
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiscovery of human TyrRS inhibitors by structure-based virtual screening, structural optimization, and bioassays†
Shenzhen
Huang‡
 a,
Xiang
Wang‡
bc,
Guifeng
Lin
a,
Jie
Cheng
a,
Xiuli
Chen
a,
Weining
Sun
d,
Rong
Xiang
b,
Yamei
Yu
a,
Linli
Li
d and
Shengyong
Yang
*a
a,
Xiang
Wang‡
bc,
Guifeng
Lin
a,
Jie
Cheng
a,
Xiuli
Chen
a,
Weining
Sun
d,
Rong
Xiang
b,
Yamei
Yu
a,
Linli
Li
d and
Shengyong
Yang
*a
aState Key Laboratory of Biotherapy and Cancer Center, West China Hospital, Sichuan University/Collaborative Innovation Center of Biotherapy, Chengdu, Sichuan 610041, China. E-mail: yangsy@scu.edu.cn
bDepartment of Clinical Medicine, School of Medicine, Nankai University, Tianjin 300071, China
cDepartment of Chemistry, Fudan University, Shanghai 200433, China
dKey Laboratory of Drug Targeting and Drug Delivery System of Ministry of Education, West China School of Pharmacy, Sichuan University, Chengdu, Sichuan 610041, China
First published on 22nd March 2019
Abstract
The human tyrosyl transfer-RNA (tRNA) synthetase (TyrRS), which is well known for its essential aminoacylation function in protein synthesis, has been shown to translocate to the nucleus and protect against DNA damage caused by external stimuli. Small molecules that can fit into the active site pocket of TyrRS are thought to affect the nuclear role. The exploitation of TyrRS inhibitors has attracted attention recently. In this investigation, we adopted a structure-based virtual screening strategy and subsequent structure–activity relationship analysis to discover new TyrRS inhibitors, and identified a potent compound 5,7-dihydroxy-6,8-bis((3-hydroxyphenyl)thio)-2-phenyl-4H-chromen-4-one (compound 11, Ki = 8.8 μM). In intact HeLa cells, this compound showed a protective effect against DNA damage. Compound 11 is a good lead compound for the further development of drugs against disorders caused by DNA damage.
Introduction
Resveratrol (3,4,5-trihydroxystilbene, RSV), a natural product, is an important component of red wine. RSV has long been thought to impart benefits in lifespan expectancy,1,2 cancer chemopreventive activity,3 cardioprotective,4 and neuroprotective effects,5 by launching a stress response.6,7 However, because it has low activity and the target of its action is not clear;8RSV is only marketed as a health care product. Human tRNA synthetase is a potent PARP1-activating effector target for RSV.Recently, Sajish et al. demonstrated that RSV could interact with human tyrosyl transfer-RNA synthetase (TyrRS) and hence play its beneficial role.7 TyrRS was originally known for its essential aminoacylation function in protein synthesis. Recent studies have found a new function for TyrRS in DNA damage protection.7,9,10 External stimuli, such as oxidative stress, can induce TyrRS to rapidly translocate from the cytosol to the nucleus. The nuclear-localized TyrRS activates transcription factor E2F1 to upregulate the expression of DNA damage repair genes such as BRCA1 and RAD51.9 In particular, RSV can also trigger the translocation of TyrRS to the nucleus by directly binding to the active site of TyrRS, leading to the activation of NAD+-dependent auto-poly-ADP ribosylation of poly(ADP ribose) polymerase 1 (PARP-1). PARP-1 is a major stress response and DNA repair factor that has a considerable impact on longevity.11 PARP-1 activated by TyrRS leads to activation of a series of protective genes, including the tumor suppressor gene p53, FOXO3A, or SIRT6.7 Therefore, TyrRS is an attractive target for developing therapeutics for diseases of the nervous system diseases and for DNA damage protection.
To date, RSV is the only small molecule compound with an inhibition constant (Ki) value in the micromolar range that has been identified as an inhibitor of TyrRS.7 However, RSV has also been identified as a modulator against many targets.12–15 Therefore, discovery of novel potent and selective TyrRS inhibitors, with scaffolds different from RSV, is desirable.
Here, we report the discovery of a new series of TyrRS inhibitors containing the scaffold 5,7-dihydroxy-2-phenyl-6,8-bis(phenylthio)-4H-chromen-4-one. These compounds were identified by a high throughput virtual screening and subsequent structure–activity relationship (SAR) analysis. The most active compound 5,7-dihydroxy-6,8-bis((3-hydroxyphenyl)thio)-2-phenyl-4H-chromen-4-one (11) was further investigated in binding mode analysis with TyrRS and bio-functional studies.7,9,10,16,17 Compound 11 is a promising lead compound for the further development of drugs for DNA damage related diseases.
Results and discussion
Structure-based virtual screening of novel hit compounds against TyrRS
The virtual screening process is schematically shown in Fig. 1A. The chemical databases included the commercial chemical library Specs and an in-house database. In the first step, compounds were filtered by Lipinski's rules and for pan-assay interference compounds (PAINS) in Pipeline Pilot 8.5, which is used for filtering pan-assay interference compounds18,19 and has been used with great success in drug discovery.18,20 Then molecular docking was carried out with the remaining compounds. Here, the GOLD program was used because it provides good docking performance.21 All the calculations were carried out on the platform of Accelrys Discovery Studio 3.1 (Accelrys Inc., San Diego, CA, USA). The crystal structure of TyrRS in complex with RSV (PDB entry 4Q93) was used for the docking studies,7 and RSV was used as a reference molecule with a root-mean-square deviation (RMSD) of 0.57 Å (Fig. S2†). Next, the consensus scoring strategy was used to score the compounds that may target TyrRS. Here, the scoring functions, GoldScore incorporated in the GOLD program and ID-Score developed by our laboratory,22 were selected for the consensus scoring strategy because they have provide good performance for scoring.18,22 Finally, 30 compounds were selected from the top ranked compounds by the consensus scoring and visual inspection.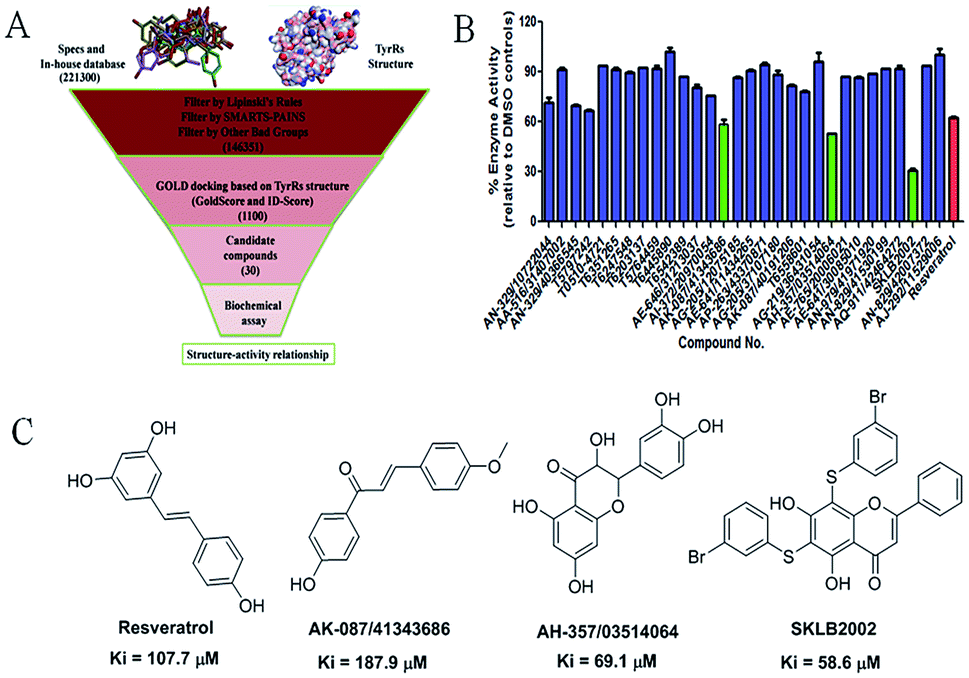 | ||
| Fig. 1 (A) Schematic diagram of the underlying workflow of the TyrRS drug discovery. (B) Enzyme activity of the 30 candidate molecules based on virtual screening at 100 μM. The red columnar bar represents the activity of the reference compound RSV. (C) The structures of the hit compounds and RSV. | ||
TyrRS enzyme inhibition assays
The 30 in silico-screened compounds were tested for inhibitory activity against TyrRS using the ATP–pyrophosphate exchange assay (Fig. 1B). From the enzyme inhibition assay, the hit compounds AK-087/41343686, AH-357/03514064, and SKLB2002 were identified, they showed an enzyme activity < 60% against TyrRS at the concentration of 100 μM and inhibited TyrRS activity in biochemical assays in a dose-dependent manner with Ki values of 187.9, 69.1, and 58.6 μM, respectively. The positive control compound RSV showed weak potency (Ki = 107.7 μM) (Fig. 1C and S1†). Because the hit compounds AH-357/03514064 and SKLB2002 have similar scaffolds, and compound SKLB2002 was the most active compound and contained the new scaffold 5,7-dihydroxy-2-phenyl-4H-chromen-4-one, further structural optimization and SAR analysis were carried out on this compound.Chemistry
All of the target compounds were prepared from the commercially available 5,7-dihydroxy-2-phenyl-4H-chromen-4-one. The general synthetic routes for compounds 1 (SKLB2002), 2–12, and 14–31 shown in Scheme 1. In route 1, intermediate 1a was prepared by a similar synthetic method as described in the literature.23 Target compounds 1–12 were then obtained by a conventional coupling reaction of various benzenethiols and intermediate 1a in DMF in yields of 40–70%. In route 2, intermediate 13a was obtained by a displacement reaction with iodomethane and commercially available 5,7-dihydroxy-2-phenyl-4H-chromen-4-one. Intermediate 13b was then obtained by a classic electrophilic substitution reaction between NIS and the previously synthesized intermediate 13a in quantitative yield. Target compounds 14–22 were synthesized from various benzenethiols and intermediate 13b in DMF. Finally, the target compounds 23–31 were obtained by demethylation of compounds 14–22 with boron tribromide.24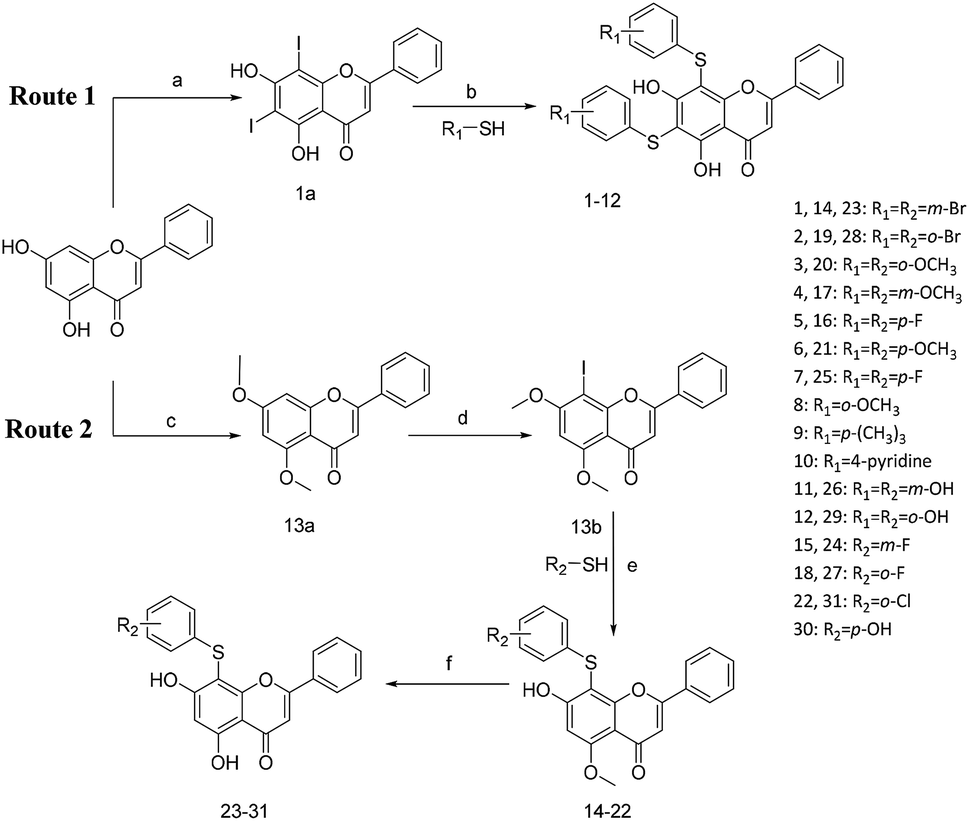 | ||
| Scheme 1 Reagents and conditions: (a) HNO3, I2, CH3COOH, RT. (b and e) Pd2(dba)3, X-phos, Cs2CO3, DMF, N2, 110 °C. (c) CH3I, K2CO3, DCM, RT. (d) NIS, CF3COOH, 80 °C. (f) BBr3, DCM, RT. | ||
SAR analysis and optimization of hit compounds
The inhibitory effects of the synthesized compounds on human TyrRS are displayed in Tables 1 and 2. As can be seen from Table 1, the introduction of methyl, methoxyl, and tert-butyl groups, that is compounds 3, 6, 8, and 9, which substantially diminished activity. The series of derivatives (1, 2, 5, 7, 11, and 12) revealed that the presence of halogen or hydroxyl groups in the skeletal structure of these molecules was desirable; compound 7 and, in particular, compound 11, displayed an inhibition rate larger than 80% at a concentration of 100 μM. Compounds 7 and 11 strongly inhibited TyrRS with Ki values of 10.6 and 8.8 μM, respectively, which were considerably higher than that shown by the positive control RSV (Ki = 107.7 μM, Fig. 2 and S1†).
|
|
|||||
|---|---|---|---|---|---|
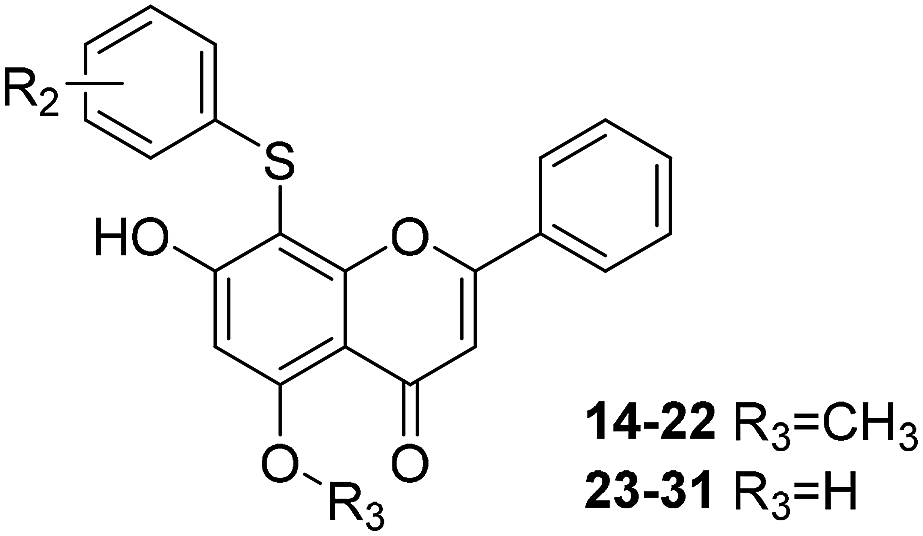
| Cpd | R2 | Enzyme activity@100 μM (%) | Cpd | R2 | Enzyme activity@100 μM (%) |
| 14 | m-Br | 86.42 | 23 | m-Br | 91.4 |
| 15 | m-F | 68.51 | 24 | m-F | 75.12 |
| 16 | p-F | 76.37 | 25 | p-F | 56.25 |
| 17 | m-OCH3 | 71.84 | 26 | m-OH | 93.16 |
| 18 | o-F | 73.76 | 27 | o-F | 87.04 |
| 19 | o-Br | 69.55 | 28 | o-Br | 72.44 |
| 20 | o-OCH3 | 79.91 | 29 | o-OH | 81.88 |
| 21 | p-OCH3 | 73.88 | 30 | p-OH | 78.55 |
| 22 | m-Cl | 77.86 | 31 | m-Cl | 87.07 |
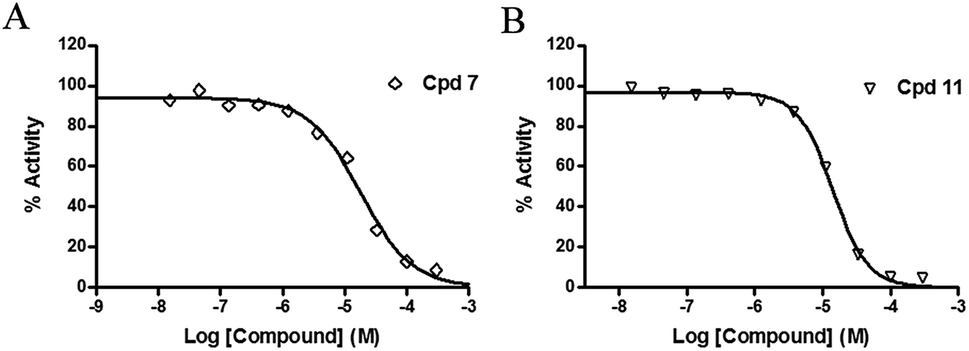 | ||
| Fig. 2 Enzyme activity data for TyrRS with compounds 7 and 11. (A and B) Determination of the Ki values of compounds 7 and 11, respectively. | ||
As can be seen from Table 2, the R2 and R3 groups were also optimized. However, deletion of the benzenethiol-substituted unit at the 18-position of in the skeletal structure of these molecules was not ideal. Compounds 14–31 showed weaker potency compared with the benzenethiol-substituted compounds. Compound 11, Ki = 8.8 μM, was the most active compound and was the most promising inhibitor in the present series of derivatives.
Differential scanning fluorimetry (DSF) and differential scanning calorimetry (DSC) assays
DSF is a good platform for screening protein stability conditions and has the advantages of being fast and inexpensive. Because of the small amount and low concentration of protein required, DSF can be used to identify low-molecular-weight ligands. A DSF assay was employed to understand the relationship between TyrRS inhibitor binding and protein construct stability25,26 (Fig. 3A and B). The impact of the binding of RSV and compound 11 on the TyrRS was explored by determining the changes in the melting temperature (Tm) of the protein in the absence and the presence of ligand. The shift in the Tm as defined by the relative midpoint of the denaturation profile assay indicated that the thermal stability was dependent on the ligand concentration. As shown in Fig. 3A, the presence of compound 11 increased the Tm value of the native TyrRS protein, showing that this class of inhibitor retained the ability to bind and stabilize the protein construct. Similarly, a significant increase in the Tm value was also observed for RSV, a specific binding ligand for this protein (Fig. 3B).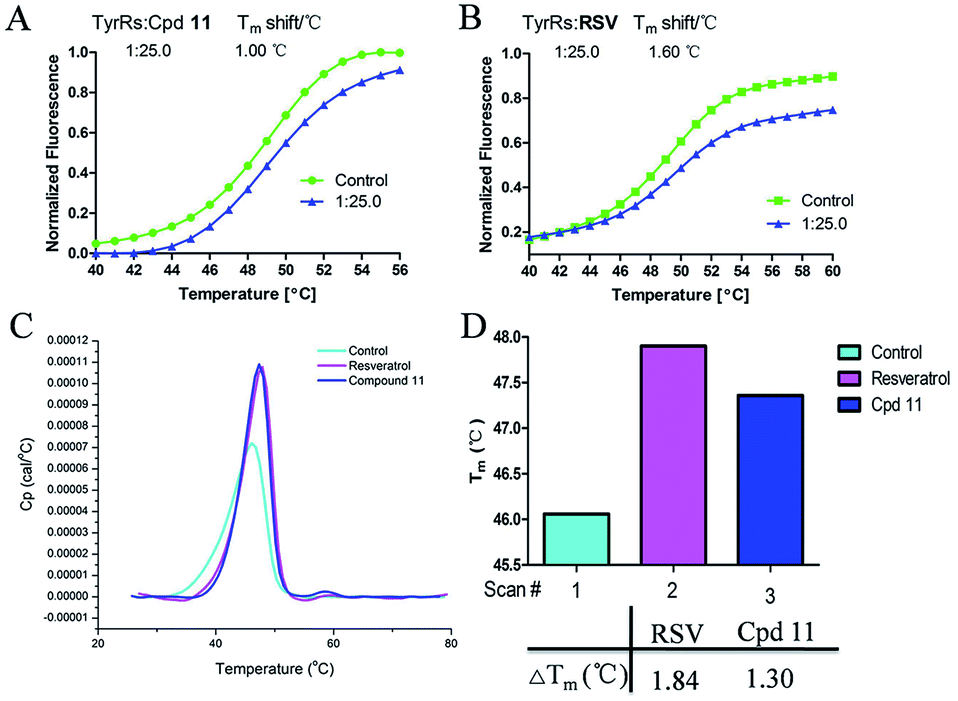 | ||
| Fig. 3 DSF and DSC were used to test the stabilization of TyrRS by RSV and compound 11. (A) The Tm value of compound 11. (B) The Tm value of RSV. Also shown are graphs of the unfolding transition of 2 μM TyrRS in the presence of 50 μM RSV or compound 11, respectively. (C and D) Representative DSC melting profiles for TyrRS with RSV or compound 11; ΔTm of RSV and compound 11 obtained from DSC. | ||
DSC, a thermal analysis method, has become an efficient experimental technique for determination of the thermodynamic properties of biological macromolecules such as proteins.27–29 A DSC assay was also performed to determine the Tm values for TyrRS. In the DSC assay, the Tm values of the TyrRS protein in the absence and presence of ligands were determined by the midpoint of the rise in each curve30,31 (Fig. 3C and D). The results obtained were comparable with the DSF method and showed the same trends. As shown in Fig. 3C and D, the presence of RSV or compound 11 shifted the native TyrRS protein Tm value by more than 1.84 or 1.30 °C higher, respectively.
Binding-mode analysis
Fig. 4 shows a possible binding mode of compound 11 with TyrRS. For comparison, RSV was also shown in the active site of TyrRS (PDB entry 4Q93). Obviously compound 11 occupies the same binding site but a larger area than RSV does. The 3-(λ1-sulfanyl)phenol group at the 6, 8-position of 5,7-dihydroxy-2-phenyl-4H-chromen-4-one is orientated with the H atom of the OH group and the S atom hydrogen-bonded to the Asp173 or Trp40 residues in the active site of TyrRS. The O atom in the 7-hydroxy branch of compound 11 formed another hydrogen-bonding interaction with the Gln170 residue. Compound 11 also formed hydrophobic interactions with residues Tyr39, Tyr52, Leu72, and Phe183 in the active site of TyrRS.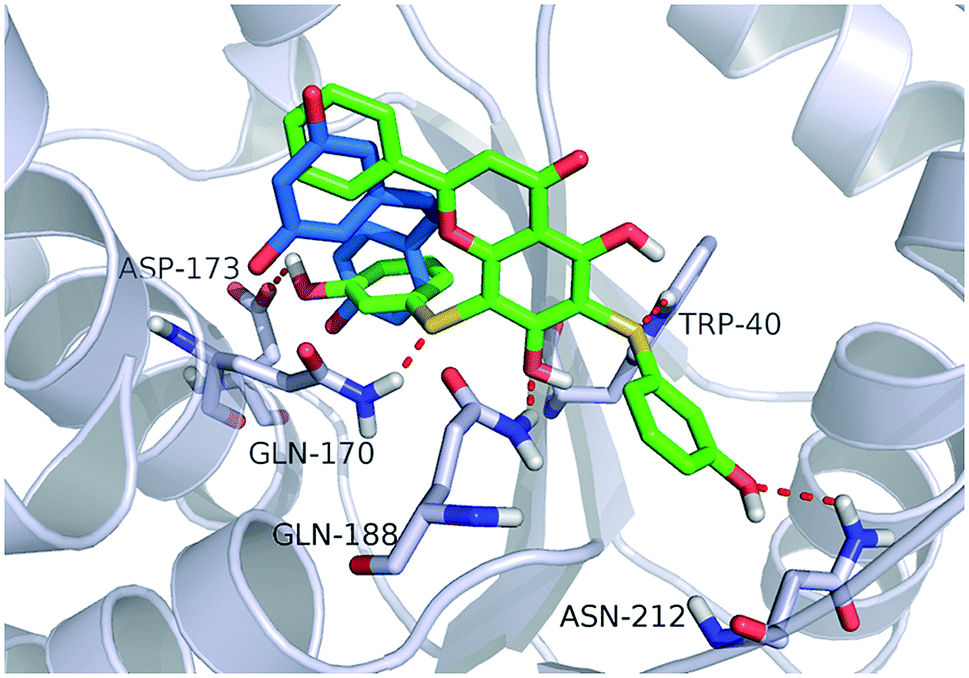 | ||
| Fig. 4 Binding modes of compound 11 and RSV with TyrRS (PDB code 4Q93). Compound 11 is shown as green sticks, and RSV is displayed as marine sticks. | ||
Protection against DNA damage
DNA damage can be induced by various stimuli, including UV radiation from the environment, oxygen produced by endogenous reactive oxygen species, and chemicals for disease treatment. DNA damage is considered a major source of gene mutations and a cause of many diseases, for example, neurodegenerative diseases, cardiovascular disease, immune dysfunction, and cancer.32,33 Even though there is a DNA damage response in cells to fight against DNA damage caused by endogenous or environmental stimulus, it is not sufficient for cells to eliminate all DNA damage. Thus, chemicals that can help cells fight against DNA damage are desirable. Serum starvation (SS, oxidative stress) and RSV have been shown to affect TyrRS nuclear localization.7,9 This observation prompted us to investigate whether compound 11 can regulate TyrRS nuclear localization in HeLa cells. By immunoblotting of the cytoplasmic and nuclear fractions from cells, we found that treatment with compound 11 dramatically promoted the nuclear localization of TyrRS in a concentration-dependent manner in HeLa cells, without the influence of TyrRS in the cytoplasm (Fig. 5).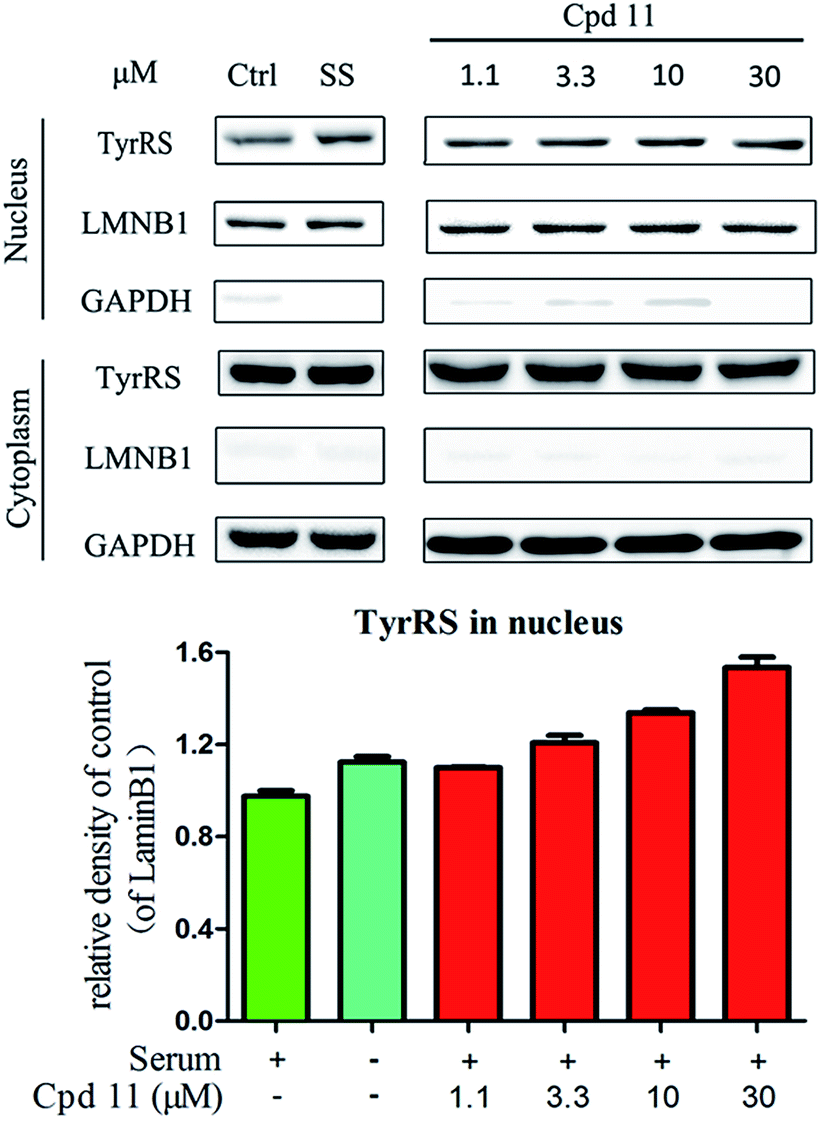 | ||
| Fig. 5 Compound 11 promoted TyrRS nuclear localization in HeLa cells. Hela cells were treated with compound 11 (1.1, 3.3, 10, or 30 μM) or DMSO for 8 h. Cells cultured in serum-free media for 8 h were under serum starvation. The cytoplasmic and nuclear fractions from cells were subjected to immunoblot assays. Quantification of immunoblots is shown below. | ||
Having demonstrated that nuclear localization of TyrRS is promoted by compound 11, we investigated the potential for TyrRS inhibitors to protect cells from DNA damage. We pre-treated HeLa cells with compound 11 for 24 h before inducing DNA damage in cells by treatment with 30 μM cisplatin for 24 h. To visualize the DNA damage level in cells, phosphorylated histone H2A.X at S139 (γ-H2A.X), used as a biomarker for DNA double-strand breaks, was detected by immunoblotting. Without cisplatin treatment, the level of γ-H2A.X was below the detection limit (Fig. 6). However, with cisplatin treatment, γ-H2A.X could be detected. We found that treatment with compound 11 dramatically decreased the level of γ-H2A.X in HeLa cells, suggesting that nuclear TyrRS had a protective effect on DNA damage. Compound 7 had a similar effect as shown in Fig. S7.†
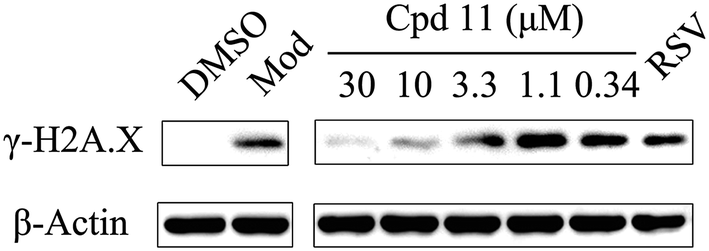 | ||
| Fig. 6 Nuclear TyrRS promoted by compound 11 protected cells from DNA damage in HeLa cells. HeLa cells were pre-treated with compound 11 (30, 10, 3.3, 1.1, or 0.37 μM), RSV (1 μM), or DMSO for 24 h. To induce DNA damage, cells were treated with cisplatin (30 μM) or DMSO in the presence of the test compounds or DMSO for 24 h. Then, whole cell lysates were subjected to immunoblot assays. | ||
Experimental
Ligand preparation
Compound libraries, including In-house and Specs databases, were used for virtual screening in this investigation. The total number of compounds is approximately 220![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000. All the compounds were used for docking, which were prepared with ‘Prepare Ligands’ module in Discovery Studio (DS) 3.1 (Accelrys Inc., San Diego, CA, USA). Parameter values for “Change Ionization, Generate Tautomers, and Generate Isomers” were set to false. Other parameters were set to their default values. The compounds were retrieved according to previous reported.18,34,35 Selected compounds were purchased in milligram quantities from chemical vendors or synthesis by our laboratory. Purity of compounds was ≥95%, as declared by the chemical vendors.
000. All the compounds were used for docking, which were prepared with ‘Prepare Ligands’ module in Discovery Studio (DS) 3.1 (Accelrys Inc., San Diego, CA, USA). Parameter values for “Change Ionization, Generate Tautomers, and Generate Isomers” were set to false. Other parameters were set to their default values. The compounds were retrieved according to previous reported.18,34,35 Selected compounds were purchased in milligram quantities from chemical vendors or synthesis by our laboratory. Purity of compounds was ≥95%, as declared by the chemical vendors.
Structure-based virtual screening of novel hit compounds against TyrRS
In this investigation, the docking method GOLD was involved in the platform of DS 3.1. Before molecular docking, the receptor protein was prepared by the DS 3.1 software package with standard preparation procedures (protein preparation protocol), which include removing water molecules, adding hydrogen atoms to the protein, and assigning force field (here the CHARMm forcefield was adopted). For GOLD, the “pre-defined generic algorithm (GA)” setting of ‘automatic’ was employed and others were set to default.General procedure for the synthesis of compounds 1–31
All commercially available starting materials, solvents, and reagents were obtained from commercial suppliers and used without further purification unless otherwise indicated. Reactions were monitored by thin layer chromatography (TLC) on Merck silica gel 60 F-254 thin layer plates. 1H NMR spectra was recorded on a Bruker AV-400 spectrometer at 400 MHz. Spin multiplicities are described as s (singlet), br. s (broad singlet), t (triplet), q (quartet), and m (multiplet). Chemical shifts (δ) are listed in parts per million (ppm) relative to tetramethylsilane (TMS) as an internal standard. Mass spectral (MS) data were acquired on a Waters Q-TOF Premier mass spectrometer (Micromass, Manchester, U.K.). The 1H NMR, 13C NMR, and MS of compounds 1–31 are listed in the ESI.†Purification of human TyrRS protein
Human TyrRS was purified as previously described.7,36,37 The plasmid of mini-TyrRS (amino acids 1–341) as a gift from Paul Schimmel. Briefly, this plasmid was transformed into E. coli Arctic Express (DE3) cells. The cells were cultured at 37 °C in fresh LB liquid medium (5 g of NaCl, 10 g of bactotrypton, and 10 g of yeast extract per litre). Isopropyl-β-D-1-thiogalactopyranoside (IPTG, 0.5 mM) was used to induce expression when attenuance D600 nm was 1, and the culture was grown for 20 h at 16 °C. Cells were collected by centrifugation at 4000 rpm min−1 for 15 min at 4 °C and then re-suspended in lysis buffer (50 mM Hepes-Na, pH 7.5, 5 mM imidazole pH 7.5, 400 mM NaCl, 1× PMSF, and 2 mM BME). Cells were lysed using a cell disrupter. After centrifugation at 14000 rpm min−1 for 25 min at 4 °C, the supernatant was loaded onto a nickel-affinity chromatography column (GE-Healthcare) pre-equilibrated with lysis buffer. The protein was eluted with a linear gradient of imidazole (0–300 mM). The desired fractions were pooled, concentrated and subjected to gel-filtration (Superdex 200; GE-Healthcare) chromatography and the protein peak corresponding to homogenous protein in buffer (20 mM Hepes-Na, pH 7.5, 50 mM NaCl, and 2 mM BME) was collected (see Fig. S3†). The quality of the protein purification was validated by SDS-PAGE analysis (see Fig. S4†).
In vitro enzymatic inhibition assays
In vitro enzymatic inhibition assays were performed using an ATP–pyrophosphate exchange assay as previously described7 and the results provided by Reaction Biology Corp, Malvern, United States. Briefly, tyrosyl adenylate synthesis was measured using the tyrosine-dependent ATP–pyrophosphate (PPi) exchange assay. A mixture containing 100 mM HEPES-Na (pH 7.5), 20 mM KCl, 2 mM ATP, 1 mM NaPPi, 2 mM DTT, 100 μM L-tyrosine (T3754-50G, Sigma), 0.02% Brij35 (w/v), 10 mM MgCl2, and approximately 0.01 mCi ml−1 Na[32P]PPi (NEX019001MC, PerkinElmer) was added to 12.5 nM purified TyrRS, pre-incubated with 0–300 μM of test compounds or 0–1000 μM of positive control RSV (ref. 7) at 4 °C for 30 min. The ATP–PPi exchange reaction mixture was incubated at room temperature for 2 h, and aliquots were removed at specified time intervals and quenched in a mixture containing 40 mM NaPPi, 1.4% (w/v) HClO4, 0.4% (w/v) HCl, and 8% (w/v) of activated charcoal. After thorough mixing, the charcoal was filtered and washed with a solution of 7% HClO4 (w/v) and 200 mM NaPPi using Spin-X Centrifuge Filters (Corning) containing 0.45 μm pore-size cellulose acetate filters. After drying, the charcoal was punched into scintillation vials and the radioactivity of the ATP bound to the charcoal mixture was measured by scintillation counting. The results of the L-tyrosine Km values and enzyme titration can be found in Fig. S5 and S6.† The Ki value was calculated by the equation Ki = IC50/(1+[S]/Km).Differential scanning calorimetry (DSC) assay
Human TyrRS (1 mg mL−1) was diluted in buffer (20 mM Hepes-Na, pH 7.4, and 400 mM NaCl) and melted using a Microcal VP Capillary DSC (GE Healthcare Life Sciences). Data were collected at 20–110 °C, with a 200 °C h−1 scan rate and 10 s filtering period. Data were analyzed in origin software. TyrRS was incubated with 200 μM RSV or compound 11 for at least 0.5 h before melting.Differential scanning fluorimetry (DSF) assay
DSF experiments were performed on a RT-PCR detection system (BIO-RAD CFX96) according to the known protocol.25 SYPRO orange (5000×, Invitrogen) was monitored using the filters of FRET at the wavelength of 492 nm for excitation and of ROX at the wavelength of 610 nm for emission. Each reaction solution containing 2 μM protein in buffer (20 mM Hepes-Na, pH 7.4, and 100 mM NaCl), 5 × SYPRO orange, and compound (10 μL) was heated from 25 to 95 °C. Fluorescence intensities were recorded every 1 °C min−1 and plotted as a function of temperature. The inflection point of the transition curve (Tm) was calculated by fitting the Boltzmann equation to the sigmoidal curve in GraphPad Prism 5.0. Each condition was tested in triplicate.Cell cultures
HeLa cells were maintained in Dulbecco's modified Eagle's medium which was supplemented with 10% (v/v) fetal bovine serum. To mimic oxidative stress in cells, serum starvation under which condition cells were cultured in serum-free media was used. Cells were grown at 37 °C and 5% CO2 in a humidified incubator.Detection of DNA damage in cells
To determine the protection of TyrRS inhibitors against DNA damage, HeLa cells pre-seeded in 6-well plate were pre-treated with compound 7 (30, 10, 3.3, 1.1, or 0.37 μM), compound 11 (30, 10, 3.3, 1.1, or 0.37 μM), RSV (1 μM), or DMSO for 24 h. To induce DNA damage, cells were treated with cisplatin (30 μM) or DMSO in the presence of test compounds or DMSO for 24 h. Then whole cell lysates were extracted with radioimmunoprecipitation assay (RIPA) lysis buffer (Beyotime Biotechnology) that contained phenylmethanesulfonyl fluoride (PMSF, Sigma) and protease inhibitor cocktail (Sigma) and subjected to immunoblot assays. Anti-Histone H2A.XS139ph (phospho Ser139) antibodies (GeneTex, GTX127340, 1:1000 dilution) were used to detect DNA damage levels in cells. Anti-β-actin antibodies (Proteintech, 60008-1-Ig, 1:1000 dilution) were used to probe β-actin as a control.
Cellular localization of TyrRS
HeLa cells were treated with compound 7 (30, 10, 3.3, and 1.1 μM), compound 11 (30, 10, 3.3, and 1.1 μM), RSV (1 μM), or DMSO for 8 h. Cells cultured in serum-free media for 8 h were under serum starvation. The cytoplasmic and nuclear fractions were separated by NE-PER® nuclear and cytoplasmic extraction reagents (Thermo Scientific, 78835) as per the manufacturer's instructions. Both fractions were then subjected to immunoblot assays. Anti-tyrosyl tRNA synthetase antibodies (Abcam, ab150429, 1:1000 dilution) were used to probe TyrRS in the nucleus and cytoplasm. Anti-GAPDH antibodies (Proteintech, 60004-1-Ig, 1:1000 dilution) and anti-Lamin B1 (Proteintech, 1298.7-1-AP, 1:1000 dilution) antibodies were used to probe GAPDH and Lamin B1 as controls for the cytoplasmic and nuclear fractions, respectively.
Immunoblot assays
Proteins from cells were resolved by 12% or 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene fluoride (PVDF) membranes (Millipore). The membranes were blocked with TBS-T blocking buffer (5% nonfat milk in TBS-T) for 1–4 h at RT and then probed with primary antibodies overnight at 4 °C with gentle shaking. After three washes with TBS-T, the membranes were incubated with horseradish peroxidase-conjugated goat anti-mouse or anti-rabbit antibody (Proteintech, 1:5000 dilution) for 1 h at 37 °C with gentle shaking. After extensive washing with TBS-T, the membranes were preserved in TBS, and the immunoblots were visualized by ECL (Abbkine). Immunoblots were quantified using Adobe photoshop software.
Conclusions
A structure-based docking strategy was adopted to screen for novel TyrRS inhibitors. After two campaigns including virtual screening and bioassays, hit compounds with the novel scaffold 5,7-dihydroxy-2-phenyl-4H-chromen-4-one were selected for structural optimization and SAR analysis. The most active compound against TyrRS was compound 11 with a Ki value of 8.8 μM. The results of orthogonal assays, including DSC and DSF assays, revealed that these compounds bind to the active site of TyrRS. Furthermore, compound 11 could protect against the DNA damage caused by cisplatin in intact HeLa cells. Therefore, these compounds represent useful probes for further research into the biological function of TyrRS.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (81573349, 81773633, and 21772130), National Science and Technology Major Project (2018ZX09711002-014-002, 2018ZX09711002-011-019, and 2018ZX09711003-003-006), and 1.3.5 project for disciplines of excellence, West China Hospital, Sichuan University.Notes and references
- J. A. Baur, K. J. Pearson and N. L. Price, Resveratrol improves health and survival of mice on a high-calorie diet, Nature, 2006, 444, 337–342 CrossRef CAS.
- S. J. Park, F. Ahmad and A. Philp, Resveratrol ameliorates aging-related metabolic phenotypes by inhibiting cAMP phosphodiesterases, Cell, 2012, 148, 421–433 CrossRef CAS.
- M. Jang, L. Cai and G. O. Udeani, Cancer chemopreventive activity of resveratrol, a natural product derived from grapes, Science, 1997, 275, 218–220 CrossRef CAS PubMed.
- A. Riba, L. Deres and B. Sumegi, Cardioprotective Effect of Resveratrol in a Postinfarction Heart Failure Model, Oxid. Med. Cell. Longevity, 2017, 2017, 6819281 Search PubMed.
- D. A. Sinclair and L. Guarente, Small-molecule allosteric activators of sirtuins, Annu. Rev. Pharmacol. Toxicol., 2014, 54, 363–380 CrossRef CAS.
- M. Viswanathan, S. K. Kim and A. Berdichevsky, A role for SIR-2.1 regulation of ER stress response genes in determining C. elegans life span, Dev. Cell, 2005, 9, 605–615 CrossRef CAS.
- M. Sajish and P. Schimmel, A human tRNA synthetase is a potent PARP1-activating effector target for resveratrol, Nature, 2015, 519, 370–373 CrossRef CAS.
- S. S. Kulkarni and C. Cantó, The molecular targets of resveratrol, BBA, Mol. Basis Dis., 2015, 1852, 1114–1123 CrossRef CAS.
- N. Wei, Y. Shi and L. N. Truong, Oxidative stress diverts tRNA synthetase to nucleus for protection against DNA damage, Mol. Cell, 2014, 56, 323–332 CrossRef CAS.
- X. Cao, C. Li and S. Xiao, Acetylation promotes TyrRS nuclear translocation to prevent oxidative damage, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 687–692 CrossRef CAS.
- X. Luo and W. L. Kraus, On PAR with PARP: cellular stress signaling through poly(ADP-ribose) and PARP-1, Genes Dev., 2012, 26, 417–432 CrossRef CAS.
- L. Pirola and S. Fröjdö, Resveratrol: One molecule, many targets, IUBMB Life, 2008, 60, 323–332 CrossRef CAS PubMed.
- F. Mao, J. Yan and J. Li, New multi-target-directed small molecules against Alzheimer's disease: a combination of resveratrol and clioquinol, Org. Biomol. Chem., 2014, 12, 5936–5944 RSC.
- R. I. Tennen, E. Michishita-Kioi and K. F. Chua, Finding a target for resveratrol, Cell, 2012, 148, 387–389 CrossRef CAS.
- M.-H. Lee, B. Y. Choi and J. K. Kundu, Resveratrol suppresses growth of human ovarian cancer cells in culture and in a murine xenograft model: eukaryotic elongation factor 1A2 as a potential target, Cancer Res., 2009, 69, 7449–7458 CrossRef CAS.
- S. H. Yang, Q. Sun and H. Xiong, Discovery of a butyrylcholinesterase-specific probe via a structure-based design strategy, Chem. Commun., 2017, 53, 3952–3955 RSC.
- C. M. Lombardo, I. S. Martinez and S. Haider, Structure-based design of selective high-affinity telomeric quadruplex-binding ligands, Chem. Commun., 2010, 46, 9116–9118 RSC.
- S. Huang, C. Song and X. Wang, Discovery of New SIRT2 Inhibitors by Utilizing a Consensus Docking/Scoring Strategy and Structure-Activity Relationship Analysis, J. Chem. Inf. Model., 2017, 57, 669–679 CrossRef CAS.
- J. Baell and M. A. Walters, Chemistry: Chemical con artists foil drug discovery, Nature, 2014, 513, 481–483 CrossRef CAS.
- K. Ding, Y. Lu and Z. Nikolovska-Coleska, Structure-based design of potent non-peptide MDM2 inhibitors, J. Am. Chem. Soc., 2005, 127, 10130–10131 CrossRef CAS.
- E. Perola, W. P. Walters and P. S. Charifson, A detailed comparison of current docking and scoring methods on systems of pharmaceutical relevance, Proteins, 2004, 56, 235–249 CrossRef CAS.
- G.-B. Li, L.-L. Yang and W.-J. Wang, ID-Score: a new empirical scoring function based on a comprehensive set of descriptors related to protein–ligand interactions, J. Chem. Inf. Model., 2013, 53, 592–600 CrossRef CAS PubMed.
- H. Park, T. T. Dao and H. P. Kim, Synthesis and inhibition of PGE2 production of 6,8-disubstituted chrysin derivatives, Eur. J. Med. Chem., 2005, 40, 943–948 CrossRef CAS.
- I. Ryu, H. Matsubara and S. Yasuda, Phase-vanishing reactions that use fluorous media as a phase screen. Facile, controlled bromination of alkenes by dibromine and dealkylation of aromatic ethers by boron tribromide, J. Am. Chem. Soc., 2002, 124, 12946–12947 CrossRef CAS.
- F. H. Niesen, H. Berglund and M. Vedadi, The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability, Nat. Protoc., 2007, 2, 2212–2221 CrossRef CAS.
- J. V. Rodrigues, V. Prosinecki and I. Marrucho, Protein stability in an ionic liquid milieu: on the use of differential scanning fluorimetry, Phys. Chem. Chem. Phys., 2011, 13, 13614–13616 RSC.
- M. M. Knopp, K. Löbmann and D. P. Elder, Recent advances and potential applications of modulated differential scanning calorimetry (mDSC) in drug development, Eur. J. Pharm. Sci., 2016, 87, 164–173 CrossRef CAS.
- P. Alam, A. S. Abdelhameed and R. K. Rajpoot, Interplay of multiple interaction forces: Binding of tyrosine kinase inhibitor nintedanib with human serum albumin, J. Photochem. Photobiol., B, 2016, 157, 70–76 CrossRef CAS.
- A. S. Abdelhameed, P. Alam and R. H. Khan, Binding of janus kinase inhibitor tofacitinib with human serum albumin: multi-technique approach, J. Biomol. Struct. Dyn., 2016, 34, 2037–2044 CrossRef CAS.
- M. Epple, U. Sazama and A. Reller, Simultaneous X-ray absorption fine-structure spectroscopy (XAFS) and differential scanning calorimetry (DSC), Chem. Commun., 1996, 1755–1756 RSC.
- R. W. Harkness, S. Slavkovic and P. E. Johnson, Rapid characterization of folding and binding interactions with thermolabile ligands by DSC, Chem. Commun., 2016, 52, 13471–13474 RSC.
- H.-m. Chow and K. Herrup, Genomic integrity and the ageing brain, Nat. Rev. Neurosci., 2015, 16, 672 CrossRef CAS.
- W. P. Roos, A. D. Thomas and B. Kaina, DNA damage and the balance between survival and death in cancer biology, Nat. Rev. Cancer, 2016, 16, 20 CrossRef CAS.
- C. A. Lipinski, F. Lombardo and B. W. Dominy, Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings, Adv. Drug Delivery Rev., 2012, 64, 4–17 CrossRef.
- D. F. Veber, S. R. Johnson and H.-Y. Cheng, Molecular properties that influence the oral bioavailability of drug candidates, J. Med. Chem., 2002, 45, 2615–2623 CrossRef CAS.
- X. L. Yang, R. J. Skene and D. E. McRee, Crystal structure of a human aminoacyl-tRNA synthetase cytokine, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 15369–15374 CrossRef CAS.
- M. Sajish, Q. Zhou and S. Kishi, Trp-tRNA synthetase bridges DNA-PKcs to PARP-1 to link IFN-gamma and p53 signaling, Nat. Chem. Biol., 2012, 8, 547–554 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: General procedure for the synthesis of compounds (1–31), Fig S1–S7, and Table S1. See DOI: 10.1039/c9ra00458k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |