
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring the mode of action of inhibitors targeting the PhoP response regulator of Salmonella enterica through comprehensive pharmacophore approaches†
Keng-Chang Tsai‡
ab,
Po-Pin Hung‡c,
Ching-Feng Chengde,
Chinpan Chen *e and
Tien-Sheng Tseng*f
*e and
Tien-Sheng Tseng*f
aNational Research Institute of Chinese Medicine, Ministry of Health and Welfare, Taipei 112, Taiwan
bThe PhD Program in Medical Biotechnology, College of Medical Science and Technology, Taipei Medical University, Taipei, Taiwan
cDivision of Infectious Disease, Department of Internal Medicine, Taipei Tzu Chi Hospital, The Buddhist Tzu Chi Medical Foundation, New Taipei City 231, Taiwan
dDepartment of Pediatrics, Taipei Tzu Chi Hospital, Buddhist Tzu Chi Medical Foundation, Taipei and Tzu Chi University, Hualien, Taiwan
eInstitute of Biomedical Sciences, Academia Sinica, Taipei 115, Taiwan
fDepartment of Research, Taipei Tzu Chi Hospital, The Buddhist Tzu Chi Medical Foundation, New Taipei City 231, Taiwan
First published on 21st March 2019
Abstract
The PhoQ/PhoP two-component system regulates the physiological and virulence functions of Salmonella enterica. However, the mode of action of known PhoP inhibitors is unclear. We systematically constructed a pharmacophore model of inhibitors to probe the interface pharmacophore model of the PhoP dimer, coupling it with Ligplot analysis. We found that these inhibitors bind on the α5-helix, altering the conformation and interfering with PhoP binding on DNA.
The development of antibiotic resistance to various infectious diseases is highly associated with the antimicrobial agents used in clinical practice. Long-term antibiotic therapy mainly causes microorganisms, initially sensitive to but gradually adapted to antibiotics, to develop resistance.1 Notably, with the increasing emergence of multi-antibiotic-resistant bacteria, the invention of antibiotics which are less subject to developing resistance is urgently needed, while the targeting of the signal transduction systems of bacteria has been demonstrated to be a feasible strategy in the development of antibiotics.2–4 The two-component system (TCS), the predominant signal transduction pathway in bacteria, senses and responds to environmental changes to enable the bacteria to thrive and survive.5–8 Generally, TCSs consist of a histidine kinase sensor and a receiver response regulator.5 The histidine kinase sensor, autophosphorylated on the conserved histidine residue, transfers the phosphoryl group to the specific aspartate residue of the response regulator. The response regulators are mainly transcription factors which are composed of an N-terminal receiver domain and a C-terminal DNA-binding domain. The phosphorylation of the response regulator modulates the transcriptional regulation of target genes.9
OmpR/PhoB, NarL/FixJ, and Ntrc/DctD are major subfamilies of response regulators, classified on the basis of the sequence similarity of the C-terminal DNA-binding domain.10 The PhoQ/PhoP two-component system, belonging to the OmpR/PhoB subfamily, plays an important role in the virulence of Salmonella enterica.11 The low Mg2+ level, antimicrobial peptides, and acidic pH activate the PhoQ/PhoP in S. enterica to modulate the physiological and virulence functions.12–15 PhoQ is autophosphorylated under a low extracellular Mg2+ level and subsequently transfers its phosphate group from histidine to the conserved aspartate of the PhoP response regulator. The phosphorylated PhoP forms a homodimer and in turn recognizes and binds to PhoP boxes in the promoters of PhoP-regulated genes.16 The receiver domain of the response regulators in the OmpR/PhoB subfamily shares a highly conserved α4–β5–α5 dimeric interface.17 It was proposed that the bacterial virulence could be disrupted by interfering with the interactions within this conserved, structural motif. The plastic α4–β5–α5 interfaces of the response regulators share almost identical hydrophobic contacts and salt-bridge interactions essential for homodimerization among different bacteria species.6,18 The crystal structure of E. coli PhoP (PDB ID: 2PKX) shows that the dimer interface has only one residue different to that of S. enterica.19 Therefore, S. enterica PhoP is an attractive target for a structure-based drug design to identify potential inhibitors for virulence regulation.
Recently, Tang et al. conducted an approach to screen inhibitors against the PhoP response regulator of S. enterica.16 Eight compounds were found to inhibit the PhoP–DNA complex formation of S. enterica. Additionally, it has been proposed that the inhibitors may prevent PhoP binding to cognate DNA by an allosteric mechanism. However, the potential binding site of these inhibitors is unclear. No atomic details of the PhoP–inhibitor complex investigated by X-ray crystallography or NMR are available. Quantitative structure–activity relationship (QSAR) is a technique to generate a pharmacophore with common features among a set of active ligands, allowing us to estimate the functional features responsible for interacting with the target site.20–22 Our previous study conducted a protein–DNA complex-guided pharmacophore approach to screen inhibitors against the response regulator, PmrA, of polymyxin B-resistant Klebsiella pneumoniae (KP). The identified lead, E1 (IC50 = 10.2 μM), targets the DNA binding domain of PmrA (KD = 1.7 μM), and restores the susceptibility of KP to polymyxin B.23 Likewise, here, unprecedentedly, we investigated and constructed a ligand/inhibitor-based pharmacophore model (LigPhar) probing into the interface pharmacophore (InfPhar) of PhoP to unveil the potential binding site of PhoP inhibitors. Meanwhile, ligand-pharmacophore mapping and Ligplot analysis were applied to disclose the detailed interactions of inhibitors with PhoP in this study.
Ligand-based common feature pharmacophore generation
In the current study, 6 representative PhoP inhibitors (Fig. 1), with concentrations covering 6 orders of magnitude (from 3.6 μM to 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 μM), were applied to train and build the common feature pharmacophore model by using the Common Feature Pharmacophore Generation module implemented in Discovery Studio 3.5 (Accelrys Software, Inc., San Diego, CA, USA). A CHARMM-like force field24 was employed to build and minimize all chemical structures. The value of the maximum conformations was limited to 255 by using the “best conformation generation” method with a 20 kcal mol−1 energy cutoff. The rest of the parameters were set as the default. Three features—hydrogen-bond donor, hydrogen-bond acceptor and hydrophobic group features—were applied to build the pharmacophore hypothesis. All calculations were performed on a Scorpio SGI Origin 3800 (NCHC, Taiwan). The results are shown in Fig. 1: the common feature pharmacophore of NSC46830 and NSC45576 was generated first, consisting of one hydrogen-bond donor, two hydrogen-bond acceptors and two hydrophobic features. Subsequently, molecules NSC88915, NSC168179, NSC9608, and NSC35489 were individually added into the pharmacophore generation. The top-ranked pharmacophore generated in each step is presented (Fig. 1) and the final pharmacophore model, LigPhar, was constructed based on all 6 compounds, consisting of one hydrogen-bond acceptor and two hydrophobic group features.
000 μM), were applied to train and build the common feature pharmacophore model by using the Common Feature Pharmacophore Generation module implemented in Discovery Studio 3.5 (Accelrys Software, Inc., San Diego, CA, USA). A CHARMM-like force field24 was employed to build and minimize all chemical structures. The value of the maximum conformations was limited to 255 by using the “best conformation generation” method with a 20 kcal mol−1 energy cutoff. The rest of the parameters were set as the default. Three features—hydrogen-bond donor, hydrogen-bond acceptor and hydrophobic group features—were applied to build the pharmacophore hypothesis. All calculations were performed on a Scorpio SGI Origin 3800 (NCHC, Taiwan). The results are shown in Fig. 1: the common feature pharmacophore of NSC46830 and NSC45576 was generated first, consisting of one hydrogen-bond donor, two hydrogen-bond acceptors and two hydrophobic features. Subsequently, molecules NSC88915, NSC168179, NSC9608, and NSC35489 were individually added into the pharmacophore generation. The top-ranked pharmacophore generated in each step is presented (Fig. 1) and the final pharmacophore model, LigPhar, was constructed based on all 6 compounds, consisting of one hydrogen-bond acceptor and two hydrophobic group features.
