
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effect of gold addition by the recharge method on silver supported catalysts in the catalytic wet air oxidation (CWAO) of phenol
Adib A. Silahua-Pavóna,
Gilberto Torres-Torres *a,
Juan Carlos Arévalo-Péreza,
Adrián Cervantes-Uribea,
Zenaida Guerra-Queac,
Adrián Cordero-Garcíaa,
Alejandra Espinosa de los Monterosa and
Jorge N. Beltraminib
*a,
Juan Carlos Arévalo-Péreza,
Adrián Cervantes-Uribea,
Zenaida Guerra-Queac,
Adrián Cordero-Garcíaa,
Alejandra Espinosa de los Monterosa and
Jorge N. Beltraminib
aUniversidad Juárez Autónoma de Tabasco, Laboratorio de Nanomateriales Catalíticos Aplicados al Desarrollo de Fuentes de Energìa y de Remediaciòn Ambiental, Centro de Investigación de Ciencia y Tecnología Aplicada de Tabasco (CICTAT), DACB, Km. 1 Carretera Cunduacán-Jalpa de Méndez AP. 24, C.P. 86690, Cunduacán, Tabasco, Mexico. E-mail: gilberto.torres@ujat.mx; torremensajes@gmail.com; Fax: +52 19143360928; Tel: +52 19143360300
bARC Centre of Excellence for Functional Nanomaterials, The Australian Institute for Bioengineering and Nanotechnology (AIBN) and School of Engineering, The University of Queensland, St. Lucia, QLD 4072, Australia
cInstituto Tecnológico de Villahermosa, Km. 3.5 Carretera, Villahermosa – Frontera, Cd. Industrial, 86010 Villahermosa, Tabasco, Mexico
First published on 9th April 2019
Abstract
Catalysts Ag/ZrO2–CeO2 and Au/ZrO2–CeO2 were synthesized by a deposition–precipitation method and Ag–Au/ZrO2–CeO2 was prepared using a recharge method for the second metal (Au). The materials were characterized by physisorption of N2, XRD, ICP, UV-vis RDS, H2-TPR, XPS and TEM. The results obtained show that the specific areas for monometallic materials were 29–37 m2 g−1 and 27–74 m2 g−1 for bimetallics. The tetragonal crystal phase of ZrO2 stabilizes when CeO2 quantity increases. Using XPS an increment in Ce3+ species abundance was determined for bimetallic catalysts in contrast to the monometallic ones; according to the Ag 3d region, this metal oxidation was observed when augmenting the content of CeO2 in the materials, and with Au the opposite effect was produced. It was determined by TEM, that the average size of the metallic particles was smaller at bimetallic catalysts due the preparation method. Catalytic activity was evaluated by CWAO of phenol, the Ag–Au/ZrO2–CeO2 catalyst with 20% wt of cerium reached a degradation of 100% within an hour, being the most active catalyst. Maleic, formic and oxalic acid were identified as reaction intermediates; and at the end of the reaction acetic acid was identified as the main by-product, because it is the most refractory and the conditions for oxidation must be more severe.
Introduction
Industrial effluent discharges are considered one of the biggest sources of pollution in water bodies, as they carry organic and inorganic compounds such as: phenols, carboxylic acids, ethers and heavy metals, which cause problems to the environment and human health.1 Phenol is one of the most common organic contaminants found in wastewaters, because it is widely used as a precursor for other substances, and for explosives, fertilizers, textiles, adhesives, pharmaceuticals, paper, soap and wood preservatives manufacture. In consequence, residual phenol causes serious damage to human health. In low concentrations, this pollutant is a strong neurotoxin that causes death, affecting vital organs such as kidneys, liver and lungs.2 Phenol in aqueous medium has been successfully treated with physical (adsorption),3 chemical (oxidation)4 and biological (biodegradation)5 methods. Among the most efficient chemical methods are the advanced oxidation processes. One of these processes with greater oxidation capacity and volume of treatment is Catalytic Wet Air Oxidation (CWAO), which allows partial or total destruction of organic contaminants in aqueous environment, with the advantage of using an effluent with high concentrations, toxicity decrease and aerobic biodegradability. Mild conditions of both pressure (0.5–10 MPa) and temperature (100–200 °C), and metal catalysts supported on inorganic oxides such as Al2O3, SiO2, TiO2, ZrO2, CeO2 and others6 are used in the process to provide positive thermal, mechanical and electronic properties under conditions of pressure and temperature, which ZrO2 owns. Also, it is known that the tetragonal phase of zirconia which is thermally stable at high temperatures plays an important role in oxidation reactions due to its high oxygen ion conductivity properties.7,8 In addition, the use of ceria increases the oxygen on the surface of the catalyst and improves the oxidation reaction of contaminates. Z. Guerra-Que et al.,9 reported a study of silver nanoparticles supported on ZrO2 modified with Ce to 15 wt% and 20 wt% using CWAO process to destroy the MTBE and they observed a better conversion and selectivity to CO2 using catalysts supported on zirconia modified with ceria.To obtain a higher catalytic efficiency by CWAO, the noble metals (Ir, Pd, Ru, Ag, Au) have been added superficially in supports as active sites, besides they have little leaching capacity in the process in comparison with the metals of transition.10 Including these metals are silver and gold. Silver has special characteristics to improve catalytic oxidation reactions. It is known to have a high chemisorption capacity of O2.11–13 Silver has been studied in the oxidation of CO,14 gasoline oxygenates (MTBE),9 p-cresol,15 phenol,16 among others. On the other hand, gold is catalytically active when dispersed as small particles in an oxide support, the preparation of gold-based catalysts has been widely studied. They are active in many reactions of industrial and environmental importance.17 Ngoc Dung Tran et al. demonstrated that Au°, is the most active gold species in the Au/CeO2 catalysts in the CWAO of carboxylic acids, in addition to showing good yields, attributed by the high mobility of oxygen in the surface of the gold/ceria system.18 Also, Au, has been studied in oxidations of volatile organic compounds (VOC such as 2-propanol, toluene and ethanol),19 CO,20 glucose,21 among others. However, it is also reported that gold does not have good stability and its periods of reuse are very short. For this reason, the addition of a second metal can influence the catalytic properties, improving activity, stability and selectivity. To achieve this, it is necessary to have a low reduction element with a high metal support interaction capable of stabilizing and dispersing the first element. In fact, bimetallic combinations such as Au–Ag exhibit significantly improved activity and stability and synergistic effects, reported by Alberto Sandoval et al. for oxidations of CO.22 This is because Ag has a greater capacity to donate electrons and modifies the electronic properties of gold by a strong interaction between Au and Ag. Another work reported using Ag–Au by photocatalysis. Zielińska-Jurek et al. shows that the catalysts of Ag–Au/TiO2 obtained a better degradation than the catalysts of Au/TiO2 and Ag/TiO2 for the degradation of 20 ppm of phenol. However, the problem in photocatalysis is the low concentration of phenol that can be degraded.23 Previous work reported Au–Ag/ZrO2–CeO2 catalysts synthesized by the redox method for MTBE oxidation where the bimetallic catalysts showed better activity than the catalysts Ag/ZrO2CeO2, on the other, the catalytic activity of Au–Ag/ZrO2–CeO2 is also shown synthesized by deposition–precipitation with urea for the oxidation of phenol by CWAO, obtaining a conversion of 61% and a TOC of 40%.24
The synthesis of bimetallic Ag–Au catalysts has been reported by different methods, such as dealloying for borohydride electro-oxidation,25 microemulsion,26 sequential deposition–precipitation with urea,24 KOH19 and NaOH,22 and redox.27 The latter offers the advantage of performing a selective deposition of a metal on another metal's surface, both in reduced state in presence of hydrogen. Electron configurations, the atomic radio of metals and low temperatures of preparation are variables that affect the synthesis of bimetallic catalysts. In this case, the second metal can be dispersed as a monolayer on the surface of a first metal forming solid solutions with comparable particle sizes.
Considering the above, in this paper bimetallic catalysts were prepared using a mixed oxide ZrO2–CeO2 as support, by sol–gel method varying the content of the second oxide. The first metal (Ag) was added by the deposition–precipitation method using NaOH as the dispersing agent, then the second metal (Au) was incorporated by the Recharge method (redox) using a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio between Ag and Au.
:1 molar ratio between Ag and Au.
Results and discussion
(a) Elemental analysis
The theoretical metal loadings of the monometallic and bimetallic catalysts were 1.4 wt% for Ag and 2.5 wt% for Au, which corresponds to molar ratio 1:1 (Ag:Au) for the bimetallics. Table 1 compares the theoretical and the measured gold and silver loadings in wt% and the Au/Ag atomic ratios for the Au–Ag samples. As expected for AgZr and AgZrCe, practically all the silver present in solution was deposited on the catalysts. In the case of AgZr, AgZrCe10 and AgZrCe20, about 93, 85, 93% was deposited on the catalysts respectively. The Au actual deposited on the monometallic were 88, 92 and 92% for 0, 10 and 20% w/w cerium respectively. These results showed that the deposit–precipitation method produces a good behavior for the deposit of Ag and Au on the simple and mixed oxide surface. In the case of bimetallic samples, the actual gold loadings are also close to the theoretical value (2.5 wt%), whereas for silver, the actual loading is always lower than the nominal loading. According with the molar ratio of AgAu was found the ratios between 0.91 and 0.95 that is very close to the theoretical ratio.
| Catalyst | Metal loading (%) | Actual Ag/Au molar ratio | |||
|---|---|---|---|---|---|
| Theoretical | Actual | ||||
| Ag | Au | Ag | Au | ||
| AgZr | 1.4 | — | 1.3 | — | — |
| AgZrCe10 | 1.4 | — | 1.2 | — | — |
| AgZrCe20 | 1.4 | — | 1.3 | — | — |
| AuZr | 2.5 | 2.2 | |||
| AuZrCe10 | 2.5 | 2.3 | |||
| Au | 2.5 | 2.3 | |||
| AgAuZr | 1.4 | 2.5 | 1.2 | 2.4 | 0.91 |
| AgAuZrCe10 | 1.4 | 2.5 | 1.2 | 2.3 | 0.95 |
| AgAuZrCe20 | 1.4 | 2.5 | 1.2 | 2.3 | 0.95 |
(b) N2 physisorption
All samples show (Fig. 1) a type IV isotherm (IUPAC classification) indicating that the catalysts are mesoporous. The hysteresis loop (H2 type) is commonly associated with the presence of ink-bottle shaped pores, which could be due to inter- or intra-particle porosity.28,29 As also observed, hysteresis indicates the presence of capillary condensation suggesting the presence of high-strength agglomerates (aggregates).30 On the other hand, it is observed in Fig. 2, the pore distribution of the monometallic and bimetallic catalysts. All the samples presented a pore distribution of the unimodal type.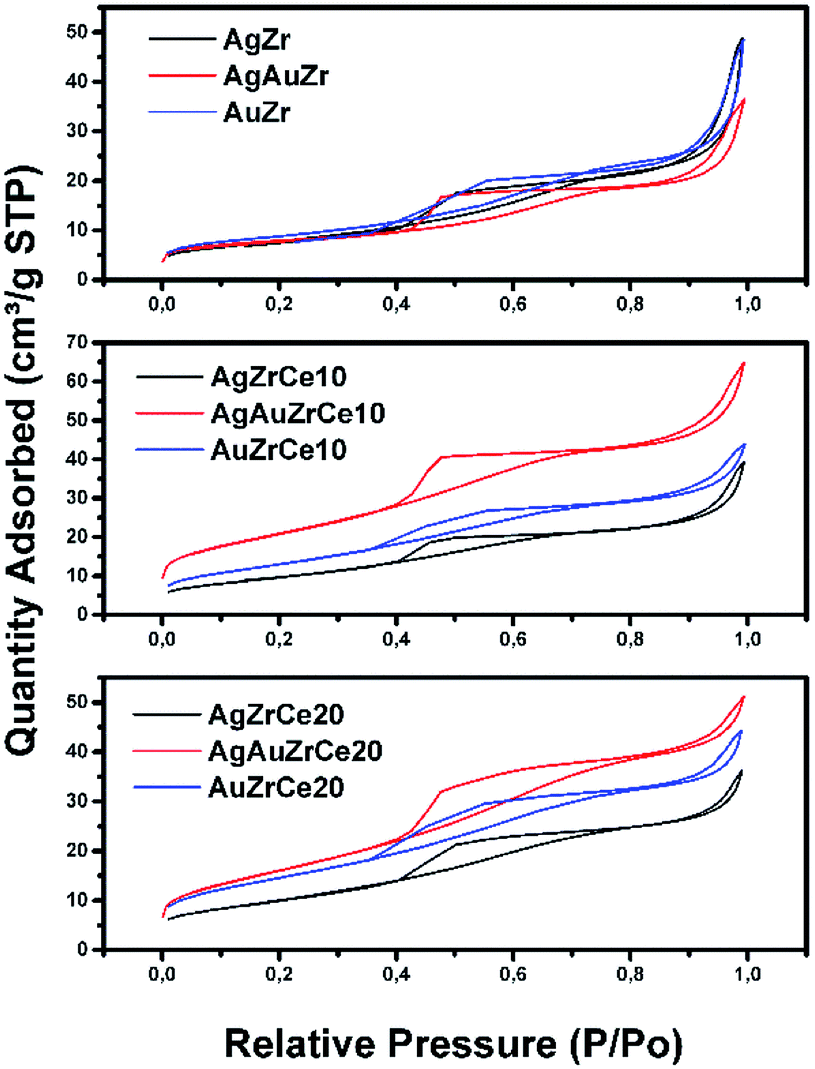 | ||
| Fig. 1 Nitrogen adsorption isotherms of Ag, Au and Au–Ag catalysts on ZrCe with 0, 10 and 20 wt% in Ce. | ||
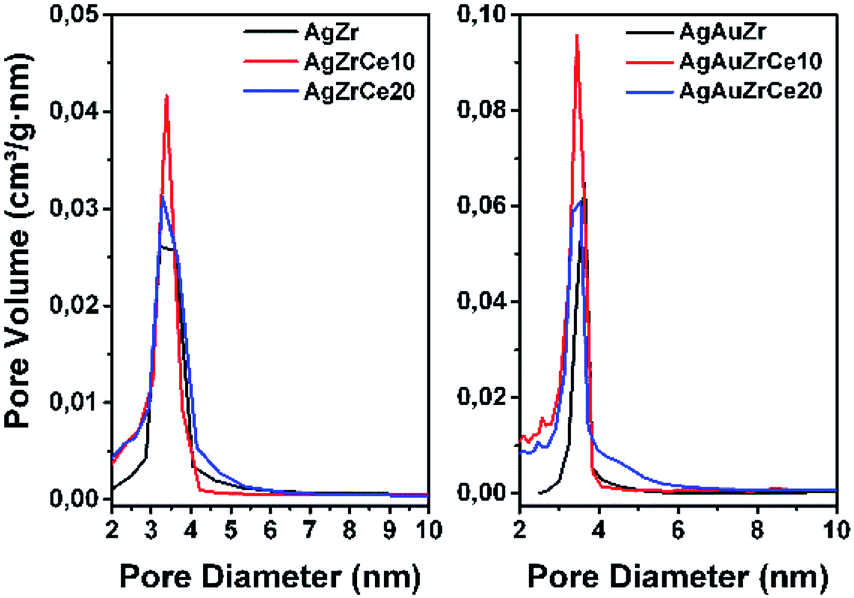 | ||
| Fig. 2 Pore distribution for monometallic and bimetallic catalysts. | ||
Table 2 reports the calculated surface areas. Regarding the surface area an increase was observed when the concentration of cerium oxide increases. Ag monometallic catalysts obtained areas of 29 to 37 m2 g−1 and the Au catalysts showed areas of 32 to 53 m2 g−1; these phenomenon's is attributed to the difference between ions of Zr4+ (0.084 nm) and Ce4+ (0.098 nm), causing Ce ions not being introduced into the ZrO2 structure; remaining on the surface of the material and therefore increasing the specific surface area.31 In addition, the surface areas are a little more in the Au than the Ag monometallic, for effect of the metal particles size. Meanwhile specific area between 27 and 74 m2 g−1 is found on bimetallic catalysts; in comparison with the monometallic catalysts, an increase in the area of about 2 times can be observed in the bimetallic catalysts containing 10 and 20% in cerium. This effect could probably be due to the acid conditions (pH = 1) of synthesis of the second metal, resulting in the redispersion of the Ce and unblocking pores on the surface of the catalyst. This effect is not observed with AgAuZr.
| Catalyst | SA (m2 g−1) | Pore size (nm) | Pore volume (cm3 g−1) | Crystal sizea (nm) | Metal particle sizeb (nm) |
|---|---|---|---|---|---|
| a Crystal size by Scherrer's equation (nm).b Metal particle size by TEM. | |||||
| AgZr | 29 | 7.9 | 0.077 | 8.8 | 10 |
| AgZrCe10 | 35 | 5.4 | 0.059 | 7.7 | 6.6 |
| AgZrCe20 | 37 | 4.7 | 0.058 | 7.5 | 6.1 |
| AuZr | 32 | 6.5 | 0.061 | 8.7 | 4.5 |
| AuZrCe10 | 48 | 6.1 | 0.061 | 8.1 | 4.3 |
| AuZrCe20 | 53 | 5.6 | 0.064 | 7.9 | 3.9 |
| AgAuZr | 27 | 6.8 | 0.057 | 8.9 | 6.8 |
| AgAuZrCe10 | 74 | 4.8 | 0.1 | 8.9 | 5.7 |
| AgAuZrCe20 | 60 | 4.4 | 0.079 | 7.6 | 4.2 |
(c) X-ray diffraction
Fig. 3 shows diffraction patterns of monometallic and bimetallic catalysts.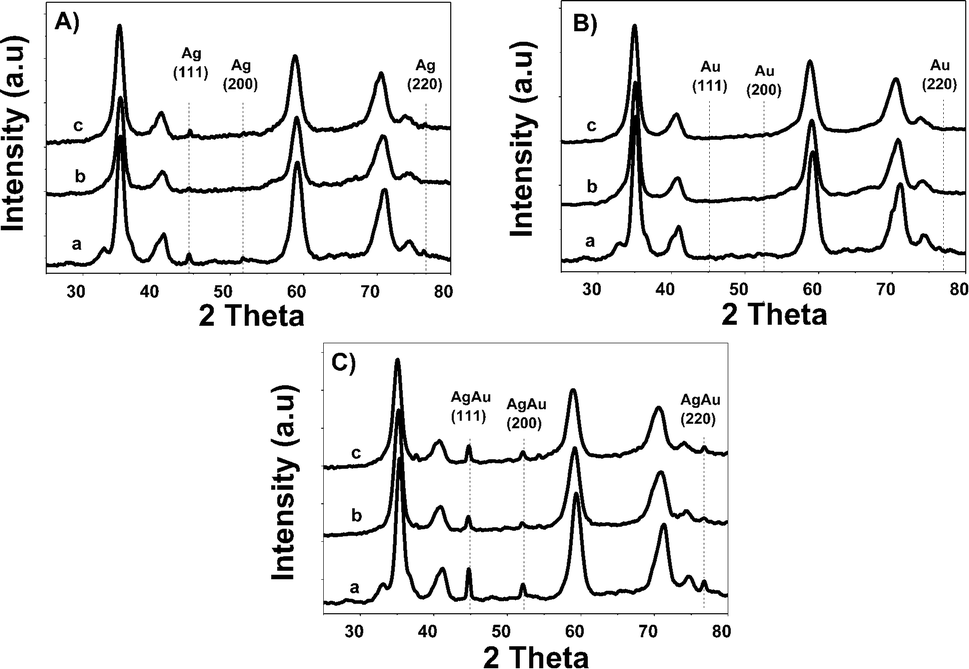 | ||
| Fig. 3 X-ray patterns of monometallic (A) Ag and (B) Au and Au–Ag bimetallic (C) catalysts, where a: Zr, b: ZrCe10 and c: ZrCe20. | ||
Phases corresponding to monoclinic and tetragonal crystal structures of zirconium oxide were identified. This, two structures coexist in the AgZr, AuZr and AuAZr catalysts with the following diffraction patterns (Fig. 3A): (a) tetragonal phase on angles 35.18, 40.03, 40.97, 59.12, 69.96, 70.14 and 74.40;32 corresponding to planes (101), (002), (110), (200), (103), (211) and (202) and (b) monoclinic phase at 32.81° of 2θ of the plane (111),33 and it is observed higher abundance of tetragonal phase. However, in catalysts with high concentrations of cerium (10–20%), only the tetragonal crystalline phase could be observed, therefore, when increasing the amount of CeO2, the material tends to stabilize to the tetragonal phase. In these synthesized catalysts it was not possible to observe the presence of the cubic phase (cerianite) characteristic of cerium oxide, this result agrees with that reported by E. Rubio-Rosas et al.34 The Ag monometallic showed a weak peak corresponding to silver phases around at 2θ = 44, 52 and 77. In addition, the intensity of peaks decreases when increase the cerium amount in the catalyst. In the Au monometallic Fig. 3B, the diffraction peaks corresponding to Au phase were not detected. It can be attributed to the small size of gold particles, indicating the high dispersion of Au via the precipitation–deposition technique, this effect showed by E. Hernández-Ramírez et al.,35 with 3 wt% over TiO2 using the same method. Concerning the bimetallic catalysts, the identified structures were tetragonal and monoclinic of zirconium oxide. Angles 38.60 and 55.73 corresponding to the planes (200) and (220) characteristic of cerium oxide cubic structure were also identified; only on catalysts with higher concentration of cerium. Recharge method re-dispersed the cerium deposited on the surface to agglomerate it in larger crystals, see Fig. 3C. As for the deposited noble metal, three diffractions were identified, characteristic of the cubic phase centred on the silver faces, at 2θ = 44.44, 51.95 and 76.49 corresponding to planes (111), (200) and (220).36 The diffractions characteristics of gold are similar to those of silver; this is because both metals have the same crystalline phase (fcc). Just an increase in the diffraction intensity was observed due to the presence of the second metal, regarding monometallic catalysts. The bimetallic catalysts AgAuZrCe10 and AgAuZrCe20 presented a lower intensity to the metallic phases in contrast to AgAuZr, this could be due to a better dispersion of gold in the catalysts with cerium. Table 2 presents the calculation of the average size of the zirconium oxide crystal for catalysts (monometallic and bimetallic), which was determined by FWHM (full width half maximum) using the Debye–Scherer equation.37 The average crystal size of ZrO2 in all synthesized materials was 7.5–8.9 nm. Materials with smaller average crystal size were the catalysts with 20% wt of cerium; Ag, Au monometallic and Au–Ag bimetallic with 7.5, 7.9 and 7.6 nm respectively.
(d) UV-vis spectroscopy
Fig. 4A shows spectra of monometallic catalysts. Absorption bands around wavelength maximum between 450 and 500 nm were observed, related with the presence of silver metal nanoparticles. These bands are due to the resonance absorption of the surface plasmon.38–40 It has been reported that bands between 230 and 260 are attributed to ions Ag+ y Ag+n related to highly dispersed silver. Furthermore, the plasmons centered between 290 and 350 are assigned to agglomerations of metallic silver Ag0n.41 These absorptions are only clear in AgZr. An increase in the range of 250–320 nm can be observed at spectra corresponding to the catalysts with cerium. Cerium oxide is an n-type semiconductor with a prohibitive energy band (bandgap) of 3.1 eV.42 It is known to have a strong absorbance in the ultraviolet range where two types of characteristic bands to 250 and 297 nm can be shown, these assigned to the transfers of charges Ce3+ ← O2− and Ce4+ ← O2− respectively.43 In relation of gold, it is reported that the characteristic bands of clusters are of 280 and 300 nm in materials with Zr/Ce > 1 and gold metal plasmon (Au°) is observed between 500 and 600 nm bands7 Au catalysts showed (Fig. 4B) in the all samples a plasmon around 550 nm, corresponding to Au°. Another hand, the Au–Ag bimetallic spectra (Fig. 4C) there is a change on the surface resonance plasmon in contrast of monometallic, moving towards the visible one; this is caused by the incorporation of Au, which is in higher concentration than Ag.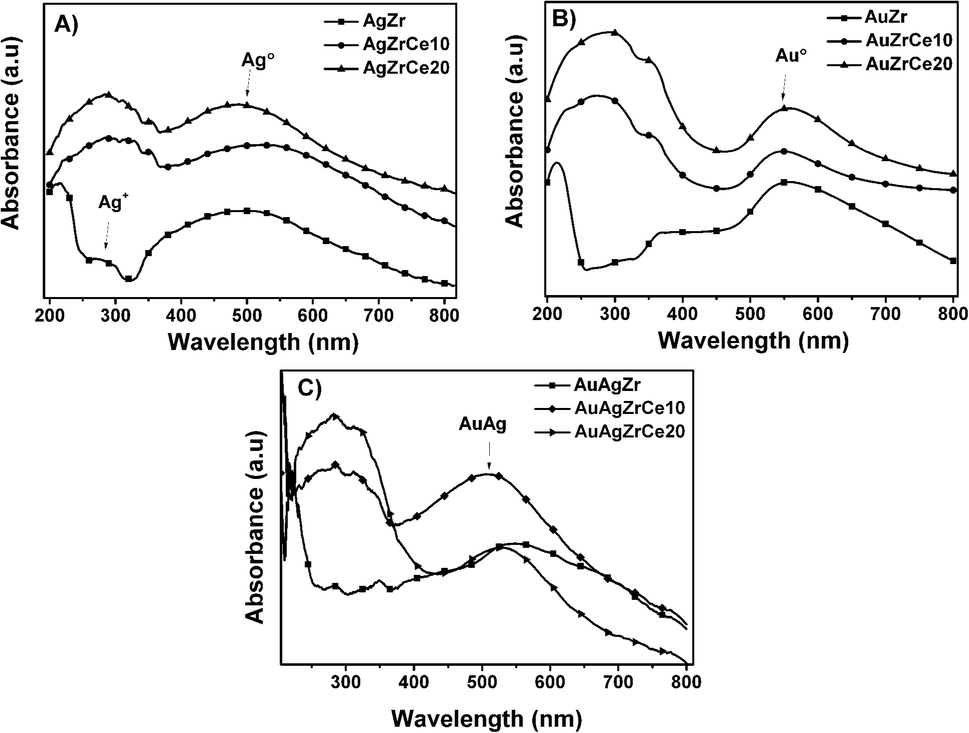 | ||
| Fig. 4 UV-vis spectra of monometallic silver (A) and gold (B) and bimetallic silver–gold (C) catalysts. | ||
(e) Temperature programmed reduction (H2-TPR)
The results of the treatment at programmed temperature are shown in Fig. 5. The reduction profile for the AgZr sample did not show hydrogen consumption in a range of analysis. However, samples with cerium, AgZrCe10 and AgZrCe20, showed a signal close to 300 °C, related to the reduction of silver oxide and another related to cerium oxide (≈370 °C); the difference between samples lies in the amount of hydrogen consumed that goes according to the amount of cerium in the samples. For gold monometallic, the AuZr sample showed no hydrogen consumption. With respect to the samples with cerium, only the reduction of the cerium oxide present was observed. The sample AgAuZr did not show reduction in the analysis performed. The reduction of cerium oxide and silver oxide was displaced at higher temperatures compared to the silver monometallic, the interaction between the metals on the surface, modify the temperature. In the three cases analyzed, the AgZr, AuZr and AgAuZr catalysts did not show hydrogen consumption, the analysis was carried out after the respective thermal treatments.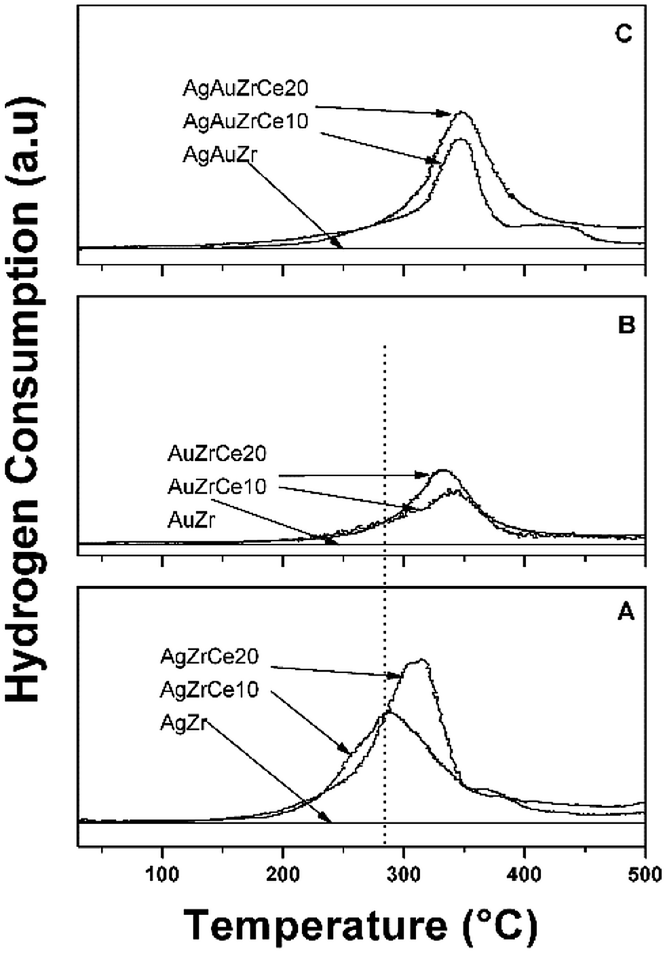 | ||
| Fig. 5 TPR profiles of catalysts: (A) Ag, (B) Au monometallic and (C) AuAg bimetallic. | ||
(f) X-ray photoelectron spectroscopy (XPS)
Fig. 6A shows de-convoluted XPS spectra for level Ag 3d5/2 of monometallic catalysts. Two picks are reported for level 3d5/2 of silver: 368.3 eV characteristic of metallic species Ag° and 367.99–367.8 eV for the oxidized species Ag+.44–46 For AgZr only the peak at 368.3 eV was observed, this indicates that the deposited silver is totally reduced. For catalysts with high concentrations of cerium (10–20%), a mixture of Ag° and Ag+ states coexists on 368.3 and between 367.97–367.88 eV respectively. Table 3 reports the relative abundances obtained. This effect in the oxidation states of Ag, on the monometallic catalysts modified with Ce, may be due to a strong electronic interaction of Ag0 with Ce4+, causing the partial oxidation of Ag0 to Ag+and the reduction of Ce4+ to Ce3+(Table 3).| Catalysts | Oxidation states | Surface atomic ratio | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Zr4+ (3d5/2) | Ce % (3d5/2) | Ag % (3d5/2) | Au% (4f7/2) | ||||||||
| Ce4+ | Ce3+ | Ag0 | Ag+ | Au0 | Au+ | O/Ce | O/Ag | O/(Au + Ag) | Au/Ag | ||
| AgZr | 182.08 | — | — | 100 | — | — | — | — | 9.48 | — | — |
| AgZrCe10 | 182.06 | 70 | 30 | 77 | 23 | — | — | 0.83 | 7.68 | — | — |
| AgZrCe20 | 181.96 | 69 | 31 | 62 | 38 | — | — | 0.91 | 8.49 | — | — |
| AuZr | 182.15 | — | — | 100 | — | — | — | — | |||
| AuZrCe10 | 182.03 | 67 | 33 | 100 | — | 0.81 | — | — | |||
| AuZrCe20 | 181.89 | 66 | 34 | 100 | — | 0.77 | — | — | |||
| AgAuZr | 182.33 | — | — | 100 | — | 100 | — | — | — | 13.74 | 0.80 |
| AgAuZrCe10 | 182.19 | 59 | 41 | 54 | 46 | 100 | — | 0.67 | — | 7.25 | 0.90 |
| AgAuZrCe20 | 181.26 | 57 | 43 | 50 | 50 | 100 | — | 0.61 | — | 6.93 | 0.92 |
On the other hand, the monometallic gold catalyst showed an abundance of 100% for all the samples for the Au metallic species. This result is corroborated by H2-TPR (Fig. 5), where no signals are observed after the reduction treatment. In the case of Zr analyzed by XPS, a signal about 182 ± 0.3 eV was found, characteristic of the oxidation state Zr4+,47 in the case of the synthesized silver monometallic catalysts, binding energies between 181.96 and 182.08 eV were found, the monometallic Au catalysts showed an effect similar that the silver catalysts with binding energy signals between 182.15 and 181.89 eV, proving that there was no change of state when incorporating cerium, gold and silver. Regarding cerium oxide, the Ce 3d spectra show a great variety of peaks, these are found in two energy levels which are Ce 3d3/2 and Ce 3d5/2, where the two oxidation states are found; Ce4+ with characteristic peaks V, V′′, V′′′, U, U′′ and U′′′, Ce3+ with V′ and U′, as seen in Fig. 6C. Presence of Ce3+ is frequently observed in mixed oxide systems of ZrO2–CeO2 and TiO2–CeO2.48–50 However, for relative abundance calculation only the de-convolved areas of level Ce 3d5/2 were analysed as reported by some authors.51,52 Table 3 reports the relative abundances obtained. In the bimetallic catalysts it is observed that when the second metal is deposited (Au), the percentage of oxidized silver increases (Ag+), this is observed in AgAuZrCe10 and AgAuZrCe20, which implies that there is an interaction between silver and gold. In the work of Zanela et al., it is mentioned that the redox potential Au3+/Au0 is greater than that of Ag+/Ag0; As a consequence, the gold precursor (HAuCl4) is capable of oxidizing silver metals.22 In addition, the reduced silver status is maintained in the AgAuZr catalyst.
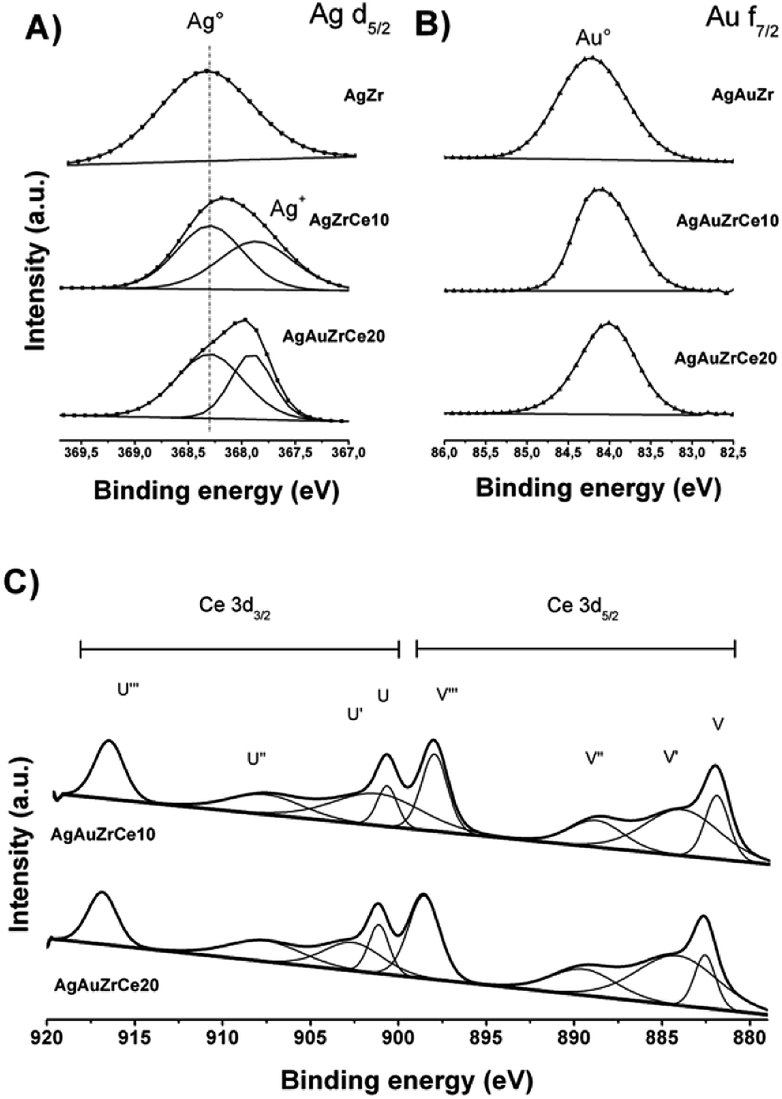 | ||
| Fig. 6 Silver XPS spectra in bimetallic catalyst (A), gold in bimetallic catalyst (B) and cerium in monometallic and bimetallic catalysts (C). | ||
Fig. 6B shows the binding energies for the Au 4f7/2 level of the bimetallic catalysts. In the case of gold, the energies in 84 eV represent Au metallic and species between 85.6–85.7 eV for the oxidized species Au+.53–55 In our work we find signals at 84.0, 84.1 and 84.2 for AgArZr, AgAuZrCe10 and AgAuZrCe20. Casaletto et al. They analyzed the Au nanoparticles in different metal oxide supports and found two gold states, Au0 and Au+. The state Au+ was shifted by Δ = 2 eV from the Au0 state.56 In our case, it moved up to Δ = 0.2 eV. For this reason, it's can affirm that gold is in a reduced state. The little displacement may be due to a strong interaction between Au and Ce.
Spectra of Ce 3d5/2 (Fig. 6C) for bimetallic catalysts indicated the presence of two oxidation states Ce3+ and Ce4+, in the same way than in monometallic catalysts, showing abundance of 59 and 57% for the state Ce4+ and 41 and 43% for Ce3+ for catalysts with AgAuCe10 and AgAuCe20 respectively, this indicates that there is a greater abundance of Ce3+ cations on the surface in contrast to the monometallic ones. The presence of the reduced species Ce3+ are associated with generation of oxygen vacancies because of charge compensation.48,49 As in other investigations, crystalline defects, such as oxygen vacancies may occur in the tetragonal structures of the ZrO2–CeO2 systems, with a particle size smaller than 15 nm.57–59
Regarding the surface oxygen ratio, monometallic catalysts showed an increase due to the presence of cerium (O/Ce). Part of the oxygen because of the contribution of cerium, and a percentage associated with oxidized silver (O/Ag). The bimetallic catalysts synthesized by recharge method favoured the formation of vacancies in the cerium oxide, this can be observed in the O/Ce ratio, as well as the reduction of gold (O/Ag + Au). The real Ag:Au atomic ratio was found between 0.8–0.92 close to the theoretical ratio proposed to the synthesis of these materials.
(g) TEM (transmission electron microscopy) and HRTEM (high resolution transmission electron microscopy)
Based on the TEM generated images, the average metal particle size of the monometallic and bimetallic catalysts was determined; Table 2 shows these results. The particle size distribution for the silver monometallic catalyst, shows 17% of particles larger than 8 nm; the highest concentration is between 4–6 nm and 45%, see Fig. 7A. After deposition of the second metal, a re-dispersion of the basic metal (Ag) was observed, causing the silver particles, smaller than 8 nm, to decrease in size; the monometallic catalysts of Au obtained an average metal size of 4.5, 4.3 and 3.9 for AuZr, AuZrCe10 and AuZrCe20 respectively, the gold catalysts showed the lowest average particle size in comparison with the other materials synthesized in this work. The result can be beneficial in the reaction of oxidation of phenol, it is known that the size of gold particles is of fundamental importance to obtain better results in oxidation reactions.60 The highest concentration of metal particles (Ag–Au) was centered between 3–5 nm with a percentage of 64%. In the bimetals, 9% of metal particles were observed between 2–3 nm, probably these are contributed by gold, because in the Ag monometallic catalysts the smallest particle quantified was 3 nm, see Fig. 6B.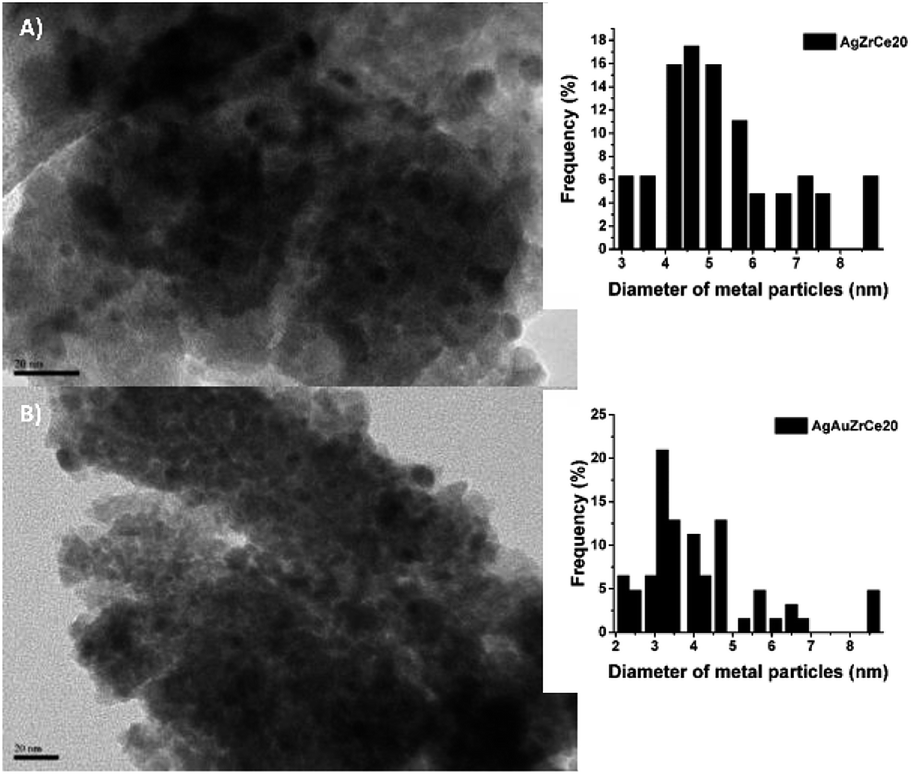 | ||
| Fig. 7 TEM images and distribution of monometallic and bimetallic catalysts particles. | ||
The HRTEM image of the AgZrCe10 sample can be seen in Fig. 8. The measurement of the interplanar distances was made and it was identified in the plane (211) of the tetragonal zirconia. It is also identical to the plane (220), corresponding to the ceramic oxide in its cubic structure. For the sample AuAgZrCe10, three planes (211), (220) and (111) were identified; Corresponding to tetragonal zirconia, ceramic oxide and silver oxide respectively. A difference in the results by X-ray diffraction, by HRTEM was possible to identify the presence of cerium oxide and silver oxide. These two species are found in the respective samples with a size that cannot be observed by X-rays. In the case of Au and Ag metallic can't be discriminated, having a very similar interplanar distance (Ag (111) d (A) = 2.359 and Au (111) d = 2.355).
 | ||
| Fig. 8 HRTEM images and distribution of monometallic and bimetallic catalysts particles. | ||
(h) Catalytic activity
Fig. 9A shows the results of the catalytic study for the oxidation of phenol using the catalysts AgZrCe and AgAuZrCe. A great difference can be observed between the Ag and Au monometallic catalysts. where 100% conversions were obtained at 7, 4 and 4 h for the AgZr and AgZrCe10 and AgZrCe20 catalysts respectively and the AuZr, AuZrCe10 and AuZrCe20 at 5, 3 and 2 h respectively, where the gold monometallic showed a better result that the ag monometallic clearly. However, the best effect to decrease the time for the phenol conversion were using the bimetallic catalysts for AgAuZr, AgAuZrCe10 and AgAuZrCe20 with 4, 2 and 1 hour respectively. According to the reaction rate (Table 4), the AgAu catalysts showed around 10 times faster than the monometallic catalysts, where the catalyst with the highest reaction rate was AgAuZrCe20 with 21.2 mmol h−1 g−1. Fig. 9B shows the behaviour to % ΔTOC. Table 4 shows the results obtained at one hour of reaction. AgAuZrCe catalysts showed the best performance to % ΔTOC with 51.8 and 63.1 for 10 and 20% in cerium respectively. Instead, Ag monometallic showed a low abatement between 2.3–6.0% and Au monometallic between 8.0–49.5%.| Catalyst | Xphenol (%) | ΔTOC (%) | SCO2 (%) | rA (mmol h−1 g−1) | TOF (h−1) |
|---|---|---|---|---|---|
| 1 h | 1h | 1 h | |||
| AgZr | 4.7 | 2.3 | 49.1 | 0.5 | 4.1 |
| AgZrCe10 | 14.7 | 6.0 | 41.1 | 2.6 | 21.1 |
| AgZrCe20 | 14.5 | 8.7 | 59.9 | 2.3 | 19.1 |
| AuZr | 16.1 | 8.0 | 49.7 | 1.7 | 12.8 |
| AuZrCe10 | 78.3 | 35.4 | 45.2 | 8.3 | 62.7 |
| AuZrCe20 | 86.2 | 49.5 | 57.2 | 9.1 | 68.9 |
| AgAuZr | 87.5 | 46.3 | 52.9 | 10.2 | 43.1 |
| AgAuZrCe10 | 98.1 | 51.8 | 52.8 | 20.7 | 90.1 |
| AgAuZrCe20 | 100 | 63.1 | 63.2 | 21.2 | 92.1 |
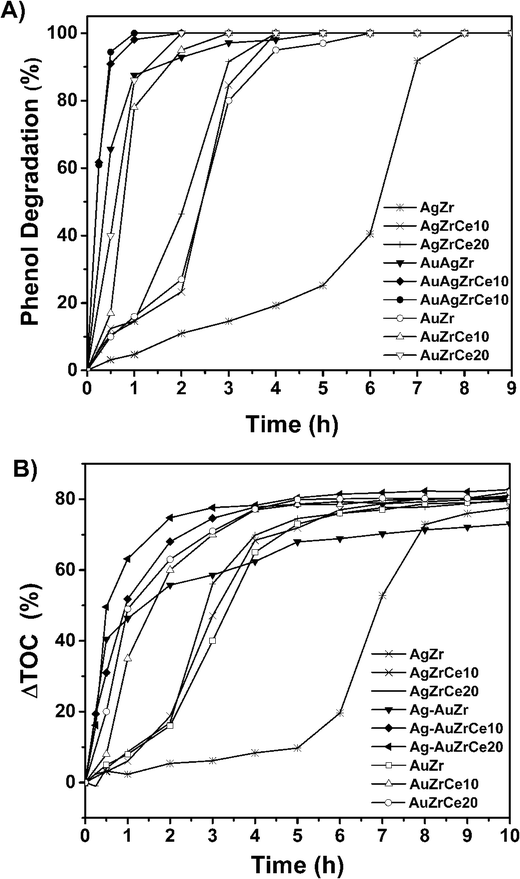 | ||
| Fig. 9 Phenol degradation in terms of time (A), total organic carbon abatement in terms of time (B). | ||
The action of cerium in the reaction is evident by producing a lower concentration in TOC abatement. This phenomenon can be explained according to the information of Table 3's data, a greater amount of Ce3+ helps to promote oxidation; this is due to the facility of the cerium oxide to pass from Ce4+ to Ce3+ state and its action as a source of oxygen for the phenol molecule oxidation.61 The turn over frequency. The turnover frequencies (TOFs) were examined for the conversion of phenol to CO2 as function of the Ag, Au and AgAu metal on catalyst ZrCe. TOF analysis is shown in Table 4. The AgAu catalysts showed a higher TOF compared to the Ag and Au catalysts with 43.1, 90.1 and 92.1 h−1 for AgAuZr, AgAuZrCe10 and AgAuZrCe20 respectively. While Ag and Au catalyst between 4.1–21.1 and 12.8–68.9 (h−1) respectively. This may be due to a saturation in the active sites due to the presence of acetic acid as an intermediate in the reaction, preventing the interaction of free phenol with the Ag sites. This is corroborated with the study of the selectivity of the reaction intermediates.
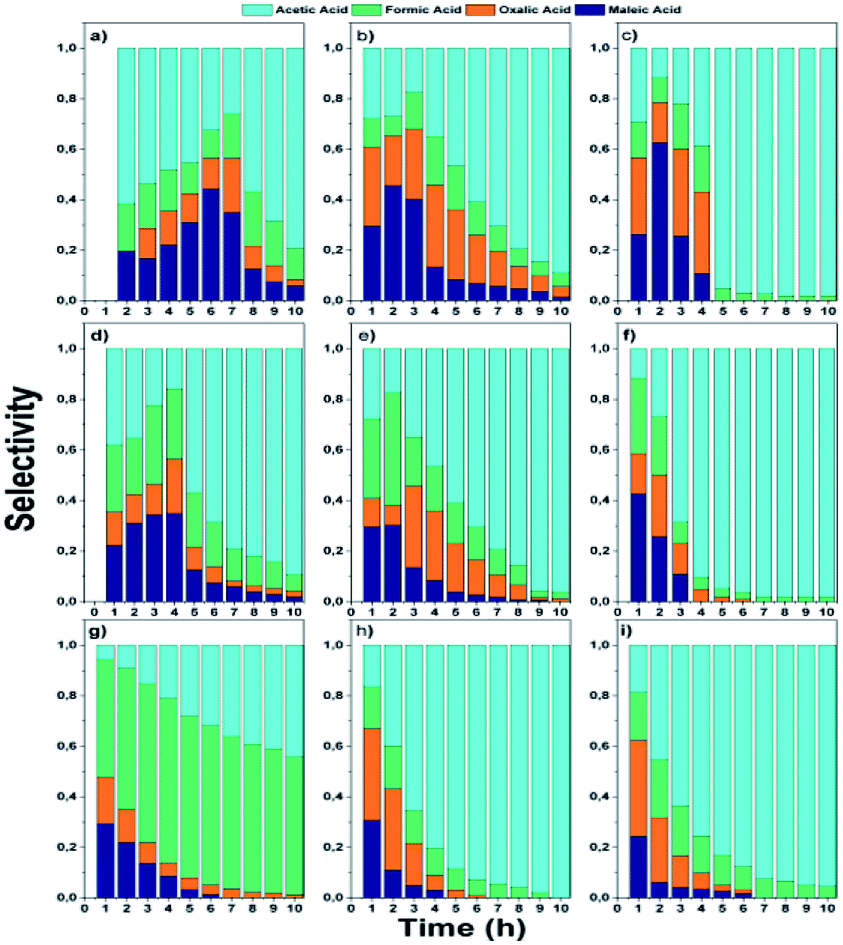 | ||
| Fig. 10 Selectivity to intermediaries from phenol oxidation: monometallic; (a) AgZr, (b) AgZrCe10, (c) AgZrCe20, (d) AuZr, (e) AuZrCe10, (f) AuZrCe20 and bimetallic catalysts (g) AgAuZr, (h) AgAuZrCe10 and (i) AgAuZrCe20. | ||
In the monometallic catalysts the identified intermediates were maleic, acetic, formic and oxalic acid. The AgZr catalyst showed a very important selectivity for acetic acid in the first 6 hours of reaction and (Fig. 10a), however, according to the selectivity behaviour, the AgZr catalyst took a long time to convert the acetic acid to CO2. Thus, some of the active sites were saturated, causing a very low reaction rate and conversion, shown in Fig. 9 and Table 4. Meanwhile, the AgZrCe catalysts with 10 and 20% in ceria showed similar behaviour to AgZr, but in a shorter time of 2 and 1 hour of reaction, accelerating the oxidation process of acetic acid (Fig. 10b and c). This is caused by the amount of cerium that contributes a greater quantity of oxygen to the surface of the catalyst, benefiting the oxidation of the intermediaries. For the monometallic gold catalysts (Fig. 10d–f), a greater preference can be observed towards the intermediates, oxalic acid and formic acid in comparison with the monometallic silver catalysts, this means that the gold catalysts have a greater preference towards the reaction route for maleic acid–oxalic acid–formic acid–CO2. In addition, it can be observed that acetic acid is a final product in the reaction. In addition, it can be observed that acetic acid is a final product in the reaction. On the other hand, the AgAuZr catalyst (Fig. 10 g) showed a greater selectivity to formic acid, since a greater selectivity was observed in comparison to the other intermediates. That is, gold provides another reaction route different from AgZr, besides significantly reducing the number of intermediaries with respect to silver at the end of the reaction, where only acetic and formic acid are observed. In Fig. 10h and 9i, the behaviour of the catalysts AgAuZrCe10 and AgAuZrCe20 respectively is observed. In comparison with AgAuZr, a high selectivity to formic acid is not observed, this is caused by the rapid selectivity of formic acid to CO2. This is proven by the increase in CO2 selectivity (Table 4). In addition, it is possible to observe a greater selectivity to oxalic acid than to maleic acid. It is reported by other authors that the route of oxidation of phenol by oxalic acid and formic acid is easier to reach CO2 and H2O than the route by acetic acid. In all catalysts the production of acetic acid was identified, which did not degrade; it is reported that for the degradation of the latter, higher reaction temperatures are required; around 200 °C,62 for this reason acetic acid could not be degraded since the reaction conditions were not the optimum, being the by-product of the reaction. After the analysis of the selectivity of intermediaries for the oxidation of phenol. In Fig. 11, the reaction route for the monometallic and bimetallic catalysts is proposed.
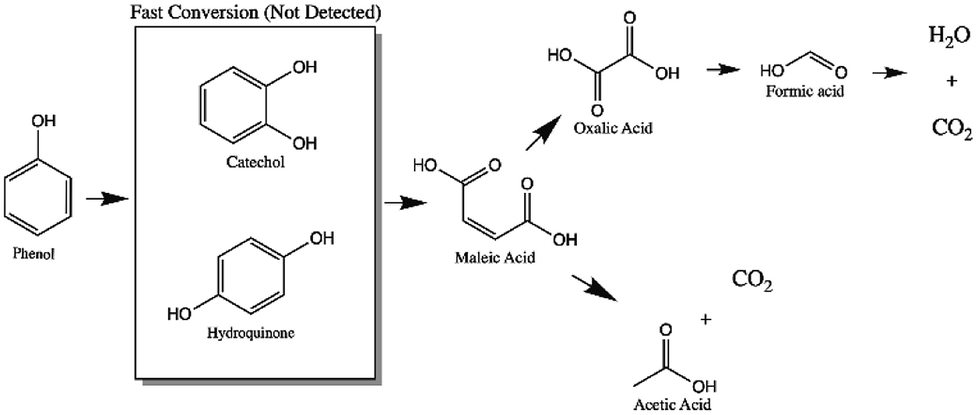 | ||
| Fig. 11 Route of the reaction proposed by CWAO of phenol using mono and bimetallic catalysts. | ||
It is of extreme importance to explore the stability and recyclability of the catalyst material as it could reduce the cost of the catalytic process and therefore results in promising catalyst. In the Fig. 11 showed the reuse cycles of the better catalyst in CWAO of phenol in this work. Moreover, AgAuZrCe20, AgZrCe20 and AuZrCe20 were compared to study the stability of the material. The reaction was carried out at 160 °C and 8 bar of O2, the reaction rate was quantified at the end of one hour of reaction. In each recycle, the catalyst was filtered and washed with methanol several times and the solvent were evaporated at a temperature of 120 °C, finally it was given a heat treatment with H2 at 400 °C for 1 hour. Fig. 12 show the results obtained. In the case of the AgZrCe20 catalyst it showed after of the third reuse a loss of reaction rate about 70% and AuZrCe20 with 22%. Meanwhile, the bimetallic catalyst showed 10% only. In such a way, the interaction gold/silver provides an improvement in stability and reusability in phenol oxidation has been showed clearly.
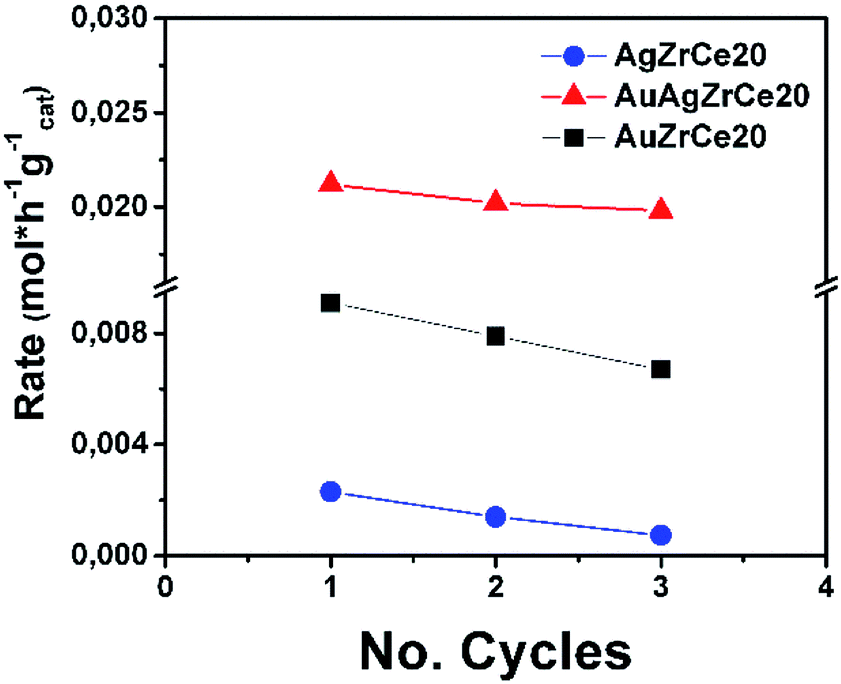 | ||
| Fig. 12 Reuse cycles for AgZrCe20 and bimetallic AgAuZrCe20 catalysts in CWAO of phenol. | ||
Experimental
(a) Supports synthesis
Zirconium oxide synthesis (ZrO2) was prepared by zirconium butoxide hydrolysis (Sigma-Aldrich, (Zr(OC4H9)4) 80% wt on 1-butanol), using butanol (Aldrich 98%) as solvent medium. The alkoxide/water ratio was 1:16 and the water/alcohol ratio was 1:8 molar. Water/butanol mixture was added in a three-necked flask, heated in recirculation at 70 °C. At the desired temperature, the zirconium butoxide was slowly incorporated for 3 hours. The pH was adjusted to 3 with acetic acid (J. T. Baker 98%) and left in recirculation for 24 hours. Once the gel was formed, it was subjected to the extraction of liquids in a rotary evaporator (Büchi-R-II) at a temperature of 60 °C.
ZrO2–CeO2 supports were synthesized using the same methodology as for ZrO2, first dissolving cerium precursor in a water–butanol mixture; which was cerium acetylacetonate (Sigma-Aldrich, Ce(C5H7O2)3·xH2O). Subsequently the zirconium butoxide was slowly added. All materials were exposed to an oxidizing atmosphere with a heating ramp of 2 °C min−1 until reaching 500 °C to stabilize the phases. The %wt of cerium was 10 and 20.
(b) Ag monometallic catalysts synthesis
The silver deposit was carried out by deposition–precipitation method, starting from silver nitrate (J. T Baker, (AgNO3), 99.7%), which was dissolved and stirred with oxide immersed in the solution for 20 minutes. Later, the temperature was warmed until 80 °C, and a solution of NaOH (J. T Baker, (NaOH) 98.1%) was slowly added, with a concentration of 0.5 M reaching a pH of 11. The mixture was left stirring for 2 hours. The sample was centrifuged to separate the phases, the solid was recovered and washed with distilled water at 50 °C, the washing process was repeated until a pH of 7 of the leached water was obtained. The material was dried at 120 °C for 12 hours. The product was oxidized in an oxidizing atmosphere, and then reduced in a reducing atmosphere (H2). Both thermal treatments were completed on a heating ramp of 2 °C min−1 and a gas flow of 60 mL min−1; final temperature was 300 °C for oxidation and 400 °C for metal reduction. The symbology of these materials is AgZr and AgZrCeX; where X is the percentage of cerium added.(c) Au monometallic catalyst synthesis
The preparation of Au monometallic sample with 2.5 wt% Au as theoretical loading was performed by deposition–precipitation with urea (DPU) in the absence of light. Briefly, the amount necessary of gold precursor, HAuCl4, and the urea (0.42 M) were dissolved in 100 mL of distilled water. Then, the support was added to this solution; thereafter, the suspension temperature was increased to 80 °C and kept constant for 16 h under stirring. After, the material was dried at 120 °C for 12 hours. Finally, the thermal treatments of the gold were similar that Ag catalyst. The symbology of these materials is AuZr and AuZrCeX; where X is the percentage of cerium added.(d) Bimetallic catalysts synthesis
Monometallic silver catalysts were modified by gold addition carried out with the recharge method reported by author J. Barbier et al.63 The molar ratio of the second metal (Au) was 1:1 regarding the present metal (Ag); using tetrachloroauric acid as precursor (Sigma-Aldrich, (HAuCl·6H2O), 99.999%).
The monometallic catalyst was introduced in a glass reactor, subsequently hydrogen was introduced by bubbling (1 h) and a further nitrogen treatment (0.5 h), allows elimination of dissolved or reversibly adsorbed hydrogen. The gold solution (HAuCI4) was introduced as an acid solution in which HCl was added to adjust the pH to 1. So freshly reduced catalyst was maintained in gold solution in the reactor and continuously stirred by a counter-current nitrogen flow (1 h). After the reaction, bimetallic catalyst prepared was dried until room temperature, then left at a temperature of 120 °C and a heating ramp of 2°C min−1 for 12 h. Finally, the catalyst was activated by hydrogen at a temperature of 400 °C for 4 h. The symbology for these materials is AgAuZr and AgAuZrCeX; where X is the percentage of cerium added.
(e) Characterization
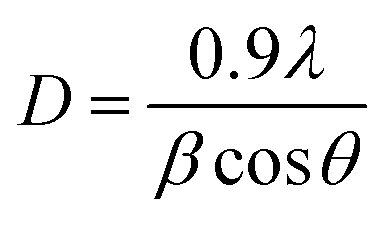
(f) Catalytic evaluation (catalytic wet air oxidation)
Wet air oxidation of phenol was performed in a stainless-steel Parr reactor with a capacity of 600 mL. The reaction was carried out at 160 °C and a partial pressure of 8 bar oxygen (ultra high purity 99.9999%) as oxidizing agent. Stirring of 1000 rpm during the reaction. A solution of 350 mL with a concentration of 1000 ppm and a catalyst ratio of 1 g L−1 with respect to the aqueous solution were introduced. Samples were taken at different times.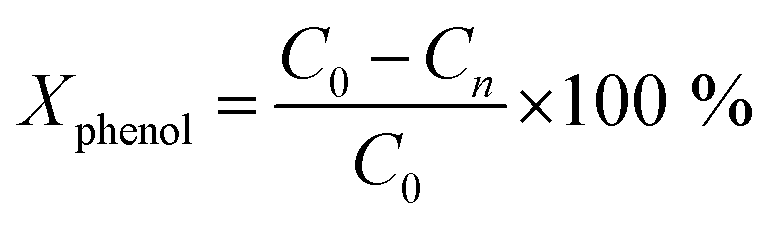
where C0 is a initial concentration of phenol and Cn is the phenol concentration to different reaction times. Ci and CT are the amount of one intermediate formed and the total amount of the other intermediates at a given time respectively. The initial rate (ri) was calculated from the phenol concentration as a function of time, using the follow equation:
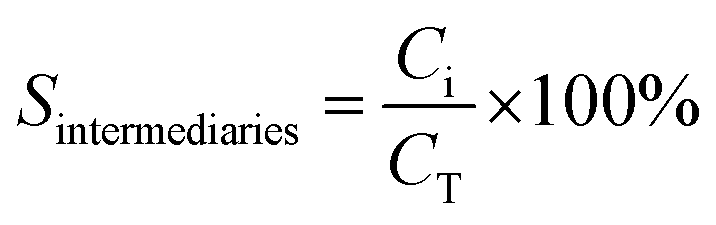
where
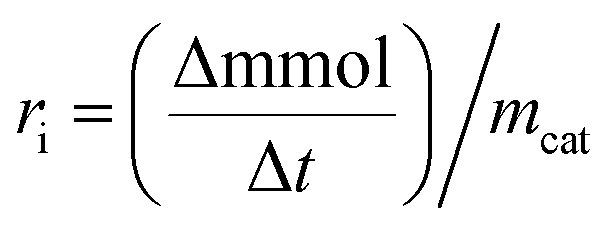
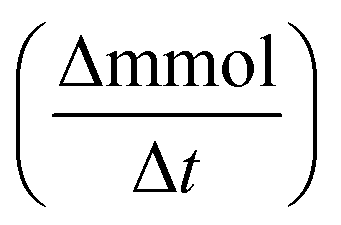 is a phenol concentration and mcat is the mass of the catalyst.
is a phenol concentration and mcat is the mass of the catalyst.where TOC0 is total organic carbon at t = 0 and TOCt is total organic carbon at different time in the oxidation reaction. The selectivity to CO2 was calculated according to follow equation:65
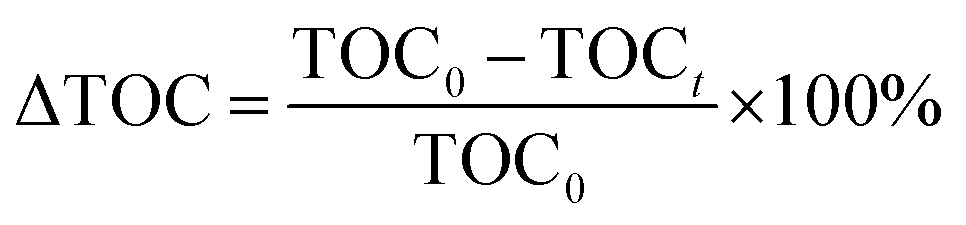
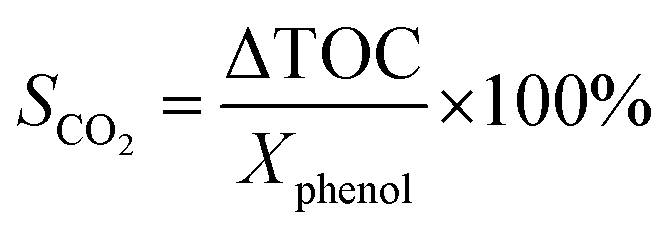
Conclusions
Strong interaction between added superficially metals (Ag–Au) with 1:1 molar ratio was found, due to the method of preparation. The prepared materials were evaluated by CWAO of phenol, in catalytic tests it was revealed that the addition of Au decreased the time of degradation. A significant effect was observed with the presence of CeO2 in the materials, which led to a lower formation of reaction intermediates and auto-oxidation of Ag and auto-reduction of Au. The presence of reduced Ce3+ species is associated with the generation of oxygen vacancies due to the charge de-compensation on the surface atoms of the catalyst. The addition of Ce caused an important TOC reduction in phenol CWAO, because of the oxide-reduction capacity of Ce4+/Ce3+.
CWAO reaction tests showed that the best catalyst was AgAuZrCe20 with 100% conversion in 1 h. Furthermore, the AuAgZrCe20 catalyst obtained a better conversion and selectivity to CO2 in comparison with previous results using the Au–Ag/ZrO2–CeO2 catalyst synthesized by deposition–precipitation precipitation with urea. In this way, the recharge method showed better effects for the synthesis of bimetallic catalyst with gold and silver metals over the mixed oxides ZrO2–CeO2. The activity may be related to several factors such as: a strong metal support interaction and the reducibility of the support; which influence the release of surface oxygen atoms during the reaction maintaining oxidized silver and gold in metallic state, being a determining factor for the catalytic activity; Fig. 13 illustrates all of the foregoing. Addition of gold changed the properties of silver monometallic catalysts by inhibiting the low formation of intermediates and changed of reaction route by formic acid to CO2 and water. In addition, the bimetallic catalyst showed in the reuse cycles the better stability in catalytic wet air oxidation of phenol. These results generate give guidelines to investigate another molar ratio for Ag–Au to improve in the phenol oxidation by CWAO.
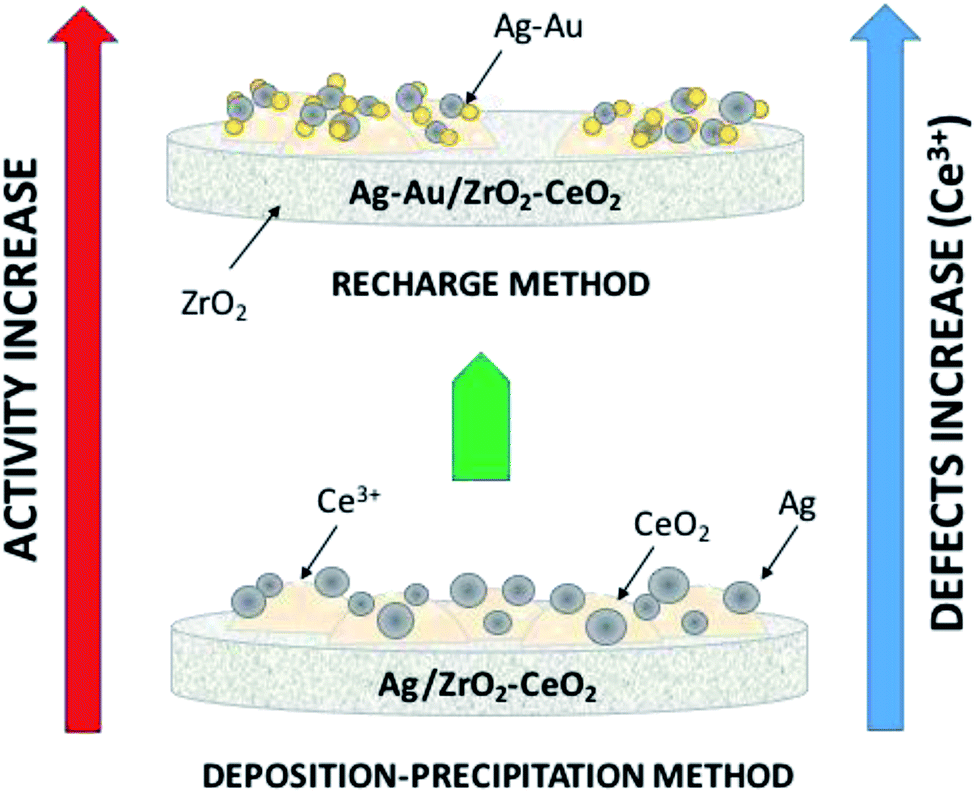 | ||
| Fig. 13 Illustration of the effects involved on catalytic activity on phenol degradation. | ||
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Adib Abiu Silahua Pavón thanks the National Council for Science and Technology (CONACYT) for Scholarship Postgraduate Studies, for financing the project 132648 and thanks the Universidad Juárez Autónoma de Tabasco for the support with PFCE Project. The ARC Centre of Excellence for Functional of Australian Institute of Bioengineering and Nanotechnology (AIBN) – University of Queensland by providing facilities and equipment for conducting characterizations during his international stay at the centre.References
- K. Pirkanniemi and M. Sillanpää, Chemosphere, 2002, 48, 1047–1060 CrossRef CAS PubMed.
- M. Abdollahi, S. Hassani and M. Derakhshani, Phenol, in Encyclopedia of Toxicology, ed. P. Wexler, Elsevier Academic Press, Amsterdam, The Netherland, 3rd edn, 2014, pp. 871–873 Search PubMed.
- W. Tang, H. Huang, Y. Gao, X. Liu, X. Yang, H. Ni and J. Zhang, Mater. Des., 2015, 88, 1191–1200 CrossRef CAS.
- A. E. de los Monteros, G. Lafaye, A. Cervantes, G. Del Angel, J. Barbier Jr and G. Torres, Catal. Today, 2015, 258, 564–569 CrossRef.
- Y. Zhang and J. H. Tay, Biochem. Eng. J., 2016, 106, 1–10 CrossRef CAS.
- J. Levec and A. Pintar, Catal. Today, 2007, 124, 172–184 CrossRef CAS.
- C. Pojanavaraphan, A. Luengnaruemitchai and E. Gulari, Int. J. Hydrogen Energy, 2013, 38, 1348–1362 CrossRef CAS.
- J. A. Montoya, E. Romero-Pascual, C. Gimon, P. Del Angel and A. Monzon, Catal. Today, 2000, 63, 71–85 CrossRef CAS.
- Z. Guerra-Que, G. Torres-Torres, H. Pérez-Vidal, I. Cuauhtémoc-López, A. E. de los Monteros, J. N. Beltramini and D. M. Frías-Márquez, RSC Adv., 2017, 7, 3599–3610 RSC.
- J. Barbier Jr, F. Delanoë, F. Jabouille, D. Duprez, G. Blanchard and P. Isnard, J. Catal., 1998, 177, 378–385 CrossRef.
- D. Tian, G. Yong, Y. Dai, X. Yan and S. Liu, Catal. Lett., 2009, 130, 211–216 CrossRef CAS.
- A. Lange de Oliveira, A. Wolf and F. Schüth, Catal. Lett., 2001, 73, 157–160 CrossRef CAS.
- H. Nakatsuji, Z.-M. Hu, H. Nakai and K. Ikeda, Surf. Sci., 1997, 387, 328–341 CrossRef CAS.
- C.-W. Yen, M.-L. Lin, A. Wang, S.-A. Chen, J.-M. Chen and C.-Y. Mou, J. Phys. Chem. C, 2009, 113, 17831–17839 CrossRef CAS.
- F. Núñez, G. Del Angel, F. Tzompantzi and J. Navarrete, Ind. Eng. Chem. Res., 2010, 50, 2495–2500 CrossRef.
- A. Izquierdo-Colorado, G. Torres-Torres, M. T. Gamboa-Rodríguez, A. A. Silahua-Pavón, J. C. Arévalo-Pérez, A. Cervantes-Uribe, A. Cordero-García and J. N. Beltramini, ChemistrySelect, 2019, 4, 1275–1284 CrossRef CAS.
- G. C. Bond and C. Louis and D. Thompson, Catalysis by gold, Imperial College Press, London, 2006 Search PubMed.
- N. D. Tran, M. Besson, C. Descorme, K. Fajerwerg and C. Louis, Catal. Commun., 2011, 16, 98–102 CrossRef CAS.
- R. Fiorenza, C. Crisafulli, G. G. Condorelli, F. Lupo and S. Scirè, Catal. Lett., 2015, 145, 1691–1702 CrossRef CAS.
- H. H. Kung, M. C. Kung and C. K. Costello, J. Catal., 2003, 216, 425–432 CrossRef CAS.
- H. Zhang and N. Toshima, Catal. Sci. Technol., 2013, 3, 268–278 RSC.
- A. Sandoval, A. Aguilar, C. Louis, A. Traverse and R. Zanella, J. Catal., 2011, 281, 40–49 CrossRef CAS.
- A. Zielińska-Jurek, E. Kowalska, J. W. Sobczak, W. Lisowski, B. Ohtani and A. Zaleska, Appl. Catal., B, 2011, 101, 504–514 CrossRef.
- Z. G. Que, J. G. T. Torres, H. P. Vidal, M. A. L. Rocha, J. C. A. Pérez, I. C. López, D. D. L. C. Romero, A. E. E. D. L. M. Reyna, J. G. P. Sosa, A. A. S. Pavón and J. S. F. Hernández, in Silver Nanoparticles - Fabrication, Characterization and Applications, ed. K. Maaz, InTech, 2018 Search PubMed.
- G. Li, X. Zhang, L. Wang, X. Song and Z. Sun, J. Electrochem. Soc., 2013, 160, F1116–F1122 CrossRef CAS.
- A. Zielińska-Jurek, E. Kowalska, J. W. Sobczak, W. Lisowski, B. Ohtani and A. Zaleska, Appl. Catal., B, 2011, 101, 504–514 CrossRef.
- E. A. Redina, O. A. Kirichenko, A. A. Greish, A. V. Kucherov, O. P. Tkachenko, G. I. Kapustin, I. V. Mishin and L. M. Kustov, Catal. Today, 2015, 246, 216–231 CrossRef CAS.
- K. A. Cychosz, R. Guillet-Nicolas, J. García-Martínez and M. Thommes, Chem. Soc. Rev., 2017, 46, 389–414 RSC.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CAS.
- A. L. Quinelato, E. Longo, E. R. Leite, M. I. B. Bernardi and J. A. Varela, J. Mater. Sci., 2001, 36, 3825–3830 CrossRef CAS.
- L. Meng, L. Liu, X. Zi, H. Dai, Z. Zhao, X. Wang and H. He, Front. Environ. Sci. Eng. China, 2010, 4, 164–171 CrossRef CAS.
- N. Igawa and Y. Ishii, J. Am. Ceram. Soc., 2001, 84, 1169–1171 CrossRef CAS.
- A. L. Quinelato, E. Longo, E. R. Leite, M. I. B. Bernardi and J. A. Varela, J. Mater. Sci., 2001, 36, 3825–3830 CrossRef CAS.
- E. Rubio-Rosas, D. Mendoza-Anaya, M. G. Salazar-Morales, P. Salas, P. R. G. Martínez and V. Rodríguez-Lugo, Mater. Manuf. Processes, 2007, 22, 301–304 CrossRef CAS.
- E. Hernández-Ramírez, J. A. Wang, L. F. Chen, M. A. Valenzuela and A. K. Dalai, Appl. Surf. Sci., 2017, 399, 77–85 CrossRef.
- W. Huang, J. Yang, C. Wang, B. Zou, X. Meng, Y. Wang, X. Cao and Z. Wang, Mater. Res. Bull., 2012, 47, 2349–2356 CrossRef CAS.
- E. W. N. U. F. F. I. E. L. D, X-ray Diffraction Methods, John Wiley and Sons Inc, New York, 1986 Search PubMed.
- A. Slistan-Grijalva, R. Herrera-Urbina, J. F. Rivas-Silva, M. Ávalos-Borja, F. F. Castillón-Barraza and A. Posada-Amarillas, Phys. E, 2005, 27, 104–112 CrossRef CAS.
- C. Brechignac, P. Houdy and M. Lahmani, Nanomaterials and Nanochemistry, Springer, Berlin, 2008 Search PubMed.
- F. Foss, Metal Nanoparticles: Synthesis, Characterization, and Applications, Marcel Dekker, Inc., New York and Basel, 2002 Search PubMed.
- Z. Qu, F. Yu, X. Zhang, Y. Wang and J. Gao, Chem. Eng. J., 2013, 229, 522–532 CrossRef CAS.
- G. Balducci, J. Kas, P. Fornasiero, M. Graziani and M. S. Islam, J. Phys. Chem. B, 1998, 102, 557–561 CrossRef CAS.
- G. Ranga Rao and H. R. Sahu, J. Chem. Sci., 2001, 113, 651–658 CrossRef.
- S. Shukla and S. Seal, Nanostruct. Mater., 1999, 11, 1181–1193 CrossRef CAS.
- D. M. Mott, D. T. N. Anh, P. Singh, C. Shankar and S. Maenosono, Adv. Colloid Interface Sci., 2012, 185–186, 14–33 CrossRef CAS PubMed.
- M. U. Niemann, S. S. Srinivasan, A. R. Phani, A. Kumar, D. Y. Goswami and E. K. Stefanakos, J. Nanomater., 2008, 1–9 Search PubMed.
- J. Sun, X. Gao, D. He, J. Chen, X. Meng, Q. Zhang, L. Shen and H. Jiao, Particuology, 2013, 11, 776–781 CrossRef CAS.
- B. Azambre, L. Zenboury, A. Koch and J. V. Weber, J. Phys. Chem. C, 2009, 113, 13287–13299 CrossRef CAS.
- S. Watanabe, H. Sugiyama, H. Adachi, H. Tanaka and M. T. Miyahara, J. Chem. Phys., 2009, 130, 164707 CrossRef PubMed.
- G. S. A. M. Theunissen, A. J. A. Winnubst and A. J. Burggraaf, J. Mater. Sci., 1992, 27, 5057–5066 CrossRef CAS.
- X. Wu, J. Fan, R. Ran and D. Weng, Chem. Eng. J., 2005, 109, 133–139 CrossRef CAS.
- S. Ricote, G. Jacobs, M. Milling, Y. Ji, P. M. Patterson and B. H. Davis, Appl. Catal., A, 2006, 303, 35–47 CrossRef CAS.
- Q. Fu, S. Kudriavtseva, H. Saltsburg and M. Flytzani-Stephanopoulos, Chem. Eng. J., 2003, 93, 41–53 CrossRef CAS.
- S. Peters, S. Peredkov, M. Neeb, W. Eberhardt and M. Al-Hada, Surf. Sci., 2013, 608, 129–134 CrossRef CAS.
- L. Makhova, Y. Mikhlin and A. Romanchenko, Nucl. Instrum. Methods Phys. Res., Sect. A, 2007, 575, 75–77 CrossRef CAS.
- M. P. Casaletto, A. Longo, A. Martorana, A. Prestianni and A. M. Venezia, Surf. Interface Anal., 2006, 38, 215–218 CrossRef CAS.
- P. Duran, M. Gonzalez, C. Moure, J. R. Jurado and C. Pascual, J. Mater. Sci., 1990, 25, 5001–5006 CrossRef CAS.
- K. Matsuo and K. Nakano, Appl. Surf. Sci., 1990, 41–42, 53–56 CrossRef.
- S. Tsunekawa, R. Sivamohan, S. Ito, A. Kasuya and T. Fukuda, Nanostruct. Mater., 1999, 11, 141–147 CrossRef CAS.
- P. Sangeetha and Y.-W. Chen, Int. J. Hydrogen Energy, 2009, 34, 7342–7347 CrossRef CAS.
- A. Shah, L. Ur Rahman, R. Qureshi and Z. Ur Rehman, Rev. Adv. Mater. Sci., 2012, 30, 133–149 CAS.
- J. Mikulová, S. Rossignol, J. Barbier, D. Mesnard, C. Kappenstein and D. Duprez, Appl. Catal., B, 2007, 72, 1–10 CrossRef.
- J. Barbier, J. C. Menezo, C. Montassier, J. Naja, G. Del Angel and J. M. Dominguez, Catal. Lett., 1992, 14, 37–43 CrossRef CAS.
- F. Lin, B. E. Cojocaru, L. S. Williams, C. A. Cadigan, C. Tian, M. N. Grecu, H. L. Xin, S. Vyas, V. I. Parvulescu and R. M. Richards, Nanoscale, 2017, 9, 9359–9364 RSC.
- S. S. Lin, D. J. Chang, C.-H. Wang and C. C. Chen, Water Res., 2003, 37, 793–800 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |