DOI:
10.1039/C9RA00683D
(Paper)
RSC Adv., 2019,
9, 10976-10982
Nitrogen doped microporous carbon nanospheres derived from chitin nanogels as attractive materials for supercapacitors
Received
25th January 2019
, Accepted 1st April 2019
First published on 9th April 2019
Abstract
N-doped porous carbon nanospheres were fabricated directly by pyrolyzing chitin nanogels, which were facilely prepared by mechanical agitation induced sol–gel transition of chitin solution in NaOH/urea solvent. The resulting carbon nanospheres displayed ordered micropores (centered at ∼0.6 nm) and high BET surface area of up to 1363 m2 g−1, which is substantially larger than that of the carbons from raw chitin (600 m2 g−1). In addition, the carbon nanospheres retained a nitrogen content of 3.2% and excellent conductivity. Consequently, supercapacitor electrodes prepared from the carbon nanospheres pyrolyzed at 800 °C showed a specific capacitance as high as 192 F g−1 at a current density of 0.5 A g−1 and impressive rate capability (81% retention at 10 A g−1). When assembled in a symmetrical two-electrode cell, N-doped porous carbon nanospheres demonstrated excellent cycling stability both in aqueous and organic electrolytes (95% retention after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles at 10 A g−1), together with outstanding energy density of 5.1 W h kg−1 at the power density of 2364.9 W kg−1. This work introduces a novel and efficient method to prepared N-doped porous carbon nanospheres directly from chitin and demonstrates the great potential of utilization of abundant polymers from nature in power storage.
000 cycles at 10 A g−1), together with outstanding energy density of 5.1 W h kg−1 at the power density of 2364.9 W kg−1. This work introduces a novel and efficient method to prepared N-doped porous carbon nanospheres directly from chitin and demonstrates the great potential of utilization of abundant polymers from nature in power storage.
Introduction
Carbon materials are promising candidates for supercapacitor electrodes due to their low cost, good conductivity, abundance and long cycle life.1 The development of carbon materials with a hierarchical porous structure, which facilitates electrolyte–ion transfer and provides a shorter diffusion pathway, is essential for the fabrication of advanced electric double-layer capacitors.2–4 Biomass wastes are attractive renewable resources for porous carbon preparation (leaves,5 beans shells,6 cuttlebones,7 wood,8 etc.). So far, a variety of carbon materials with tailored porous structures have been prepared from natural precursors, such as protein,9 cellulose,10–13 alginate14,15 and chitin.16–20 Among them, porous carbon from chitin has attracted special attention due to its high nitrogen content, allowing direct fabrication of N-enriched carbon without additional nitrogen sources. The faradaic reactions between nitrogen and electrolyte contribute pseudocapacitance, which could substantially improve the performance of the supercapacitor.
Chitin is mainly extracted from aquatic biomass waste such as the shells of shrimp and crab. It is β-(1-4)-linked 2-acetamido-2-deoxy-D-glucopyranose with a nitrogen amount of 6.9 wt%. N-doped porous carbon can be facilely obtained by pyrolyzing chitin and has been developed for high-performance capacitor.21 However, direct carbonization of bulk chitin does not offer sufficient control over porosity and microstructure. Recently, chitin microspheres,19,22 nanofibers and aerogels18,20 were selected as precursors for N-doped carbon, which demonstrated large surface area, hierarchical porosity and high electrochemical performance. Importantly, chitin derived carbon showed favorable retention of morphology after pyrolyzation due to the stiffness of chitin chains.19 Therefore, a preformed chitin precursor with well define nanostructure provides great opportunities for new nanocarbon materials.
Recent work revealed the advantages of carbon nanospheres23–25 as high performance supercapacitor electrode when applied in supercapacitors.26,27 Particularly, carbon nanospheres with hierarchical porous architectures demonstrated maximized specific surface area while minimized electron and ion transport distances.28 The methodology to prepare carbon nanospheres generally includes either a hard-template method or a soft template method.28–30 Organic polymers29,31,32 and silica33,34 were commonly used for the fabrication of carbon nanospheres. Direct carbonization of preformed natural polymeric nanoparticles provides a facile and environmentally friendly method for the preparation of carbon nanospheres, though few reports have been devoted to this effort.
Herein, we report the preparation of carbon nanospheres directly from chitin nanogels for the first time. Chitin nanogels with a diameter of 20–30 nm were prepared by a mechanical agitation induced sol–gel transition method with high efficiency. Interconnected porous framework of N-doped carbon nanospheres (nitrogen content of 3.2%) was obtained by directly pyrolyzing the chitin nanogels. The as-prepared typical carbon had a high BET surface area (up to 1363 m2 g−1) and a regular micropore peaked at 0.6 nm, which benefit the fast transportation and diffusion of electrolyte ions. When used for supercapacitor, the carbon nanospheres showed a specific capacitance of 192 F g−1 at 0.5 A g−1 in 1 M H2SO4 electrolyte. Besides, the chitin nanospheres could be used in organic electrolyte, demonstrating a specific capacitance of 107 F g−1 at 1.0 A g−1 in TEABF4-AN electrolyte and an excellent cycling stability, with a capacitance retention of 95% after 10000 cycles at 10 A g−1. The present study provides a simple and highly efficient route to fabricate N-doped carbon nanospheres from chitin and highlights the great potential of well-structured carbon nanospheres derived from chitin for high-performance supercapacitors.
Experimental
Materials
The raw chitin power was purchased from Yun-Zhou Biochemical Co. Ltd. (Shandong, China). Tetraethylammonium tetrafluoroborate salt (TEABF4), polytetrafluoroethylene (PTFE) (60 wt% dispersion in ethanol), acetylene black were purchased from Aladdin Reagent Co. Ltd. The titanium mesh was supplied by Gaoss Union Co. Ltd. All of the chemical reagents were of analytical grade and used without further purification.
Preparation of chitin nanogels
According to previously reported method,35 the purchased chitin was purified by treating with 5 wt% NaOH aqueous solution, 7% (v/v) HCl aqueous solution and 1.7 wt% NaClO2 in 0.3 M CH3COONa buffer to remove impurities such as the protein, mineral and pigments. Purified chitin powder (2 g) was dispersed in a solvent of NaOH, urea, and distilled water (98 g, weight ratio 8:4:88). Subsequently, the mixed suspension was refrigerated at −30 °C for 4 h, later thawed at room temperature. After two freezing–thawing cycles, a transparent chitin solution was obtained.36 The chitin solution was stirred with a homogenizer to form a milky dispersion. Then the dispersion was dialyzed against deionized water to remove residual inorganic salts. After that the chitin nanogels dispersion was obtained. The resulted chitin nanogels were pre-frozen in liquid nitrogen and then dried in a freeze dryer to get the precursor.
Preparation of carbon nanospheres
The resulted chitin nanogels were carbonized under 800 °C or 900 °C for 2 h, under an Ar atmosphere with a ramp rate of 3 °C min−1 in a tubular furnace. The obtained products were coded as CNC-800 and CNC-900, respectively.
Material characterization
Surface morphology of the samples was observed by field emission scanning electron microscope (FE-SEM, Zeiss, SIGMA) under an accelerating voltage of 5 kV, high resolution transmission electron microscope (HR-TEM, JEM-2100) under an acceleration voltage of 200 kV and atomic force microscope (AFM, Cypher ES, Asylum Research) in AC mode at 25 °C. Crystallographic structure was measured on an X-ray diffraction system (XPert Pro) with Cu Kα radiation (λ = 0.15406 nm) at 40 kV and 40 mA, and the data was collected in the 2θ range of 10–80°. Raman spectrum was employed by confocal Raman microspectroscopy (Renishaw, RM-1000) at an excitation wavelength of 514.5 nm from Ar+ laser. The surface chemistry analysis was conducted by X-ray photoelectron spectrometer (Thermo Fisher Scientific, ESCALAB250Xi) with monochromatic Al Kα radiation. The N2 adsorption/desorption measurement was performed on an automatic surface area analyzer (Micromeritics, ASAP-2460, USA) at 77 K. Before the experiment, the samples were degassed at 150 °C for 12 h under vacuum to remove certain adsorbed species. The specific surface area and pore size distribution were analyzed through the Brunauer–Emmett–Teller (BET) method and Density Functional Theory (DFT) model.
Electrochemical measurements
The electrochemical measurements were performed on an Autolab PGSTAT302N at room temperature. In a three-electrode cell, a platinum plate acted as the counter electrode, and a saturated calomel electrode (SCE) acted as the reference electrode connecting by a Luggin capillary filled with saturated KCl solution. The working electrode was consisted of 80 wt% as-prepared N-doped carbon nanospheres, 10 wt% acetylene black and 10 wt% polytetrafluoroethylene (PTFE) dispersed in ethanol. The mixture was homogenized by ultrasound and dried until ethanol was evaporated to form viscous slurry, which was roll-pressed into a thin film on a roller machine. The film was cut into pieces of 1 cm2 surface area and the mass of each piece was around 2 mg. Then this film was pressed onto a titanium mesh current collector. Before measurements, the prepared electrode was immersed in 1 M H2SO4 aqueous electrolyte overnight. The cyclic voltammetry (CV) was measured at different scan rates (20–200 mV s−1) with the potential window of −0.4 to 0.6 V vs. SCE. The galvanostatic charge/discharge (GCD) test was carried out at various current densities (0.5–10 A g−1) in the potential range of −0.4 to 0.6 V. The specific capacitance (C, F g−1) of the electrode material was calculated according to eqn (1)32 from GCD curve.| |
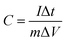 | (1) |
where I is current (A); Δt is the discharge time (s); m represents the mass of the active material in an electrode (g); and ΔV is the discharge potential range (V).
In addition, to further investigate the electrochemical performance, symmetrical two-electrode systems were installed in 1 M H2SO4 aqueous electrolyte and 1 M TEABF4-AN organic electrolyte, respectively. In aqueous electrolyte, CV and GCD were tested with the potential window of 0–1 V. Electrochemical impedance spectroscopy (EIS) was recorded over a frequency range from 0.01 Hz to 100 kHz with an alternating current voltage of 5 mV amplitude. In organic electrolyte, the potential window increased to 0–2.5 V. Under this condition, CV, GCD and cycling stabilities measurements were conducted. The specific capacitance of the symmetric electrodes (Cs, F g−1), the energy density (E, W h kg−1) and power density (P, W kg−1) of the symmetrical electrode system were calculated as follows:37–39
| |
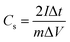 | (2) |
| |
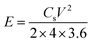 | (3) |
| |
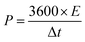 | (4) |
where
V is the cell operating voltage (excluding the IR drop).
Results and discussion
The preparation of chitin-derived carbon nanospheres includes the formation of chitin nanogels and its subsequent carbonization, as demonstrated in Fig. 1. Firstly, transparent chitin solution was obtained by dissolution of chitin powder in a solvent of NaOH, urea, and distilled water (weight ratio 8:4:88) through freezing–thawing cycles.36 The strong intermolecular hydrogen bonds were destroyed by NaOH/urea solvent and led to complete dissolution of chitin chains. Secondly, chitin nanogels were fabricated by mechanical agitation induced sol–gel transition. The chitin solution is unstable and sensitive to temperature increase. The sol–gel transition was induced by the in situ heat generated during high-speed stirring (10000 rpm). Thirdly, the resulted chitin nanogels were freeze-dried and carbonized for 2 h under an Ar atmosphere to obtain N-doped carbon nanospheres. The dark carbon nanospheres calcined under 800 °C and 900 °C were coded as CNC-800 and CNC-900 and the yields of CNC-800 and CNC-900 were 31.1% and 15.2% respectively. By comparison, carbons were also obtained by calcination of raw chitin powder at 800 °C and 900 °C, coded as chitin-800 and chitin-900.
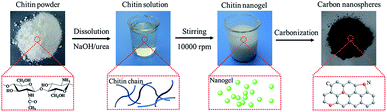 |
| | Fig. 1 Graphical representation of the formation process for chitin-derived nanospheres. | |
The morphology of chitin nanogels was firstly observed by TEM and AFM (Fig. 2a and b). Well shaped nanospheres with a diameter of 20–30 nm were clearly observed. Freeze drying of the chitin nanogels resulted in a porous sponge, which was further observed by SEM. The chitin sponge exhibited interconnected network with randomly opened pores (Fig. 2c) and the pore walls demonstrated a rough surface consisting of nanoparticles (Fig. 2d). N-doped carbon nanospheres were obtained by direct carbonization of the chitin nanogels. After carbonization, the original porous structure of the chitin nanogels was still maintained as shown by the low magnification images in Fig. 3a and c. The zoom in images demonstrated the carbon nanospheres were assembled into an interconnected framework and nanopores were clearly observed (Fig. 3b and d), especially in CNC-800. By comparison, the SEM images of raw chitin calcined at 800 °C (Fig. 3e) and 900 °C (Fig. 3f) demonstrated more compact structure. TEM and HRTEM images (Fig. 4) further confirmed the spherical morphology of CNC-800 and CNC-900 with the mean size of 3.3 nm and 5.4 nm, respectively. The HRTEM micrographs (Fig. 4c and f) showed the carbon nanosphere had partly graphitized structure with an adjacent interlayer distance of ∼0.3 nm.
 |
| | Fig. 2 Structural characterization of chitin nanogels: TEM image (a), AFM image (b) and SEM images (c and d). | |
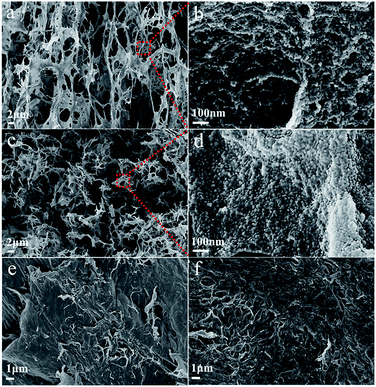 |
| | Fig. 3 Structural characterization of chitin-derived carbons: the SEM images of CNC-800 (a and b), CNC-900 (c and d), chitin-800 (e), chitin-900 (f). | |
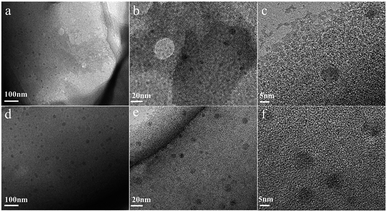 |
| | Fig. 4 TEM images of CNC-800 (a) and CNC-900 (d), high-resolution TEM images of CNC-800 (b and c), CNC-900 (e and f). | |
In order to better understand the detailed pore structure of resultant chitin-derived carbon nanospheres, the nitrogen adsorption/desorption experiments were conducted. According to IUPAC classification, the isotherms of chitin-derived carbon nanospheres corresponded to the Type I, while the isotherms of carbons from raw chitin were the combined Type I and Type IV (Fig. 5a). All the curves exhibited sharp increase in N2 adsorption at relative low pressure (P/P0), which was caused by the enhanced interaction between adsorbent–adsorbate in the narrow micropores.40 The H4 hysteresis loops were observed in the isotherms of raw chitin-derived carbon at relative pressure in the range of 0.4–0.8, which were associated with the capillary condensation in mesopores.41 In addition, according to the corresponding DFT pore size distribution curves (Fig. 5b), most pores in CNC-800 and CNC-900 felt in the ultramicroporous region (pore size < 0.7 nm) and peaked at 0.6 nm, a few pores were in the domain of supermicropores (0.7–2 nm) and narrow mesopores (2–4 nm), whereas the chitin-800 and chitin-900 showed large portion of mesopores. Recent study suggests that the existence of ultramicropores (<0.7 nm) close to the ion size is advantageous for electrochemical performance of carbon.42–44 The hydration sheaths could be removed as the solvated ions squeezed through the ultramicropores.42,45 As a consequence, the distance from the ion center to the electrode surface was closer, leading to enhanced capacitor performance. More structural parameters were listed in Table 1. CNC-800 and CNC-900 showed high BET surface areas of 1031 m2 g−1 and 1363 m2 g−1, much higher than the carbons derived from the direct carbonization of chitin (626 m2 g−1 and 600 m2 g−1 respectively). The high surface area and narrow micropore size distribution of chitin derived carbon nanospheres are beneficial to capacitance performance.
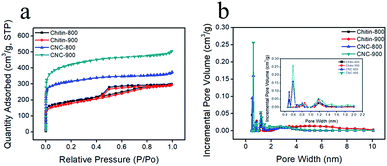 |
| | Fig. 5 Nitrogen adsorption/desorption isotherms. | |
Table 1 Surface area and porous size distribution
| Sample |
SBET (m2 g−1) |
Smicro (m2 g−1) |
Vtotal (cm3 g−1) |
Vmicro (cm3 g−1) |
Vmeso (cm3 g−1) |
Da (nm) |
| Chitin-800 |
626 |
312 |
0.46 |
0.16 |
0.30 |
2.95 |
| Chitin-900 |
600 |
262 |
0.45 |
0.13 |
0.32 |
3.02 |
| CNC-800 |
1031 |
706 |
0.57 |
0.35 |
0.22 |
2.22 |
| CNC-900 |
1363 |
877 |
0.78 |
0.44 |
0.34 |
2.29 |
XRD patterns of chitin-derived carbon nanospheres as well as carbons from raw chitin are shown in Fig. 6a. All the XRD patterns showed a broad peak at ∼24° and a weak peak at ∼43°, which corresponded to the (002) diffraction and (100) diffraction,5 indicating a partly graphitized structure and a disorder phase.21,46 This structure feature was also confirmed by Raman spectroscopy (Fig. 6b). The G band at ∼1606 cm−1 and D band at ∼1355 cm−1 are associated with the graphitic layers and disordered carbons or defective graphitic structures, respectively.7,43,47 The intensity ratio of these two peaks partially depends on the degree of graphitization.19 The ID/IG ratio for chitin-800, chitin-900, CNC-800, CNC-900 is 0.83, 0.81, 0.88, 0.87, respectively. The slightly increase of ID/IG ratios for chitin-derived carbon nanospheres suggested the existence of larger amounts of disordered carbon or graphitic defects with a low graphitization degree.
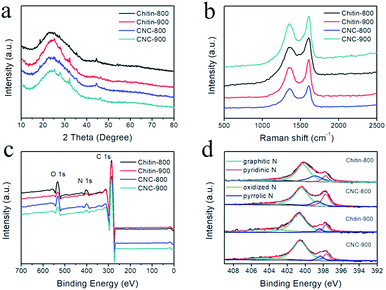 |
| | Fig. 6 The structural characterization: XRD patterns (a), Raman spectra (b), full-scale XPS spectra (c), high-resolution N 1s spectra (d). | |
Due to the presence of acetyl amino groups on chitin chains, the calcination of chitin nanogels leads to N-doping in the carbon nanospheres. The full survey spectra of XPS confirmed the existence of C, N, and O elements in the resulted carbons (Fig. 6c). High resolution spectrum of N 1s can be divided into four peaks (Fig. 6d), centered at 397.7 eV (pyridinic N), 398.7 eV (pyrrolic N), 400.6 eV (graphitic N) and 403.5 eV (oxidized-N).19 From quantitative analysis in Table 2, we can see the carbons carbonized at 800 °C possessed more pyridinic and pyrrolic types of nitrogen, which can create defects and electrochemically active sites contributing to the pseudocapacitance contents.19 Furthermore, it has been reported that nitrogen doping can enhance the conductivity of the carbon materials and improves the charge transfer property.16 The nitrogen doping also increases the wettability of the carbon surface, resulting in improved ion adsorption and increased capacitance.48
Table 2 Atomic content and percentage of different types of nitrogen based on XPS survey spectra
| Samples |
C% |
O% |
N% |
% of total N 1s |
| N-Q |
N-5 |
N-6 |
Oxidized-N |
| Chitin-800 |
88.3 |
7.6 |
4.1 |
57.8 |
9.5 |
20.8 |
11.9 |
| Chitin-900 |
93.9 |
4.4 |
1.7 |
58.7 |
4.3 |
9.3 |
27.7 |
| CNC-800 |
91.5 |
5.3 |
3.2 |
57 |
11.8 |
21.5 |
9.7 |
| CNC-900 |
95.6 |
2.8 |
1.6 |
66.5 |
7.2 |
9.8 |
16.5 |
Cyclic voltammetry (CV) and charge/discharge measurements (GCD) were performed to evaluate the electrochemical characteristics of the obtained chitin-derived carbon nanospheres. Fig. 7a showed the CV curves of different carbons at a scan rate of 20 mV s−1 in 1 M H2SO4 aqueous electrolyte using a three-electrode system. All the CV curves displayed quasi-rectangular shapes, suggesting a typical electrical double layer capacitive behavior. Slight humps occurred due to the pseudocapacitance provide by the doped nitrogen. The integrated area of the rectangles for CNCs was larger than the carbons from raw chitin (chitin-900 < chitin-800 < CNC-900 < CNC-800), indicating higher capacitance of the carbon nanospheres, which may be related to more defects and larger specific surface area. Fig. 7b showed the GCD curves of different carbons at a current density of 1 A g−1. The GCD curves presented almost symmetrical triangles with slight tilts, confirming the good capacitive performance and a small fraction of pseudocapacitance nature. The specific capacitance values from GCD curves were shown in Fig. 7c. CNC-800 exhibited superior specific capacitance of 192 F g−1 at 0.5 A g−1, 179 F g−1 at 1 A g−1, which was comparable to several previously reported bio-carbons but lower than polymer precursor-derived carbons (Table 3).6,39,49–52 This may be caused by a more optimal structure and higher nitrogen content for polymer precursor-derived carbons. When the current density increased to 10 A g−1, the specific capacitance decreased to 155 A g−1 in virtue of the diffusion limitation of ions during fast charging–discharging,53 with capacitance retention of 81%, indicating good rate capability.
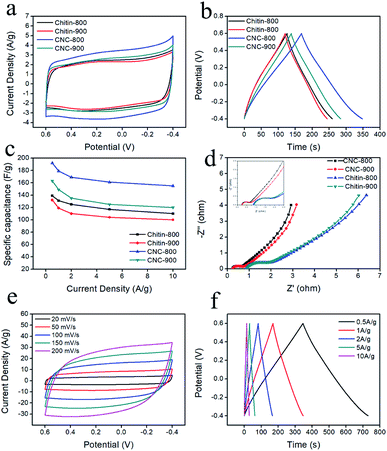 |
| | Fig. 7 CV curves (a) of carbons at a scan rate of 20 mV s−1; galvanostatic charge/discharge curves (b) at a current density of 1 A g−1; specific capacitance versus current density (c); EIS analysis (d); CV curves (e) of CNC-800 at different scan rates in 1 M H2SO4 aqueous solution; galvanostatic charge/discharge curves (f) at different current densities. | |
Table 3 Comparison of specific capacitance of different carbon materials from recent literature
| Material |
Precursor |
Capacitance at 1 A g−1 (F g−1) |
Electrolyte |
Ref. |
| N-doped carbon nanospheres |
Melamine–phenolic–formaldehyde (MPF) resin |
432 |
6 M KOH |
39 |
| Hierarchical N-doped porous carbon |
Endothelium corneum gigeriae galli |
198 |
6 M KOH |
49 |
| Sulfur and nitrogen dual-doping carbon |
Shell of broad beans |
174 |
6 M KOH |
6 |
| N-doped carbon double-shell nanoparticles |
Polydopamine |
184 |
1 M H2SO4 |
50 |
| Activated carbon |
Coconut kernel |
173 |
1 M H2SO4 |
51 |
| N-doped hollow carbon nanospheres |
Glucose and glucosamine |
205 |
1 M H2SO4 |
52 |
| N-doped microporous carbon nanospheres |
Chitin |
179 |
1 M H2SO4 |
Our work |
EIS measurements (Fig. 7d) were also conducted to further investigate the conductivity and ion transfer. The Nyquist plots consist of a semicircle in the high frequency range and a straight line in the low frequency range. The intercept of the semicircle with the real axis represents the equivalent series resistance (ESR), including the resistance of the electrolyte (Rs), the internal electrode resistance and the contact resistance between the electrode and current collector,8,42 among which the electrolyte resistance (Rs) is the dominant part.54 The Rs values of chitin-900, chitin-800, CNC-800, CNC-900 were 0.93, 0.86, 0.25 and 0.26 Ω, respectively, indicating that the carbon nanospheres have low resistance and high conductivity. The diameter of semicircle refers to the interfacial charge transfer resistance (Rct),55 the CNC-800 and CNC-900 have smaller semicircle diameters, revealing a rapid charge transference. CNC-800 was further characterized with CV at different scan rates and GCD at various current densities. As shown in Fig. 7e and f, the curves all exhibited ideal shapes, indicating good capacitive characteristic and electrochemical reversibility.
In order to further evaluate the applicability of the chitin derived carbon nanospheres, the electrochemical performance of CNC-800 was tested with a symmetrical two-electrode cell both in aqueous and organic electrolytes. Fig. 8a showed the CV curves of the aqueous device, which presented perfect symmetrical rectangular shapes even under a high scan rate, indicating a high rate capability. The GCD curves (Fig. 8b) also maintained symmetric triangular shapes at all current densities. The specific capacitance at current density of 1 A g−1 was 180 F g−1, being comparable to that in three-electrode system. Ragone plot was recorded in Fig. 8c. The energy density and power density were calculated from the GCD curves. The maximal energy density was 6.4 W h kg−1 at a power density of 125 W kg−1, while the energy density remained as high as 5.1 W h kg−1 with a power density of 2364.9 W kg−1, indicating good energy storage property and fast charge/discharge performance. Fig. 8d illustrated the CV curves of organic device. Interestingly, the CV curves still maintained rectangular-like shapes as the scan rate increased to 700 mV s−1. The specific capacitance from Fig. 8e was 107 F g−1 at current density of 1 A g−1. The energy density in organic electrolyte was 23.4 W h kg−1 with a power density of 313.9 W kg−1, 20.6 W h kg−1 with a power density of 6035.6 W kg−1, which was comparable with other literature.56–58 And as shown in Fig. 8f, the capacitance retention still retained 95% after 10000 cycles at high current density of 10 A g−1, manifesting an outstanding cycling stability. The advanced performance of CNC-800 in both organic and aqueous electrolytes confirms the versatile applicability of chitin derived carbon nanospheres for energy storage.
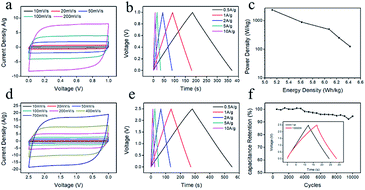 |
| | Fig. 8 CV curves (a) at different scan rates, galvanostatic charge/discharge curves (b) at different current densities, Ragone plots (c) of CNC-800 in symmetrical two-electrode cell in 1 M H2SO4 aqueous solution; CV curves (d) at different scan rates, galvanostatic charge/discharge curves (e) at different current densities, cycling stabilities at galvanostatic current density of 10 A g−1 for 10000 cycles (f) of CNC-800 in symmetrical two-electrode cell in 1 M TEABF4-AN. | |
Conclusion
In summary, N-doped carbon nanospheres with ordered microporous structure were fabricated from chitin nanogels. The nanospheres had a diameter of 20–30 nm and demonstrated large BET surface area of 1363 m2 g−1. CNC-800 showed high electrochemical capacitance of 192 F g−1 at 0.5 A g−1 in 1 M H2SO4 and 107 F g−1 in 1 M TEABF4-AN organic electrolyte. Moreover, CNC-800 showed outstanding cycling stability over 10000 cycles and good retention capability of 95% at a current density of 10 A g−1. Therefore, the facile preparation N-doped carbon nanospheres with outstanding capability provides a novel method to construct attractive energy storage materials from chitin wastes.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by National Natural Science Foundation of China (51373124, 21334005), Fundamental Research Funds for the Central Universities (2042016kf0145), Natural Science Foundation of Hubei Province of China (Team Project, No. 2015CFA017).
Notes and references
- B. Shan, Y. Cui, W. Liu, Y. Zhang, S. Liu, H. Wang, L. Sun, Z. Wang and R. Wu, ACS Sustainable Chem. Eng., 2018, 6, 14989–15000 CrossRef CAS.
- C. X. Guo and C. M. Li, Energy Environ. Sci., 2011, 4, 4504–4507 RSC.
- J. Deng, T. Y. Xiong, F. Xu, M. M. Li, C. L. Han, Y. T. Gong, H. Y. Wang and Y. Wang, Green Chem., 2015, 17, 4053–4060 RSC.
- G. P. Xiong, P. G. He, Z. P. Lyu, T. F. Chen, B. Y. Huang, L. Chen and T. S. Fisher, Nat. Commun., 2018, 9, 790 CrossRef PubMed.
- E. Hao, W. Liu, S. Liu, Y. Zhang, H. Wang, S. Chen, F. Cheng, S. Zhao and H. Yang, J. Mater. Chem. A, 2017, 5, 2204–2214 RSC.
- G. Xu, J. Han, B. Ding, P. Nie, J. Pan, H. Dou, H. Li and X. Zhang, Green Chem., 2015, 17, 1668–1674 RSC.
- Y. Guo, W. Liu, R. Wu, L. Sun, Y. Zhang, Y. Cui, S. Liu, H. Wang and B. Shan, ACS Appl. Mater. Interfaces, 2018, 10, 38376–38386 CrossRef CAS PubMed.
- B. H. Lu, L. Y. Hu, H. Y. Yin, W. Xiao and D. H. Wang, RSC Adv., 2016, 6, 106485–106490 RSC.
- Y. X. Wang, J. Q. Yang, R. B. Du and L. Y. Chen, ACS Appl. Mater. Interfaces, 2017, 9, 23731–23740 CrossRef CAS PubMed.
- C. Wang, X. F. Wang, H. Lu, H. L. Li and X. S. Zhao, Carbon, 2018, 140, 139–147 CrossRef CAS.
- L. F. Chen, Z. H. Huang, H. W. Liang, W. T. Yao, Z. Y. Yu and S. H. Yu, Energy Environ. Sci., 2013, 6, 3331–3338 RSC.
- C. L. Long, D. P. Qi, T. Wei, J. Yan, L. L. Jiang and Z. J. Fan, Adv. Funct. Mater., 2014, 24, 3953–3961 CrossRef CAS.
- Y. Yang, R. C. Sun and X. H. Wang, Mater. Lett., 2017, 189, 248–251 CrossRef CAS.
- Q. H. Bai, Q. C. Xiong, C. Li, Y. H. Shen and H. Uyama, Appl. Surf. Sci., 2018, 455, 795–807 CrossRef CAS.
- Z. Q. Ye, F. J. Wang, C. Jia and Z. Q. Shao, J. Mater. Sci., 2018, 53, 12374–12387 CrossRef CAS.
- C. J. Raj, M. Rajesh, R. Manikandan, K. H. Yu, J. R. Anusha, J. H. Ahn, D. W. Kim, S. Y. Park and B. C. Kim, J. Power Sources, 2018, 386, 66–76 CrossRef CAS.
- T. D. Nguyen, K. E. Shopsowitz and M. J. MacLachlan, J. Mater. Chem. A, 2014, 2, 5915–5921 RSC.
- B. B. Ding, S. S. Huang, K. Pang, Y. X. Duan and J. M. Zhang, ACS Sustainable Chem. Eng., 2018, 6, 177–185 CrossRef CAS.
- B. Duan, X. Gao, X. Yao, Y. Fang, L. Huang, J. Zhou and L. N. Zhang, Nano Energy, 2016, 27, 482–491 CrossRef CAS.
- J. You, M. J. Li, B. B. Ding, X. C. Wu and C. X. Li, Adv. Mater., 2017, 29, 1606895 CrossRef PubMed.
- J. Zhou, L. Bao, S. J. Wu, W. Yang and H. Wang, Carbohydr. Polym., 2017, 173, 321–329 CrossRef CAS PubMed.
- L. Gao, L. Xiong, D. Xu, J. Cai, L. Huang, J. Zhou and L. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 28918–28927 CrossRef CAS PubMed.
- A. Chen, Y. Yu, T. Xing, R. Wang, Y. Zhang and Q. Li, J. Mater. Sci., 2015, 50, 5578–5582 CrossRef CAS.
- D. Guo, X. a. Chen, Z. Fang, Y. He, C. Zheng, Z. Yang, K. Yang, Y. Chen and S. Huang, Electrochim. Acta, 2015, 176, 207–214 CrossRef CAS.
- Q. Zhang, L. Li, Y. Wang, Y. Chen, F. He, S. Gai and P. Yang, Electrochim. Acta, 2015, 176, 542–547 CrossRef CAS.
- M. H. Naveen, K. Shim, M. S. A. Hossain, J. H. Kim and Y.-B. Shim, Adv. Energy Mater., 2017, 7, 1602002 CrossRef.
- S. Liu, Y. Cai, X. Zhao, Y. Liang, M. Zheng, H. Hu, H. Dong, S. Jiang, Y. Liu and Y. Xiao, J. Power Sources, 2017, 360, 373–382 CrossRef CAS.
- S.-K. Kim, E. Jung, M. D. Goodman, K. S. Schweizer, N. Tatsuda, K. Yano and P. V. Braun, ACS Appl. Mater. Interfaces, 2015, 7, 9128–9133 CrossRef CAS PubMed.
- J. Liu, T. Y. Yang, D. W. Wang, G. Q. M. Lu, D. Y. Zhao and S. Z. Qiao, Nat. Commun., 2013, 4, 2798 CrossRef.
- Z. Lei, N. Christov and X. S. Zhao, Energy Environ. Sci., 2011, 4, 1866–1873 RSC.
- Y. Fang, D. Gu, Y. Zou, Z. X. Wu, F. Y. Li, R. C. Che, Y. H. Deng, B. Tu and D. Y. Zhao, Angew. Chem., Int. Ed., 2010, 49, 7987–7991 CrossRef CAS PubMed.
- Z. B. Wang, H. W. Qiang, Z. H. Zhu, J. P. Liu, C. N. Chen and D. W. Zhang, ChemElectroChem, 2018, 5, 2242–2249 CrossRef CAS.
- S. E. Bae, K. J. Kim, I. H. Choi and S. Huh, Carbon, 2016, 99, 8–16 CrossRef CAS.
- G. Wang, Y. H. Sun, D. B. Li, H. W. Liang, R. H. Dong, X. L. Feng and K. Mullen, Angew. Chem., Int. Ed., 2015, 54, 15191–15196 CrossRef CAS PubMed.
- B. Duan, C. Y. Chang, B. B. Ding, J. Cai, M. Xu, S. C. Feng, J. Z. Ren, X. W. Shi, Y. M. Du and L. N. Zhang, J. Mater. Chem. A, 2013, 1, 1867–1874 RSC.
- B. Duan, X. Zheng, Z. Xia, X. Fan, L. Guo, J. Liu, Y. Wang, Q. Ye and L. Zhang, Angew. Chem., Int. Ed., 2015, 54, 5152–5156 CrossRef CAS PubMed.
- J. Zhao, H. Lai, Z. Lyu, Y. Jiang, K. Xie, X. Wang, Q. Wu, L. Yang, Z. Jin, Y. Ma, J. Liu and Z. Hu, Adv. Mater., 2015, 27, 3541–3545 CrossRef CAS PubMed.
- F. Sun, Z. Qu, J. Gao, H. B. Wu, F. Liu, R. Han, L. Wang, T. Pei, G. Zhao and Y. Lu, Adv. Funct. Mater., 2018, 28, 1804190 CrossRef.
- F. Sun, J. Gao, X. Pi, L. Wang, Y. Yang, Z. Qu and S. Wu, J. Power Sources, 2017, 337, 189–196 CrossRef CAS.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CAS.
- Y. Gao, W. Zhang, Q. Yue, B. Gao, Y. Sun, J. Kong and P. Zhao, J. Power Sources, 2014, 270, 403–410 CrossRef CAS.
- A. Ghosh and Y. H. Lee, ChemSusChem, 2012, 5, 480–499 CrossRef CAS PubMed.
- J. S. Zhou, J. Lian, L. Hou, J. C. Zhang, H. Y. Gou, M. R. Xia, Y. F. Zhao, T. A. Strobel, L. Tao and F. M. Gao, Nat. Commun., 2015, 6, 8503 CrossRef CAS PubMed.
- J. Chmiola, C. Largeot, P. L. Taberna, P. Simon and Y. Gogotsi, Angew. Chem., Int. Ed., 2008, 47, 3392–3395 CrossRef CAS PubMed.
- C. Vix-Guterl, E. Frackowiak, K. Jurewicz, M. Friebe, J. Parmentier and F. Beguin, Carbon, 2005, 43, 1293–1302 CrossRef CAS.
- B. Lu, J. Zhou, Y. Song, H. Wang, W. Xiao and D. Wang, Faraday Discuss., 2016, 190, 147 RSC.
- F. C. Zheng, Y. Yang and Q. W. Chen, Nat. Commun., 2014, 5, 5261 CrossRef CAS PubMed.
- Y. Deng, Y. Xie, K. Zou and X. Ji, J. Mater. Chem. A, 2016, 4, 1144–1173 RSC.
- X. Hong, K. S. Hui, Z. Zeng, K. N. Hui, L. Zhang, M. Mo and M. Li, Electrochim. Acta, 2014, 130, 464–469 CrossRef CAS.
- J. Yun, J. Jun, J. Lee, J. Ryu, K. Lee, H. Yu and J. Jang, RSC Adv., 2017, 7, 20694–20699 RSC.
- B. Kishore, D. Shanmughasundaram, T. R. Penki and N. Munichandraiah, J. Appl. Electrochem., 2014, 44, 903–916 CrossRef CAS.
- H. Qu, X. Zhang, J. Zhan, W. Sun, Z. Si and H. Chen, ACS Sustainable Chem. Eng., 2018, 6, 7380–7389 CrossRef CAS.
- B. H. Lu, L. Y. Hu, H. Y. Yin, X. H. Mao, W. Xiao and D. H. Wang, Int. J. Hydrogen Energy, 2016, 41, 18713–18720 CrossRef CAS.
- Z. Li Li and X. S. Zhao, Chem. Soc. Rev., 2009, 38, 2520–2531 RSC.
- S. Biswas and L. T. Drzal, Chem. Mater., 2010, 22, 5667–5671 CrossRef CAS.
- F. Sun, H. Wu, X. Liu, F. Liu, H. Zhou, J. Gao and Y. Lu, Nano Res., 2016, 9, 3209–3221 CrossRef CAS.
- X. M. Fan, C. Yu, J. Yang, Z. Ling, C. Hu, M. D. Zhang and J. S. Qiu, Adv. Energy Mater., 2015, 5, 7 Search PubMed.
- Z. Lu, C. Hui, S. Zhu, L. Hou and C. Yuan, Green Chem., 2015, 17, 2373–2382 RSC.
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Chuang Peng* and
Dihua Wang
*,
Chuang Peng* and
Dihua Wang