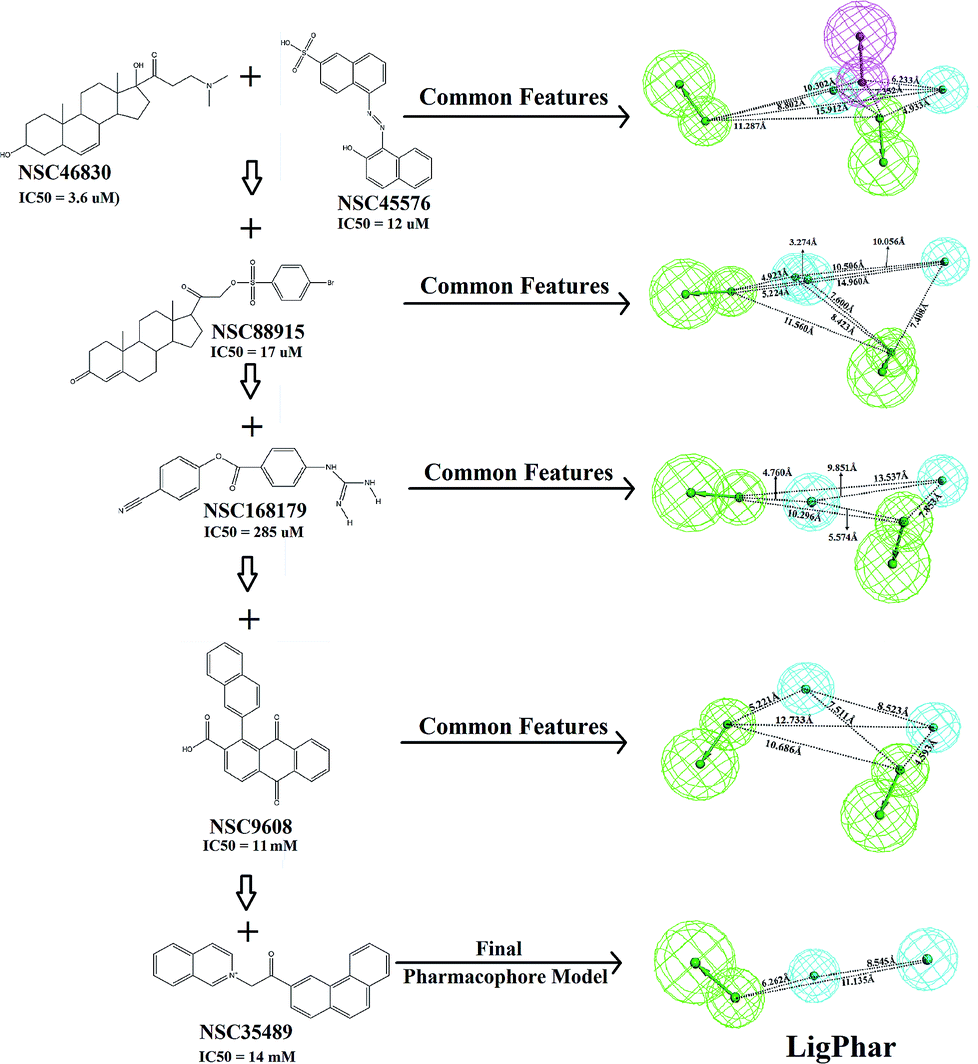 | ||
| Fig. 1 The chemical structures of 6 PhoP inhibitors and the scheme of ligand-based pharmacophore generation. (Pharmacophore features are colored as follows: hydrogen-bond acceptor, green; hydrogen-bond donor, magenta; hydrophobic group, cyan.) | ||
Receptor–ligand pharmacophore generation
The homodimerization of PhoP is essential for recognizing and binding to DNA. According to Garland's study, it was suggested that the inhibitors potentially dissociate PhoP dimerization.16 Thus, an understanding of the residue interaction network within the dimer interface of PhoP is important and helpful for identifying the potential binding site of inhibitors. A set of hydrophobic and charge residues, contributing to homodimerization by van der Waals contacts and salt-bridges, were observed at the α4–β5–α5 interface of PhoP (Fig. 2). These crucial residues are functionally complementary so they interact with each other, maintaining and stabilizing the PhoP homodimer. Therefore, we carried out receptor–ligand pharmacophore generation to construct the interface pharmacophore of PhoP, which reveals the functionally essential features for homodimerization. The whole process is illustrated in Fig. 3. Firstly, chain B of PhoP (PDB ID: 2PKX) was regarded as a receptor, and the interface residues of chain A (residues: 84–119) were considered as ligands. These two chains were further subjected to Receptor–ligand pharmacophore generation (implemented in Discovery Studio 3.5) to build an interface pharmacophore, InfPhar. The minimum and maximum features were set to 4 and 20, respectively. The default setting was employed for the rest of the parameters. The receptor–ligand pharmacophore generation produced 10 pharmacophore hypotheses and the top-ranked model, InfPhar, which contains two hydrogen-bond acceptors, four hydrogen-bond donors, two hydrophobic, one negative ionizable, two positive ionizable, and one ring aromatic features (Fig. 3), was used for further study.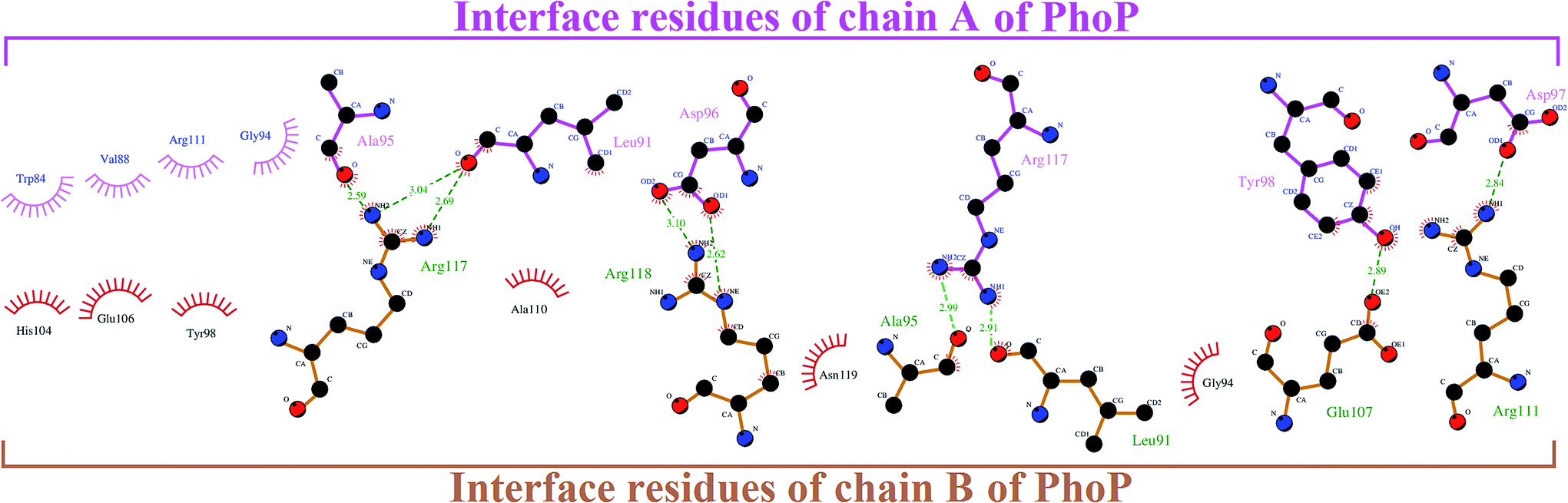 | ||
| Fig. 2 The interface interaction network within the PhoP homodimer analyzed by Ligplot.25,26 | ||
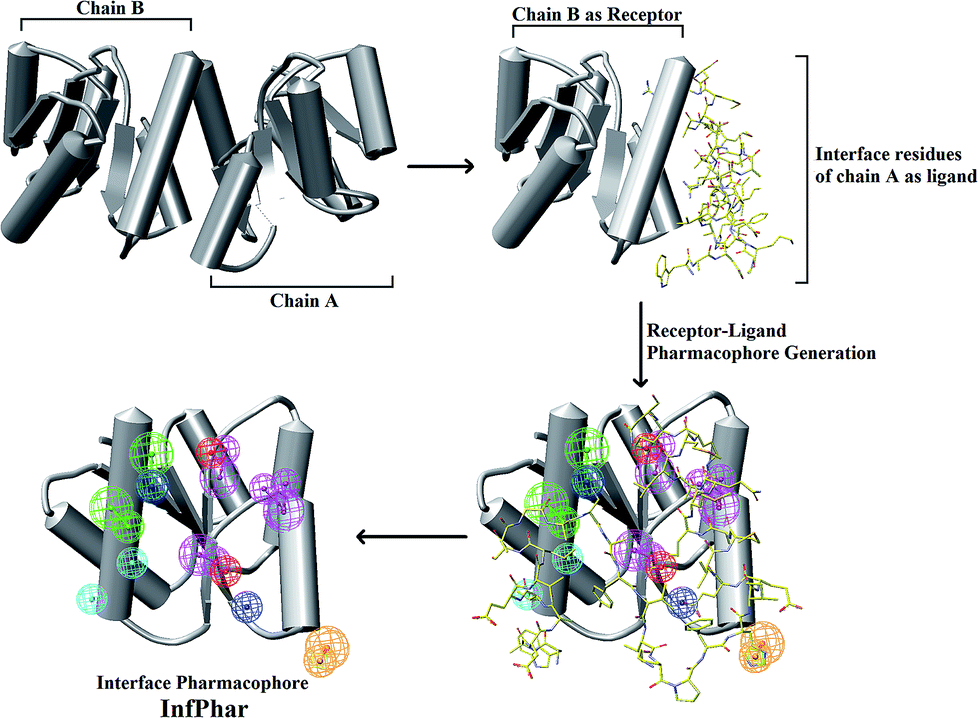 | ||
| Fig. 3 A flowchart of the interface pharmacophore (InfPhar) generation process. (Pharmacophore features are colored as follows: hydrogen-bond acceptor, green; hydrogen-bond donor, magenta; hydrophobic group, cyan; negative ionizable, red; positive ionizable, blue; one ring aromatic, orange) | ||
Pharmacophore probing reveals the potential binding site of PhoP inhibitors
To find out the potential binding site of PhoP inhibitors, the built ligand-based pharmacophore model, LigPhar, was used to probe further into the interface pharmacophore model, InfPhar. The results showed that three features (one hydrogen-bond acceptor and two hydrophobic groups) in the left-hand corner of InfPhar (Fig. 4B) overlapped well with the LigPhar (RMSD = 1.06 Å), demonstrating high similarity in terms of the steric arrangement and functional properties existing among the probed and targeted features and revealing the potential binding site of those inhibitors on PhoP. Consequently, ligand-pharmacophore mapping was applied to verify and investigate the binding modes of the inhibitors on the targeted features (one hydrogen-bond acceptor and two hydrophobic groups). The inhibitors fitted onto the targeted features are shown in Fig. 5 and their fitted values present statistically significant correlation with the pIC50 values (r ≥ 0.6). Moreover, the models of the inhibitor complexed with PhoP were analyzed by Ligplot25,26 to understand the interactions in detail, as shown in Fig. 5. All the inhibitors bind at the α5-helix and display hydrogen-bond interaction with Arg117(B). Meanwhile, Ala110(B), Arg111(B), Glu113(B), and Ala114(B) are the most common residues exhibiting hydrophobic contacts with the inhibitors. Moreover, molecular dynamics (MD) simulations (please see the ESI† for details) were performed to observe the conformations of the docked poses of PhoP inhibitors under well-equilibrated conditions. The results showed that the docked inhibitors of PhoP exhibited minor or very little change in their orientations and conformations after the MD simulations. The RMSD values of the superimposed inhibitors (before and after MD simulations) are shown in Fig. S1.† The inhibitors NSC45576 and NSC88915 displayed minor deviations in their conformations with RMSD values of 1.68 and 1.74, respectively. The inhibitors NSC168179, NSC48630, NSC35498, and NSC9608 showed very little change in their conformations with RMSD values of 1.01, 1.0, 0.81, and 0.58, respectively. This indicated that the docked poses of inhibitors of PhoP from pharmacophore mapping are reliable under conditions close to well-equilibrated systems. This result could also validate the observed interactions of the identified inhibitors at the binding site on PhoP.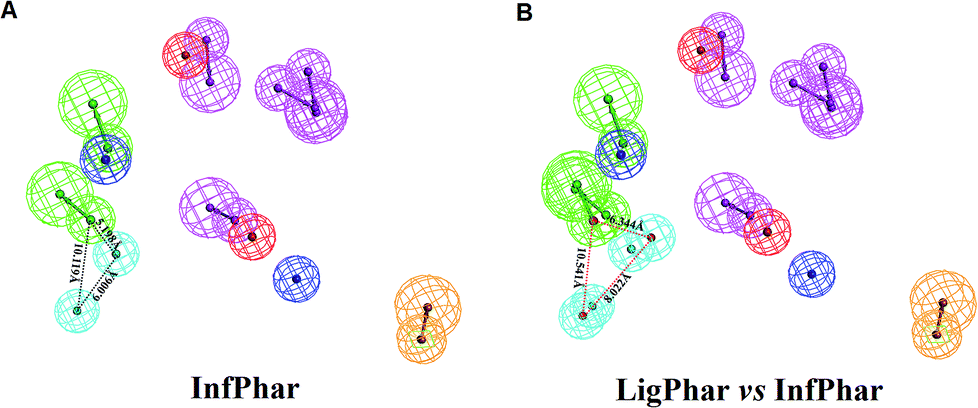 | ||
| Fig. 4 A diagrammatic representation of the result of pharmacophore probing. (A) The targeted pharmacophore features (one hydrogen-bond acceptor and two hydrophobic groups) on the interface pharmacophore model, InfPhar. The distances between these features were measured and labelled by black dashed lines. (B) The LigPhar was superimposed onto InfPhar and the RMSD was 1.06 Å. The red dashed lines denote the distances of the features in LigPhar. | ||
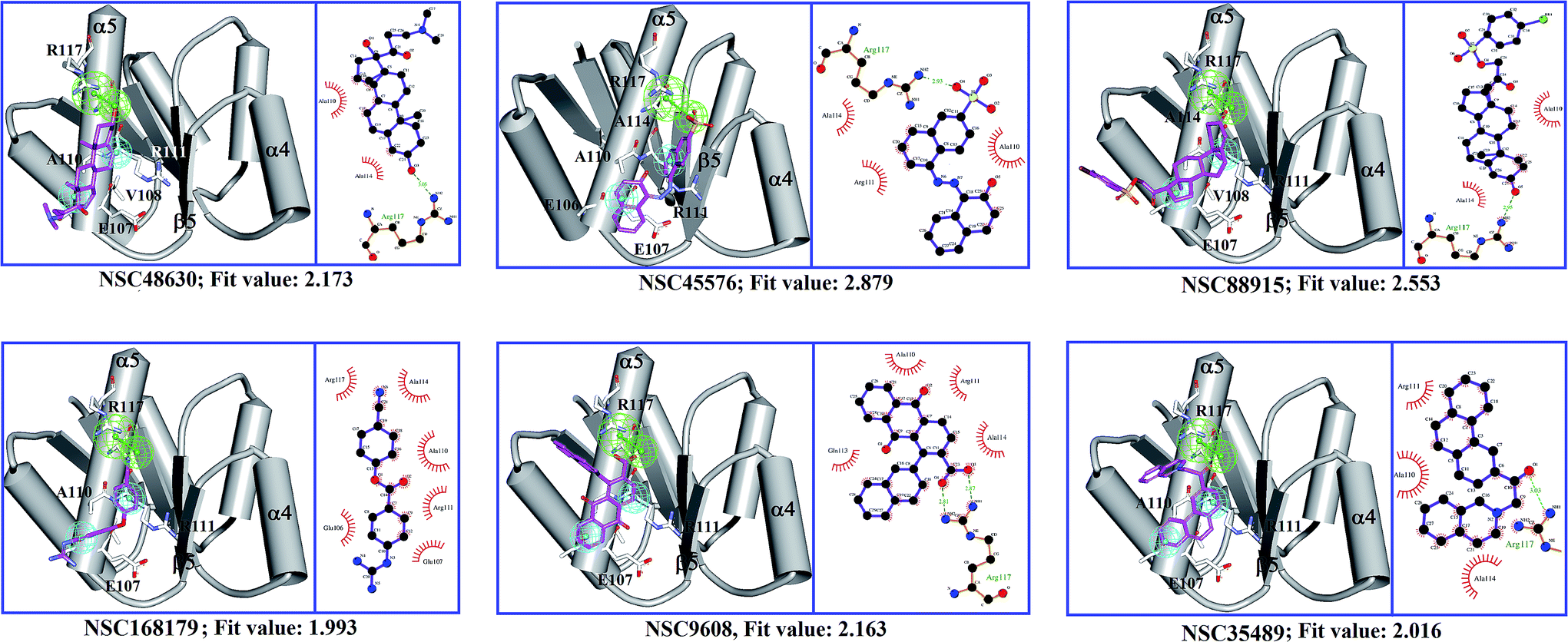 | ||
| Fig. 5 A diagrammatic representation of the results of ligand-pharmacophore mapping and Ligplot analysis of each PhoP–inhibitor complex model. Chain B of PhoP (PDB ID: 2PKX) is shown in schematic style, colored in gray. The PhoP inhibitors are presented as sticks in magenta. Residues of PhoP interacting with inhibitors are shown as white sticks. | ||
It has been reported that phosphorylation of PhoP presumably induces a conformational change to mediate homodimerization for DNA binding.16,27–29 However, a recent study reveals that dimerization of PhoP is phosphorylation independent.30 Sangaralingam et al., 2005, reported that a PhoP monomer sequentially binds to a canonical PhoP box—the first monomer binds with higher affinity followed by weak binding, but the rate-limiting second PhoP monomer, and the increasing concentration of PhoP promote (PhoP)2–DNA complex formation.31 Importantly, the phosphorylation has a favorable effect on the kinetics of PhoP–DNA interaction by overcoming the rate-limiting step. In a previous study by Garland et al., in 2012, eight inhibitors were discovered to disrupt PhoP–DNA complex formation by an allosteric mechanism binding to the plastic α4–β5–α5 interface.16 In the current study, we applied those inhibitors to build a ligand-based pharmacophore model (LigPhar) probing the receptor–ligand pharmacophore model (InfPhar) to reverse trace the potential binding site of PhoP inhibitors. Our study found that inhibitors have great potential to bind at the α5-helix of monomeric PhoP, interacting with Arg117(B) by hydrogen-bonding and contacting Ala111(B), Arg111(B), and Glu111(B) by hydrophobic interactions. At the plastic α4–β5–α5 interface of PhoP, Arg117(B) forms hydrogen-bonds with Ala95(A) and Leu91(A). In addition, Ala110b, Arg111(B), and Glu113(B), interact with the interface residues of chain A by hydrophobic contacts (Fig. 2). These interactions are not only essential for the homodimerization of PhoP but contribute to its stability. Thus, the binding of inhibitors on monomeric PhoP could interfere with the interaction networks of the α4–β5–α5 interface of dimeric PhoP. Notably, PhoP is in monomer–dimer equilibrium after phosphorylation. Thus, upon the binding of inhibitors, the structural property and conformation of monomeric PhoP could somehow be changed. This may have little or no impact on the homodimerization, as observed by Garland's study, but it could significantly affect the binding of either the first or the rate-limiting second monomeric PhoP on DNA.
Conclusions
In summary, our study presents a unique way to systematically and rationally reveal the potential and reliable binding site of PhoP inhibitors by comprehensively analyzing and organizing the information about PhoP inhibitors to construct the common, pharmacophore, LigPhar. We employed this probe to reverse trace the same functional features on the interface pharmacophore, InfPhar. We unveiled that the inhibitors bind on the α5-helix by interacting with Arg111(B), Ala110(B), Arg111(B), and Glu113(B) and these observations are in accordance with the original virtual screening setting in Garland's study—the active site center is surrounded by Arg111(B) and Arg118(B). Based on our finding and previous reports,16,30,31 we proposed a possible mode of action of the PhoP inhibitors—the binding of inhibitors on the α5-helix may interfere with the interaction of either the first or the second, rate-limiting, monomeric PhoP on DNA by altering the structural properties or conformation of monomeric PhoP. Here, we demonstrated a novel strategy to trace the potential binding site of inhibitors by using ligand-based and receptor–ligand pharmacophore generation techniques. We believe that this strategy will work and be applicable in other cases.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The study was supported by a grant from the Taipei Tzu Chi Hospital, Buddhist Tzu Chi Medical Foundation (TCRD-TPE-108-67(1/2)) and the Ministry of Science and Technology, Taiwan, RO (MOST 105-2320-B-001-019-MY3).Notes and references
- A. Giedraitiene, A. Vitkauskiene, R. Naginiene and A. Pavilonis, Medicina, 2011, 47, 137–146 CrossRef PubMed.
- D. A. Rasko, C. G. Moreira, R. Li de, N. C. Reading, J. M. Ritchie, M. K. Waldor, N. Williams, R. Taussig, S. Wei, M. Roth, D. T. Hughes, J. F. Huntley, M. W. Fina, J. R. Falck and V. Sperandio, Science, 2008, 321, 1078–1080 CrossRef CAS PubMed.
- E. A. Shakhnovich, D. T. Hung, E. Pierson, K. Lee and J. J. Mekalanos, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2372–2377 CrossRef CAS PubMed.
- D. T. Hung, E. A. Shakhnovich, E. Pierson and J. J. Mekalanos, Science, 2005, 310, 670–674 CrossRef CAS PubMed.
- A. M. Stock, V. L. Robinson and P. N. Goudreau, Annu. Rev. Biochem., 2000, 69, 183–215 CrossRef CAS PubMed.
- R. Gao, T. R. Mack and A. M. Stock, Trends Biochem. Sci., 2007, 32, 225–234 CrossRef CAS PubMed.
- R. Gao and A. M. Stock, Annu. Rev. Microbiol., 2009, 63, 133–154 CrossRef CAS PubMed.
- R. Gao and A. M. Stock, Curr. Opin. Microbiol., 2010, 13, 160–167 CrossRef CAS PubMed.
- A. M. Stock and A. H. West, Response regulator proteins and their interactions with histidine protein kinases, Academic Press, New York, 2003 Search PubMed.
- J. B. Stock, A. J. Ninfa and A. M. Stock, Microbiol. Rev., 1989, 53, 450–490 CAS.
- E. A. Groisman, J. Bacteriol., 2001, 183, 1835–1842 CrossRef CAS PubMed.
- S. F. E. Vescovi and E. Groisman, Cell, 1996, 165–174 CrossRef.
- E. Choi, E. A. Groisman and D. Shin, J. Bacteriol., 2009, 191, 7174–7181 CrossRef CAS PubMed.
- A. Kato and E. A. Groisman, Adv. Exp. Med. Biol., 2008, 631, 7–21 CrossRef CAS PubMed.
- Y. Shi, M. J. Cromie, F. F. Hsu, J. Turk and E. A. Groisman, Mol. Microbiol., 2004, 53, 229–241 CrossRef CAS PubMed.
- Y. T. Tang, R. Gao, J. J. Havranek, E. A. Groisman, A. M. Stock and G. R. Marshall, Chem. Biol. Drug Des., 2012, 79, 1007–1017 CrossRef CAS PubMed.
- L. J. Kenney, Curr. Opin. Microbiol., 2002, 5, 135–141 CrossRef CAS PubMed.
- A. Toro-Roman, T. R. Mack and A. M. Stock, J. Mol. Biol., 2005, 349, 11–26 CrossRef CAS PubMed.
- P. Bachhawat and A. M. Stock, J. Bacteriol., 2007, 189, 5987–5995 CrossRef CAS PubMed.
- R. Perkins, H. Fang, W. Tong and W. J. Welsh, Environ. Toxicol. Chem., 2003, 22, 1666–1679 CrossRef CAS PubMed.
- W. Tong, W. J. Welsh, L. Shi, H. Fang and R. Perkins, Environ. Toxicol. Chem., 2003, 22, 1680–1695 CrossRef CAS PubMed.
- J. Verma, V. M. Khedkar and E. C. Coutinho, Curr. Top. Med. Chem., 2010, 10, 95–115 CrossRef CAS PubMed.
- T. S. Tseng, I. F. Tu, H. T. Chen, L. C. Lin, K. C. Tsai, S. H. Wu and C. Chen, Chem. Commun., 2018, 54, 6372–6375 RSC.
- D. J. S. Bernard, R. Brooks Robert, E. Bruccoleri Barry, D. Olafson, S. Swaminathan and M. Karplus, J. Comput. Chem., 1983, 4, 187–217 CrossRef.
- R. A. Laskowski and M. B. Swindells, J. Chem. Inf. Model., 2011, 51, 2778–2786 CrossRef CAS PubMed.
- A. C. Wallace, R. A. Laskowski and J. M. Thornton, Protein Eng., 1995, 8, 127–134 CrossRef CAS PubMed.
- F. C. Soncini, E. G. Vescovi and E. A. Groisman, J. Bacteriol., 1995, 177, 4364–4371 CrossRef CAS PubMed.
- S. I. Miller, Mol. Microbiol., 1991, 5, 2073–2078 CrossRef CAS PubMed.
- K. Yamamoto, H. Ogasawara, N. Fujita, R. Utsumi and A. Ishihama, Mol. Microbiol., 2002, 45, 423–438 CrossRef CAS PubMed.
- P. Perron-Savard, G. De Crescenzo and H. Le Moual, Microbiology, 2005, 151, 3979–3987 CrossRef CAS PubMed.
- V. Singh, M. K. Ekka and S. Kumaran, Biochemistry, 2012, 51, 1346–1356 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra00620f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |