
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModified SiO hierarchical structure materials with improved initial coulombic efficiency for advanced lithium-ion battery anodes†
Lizhao Xie ,
Hui Liu,
Shaoxiong Lin,
Xulai Yang,
Meizhou Qi,
Lili Zhu,
Yujing Guo and
Guilue Guo*
,
Hui Liu,
Shaoxiong Lin,
Xulai Yang,
Meizhou Qi,
Lili Zhu,
Yujing Guo and
Guilue Guo*
Hefei Guoxuan High-Tech Power Energy Co., Ltd., Hefei, P. R. China. E-mail: gguo002@e.ntu.edu.sg
First published on 12th April 2019
Abstract
Silicon-based anode materials are indispensable components in developing high energy density lithium-ion batteries, yet their practical application still faces great challenges, such as large volume change during the lithiation and delithiation process that causes the pulverization of silicon particles, and continuous formation and reformation of the solid electrolyte interfaces (SEI) which results in a low initial coulombic efficiency. As an endeavor to address these problems, in this study, Si/SiO/Li2SiO3@C structures were prepared via a facile method using SiO, pitch powder and Li2CO3/PVA solution followed by annealing treatment. The Si/SiO/Li2SiO3@C composite shows a great improvement in lithium storage where a high discharge capacity of 1645.47 mA h g−1 was delivered with the 1st C.E. of 69.05% at 100 mA g−1. These results indicate that the designed method of integrating prelithiation and carbon coating for SiO and the as-prepared macro scale Si/SiO/Li2SiO3@C structures are practical for implementation in lithium-ion battery technology.
Introduction
During the last five decades, exacerbating air pollution together with the concerns of severe fossil fuel depletion have drawn public awareness of accelerating the electrification of our transportation systems.1,2 The ever-increasing demand for electric buses (E-bus) and electric vehicles (EVs) has thus stimulated the development of high performance lithium ion batteries (LIBs). However, graphite, the primary anode material, which has a theoretical capacity of 372.22 mA h g−1, cannot satisfy the high energy density requirement of the EVs and is not usually considered as an anode material in these types of LIBs, e.g. 300 W h kg−1 battery packs.3–5 Among the various types of candidate anodes proposed for LIBs, Si-based materials have high theoretical capacities, e.g., Si with a capacity of 4211.52 mA h g−1 (Li4.4Si) or 3589.28 mA h g−1 (Li3.75Si) and silicon monoxide (SiO) of 2322.15 mA h g−1, which have attracted intensive research efforts in both academic and industrial area.6–9 However, Si-based materials still suffer from huge volume expansion and pulverization issues, which inevitably lead to the rapid failure of the battery.10–13So far, various methods have been proposed and employed to modify Si-based materials in order to improve their lithium storage performance.14,15 For example, constructing core/shell structure nanoparticles/nanowires where Si acts as core materials and SiO/SiO2 serves as shell layers have been realized through solution methods or physical/chemical vapor deposition. Such one-dimensional core/shell nanostructure allows the delivery of high capacity and maintains high capacity retention.16–18 Polymer coating on Si-based materials has also led to significant improvement in specific capacity and cycling performance because of the prevention of Si particles aggregation as well as accommodation of their volume expansion.19–21 Fabrication of various Si-based composite materials, e.g. Si–carbon nanotubes,22,23 metal fibril mat-supported silicon anode,24 etc. is also demonstrated for high performance lithium storage. In addition, surface coating with carbon or metal layer on Si-based materials to avoid direct contact between Si/SiO and electrolytes is also proved to be effective in promoting stable solid electrolyte interphase (SEI) and increasing the electric conductivity to maximize the overall electrochemical performance.25–27
Compared with the huge volume change of pure Si during lithiation, the volume expansion issue of SiO is less severe than that of Si, thus SiO is currently more preferable in integrating into EVs batteries anodes.28,29 However, SiO encounters several drawbacks. One is the low intrinsic electrical conductivity, which lowers the electrochemical activity; another is the low initial coulombic efficiency (1st C.E.) because the oxygen component in SiO can irreversibly consume some Li during the first cycle.30,31 These consumed Li need to be compensated so that the 1st C.E. of the anode could be close to those of cathodes before it is widely used.32,33 Benefit from the investigation of Si anodes, great advancement has been achieved in solving the pulverization of SiO.34–36 Recently, some research attention is directly paid to solving the low 1st C.E. problem. Their attempts mainly focus on converting the O atoms in the SiO to a buffer layer like SiAl0.2O37 or lithium silicates38 during materials synthesis stage, but no significant progress has been available. Furthermore, the nanosized LIBs electrode materials provided in these studies is not yet practical for industrial application. It is of extremely urgent and meaningful to develop an effective and efficient method to tackle these issues so as to pave the way for the practical implementation of SiO.
In this work, a novel method was developed to prepare carbon-coated prelithiated micron-sized SiO anode materials for lithium storage, which is displayed in Fig. 1. Hierarchical free-standing SiO–Li2CO3/PVA sponges were first prepared through solution method, followed by annealing in N2 flow to generate macroscale Si/SiO/Li2SiO3@C hierarchical structures. Such composites combining the advantages of carbon-coating and prelithiation are desirable in lithium storage. When tested as lithium-ion battery anode, a high capacity of 1645.47 mA h g−1 was obtained with a high 1st C.E. of 69.05% compared to 12.29% of the pristine SiO-based anode at 100 mA g−1. Rate capability testing shows that when 100, 200, 500, 1000 and 2000 mA g−1 current densities were applied, the delivered capacities are 748.59, 609.68, 487.13, 311.52 and 197.80 mA h g−1, respectively.
