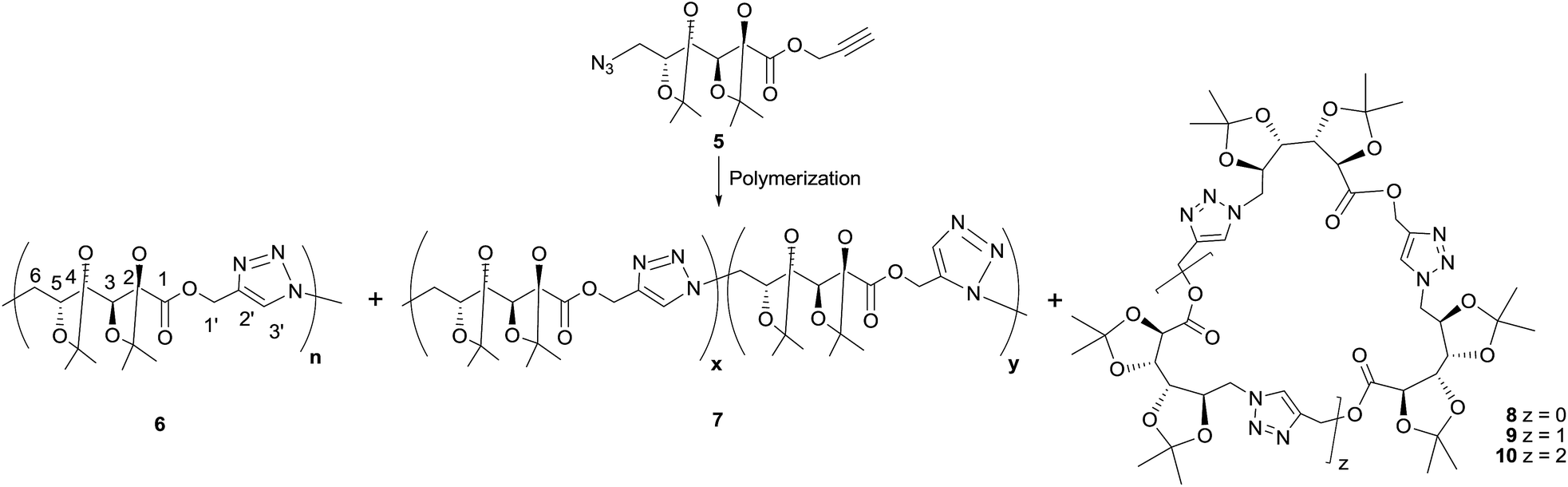 | ||
| Scheme 2 Poly(ester-triazole) synthesis. | ||
The polymers showed characteristic NMR resonances at 7.81 and 5.32 ppm, due to triazole-CH (H-3′) and OCH2 (H-1′), respectively. In polymerizations carried out without catalyst (entries 2 and 3), the triazole-CH signal at 7.81 ppm was accompanied by additional resonances at 7.78, 7.76, and 7.70 ppm. The signal at 7.70 ppm was assigned to the CH of the 1,5-disubstituted triazole ring15 (the y fragment in polymer 7), due to the absence of Cu(I) catalyst. The two remaining signals (7.78 and 7.76 ppm) were attributed to macrocyclic oligomers formed as by-products, as a result of the flexibility of the monomer chain. The random distribution of 1,4- and 1,5-disubstituted triazole units in the backbone of 7 led to NMR spectra of increased complexity. The low yield in entry 2 was probably caused by the high temperature in the MW reactor, but this was overcome by reducing the temperature and irradiation time (entry 3).
When CuAAC polymerizations were carried out in MeOH (entries 4–6), under MW irradiation for 1 h (entry 4) the broad resonance for triazole-CH at 7.81 ppm was more complex, and the additional signals at 7.73, 4.81, and 3.78 ppm (Fig. S13 in ESI†) were assigned to polymer chains modified by transesterification with the solvent. This side-reaction on the terminal propargyl ester would prevent further polymerization, and methanolysis of the ester groups included in the backbone (Scheme 3) led to splitting of the main chain in two segments, one containing the methyl ester (11) and the other with a terminal hydroxymethylene group linked to the 1,4-disubstituted triazole (12), which accounted for the resonances at 7.73 and 4.81 ppm. The signals of polymer 6 and the terminal triazole-CH in the ester by-products indicated an approximately 1:1 molar ratio, revealing a high degree of transesterification in rather short-length oligomers. With the aim of reducing this side-reaction, shorter reaction times (entry 5) and lower temperatures (entry 6) were tried. The methanolysis was reduced but not fully suppressed; therefore, MeOH was not a convenient solvent.
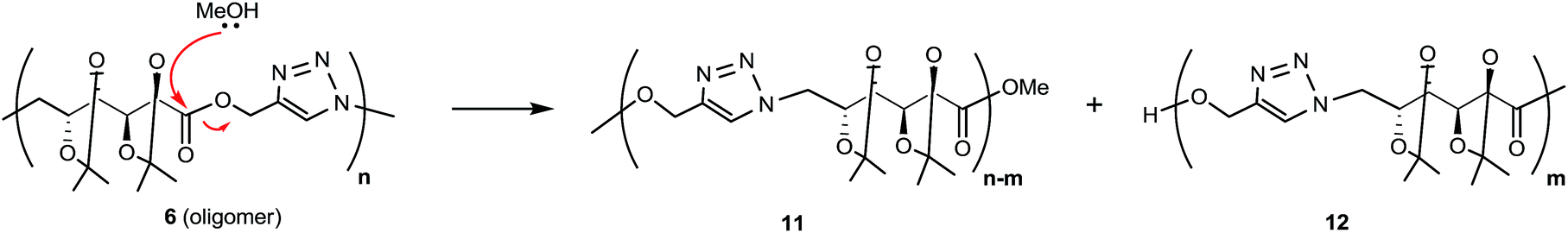 | ||
| Scheme 3 Transesterification products formed during polymerization in methanol. | ||
For polymerizations carried out in THF (entries 7–10) the best conditions (in terms of yield, molecular weight, and selectivity in triazole substitution) were achieved for entries 9 and 10, but the proportion of cyclic oligomers was high.
Polymerizations in DMF (entries 11–18) led to poly(ester-triazole)s in good yields but with variable proportions of cyclic by-products. CuAAC polymerization under conventional heating for a long period (entry 16) led a low content of the macrocycles. These compounds were analysed by tlc (Rf 0.52 and 0.44 in EtOAc) and were extracted by washing the polymeric material with hexane. The NMR spectra of the hexane-soluble fraction showed absence of terminal azide or alkynyl end groups, in accordance with their cyclic nature.
The formation of the cyclic products was favoured by CuAAC reactions in diluted solutions (entry 17). Fractionation by column chromatography gave a first fraction, which appeared as a single spot on tlc (Rf 0.52) but was, in fact, a mixture of two compounds, later identified as the cyclic dimer 8 and trimer 9 (in a 0.6:1.0 ratio respectively). Further fractions afforded the cyclic tetramer 10. The number of repeating units constituent of these cyclic compounds was determined by HRMS. The early eluting fraction gave two pairs of signals (m/z 679.2899, 701.2740 and 1018.4313, 1040.3821), corresponding to [M + H]+ and [M + Na]+ for dimer 8 and trimer 9, respectively; while the other fraction showed only m/z 1357.5703 and 1379.5000 corresponding to the cyclotetramer 10. In addition, the NMR spectra (Fig. 1) confirmed that triazole-CH gave signals at 7.76 and 7.78 ppm for trimer 9 and tetramer 10, respectively.
As expected, the polymerization with higher concentration of monomer 5 (3.0 M) led mostly to poly(ester-triazole) 6 (66%, entry 17), which upon purification was practically free of cycles 8–10. However, the molecular weight was lower than that recorded for entry 16. Probably, the higher concentration of 5 would promote precipitation of the polymer in earlier polymerization steps, lowering the molecular weight. Therefore, the CuAAC polymerization of 5 was carried out on preparative scale under the conditions of entry 16, which afforded polymer 6 in high yield (90%).
The poly(ester-triazole)s were soluble in many organic solvents, such as chloroform, dichloromethane, DMSO, and DMF, but were insoluble in water, MeOH, EtOAc, THF, acetone or hexane. The higher solubility of the polymers, in comparison with the poly(amide-triazole)s previously synthesized in our laboratory,15 could be attributed to the fact that the ester linkage, in contrast to the amide NH, cannot participate as hydrogen linkage donor. Also, the solubility could be improved by the more flexible backbone given by the O-isopropylidene protecting groups, in comparison with the fused 1,3-dioxane ring.
The molecular weights were estimated by GPC with polystyrene standards. Some samples gave bimodal distributions, with one peak centred at extremely high molecular weight values (300–500 kDa). As this unexpected molecular weight was attributed to aggregation, the samples were analysed using DMF with 0.05% LiBr and 100 mM ethylenediamine as the mobile phase, which led to a pronounced decrease in these peaks.
The poly(ester-triazole)s were also analysed by MALDI-TOF mass spectrometry using a wide variety of matrices and additives (salt dopants). As an example, the MALDI-TOF spectra of the poly(ester triazole) 6 obtained by CuAAC polymerization (Table 1, entry 13) was recorded under varied conditions, and the main peaks are listed in Table 2 (Fig. S19 in ESI†). When DHB was used as MALDI matrix, with or without addition of NaCl dopant, the [Mn + Na]+ adducts were detected up to m/z 3500 (n = 10, where n is the polymerization degree). By sample doping with of AgNO3, the [Mn + Ag]+ ions were detected again up to n = 10, together with [Mn + Na]+ minor signals. The efficiency of desorption/ionization for these oligomers was lower with CuOAc, as the main signals observed were [Mn + Cu]+ with n = 2–5. Additionally, when nHo·HCl was used as matrix in positive and negative ion modes, only high intense signals were registered for oligomers with n = 2–4. In positive ion mode, the [M + H]+ species appeared together with the corresponding [M + Na]+ and [M + K]+; while the negative ion mode spectrum showed [M + Cl]− peaks. All these results (tlc, NMR spectroscopy, ESI and MALDI MS) confirmed that compounds 8–10 (Scheme 2) were cyclic, whereas the oligomers with higher molecular weights (n > 5) were linear. The intensities of the ions with n = 2–4 in the MS recorded with nHo·HCl in positive and negative ion mode were also in agreement with their cyclic nature, since the cyclic members are usually more efficiently desorbed/ionized by MALDI than the corresponding linear isomers within different families of compounds (polysaccharides, proteins, polypeptides, and lipopeptides, among others).19,21
| Mn | Chemical formula | [M + H]+ | [M + Na]+ | [M + Ag]+ | [M + Cu]+ | [M + Cl]− | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Calcd | Exp | Calcd | Exp | Calcd | Exp | Calcd | Exp | Calcd | Exp | ||
| 2 | C30H43N6O12 | 680.30 | n.d. | 702.29 | 701.80 | 786.20 | 786.05 | 742.23 | 741.12 | 714.27 | 714.38 |
| 3 | C45H64N9O18 | 1019.44 | 1018.77 | 1041.43 | 1041.14 | 1125.34 | 1126.90 | 1081.37 | 1080.78 | 1053.41 | 1053.16 |
| 4 | C60H85 N12O24 | 1358.59 | 1357.95 | 1380.57 | 1380.51 | 1464.49 | 1465.35 | 1420.51 | 1420.00 | 1392.55 | 1391.66 |
| 5 | C75H106 N15O30 | 1697.73 | 1697.07 | 1719.71 | 1719.85 | 1803.73 | 1804.95 | 1759.65 | 1758.90 | 1731.69 | 1730.07 |
| 6 | C90H127 N18O36 | 2036.87 | 2036.65 | 2058.86 | 2059.01 | 2142.77 | 2143.97 | 2098.80 | n.d. | 2070.83 | n.d. |
| 7 | C105H148N21O42 | 2376.02 | 2376.24 | 2398.00 | 2398.50 | 2481.91 | 2482.41 | 2437.94 | n.d. | 2409.98 | n.d. |
| 8 | C120H169N24O48 | 2715.16 | n.d. | 2737.14 | 2738.86 | 2821.06 | 2822.76 | 2777.08 | n.d. | 2749.12 | n.d. |
| 9 | C135H190N27O54 | 3054.30 | n.d. | 3076.28 | 3077.03 | 3160.20 | 3161.97 | 3116.22 | n.d. | 3088.26 | n.d. |
| 10 | C150H211N30O60 | 3393.44 | n.d. | 3415.42 | n.d | 3499.34 | 3500.19 | 3455.36 | n.d. | 3427.40 | n.d. |
MALDI-MS gave also structural information on the species obtained by transesterification when MeOH was used as solvent (Table 1, entry 4, and Scheme 3). Now, the expected oligomers were detected as [Mn + H]+ and [Mn + Na]+ species when DHB was used as matrix (with or without addition of NaCl dopant), up to m/z 2500 (n = 6, Fig. S1 in ESI†). The adducts [Mn + Ag]+ and [Mn + Cu]+ (with n up to 6) were detected after the addition of AgNO3 or CuOAc, respectively. With nHo·HCl as matrix, the species [Mn + H]+ and [Mn + Na]+ were detected in positive ion mode while [Mn + Cl]− ions were detected in negative ion mode. The assignment of the main peaks is listed in Table S1 (ESI†).
Finally, in the spectrum obtained with DHB for the poly(ester-triazole) 7 prepared without addition of copper as catalyst (Table 1, entry 11), adducts were detected up to m/z 3500 (n = 10, [M10 + Na]+, Fig. S22 in ESI†). The recorded molecular weights and the corresponding molecular formula (Table S2, ESI†) were the same as those for the poly(ester-triazole) 6 prepared with copper catalysis (Table 1, entry 13; Fig. 2), in agreement with the expected regioisomerism.
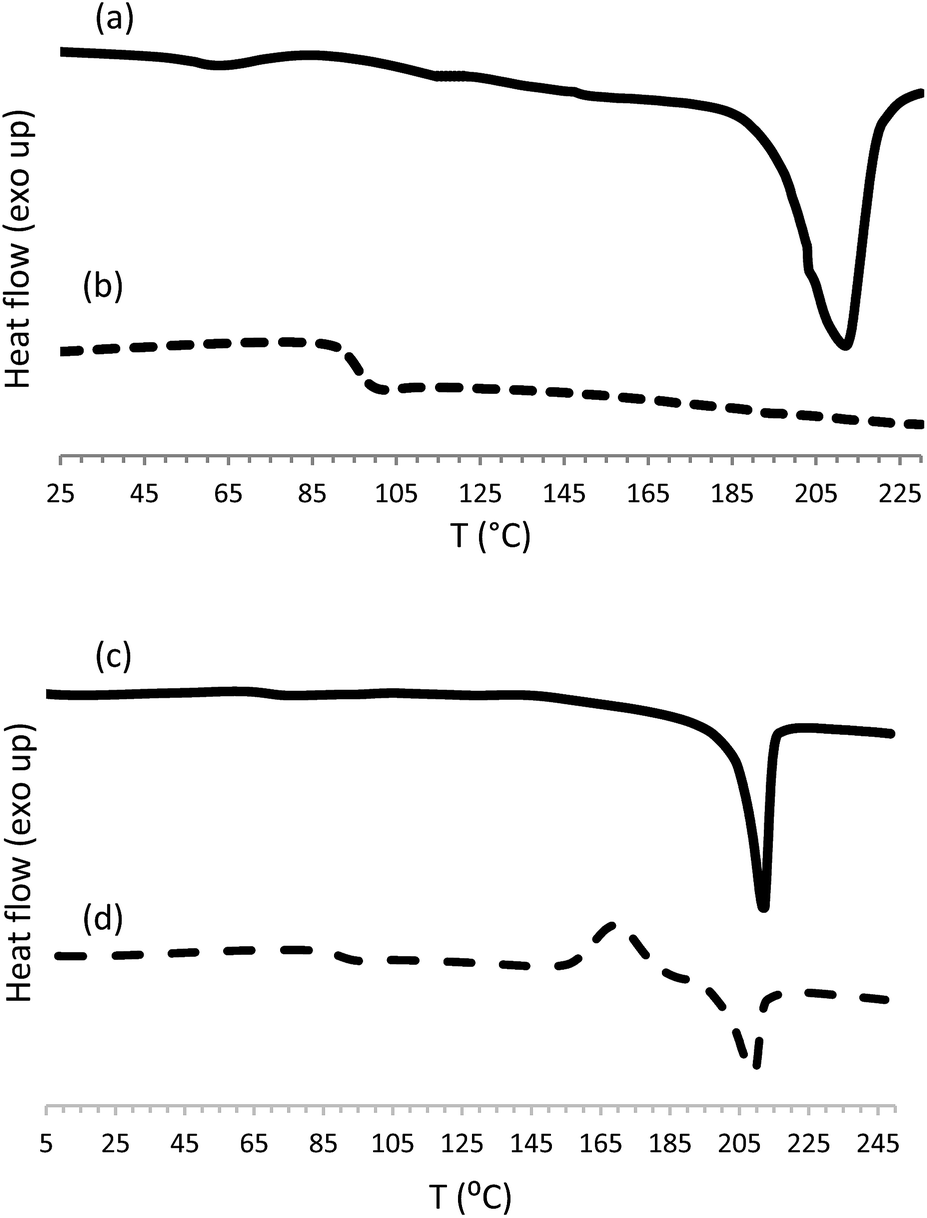 | ||
| Fig. 2 DSC curves for poly(ester-triazole) 6 obtained according to Table 1, entry 1, (a) first heating, (b) second heating; and cyclic tetramer 10 (c) first heating, (d) second heating. | ||
Comparison of the results obtained by MALDI MS and GPC indicated that only the lower linear oligomers were detected by the MS technique. This was consistent with the mass discrimination effect usually observed in polydisperse polymers, owing to a combination of factors related to desorption/ionization and detection processes. This mass discrimination in MALDI MS has been found to underestimate the polymer molecules of higher mass.22–24
Thermal analysis
The thermal stabilities of poly(ester-triazole)s were evaluated by TGA. The first weight loss (less than 10%), near to 100 °C, was attributed to the loss of residual water. Then, decomposition took place in two stages. The first one (305–335 °C), which accounted for the major mass loss (60–77%), could be related to the degradation of the carbohydrate residue; while the remaining triazole moiety was expected to be degraded above 490 °C (492–524 °C, 20–33% mass loss). All the polymers were stable below 250 °C (Table 3).| Entrya | Solvent | Cu | Tm (°C) | ΔHm (J g−1) | Tg (°C) |
|---|---|---|---|---|---|
| a Relative to Table 1.b Polymerization with Cu(I) catalyst.c Polymerization without catalyst.d Not observed. | |||||
| 1 | Bulk | +b | 212 | 30.4 | 96 |
| 2 | Bulk | −c | n.o.d | — | 92 |
| 3 | Bulk | − | n.o. | — | 98 |
| 4 | MeOH | + | 173 | 8.7 | 46 |
| 5 | MeOH | + | 190 | 14.0 | 66 |
| 6 | MeOH | + | 188 | 25.2 | 75 |
| 7 | THF | − | 167 | 8.9 | 75 |
| 8 | THF | + | 200 | 10.8 | 96 |
| 9 | THF | + | 162 | 21.8 | 96 |
| 10 | THF | + | 204 | 19.2 | 102 |
| 11 | DMF | − | 192 | 5.6 | 94 |
| 12 | DMF | + | 194 | 11.9 | 95 |
| 13 | DMF | + | 192 | 2.1 | 102 |
| 14 | DMF | + | 183 | 1.3 | 103 |
| 15 | DMF | + | 197 | 4.0 | 98 |
| 16 | DMF | + | 194 | 15.9 | 95 |
Monomer 5 was analysed by differential scanning calorimetry (DSC). In the first heating cycle a melting process was observed at 62 °C (ΔHm 96.8 J g−1) followed by a wide exothermic peak centred at 143 °C (ΔHr 637 J g−1) that was associated to the thermal cycloaddition (Fig. 2).
The polymers were also analysed by DSC (Table 3); a representative example is shown in Fig. 2. Some CuAAC poly(ester-triazole)s showed, in the first heating cycle, an endothermic phase transition associated to a melting process, in the range 194–212 °C.
In the second heating cycle, glass transitions (Tg) were observed for all the poly(ester-triazole)s at well-defined temperatures, with values related to the polymerization conditions employed. The lowest Tg (46.4–75.5 °C) were recorded for products synthesized in MeOH (entries 4–6) and in THF without catalyst (entry 7). This result was in accordance with the low molecular weight and less regular structure of the products (presence of randomly distributed triazole 1,5-disubstituted residues). For bulk polymerizations (entries 1–3) the Tg values increased to 92.3–97.8 °C, either with or without copper catalysis. A similar average Tg (∼98 °C) was obtained for CuAAC polymerizations carried out in THF (entries 8–10); while CuAAC polymerizations carried out in DMF (entries 11–18) gave the highest Tg (up to 103 °C).
Finally, the cyclotetramer isolated from the reaction mixture (Table 1, entry 17) exhibited in the first heating cycle a melting process at 212 °C (ΔHm 52.6 J g−1). The second heating cycle showed Tg (90 °C), crystallization (170 °C, ΔHc 29.5 J g−1) and melting (209 °C, ΔHm 31.1 J g−1) processes (Fig. 3). In accordance with its regular structure, compound 10 crystallized rapidly from the melt, to give a melting point similar to that observed in the first heating cycle.
 | ||
| Fig. 3 SEM images of poly(ester-triazole)s from Table 1: (a) and (b) entry 1; (c) and (d) entry 2; (e) and (f) entry 9; (g) and (h) entry 11. | ||
The poly(amide-triazole)s previously synthesized, derived from D-gluconolactone and having fused methylidene protecting groups in the repeating unit, gave Tg above 188 °C.13 The poly(ester-triazole)s prepared herein comprise more flexible repeating units, with isolated isopropylidene acetal rings, and the ester linkage could only behave as acceptor for hydrogen bonding. Because of these facts lower Tg values were observed, but they are still relatively high values in comparison with those of other carbohydrate-derived polytriazoles.7,8,12
SEM characterization of poly(ester-triazole)s
The microstructure of the polymers was examined by SEM (Fig. 3), which showed varied types of ordered structures. A highly entangled fibrillar network was registered for the polymer of entry 1, the sample with the highest Tm detected by DSC. The smaller fibrils (ca. 20 nm diameter) were bundled in rope like arrangements of 150 nm width and more than 1.7 μm length. On the other hand, the poly(ester-triazole) from entry 2, obtained in bulk and without catalyst, exhibited a morphology of stacked layers of ∼1 μm height. The sample of entry 9, obtained by CuAAC polymerization in THF, gave a tile like arrangement (80 nm depth, 170 nm width, 1000 nm length). Finally, the poly(ester-triazole) from entry 11 showed woven lamellae, with segments of 150–220 nm width.Hydrolytic degradation of poly(ester-triazole) 6
The hydrolytic degradation of poly(ester-triazole) 6 was carried out by subjecting samples in aqueous buffer solutions at pH 2, 7.4, or 10 at room temperature or 80 °C (Table 4, Scheme 4). The degradation was followed by NMR spectroscopy of the filtered supernatant (in D2O) and the remaining solid (in CDCl3). In acid buffered solution (pH 2), the material was stable at room temperature after 6 months. The polymer showed also to be stable at pH 2 and 7.4 at 80 °C for 4 days. The dissolution of the material was indicative of the degradation. Thus, polymer 6 dissolved completely when heated in a basic buffer (pH 10) at 80 °C during 4 days. The hydrolysed mixture was concentrated and analysed by NMR spectroscopy in D2O (Fig. 4). The spectrum showed formation of a single degradation product, with the isopropylidene and triazole groups (triazole-CH at 8.06 ppm) remaining. In addition, the upfield shifting (4.73 ppm) of the hydroxymethyl triazole (H-1′) revealed the hydrolysis of the ester linkage. This was confirmed by the downfield shifting for C-1 from 170.7 ppm (in the polymer) to 176.8 ppm in the 13C NMR spectrum of the product, characteristic of ester/acid conversion. Therefore, the degradation product was the ω-hydroxyacid 13. Under strong acid conditions (0.5 M TFA) at 65 °C for 16 h, the polymer was completely hydrolysed and deprotected to give the ω-hydroxyacid 13, as the spectrum showed the signal of H-1′ at 4.51 ppm (C-1′at 175.4 ppm) and the absence of isopropylidene methyl groups. The resulting material was insoluble in either CDCl3 or D2O, but soluble in DMSO-d6. | ||
| Scheme 4 Products of hydrolytic degradation of poly(ester-triazole) 6. | ||
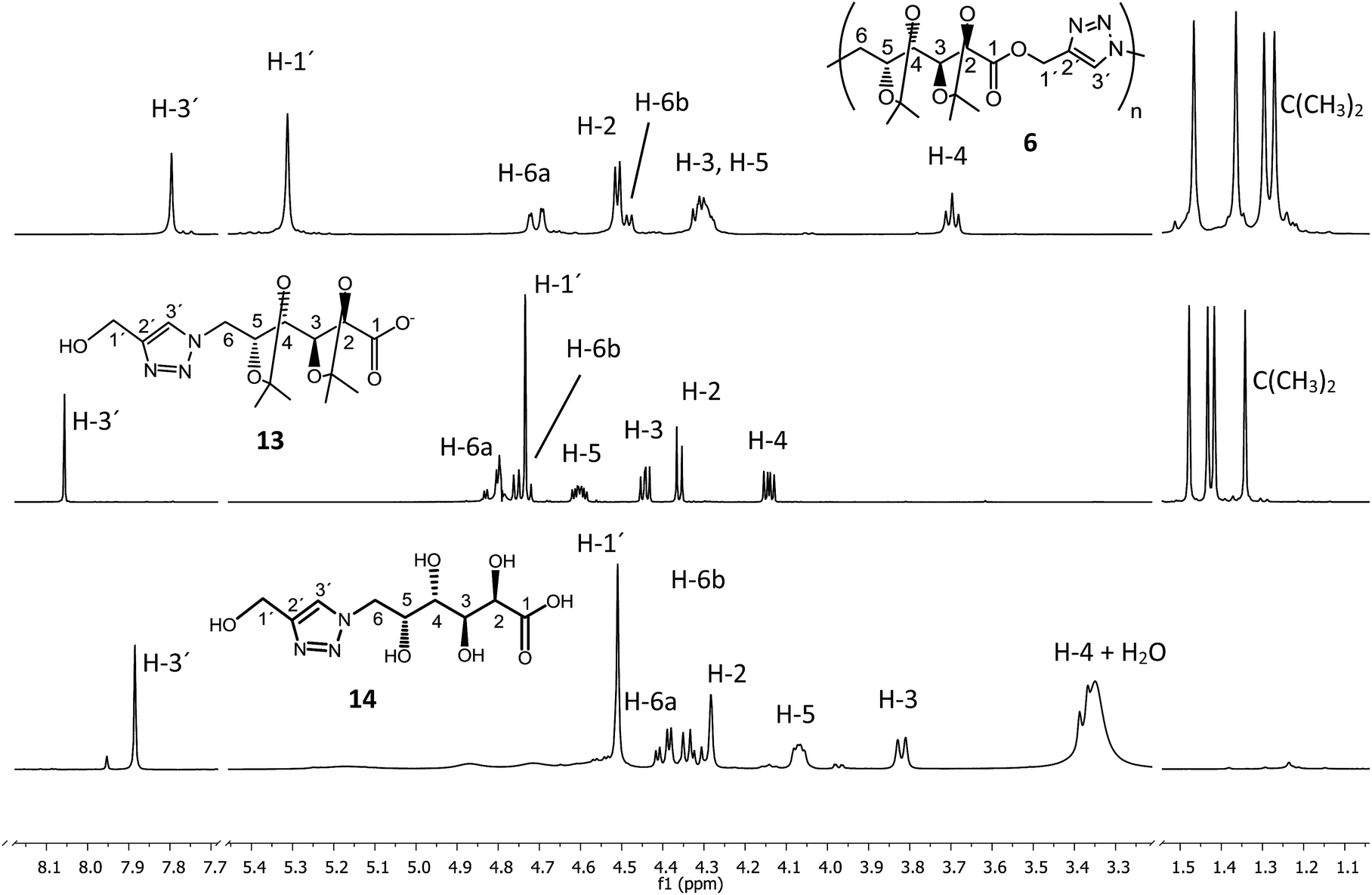 | ||
| Fig. 4 1H NMR (500 MHz) spectra of (a) poly(ester-triazole) 6 (in CDCl3) and degradation products (b) 13 at 25 °C (in D2O) or (c) 14 at 80 °C (in DMSO-d6). | ||
Conclusions
An α-azide-ω-alkynyl ester was synthesized in four steps from D-galactose with a very good overall yield (68%). This monomer showed to be a convenient precursor of carbohydrate-derived poly(ester-triazole)s by the green CuAAC or thermal polymerization. High molecular weight polymers (Mw in the range 35–85 kDa) were obtained either in solution or in bulk. The materials were soluble in chloroform, dichloromethane, DMSO, and DMF, but insoluble in water, methanol, ethyl acetate, THF or hexane. NMR and MALDI-MS analysis of the materials revealed that cyclic oligomers were also formed. These byproducts could be readily detected by MS using an appropriate combination of matrix + dopant (e.g., DHB + CuOAc) in positive ion mode, and especially with nHo·HCl in negative ion mode. Linear polymer formation prevailed under optimized polymerization conditions. The thermal analysis revealed that the materials were stable up to 300 °C, and they showed glass transition values in the range 90–100 °C. The polymers underwent degradation in either a basic (pH 10, 80 °C for 4 days) or acid media (0.5 M TFA at room temperature for 16 h), to give respectively an acetal protected ω-hydroxyacid (6-deoxy-6-(4′-(hydroxymethyl)-1′H-1′,2′,3′-triazol-1′-yl)-2,3:4,5-di-O-isopropylidene-D-galactonic acid), or a fully deprotected ω-hydroxyacid (6-deoxy-6-(4′-(hydroxymethyl)-1′H-1′,2′,3′-triazol-1′-yl)-D-galactonic acid).Despite the great achievement of CuAACP, one of the main drawbacks is the presence of copper residues in the resultant materials, which are difficult to be completely removed because of the coordination of Cu species with triazole rings. However, in this work, the residual Cu could be efficiently removed as the stability and high insolubility of the polymers in water facilitate the washings. These polymers could be useful for the formation of nanoparticles that may be hydrolysed in appropriate acid conditions. These investigations are underway.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the National Research Council of Argentina (CONICET, Project PIP 11220150100443CO, Project PUE 22920160100068CO), the National Agency of Science and Technology Promotion (ANPCYT, PICT 2016-0130) and the University of Buenos Aires (UBA, Projects 20020170100403BA and 20020170100110BA) is gratefully acknowledged. MVR is a fellow from CONICET. GP, REB, AAK and OV are research members from CONICET. The Ultraflex II (Bruker) TOF/TOF mass spectrometer was supported by a grant from ANPCYT, PME 2003-125.References
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 (Angew. Chem., 2002, 114, 2708) CrossRef CAS PubMed; C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057 CrossRef.
- M. Meldal, Macromol. Rapid Commun., 2008, 29, 1016 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 (Angew. Chem., 2001, 113, 2056) CrossRef CAS PubMed.
- D. Dheer, V. Singh and R. Shankar, Bioorg. Chem., 2017, 71, 30 CrossRef CAS PubMed.
- R. Huisgen, G. Szeimies and L. Möbius, Chem. Ber., 1967, 100, 2494 CrossRef CAS.
- V. K. Tiwari, B. B. Mishra, K. B. Mishra, N. Mishra, A. S. Singh and X. Chen, Chem. Rev., 2016, 116, 3086 CrossRef CAS PubMed.
- H. Uyama, Polym. J., 2018, 50, 1003 CrossRef CAS; A. Gandini, T. M. Lacerda, A. J. F. Carvalho and E. Trovatti, Chem. Rev., 2016, 116, 1637 CrossRef PubMed; A. Gandini and T. M. Lacerda, Prog. Polym. Sci., 2015, 48, 1 CrossRef.
- V. E. Manzano, A. A. Kolender and O. Varela, Synthesis and applications of carbohydrate-based polyurethanes, in Industrial Applications of Renewable Biomass Products. Past, Present and Future, ed. S. N.Goyanes and N. B.D'Accorso, Springer Science, 2017, pp. 1–43 CrossRef CAS PubMed; J. A. Galbis, M. G. García-Martín, M. V. de Paz and E. Galbis, Chem. Rev., 2016, 116, 1600 CrossRef CAS PubMed.
- C. Besset, S. Binauld, M. Ibert, P. Fuertes, J. P. Pascault, E. Fleury, J. Bernard and E. Drockenmuller, Macromolecules, 2010, 43, 17 CrossRef CAS.
- P. D. Raytchev, C. Besset, E. Fleury, J. P. Pascault, J. Bernard and E. Drockenmuller, Pure Appl. Chem., 2013, 85, 511 CAS.
- I. Molina-Pinilla, M. Bueno-Martínez, K. Hakkou and J. A. Galbis, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 629 CrossRef CAS.
- M. Bueno-Martínez, I. Molina-Pinilla, K. Hakkou and J. A. Galbis, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 413 CrossRef.
- M. Bueno, I. Molina and J. A. Galbis, Polym. Degrad. Stab., 2012, 97, 1662 CrossRef CAS.
- K. Hakkou, M. Bueno-Martínez, I. Molina-Pinilla and J. A. Galbis, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 2481 CrossRef CAS.
- D. M. Fidalgo, A. A. Kolender and O. Varela, Mater. Today Commun., 2015, 2, 70 CrossRef.
- P. T. Anastas and J. C. Warner Green Chemistry: Theory and Practice, Oxford University Press, New York 1998, p. 30 Search PubMed.
- H. Ye, K. Zhang, D. Kai, Z. Li and X. J. Loh, Chem. Soc. Rev., 2018, 47, 4545 RSC; H. Nakajima, P. Dijkstra and K. Loos, Polym., 2017, 9, 18 Search PubMed; R. P. Brannigan and A. P. Dove, Biomater. Sci., 2017, 5, 9 RSC; M. Murariu and P. Dubois, Adv. Drug Delivery Rev., 2016, 107, 17 CrossRef CAS PubMed.
- M. V. Rivas, A. A. Kolender, G. Duhirwe and O. Varela, in Carbohydrate Chemistry: Proven Synthetic Methods, ed. C. Vogel and P. Murphy, CRC Press, London, UK, 2017, ch. 27, vol. 4 Search PubMed.
- H. Nonami, S. Fukui and R. Erra-Balsells, J. Mass Spectrom., 1997, 32, 287 CrossRef CAS.
- D. P. Suhas, H. M. Jeong, T. M. Aminabhavi and A. V. Raghu, Polym. Eng. Sci., 2014, 54, 24 CrossRef CAS; A. V. Raghu, G. S. Gadaginamath, H. M. Jeong, N. T. Mathew, S. B. Halligudi and T. M. Aminabhavi, J. Appl. Polym. Sci., 2009, 113, 2747 CrossRef.
- M. L. Salum, L. M. Itovich and R. Erra-Balsells, J. Mass Spectrom., 2013, 48, 1150 CrossRef PubMed.
- D. C. Schriemer and L. Li, Anal. Chem., 1996, 68, 2721 CrossRef CAS PubMed.
- S. F. Macha and P. A. Limbach, Curr. Opin. Solid State Mater. Sci., 2002, 6, 213 CrossRef CAS.
- M. S. Engler, S. Crotty, M. J. Barthel, C. Pietsch, U. S. Schubert and S. Böcker, Rapid Commun. Mass Spectrom., 2016, 30, 1233 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra00398c |
| This journal is © The Royal Society of Chemistry 2019 |