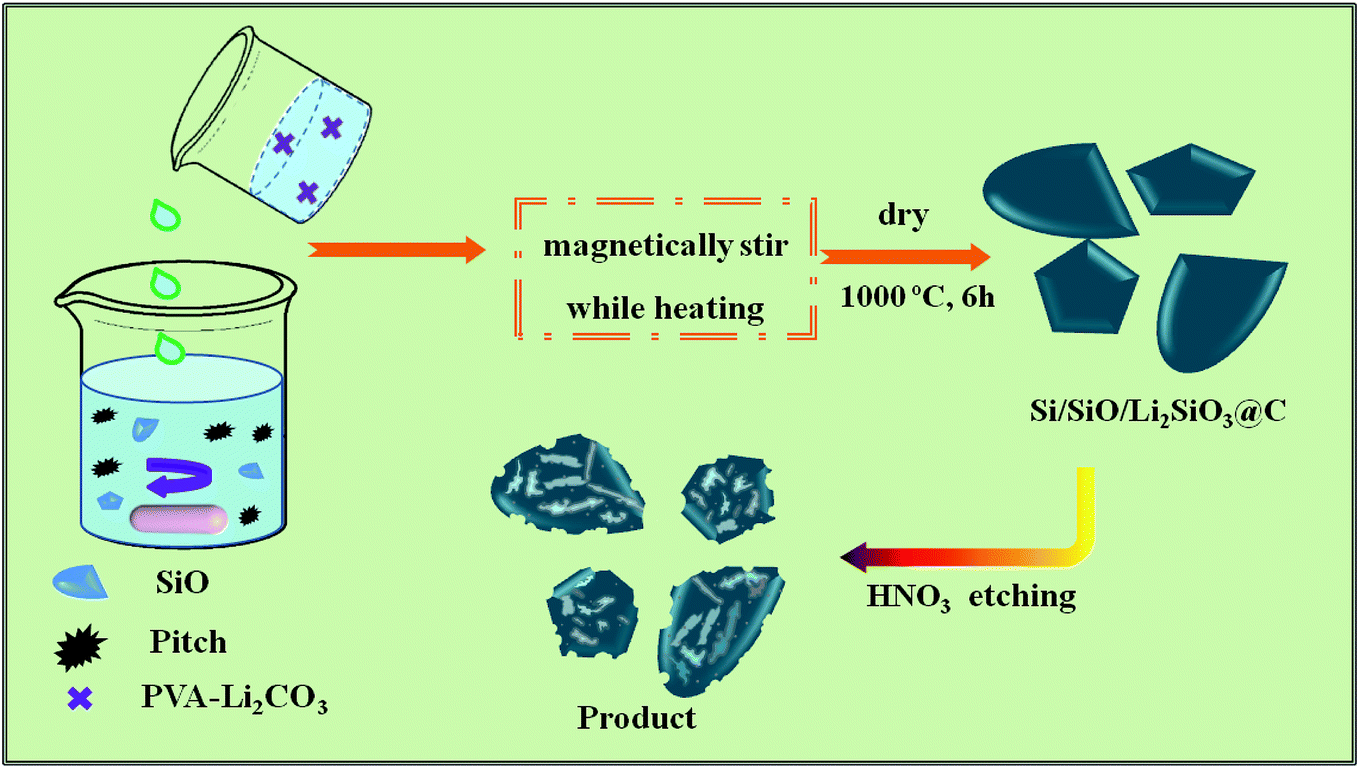 | ||
| Fig. 1 Schematic synthesis procedures of the Si/SiO/Li2SiO3@C composites. | ||
Methods
Materials preparation
In a typical synthesis, 10.0 g polyvinyl alcohol (average Mw: 31![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, Sigma Aldrich, 99.9%) were first dissolved in 180 g 80 °C DI water to form a transparent solution through continuous stirring, after which 18.0 g lithium carbonate (Li2CO3, Sigma Aldrich, 99.9%) was added to form the PVA–Li2CO3 solution. To prepare the SiO–Li2CO3–PVA sponge, 2.62 g silicon monoxide (SiO, Sigma Aldrich, 99.9%, D50 = 5.07 μm) were mixed with 50 mL PVA–Li2CO3 solution under stirring for 10 min, then 10 mL alcohol (Sigma Aldrich, 99.5%) where 0.8 g pitch powder was previously dissolved was rapidly dropped into the above mixture solution to form the SiO–Li2CO3–Pitch-PVA sponge. The final mole ratio of SiO:Li2CO3 in the sponge was adjusted to be 8:1. The obtained sponges were then kept in a 100 °C vacuum oven for 3 hours to evaporate the remained water and alcohol. The dry sponges were then transferred into a tube furnace and annealed at 1000 °C under nitrogen flow for 6 hours to form the Si/SiO/Li2SiO3@C structures. The Si/SiO/Li2SiO3@C structures were further treated by 2.0 M HNO3 for 60 minutes to remove some of the Li2SiO3 as well as introduce some pore structures. Carbon-coated SiO (SiO@C) were also prepared through carbonation of the sponge obtained from a mixture of pristine SiO–PVA solution and ethanol-pitch powder solution.
000, Sigma Aldrich, 99.9%) were first dissolved in 180 g 80 °C DI water to form a transparent solution through continuous stirring, after which 18.0 g lithium carbonate (Li2CO3, Sigma Aldrich, 99.9%) was added to form the PVA–Li2CO3 solution. To prepare the SiO–Li2CO3–PVA sponge, 2.62 g silicon monoxide (SiO, Sigma Aldrich, 99.9%, D50 = 5.07 μm) were mixed with 50 mL PVA–Li2CO3 solution under stirring for 10 min, then 10 mL alcohol (Sigma Aldrich, 99.5%) where 0.8 g pitch powder was previously dissolved was rapidly dropped into the above mixture solution to form the SiO–Li2CO3–Pitch-PVA sponge. The final mole ratio of SiO:Li2CO3 in the sponge was adjusted to be 8:1. The obtained sponges were then kept in a 100 °C vacuum oven for 3 hours to evaporate the remained water and alcohol. The dry sponges were then transferred into a tube furnace and annealed at 1000 °C under nitrogen flow for 6 hours to form the Si/SiO/Li2SiO3@C structures. The Si/SiO/Li2SiO3@C structures were further treated by 2.0 M HNO3 for 60 minutes to remove some of the Li2SiO3 as well as introduce some pore structures. Carbon-coated SiO (SiO@C) were also prepared through carbonation of the sponge obtained from a mixture of pristine SiO–PVA solution and ethanol-pitch powder solution.
Physical characterization
The morphologies of the pristine SiO, SiO@C and Si/SiO/Li2SiO3@C were visualized using field-emission electron microscopy (FESEM, FEI, Nova NanoSEM 450 operated at 1 kV). The phases of these samples were identified using X-ray diffraction (XRD, Rigaku SmartLab 3 kW, 40 kV) with Cu Kα irradiation.Electrochemical evaluation
For the preparation of working electrodes, 80 wt% active materials, 10 wt% super P carbon and 10 wt% LA133 binder (5 wt% in H2O, purchased from Chengdu Indigo Power Sources Co., Ltd) were mixed with DI water and stirred to form a slurry. The black slurry thus obtained was coated on Cu foil by an automated doctor blade and followed by drying under a vacuum environment at 90 °C for 12 h to remove the solvent. The dry electrodes were calendared and the total material loading was ∼6 mg cm−2. The electrodes were assembled into CR2016 coin cell in an argon-filled glove box configuration with lithium metal foil as the counter/reference electrode, polypropylene separator (Celgard 2400), and electrolyte solution obtained by dissolving 1 M LiPF6 and 5% FEC in a mixture of ethylene carbonate (EC) and dimethyl carbonate (DMC) (EC/DMC, 1:1, v/v). The charge/discharge tests were performed using a NEWARE battery tester in a voltage window of 0.005–1.5 V. Cyclic voltammetry characterizations (0.005 V to OCV) were performed using an electrochemical workstation (Solartron analytical equipment, 1287 Electrochemical Interface). EIS measurement was carried out on the same equipment as CV with a frequency ranging from 100 kHz to 0.01 Hz (1260 Impedance/Gain-Phase Analyzer). Before the EIS tests, the coin cells were fully discharged and then charged to 50% SOC.
Results and discussion
Fig. 1 illustrates the synthesis procedure of the Si/SiO/Li2SiO3@C composites where more details are available in the experiment section.The XRD patterns of the pristine SiO, carbon coated-SiO (SiO@C) and Si/SiO/Li2SiO3@C are displayed in Fig. 2. For the pristine SiO, a broad peak around 20–30° was observed, corresponding to the amorphous nature of SiO, which has been revealed to be composed of silicon nanocrystals, SiO2 and amorphous SiOx (0 < x < 2) interfaces. SiO@C exhibits two peaks, where the broad peak around 20–30° was SiO and the peak at 28.5° belongs to nanosized Si generated from the disproportion of SiO during the carbonization process. Diffraction patterns corresponding to Li2SiO3 (PDF no. 29–0828), nano-sized Si (PDF no. 27-1402) and the remained SiO are observed in the XRD profile of Si/SiO/Li2SiO3@C, which suggests the successful synthesis of Si/SiO/Li2SiO3 composites.
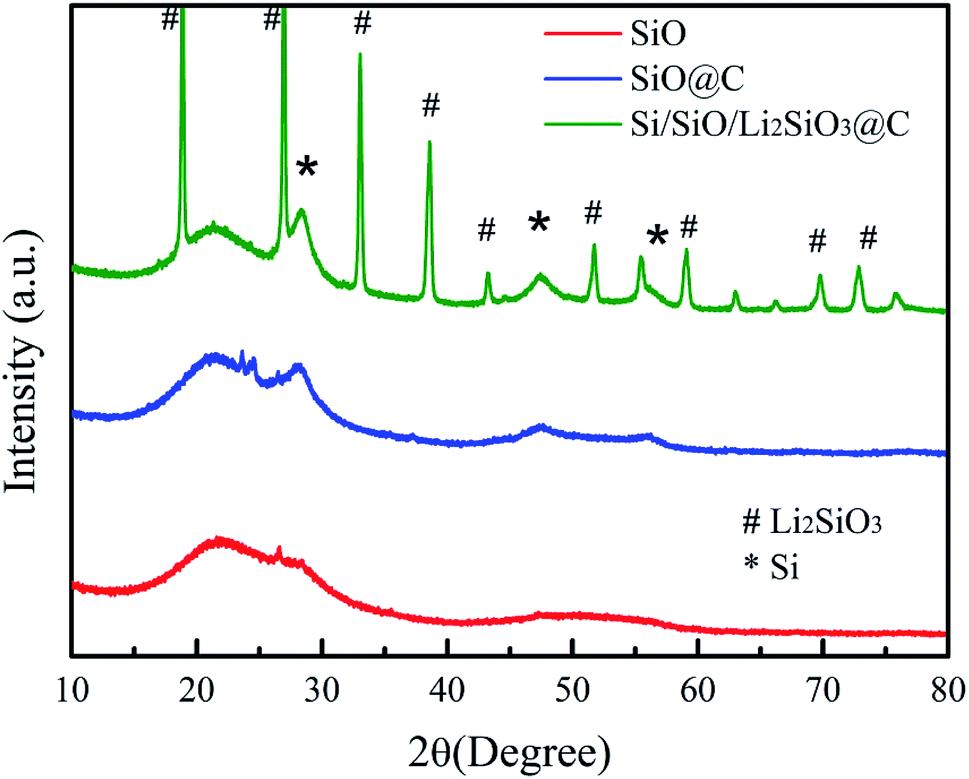 | ||
| Fig. 2 XRD patterns of SiO, SiO@C and Si/SiO/Li2SiO3@C. | ||
The morphologies of the SiO, SiO@C and Si/SiO/Li2SiO3@C composites are displayed in Fig. 3. As shown in Fig. 3a, the pristine SiO particles are 1–5 μm in diameter which have irregular shapes and smooth surfaces. The SiO@C shares similar dimensions with the pristine SiO particles while the surfaces of each particle are covered by a layer of carbon derived from the carbonization of PVA and pitch powder as indicated in Fig. 3b. The Si/SiO/Li2SiO3@C composites have the same morphology with the carbon-coated SiO where layers of carbon were found to be coated on the surfaces or intercalated among different particles. After further acid treatment of the Si/SiO/Li2SiO3@C, pore structures were generated in the composites due to the partial dissolution of the Li2SiO3 phases. The reaction was revealed to be Li2SiO3 + 2HNO3 → 2LiNO3 + H2SiO3, which was confirmed by the XRD pattern of the solute of the supernatant obtained from acid treatment and the material inside was proved to be pure LiNO3 (ESI, Fig. S1†). Higher magnification SEM image (Fig. 3d and S2 in the ESI†) further visualizes the porous structure of the composites. The surface area (SA) of pristine SiO and SiO@C are measured to be 2.01 and 3.53 m2 g−1. Dramatic increase in SA is observed in the Si/SiO/Li2SiO3@C composites which reaches 14.04 m2 g−1 due to the introduction of pore structures. Such as-prepared interconnected porous Si/SiO/Li2SiO3@C composites are desirable in high performance lithium storage application where the porous structures provide not only free space for volume expansion accommodation but also efficient channels for Li+ diffusion.
 | ||
| Fig. 3 Morphologies of (a) pristine SiO, (b) carbon-coated SiO, (c and d) Si/SiO/Li2SiO3@C composites. Scale bar: (a–c) 5 μm and (d) 3 μm. | ||
The electrochemical properties of the pristine SiO, SiO@C and Si/SiO/Li2SiO3@C composites were investigated in half-cell batteries. The CV responses of the coin-cells using these anode materials are shown in Fig. 4. Different CV responses are observed from the three types of coin cells. For the pristine SiO (Fig. 4a), in the first cycle, the broad peak ranging from 0.2–0.5 V and the shape peak followed at around 0.05–0.2 V can be attributed to the conversion and lithiation process corresponding to the lithiation of Si and irreversible formation of Li2SiO3.39,40 During the anodic scan, peaks at around 0.6 V correspond to the delithiation of Li–Si alloys. In the following cycles, the lithiation and delithiation peaks all slightly shift to the lower potential, which demonstrates the dominant process is the lithiation and delithiation of Si as SiO was converted into Si and Li2SiO3 during the first cycle. It is noted that the anodic peak in the 2nd cycle is the strongest, suggesting an activation process and in the 3rd cycle, its amplitude weakens, indicating a stable state is achieved or a possible degradation may have occurred, as pristine SiO is fragile upon cycling. The CV profiles of SiO@C are far more complicate compared with that of SiO and are exhibited in Fig. 4b. In the first cathodic scan, a broad peak centered at around 0.65 V appears which is related to the formation of the solid-electrolyte interface (SEI) layer. In the 2nd and 3rd cycle, this peak shifts to around 1.0 V and the intensity is weaker. The lithiation peaks below 0.2 V turn broader and their intensities increase as cycle number increases, which is contrary to that of pristine SiO. These results suggest a different SEI formation mechanism as well as a gradual utilization of SiO after carbon coating. The anodic scan also show a gradual increase of peak intensities upon cycling and the peaks slightly shift to the higher potential. For the Si/SiO/Li2SiO3@C, similar CV responses as SiO@C are observed where broader cathodic and anodic peaks compared with SiO@C are centered at around 0.1 and 0.2 V, respectively, as shown in Fig. 4c.
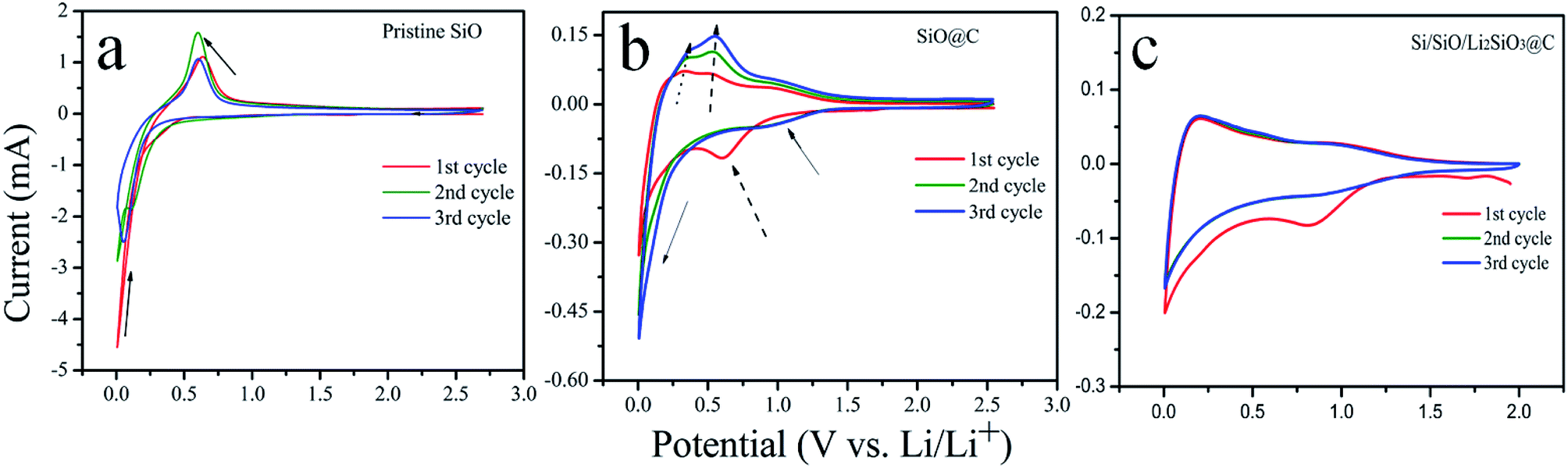 | ||
| Fig. 4 CV curves of SiO, SiO@C and Si/SiO/Li2SiO3@C at a scan rate of 0.1 mV s−1. | ||
Fig. 5a shows the first charge and discharge curves of the coin-cells using the above mentioned anode materials at a current density of 100 mA g−1. The discharge voltage of pristine SiO is 0.1–0.2 V higher than those of SiO@C and Si/SiO/Li2SiO3@C composites. It may be caused by the differences in reaction kinetics and hindrance where in pristine SiO, SiO directly contacted with electrolyte, while in the later two cases, carbon layers surrounded the particles resulting in different SEI structures. Another reason is the disproportion of SiO during the carbonization process in the SiO@C and Si/SiO/Li2SiO3@C composites and the different discharge–charge curves between nano-sized Si and micron-sized SiO.41,42 The charge and discharge curves of SiO@C and Si/SiO/Li2SiO3@C composites almost overlay with each other except for the end of charge and discharge, which indicates similar conversion and alloying reactions with different degrees of sluggish in kinetics. For the pristine SiO, the initial discharge and charge capacity are 1787.89 and 219.88 mA h g−1 respectively, in which the 1st C.E. is as low as 12.29%. It is also noted that the delivered capacities rapidly fade for pristine SiO and at the 5th cycle, dropping to only 95.03 and 89.28 mA h g−1, which evidently show that pristine SiO is not suitable to be applied in practical battery cells (Fig. 5b). When SiO was coated with 5% of carbon (SiO@C), the initial discharge and charge capacities increase to 1759.46 and 1021.02 mA h g−1 with a 1st C.E. of 58.03%, which is a significant improvement compared with that of pristine SiO. The mechanisms under such improvements in specific capacities and 1st C.E. can be attributed to both the enhancement of electric conductivity and prevention of direct contact of SiO with the electrolyte to avoid SEI formation on SiO by the introduction of the exterior carbon layer.43 Although improved 1st cycle performance was achieved, the delivered capacities at the following cycles behave similarly as the pristine SiO (Fig. 5b). As some SiO particles underwent huge volume expansion and cracked during discharge, these particles were not recovered during the following delithiation process and the interior lithiation parts were further electronically isolated due to the shrinkage of the delithiation portion, which inevitably exacerbates the capacity fading of these two kinds of materials. The delivered capacities of the Si/SiO/Li2SiO3@C composites in the first cycle are 1645.47 and 1136.18 mA h g−1, corresponding to a 1st C.E. of 69.05%. The 2nd and 3rd cycle charge capacities are stable at around 800 mA h g−1. The rate performance of the Si/SiO/Li2SiO3@C composites was also investigated. At the current density of 100, 200, 500, 1000 and 2000 mA g−1, the delivered capacities at the corresponding 10th cycle are 748.59, 609.68, 487.13, 311.52 and 197.80 mA h g−1, respectively. When the current density was switched to 200 mA g−1, the capacities restore to 601.10 mA h g−1, which demonstrates a good rate capability of the Si/SiO/Li2SiO3@C composites. When cycling at 100 mA g−1, the 2nd discharge capacity is 731.51 mA h g−1 and at the 35th cycle, this value is 573.99 mA h g−1, corresponding to 78.47% capacity retention (Fig. 5d).
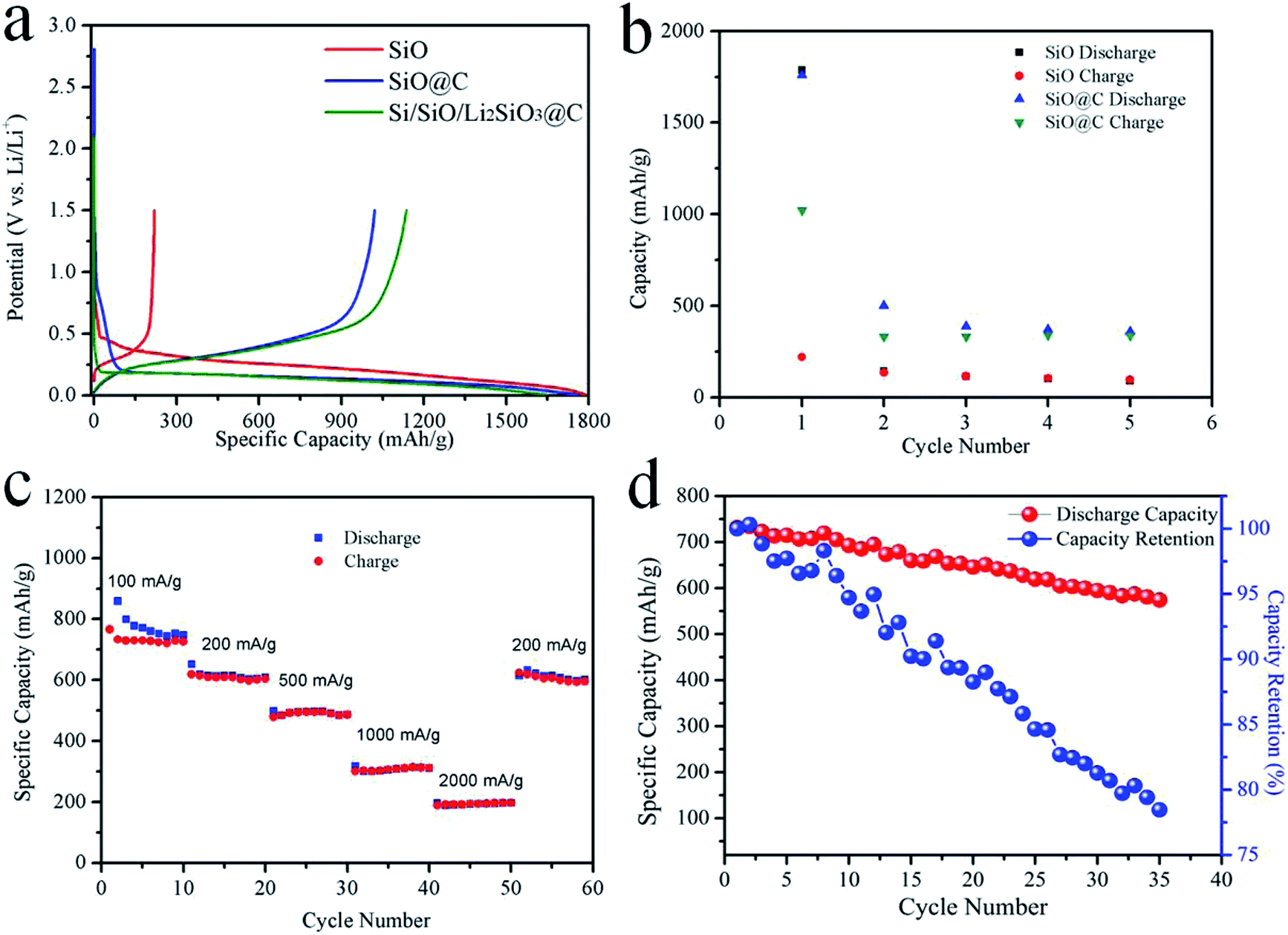 | ||
| Fig. 5 (a) The charge/discharge voltage profiles of pristine SiO, SiO@C and Si/SiO/Li2SiO3@C composites at the 1st cycle, (b) capacities of SiO and SiO@C in the initial 5 cycles, (c) rate capability and (d) cycle performance of the Si/SiO/Li2SiO3@C composites. | ||
Fig. 6 illustrates the impedance spectra of the pristine SiO, carbon-coated SiO and Si/SiO/Li2SiO3@C anodes. All the spectra were well fitted by an equivalent circuit model shown in Fig. 6c–e, in which Rs (R1) stands for the resistance of cell bulk including the electrolyte, electrode and separator. Rct (R3 & R4) is the charge-transfer resistance and W is the Warburg resistance. The Rs for the pristine SiO, SiO@C and Si/SiO/Li2SiO3@C composites are 1.6, 1.5, 1.7 Ω and the Rct (R3 & R4) are 113.5, 6.6, 17.2 Ω, respectively. These results demonstrate that improved conductivity and kinetics are achieved through the introduction of surface carbon coating when compared with pristine SiO.44 However, due to the insulating nature of Li2SiO3 which distribute around the active Si and SiO domains, the Rct for the Si/SiO/Li2SiO3@C composites slightly increased by around 10 Ω when compared with that of SiO@C. This disadvantage, on the other hand, is compensated by the improvement of the structural stability. Thus, the improvement in these aspects as well as the hierarchical design contributes to the high performance of the Si/SiO/Li2SiO3@C composites. It is also noted that for SiO@C and Si/SiO/Li2SiO3@C composites, R3//CPE2 both appear. The carbon coating (and possibly Li2SiO3 in the Si/SiO/Li2SiO3) may be responsible for this as it generates another interface between the carbon layers and the active materials of SiO or Si/SiO/Li2SiO3. To further elucidate these phenomena, coin cells of the three different anode materials were charged and discharged at 50 μA. Under such small current, the polarizations, especially kinetic aspects, which occurred during the charge and discharge processes in the coin-cells, can be minimized. From Fig. S3,† we can observe that when comparing the discharge and charge profiles (specific capacities < 100 mA h g−1), the discharge potential of pristine SiO is 0.3–0.4 V higher than those of SiO@C and Si/SiO/Li2SiO3@C composites, which delivers the largest resistance polarization. The SiO@C sample shows the lowest overpotential and at the beginning of the discharge, a typical voltage hysteresis can be observed, revealing a gradual activation of the SiO materials that is protected by the carbon coating layer. The charge and discharge curves of Si/SiO/Li2SiO3@C composites fall within the other two samples which indicates the improved conductivity of electrons or increase of lithium ions transfer hindrance from carbon coating or introduction of lithium silicates which optimizes the ionic conductivity and enhance the resistance to volume expansion, leading to an overall cycle stability.
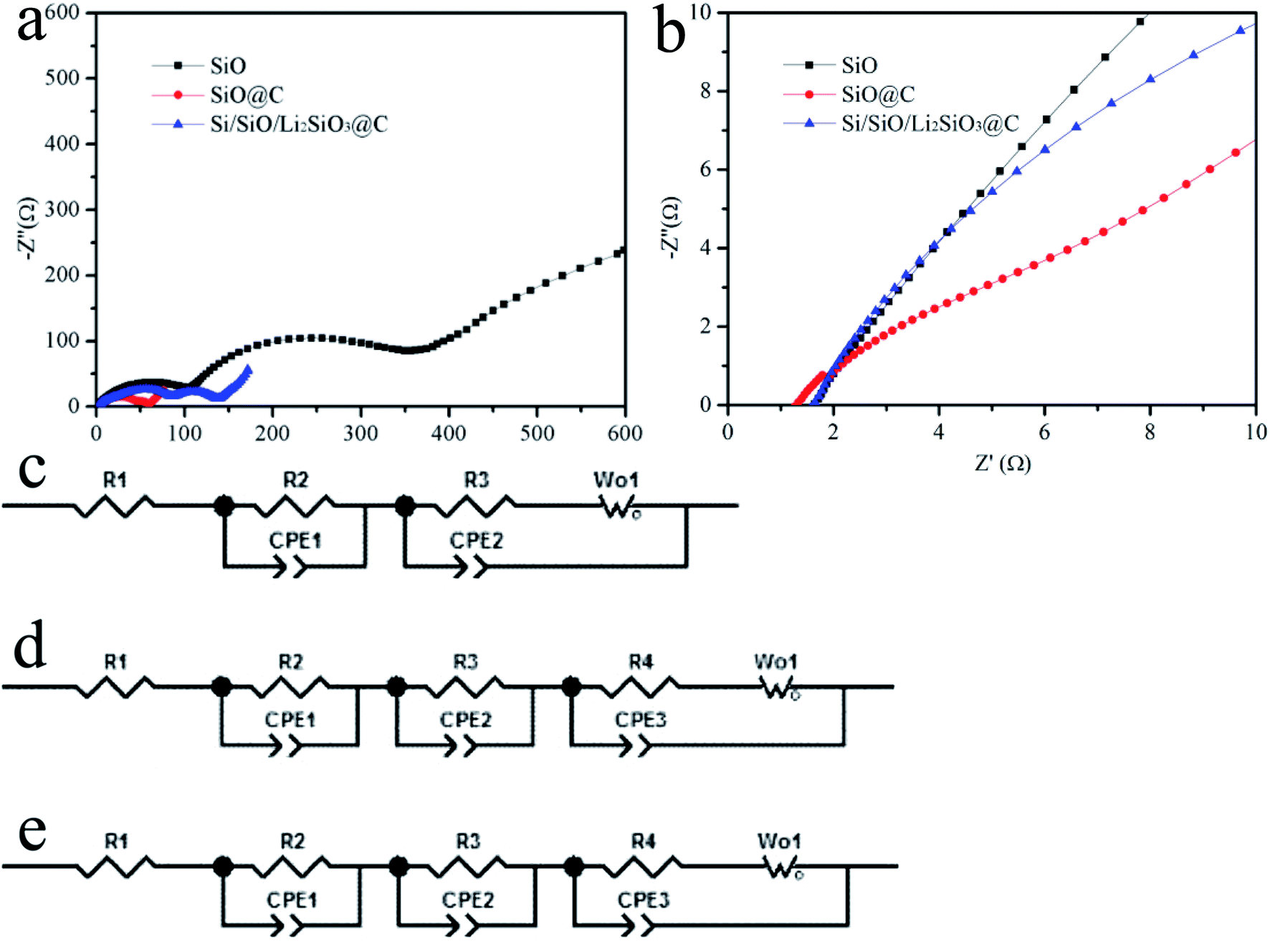 | ||
| Fig. 6 (a and b) Electrochemical impedance spectra of the SiO, SiO@C and Si/SiO/Li2SiO3@C, (c–e) are the equivalent circuits of the EIS spectra of SiO, SiO@C and Si/SiO/Li2SiO3@C. | ||
Conclusions
In summary, we have developed a facile method to construct the hierarchical Si/SiO/Li2SiO3@C composites for advanced LIBs anodes. The as-prepared composites not only have a high 1st C.E. of 69.05% but also show improved cycling stability. Benefited from the partial prelithiation of SiO and carbon coating, the Si/SiO/Li2SiO3@C anode has a superior electrochemical performance, with a prolonged cycle performance of 78.46% capacity retention over 35 cycles at 100 mA g−1 and an excellent rate property of 197.8 mA h g−1 at a high current density of 2000 mA h g−1. Such a design not only mitigates the volume expansion issue but also facilitates electron transport and guarantees structural robustness which pave the way for fabricate durable Si-based anode materials for lithium storage.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by National Key R&D Program of China (2016YFB0100306). The authors thanks to the technicians from the Gotion Validation Engineering institute for the support in the FESEM, XRD and electrochemical characterizations.References
- E. Boisselier and D. Astruc, Gold Nanoparticles in Nanomedicine: Preparations, Imaging, Diagnostics, Therapies and Toxicity, Chem. Soc. Rev., 2009, 38, 1759–1782 RSC.
- J. B. Goodenough and Y. Kim, Challenges for Rechargeable Li Batteries, Chem. Mater., 2010, 22, 587–603 CrossRef CAS.
- M. N. Obrovac and V. L. Chevrier, Alloy Negative Electrodes for Li-ion Batteries, Chem. Rev., 2014, 114, 11444–11502 CrossRef CAS PubMed.
- J. Cabana, L. Monconduit, D. Larcher and M. R. Palacín, Beyond Intercalation-Based Li-Ion Batteries: The State of the Art and Challenges of Electrode Materials Reacting Through Conversion Reactions, Adv. Mater., 2010, 22, E170–E192 CrossRef CAS PubMed.
- Z. Wang, L. Zhou and X. W. David Lou, Metal Oxide Hollow Nanostructures for Lithium-ion Batteries, Adv. Mater., 2012, 24, 1903–1911 CrossRef CAS PubMed.
- X. Su, Q. Wu, J. Li, X. Xiao, A. Lott, W. Lu, B. W. Sheldon and J. Wu, Silicon-Based Nanomaterials for Lithium-Ion Batteries: A Review, Adv. Energy Mater., 2014, 4, 1300882 CrossRef.
- J. O. Besenhard, J. Yang and M. Winter, Will Advanced Lithium-Alloy Anodes Have a Chance in Lithium-Ion Batteries, J. Power Sources, 1997, 68, 87–90 CrossRef CAS.
- C. K. Chan, H. Peng, G. Liu, K. McIlwrath, X. F. Zhang, R. A. Huggins and Y. Cui, High-performance Lithium Battery Anodes using Silicon Nanowires, Nat. Nanotechnol., 2008, 3, 31–35 CrossRef CAS PubMed.
- S. Ohara, J. Suzuki, K. Sekine and T. Takamura, A Thin Film Silicon Anode for Li-Ion Batteries Having a Very Large Specific Capacity and Long Cycle Life, J. Power Sources, 2004, 136, 303–306 CrossRef CAS.
- R. A. Huggins, Lithium Alloy Negative Electrodes Formed from Convertible Oxides, Solid State Ionics, 1998, 113, 57–67 CrossRef.
- N. Liu, Z. Lu, J. Zhao, M. T. McDowell, H. Lee, W. Zhao and Y. Cui, A Pomegranate-Inspired Nanoscale Design for Large-Volume-Change Lithium Battery Anodes, Nat. Nanotechnol., 2014, 9, 187–192 CrossRef CAS.
- M. Yoshio, H. Wang, K. Fukuda, T. Umeno, N. Dimov and Z. Ogumi, Carbon-Coated Si as a Lithium-Ion Battery Anode Material, J. Electrochem. Soc., 2002, 149, A1598–A1603 CrossRef CAS.
- X. Huang, J. Yang, S. Mao, J. Chang, P. B. Hallac, C. R. Fell, B. Metz, J. Jiang, P. T. Hurley and J. Chen, Controllable Synthesis of Hollow Si Anode for Long-Cycle-Life Lithium-Ion Batteries, Adv. Mater., 2014, 26, 4326–4332 CrossRef CAS PubMed.
- J. Liu, P. Kopold, P. A. van Aken, J. Maier and Y. Yu, Energy Storage Materials from Nature through Nanotechnology: A Sustainable Route from Reed Plants to a Silicon Anode for Lithium-Ion Batteries, Angew. Chem., Int. Ed., 2015, 54, 9632–9636 CrossRef CAS PubMed.
- M. N. Obrovac, L. Christensen, D. B. Le and J. R. Dahn, Alloy Design for Lithium-Ion Battery Anodes, J. Electrochem. Soc., 2007, 154, A849–A855 CrossRef CAS.
- T. Cetinkaya, M. Uysal and H. Akbulut, Highly Reversible Silicon/Carbon Nanofiber/Carbon Nanotube Nanocomposite Anodes for Lithium Ion Batteries, ECS Trans., 2014, 63, 23–29 CrossRef CAS.
- X. J. Li and W. F. Jiang, Enhanced Field Emission from a Nest Array of Multi-Walled Carbon Nanotubes Grown on a Silicon Nanoporous Pillar Array, Nanotechnology, 2007, 18, 065203 CrossRef.
- H. Kim, T. Hwang, K. Kang, J. Pichler-Nagl, D. So, S. Park and H. Huh, Preparation of Silicon Nanoball Encapsulated with Graphene Shell by CVD and Electroless Plating Process, J. Ind. Eng. Chem., 2017, 50, 115–122 CrossRef CAS.
- J. Zhou, T. Qian, M. Wang, N. Xu, Q. Zhang, Q. Li and C. Yan, Core–Shell Coating Silicon Anode Interfaces with Coordination Complex for Stable Lithium-Ion Batteries, ACS Appl. Mater. Interfaces, 2016, 8, 5358–5365 CrossRef CAS PubMed.
- B. Jiang, S. Zeng, H. Wang, D. Liu, J. Qian, Y. Cao, H. Yang and X. Ai, Dual Core-Shell Structured Si@SiOx@C Nanocomposite Synthesized via a One-Step Pyrolysis Method as a Highly Stable Anode Material for Lithium-Ion Batteries, ACS Appl. Mater. Interfaces, 2016, 8, 31611–31616 CrossRef CAS PubMed.
- M. Thakur, R. B. Pernites, N. Nitta, M. Isaacson, S. L. Sinsabaugh, M. S. Wong and S. L. Biswal, Freestanding Macroporous Silicon and Pyrolyzed Polyacrylonitrile as a Composite Anode for Lithium Ion Batteries, Chem. Mater., 2012, 24, 2998–3003 CrossRef CAS.
- Q. Si, K. Hanai, T. Ichikawa, A. Hirano, N. Imanishi, Y. Takeda and O. Yamamoto, A High Performance Silicon/Carbon Composite Anode with Carbon Nanofiber for Lithium-Ion Batteries, J. Power Sources, 2010, 195, 1720–1725 CrossRef CAS.
- S. Song, S. W. Kim, D. J. Lee, Y. Lee, K. M. Kim, C. Kim, J. Park, Y. M. Lee and K. Y. Cho, Flexible Binder-Free Metal Fibril Mat-Supported Silicon Anode for High-Performance Lithium-Ion Batteries, ACS Appl. Mater. Interfaces, 2014, 6, 11544–11549 CrossRef CAS PubMed.
- J. Suk, D. Y. Kim, D. W. Kim and Y. Kang, Electrodeposited 3D Porous Silicon/Copper Films with Excellent Stability and High Rate Performance for Lithium-Ion Batteries, J. Mater. Chem. A, 2014, 2, 2478–2481 RSC.
- J. Su, J. Zhao, L. Li, C. Zhang, C. Chen, T. Huang and A. Yu, Three-Dimensional Porous Si and SiO2 with in situ Decorated Carbon Nanotubes as Anode Materials for Li-ion Batteries, ACS Appl. Mater. Interfaces, 2017, 9, 17807–17813 CrossRef CAS PubMed.
- N. Dimov, S. Kugino and M. Yoshio, Carbon-Coated Silicon as Anode Material for Lithium Ion Batteries: Advantages and Limitations, Electrochim. Acta, 2003, 48, 1579–1587 CrossRef CAS.
- S. J. Lee, H. J. Kim, T. H. Hwang, S. Choi, S. H. Park, E. Deniz, D. S. Jung and J. W. Choi, Delicate Structural Control of Si-SiOx-C Composite via High-Speed Spray Pyrolysis for Li-Ion Battery Anodes, Nano Lett., 2017, 17, 1870–1876 CrossRef CAS PubMed.
- E. San Andrés, A. Del Prado, I. Mártil, G. G. Díaz, F. L. Martinez, D. Bravo and F. J. López, Rapid Thermal Annealing Effects on Plasma Deposited SiOx:H Films, Vacuum, 2002, 67, 531–536 CrossRef.
- S. W. Hwang, J. K. Lee and W. Y. Yoon, Electrochemical Behavior of Carbon-Coated Silicon Monoxide Electrode with Chromium Coating in Rechargeable Lithium Cell, J. Power Sources, 2013, 244, 620–624 CrossRef CAS.
- C. M. Park, W. Choi, Y. Hwa, J. H. Kim, G. Jeong and H. J. Sohn, Characterizations and Electrochemical Behaviors of Disproportionated SiO and Its Composite for Rechargeable Li-ion Batteries, J. Mater. Chem., 2010, 20, 4854–4860 RSC.
- J. H. Kim, C. M. Park, H. Kim, Y. J. Kim and H. J. Sohn, Electrochemical Behavior of SiO Anode for Li Secondary Batteries, J. Electroanal. Chem., 2011, 661, 245–249 CrossRef CAS.
- K. W. Kim, H. Park, J. G. Lee, J. Kim, Y. Kim, J. H. Ryu, J. J. Kim and S. M. Oh, Capacity Variation of Carbon-Coated Silicon Monoxide Negative Electrode for Lithium-Ion Batteries, Electrochim. Acta, 2013, 103, 226–230 CrossRef CAS.
- J. K. Lee, J. H. Lee, B. K. Kim and W. Y. Yoon, Electrochemical Characteristics of Diamond-Like Carbon/Cr Double-Layer Coating on Silicon Monoxide-Graphite Composite Anode for Li-Ion Batteries, Electrochim. Acta, 2014, 127, 1–6 CrossRef.
- J. L. Gomez-Ballesteros and P. B. Balbuena, Reduction of Electrolyte Components on a Coated Si Anode of Lithium-Ion Batteries, J. Phys. Chem. Lett., 2017, 8, 3404–3408 CrossRef CAS PubMed.
- J. Liang, X. Li, Z. Hou, W. Zhang, Y. Zhu and Y. Qian, A Deep Reduction and Partial Oxidation Strategy for Fabrication of Mesoporous Si Anode for Lithium Ion Batteries, ACS Nano, 2016, 10, 2295–2304 CrossRef CAS.
- D. T. Ngo, H. T. Le, X. M. Pham, C. N. Park and C. J. Park, Facile Synthesis of Si@SiC Composite as an Anode Material for Lithium-Ion Batteries, ACS Appl. Mater. Interfaces, 2017, 9, 32790–32800 CrossRef CAS PubMed.
- G. Jeong, Y. Kim, S. A. Krachkovskiy and C. K. Lee, A Nanostructured SiAl0.2O Anode Material for Lithium Batteries, Chem. Mater., 2010, 22, 5570–5579 CrossRef CAS.
- I. W. Seong, K. T. Kim and W. Y. Yoon, Electrochemical Behavior of a Lithium-pre-doped Carbon-Coated Silicon Monoxide Anode Cell, J. Power Sources, 2009, 189, 511–514 CrossRef CAS.
- Y. Liu, Z. Y. Wen, X. Y. Wang, A. Hirano, N. Imanishi and Y. Takeda, Electrochemical Behaviors of Si/C Composite Synthesized from F-containing Precursors, J. Power Sources, 2009, 189, 733–737 CrossRef CAS.
- X. Feng, J. Yang, P. Gao, J. Wang and Y. Nuli, Facile Approach to an Advanced Nanoporous Silicon/Carbon Composite Anode Material for Lithium Ion Batteries, RSC Adv., 2012, 2, 5701–5706 RSC.
- Z. H. Liu, Y. L. Zhao, R. H. He, W. Luo, J. S. Meng, Q. Yu, D. Y. Zhao, L. Zhou and L. Q. Mai, Yolk@Shell SiOx/C Microspheres with Semi-Graphitic Carbon Coating on the Exterior and Interior Surfaces for Durable Lithium Storage, Energy Storage Materials, 2018 DOI:10.1016/j.ensm.2018.10.011.
- C. S. Shan, K. F. Wu, H. J. Yen, C. N. Villarrubia, T. Nakotte, X. J. Bo, M. Zhou, G. Wu and H. L. Wang, Graphene Oxides Used as a New “Dual Role” Binder for Stabilizing Silicon Nanoparticles in Lithium-Ion Battery, ACS Appl. Mater. Interfaces, 2018, 10, 15665–15672 CrossRef CAS.
- T. Tan, P. K. Lee and D. W. Yu, Probing the Reversibility of Silicon Monoxide Electrodes for Lithium-Ion Batteries, J. Electrochem. Soc., 2019, 166, A5210–A5214 CrossRef.
- L. Y. Shen, Z. X. Wang and L. Q. Chen, Carbon-coated hierarchically porous silicon as anode material for lithium ion batteries, RSC Adv., 2014, 4, 15314–15318 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: The XRD pattern of the solute from the wash supernatant, the morphology of the Si/SiO/Li2SiO3@C composites after acid treatment, charge and discharge profiles of three samples at 50 μA. The electrochemical performance of BTR material and Si/SiO/Li2SiO3@C composite. See DOI: 10.1039/c9ra00778d |
| This journal is © The Royal Society of Chemistry 2019 |