DOI:
10.1039/C9RA00788A
(Paper)
RSC Adv., 2019,
9, 10289-10296
ZnO decorated Sn3O4 nanosheet nano-heterostructure: a stable photocatalyst for water splitting and dye degradation under natural sunlight
Received
29th January 2019
, Accepted 17th March 2019
First published on 2nd April 2019
Abstract
Herein, a facile hydrothermally-assisted sonochemical approach for the synthesis of a ZnO decorated Sn3O4 nano-heterostructure is reported. The phase purity of the nano-heterostructure was confirmed by X-ray diffraction and Raman spectroscopy. The morphological analysis demonstrated a nanosheet-like structure of Sn3O4 with a thickness of 20 nm, decorated with ZnO. The optical band gap was found to be 2.60 eV for the ZnO@Sn3O4 nano-heterostructure. Photoluminescence studies revealed the suppression of electron–hole recombination in the ZnO@Sn3O4 nano-heterostructure. The potential efficiency of ZnO@Sn3O4 was further evaluated towards photocatalytic hydrogen production via H2O splitting and degradation of methylene blue (MB) dye. Interestingly, it showed significantly superior photocatalytic activity compared to ZnO and Sn3O4. The complete degradation of MB dye solution was achieved within 40 min. The nano-heterostructure also exhibited enhanced photocatalytic activity towards hydrogen evolution (98.2 μmol h−1/0.1 g) via water splitting under natural sunlight. The superior photocatalytic activity of ZnO@Sn3O4 was attributed to vacancy defects created due to its nano-heterostructure.
1 Introduction
Recently, the discharge of industrial wastewater containing organic contaminants has led to serious environmental hazards. Commonly, biologically non-degradable waste water effluents contain synthetic pigments, organic dyes and phenolic contaminants, which cause a serious problem to aquatic life. Moreover, the higher stability of modern synthetic organic contaminants hampers orthodox biological treatments/methods and results in ineffectual degradation for the removal of these organic contaminants.1,2 Broadly, there are two approaches for the removal of toxic contaminants from waste water. (i) Contaminants are degraded and broken down into harmless moieties.3 (ii) Contaminants are removed via an adsorption technique.4 The first approach is more effective, in which reactive oxygen species are generated using a suitable semiconducting photocatalyst by advanced oxidation processes (AOP).5–7 TiO2 and ZnO are the most commonly used semiconductor photocatalysts due to their high efficiency in the degradation of organic pollutants under UV irradiation.8–10 However, these photocatalysts also possess some limitations, such as being excited only by ultraviolet light or the recombination rates of photogenerated holes and electrons being high, which consequently restrict their application due to their low utilization of solar energy.11,12 Therefore, several ideas have been put forward by researchers to promote the photocatalytic activity by the effective utilization of solar light energy. These efforts include increasing the hole concentration through doping, varying the particle size to enhance the surface area, mixing the semiconductor catalyst with suitable metal/metal oxide nanomaterials and the development of new photocatalysts with a narrow band gap.13–17 Among these techniques, the fabrication of composite materials based on two different metal oxides with variable band gap energies provides an effective way to tackle the above-mentioned issues.18–22 In this regard, there are various reports available on different coupled metal oxides, such as SnO2–ZnO, TiO2–WO3, CZTS–TiO2, Bi2O3/Bi5O7NO3, ZnO–SnO2 and TiO2–SnO2.23–28
Recently, heterovalent tin oxide (Sn3O4) with various nanomorphs, including nanosheets, nanoflowers, nanobelts, nanolayers, etc., has been used as a potential catalyst due to its narrow band gap and chemical stability.29–32 The photocatalytic performance of pristine Sn3O4 is believed to be due to the variable oxidation state of Sn: i.e. Sn2+ and Sn4+.32 Reports in the literature have demonstrated the moderate photocatalytic activity of Sn3O4 materials for H2 evolution and dye degradation under visible light. However, to achieve efficient H2 evolution under visible light, Sn3O4 materials are often utilized along with a noble metal as a co-catalyst.29 This limits their widespread use as low-cost materials. Thus, facile synthetic methods operating under milder reaction conditions for the fabrication of Sn3O4-based nanomaterials that operate under sunlight/visible light irradiation are highly desirable. ZnO with its wide band gap (3.2 eV) could be a good alternative for band coupling with Sn3O4 to realize the effective separation and transfer of photo-generated h+ and e−. Considering the above facts, we are reporting a combined hydrothermal and sonochemical method for the synthesis of a ZnO@Sn3O4 composite for the first time. To the best of our knowledge, this is the first report highlighting a ZnO@Sn3O4 composite operable under visible light.
We have synthesized pristine ZnO, Sn3O4 and ZnO@Sn3O4 composites. The as-synthesized samples were characterized using different physicochemical techniques and further utilized for their photocatalytic performance for the degradation of methylene blue (MB) dye and H2 evolution under sunlight irradiation.
2 Experimental
Chemicals
All of the chemicals were of analytical grade and used as received. Stannous oxalate (SnC2O4) was purchased from Otto Chemicals; disodium succinate hexahydrate was purchased from Central Drug House (P) Ltd; zinc acetate dihydrate (Zn(CH3COO)2·2H2O), sodium hydroxide (NaOH) and absolute ethanol were purchased from Merck (India).
Synthesis of nanocrystalline Sn3O4
In the synthesis of Sn3O4, initially, aqueous solutions of 10 mL (0.01 M) of stannous oxalate and 10 mL (0.02 M) of disodium succinate hexahydrate were mixed together using a magnetic stirrer to form a homogeneous mixture. To this solution, aq. NaOH solution (0.5 M) was added dropwise over a period of 30 min to attain a final pH of ∼12. After complete addition, a white precipitate was formed. This precipitate was then transferred to a Teflon-lined stainless-steel autoclave and hydrothermally treated at 180 °C for 24 h. After 24 h, the autoclave was cooled down naturally to room temperature. The final yellow product in the form of a precipitate was centrifuged and washed with ethanol and water several times and then dried at 60 °C in an oven for 12 h.
Synthesis ZnO@Sn3O4
0.15 g of as-synthesized Sn3O4 was well dispersed homogeneously in a (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) water:ethanol mixture. To this dispersion, 0.05 g of zinc acetate dihydrate was added to form a homogeneous solution. This was followed by the addition of 0.2 M NaOH solution to adjust the final pH to 10. The resulting solution was then sonochemically treated with an ultrasound probe (Sonics Vibra-Cell, USA) for 4 h (operated with 5 s pulse ON and 2 s pulse OFF at an amplitude of 35%). The obtained precipitate was then centrifuged, washed and dried at 60 °C for 12 h. The %wt compositions of the obtained nano-heterostructure material were 15% ZnO and 85% Sn3O4. For comparison, pure ZnO was also synthesized by following the same procedure as above without the addition of Sn3O4.
:1) water:ethanol mixture. To this dispersion, 0.05 g of zinc acetate dihydrate was added to form a homogeneous solution. This was followed by the addition of 0.2 M NaOH solution to adjust the final pH to 10. The resulting solution was then sonochemically treated with an ultrasound probe (Sonics Vibra-Cell, USA) for 4 h (operated with 5 s pulse ON and 2 s pulse OFF at an amplitude of 35%). The obtained precipitate was then centrifuged, washed and dried at 60 °C for 12 h. The %wt compositions of the obtained nano-heterostructure material were 15% ZnO and 85% Sn3O4. For comparison, pure ZnO was also synthesized by following the same procedure as above without the addition of Sn3O4.
Physicochemical characterization
The structural analysis and phase purity of the ZnO, Sn3O4 and ZnO@Sn3O4 samples were carried out using powder X-ray diffraction analysis (XRD) (Rigaku Miniflex X-ray diffractometer) with Cu Kα irradiation at λ = 1.5406 Å. Raman spectra were recorded on a Jobin Yvon T64000 triple grating spectrometer equipped with a liquid nitrogen cooled charge coupled device. The photoluminescence (PL) spectra were recorded on an F-2500 fluorescence spectrophotometer. For the PL measurements, water suspensions of the samples were excited at a wavelength of 350 nm. The surface morphologies of the as-prepared samples were investigated by field emission scanning electron microscopy (FESEM) using a JEOL-JSM Model 6700F. The field emission transmission electron microscopy (FETEM) images of the as-synthesized products were obtained on a JEOL, JEM-2200 FS transmission electron microscope equipped with a field emission gun. The particle size distribution measurements of the as-prepared samples were done using a PSS-NICOMP particle size analyser (Santa Barbara, California, USA). UV-visible absorption spectra were obtained by a JASCOV-570 UV-visible spectrophotometer. The spectral changes in the concentration of MB during photocatalytic degradation were also studied using the same spectrophotometer.
Photocatalytic H2 generation from water
The as-synthesized photocatalysts were tested for H2 evolution from the splitting of water. Typical experiments were carried using a 250 mL round bottomed flask containing 100 mL of double distilled water and 25 mL of methanol as a sacrificial reagent. Then, 100 mg of catalyst was added to this solution. The dissolved oxygen was removed from the reaction mixture by purging with argon. The photocatalytic H2 evolution was studied under solar light irradiation. The progress of the reaction was monitored by measuring evolved gas with the help of a eudiometer connected to a round bottomed flask. During experiments, the amount of gas evolved as a function of time was noted and the data was used for further calculations. The quantification of the hydrogen gas evolved was analyzed by gas chromatography operated using a 5 Å capillary column and a thermal conductivity detector.
Photocatalytic degradation of MB dye
The photocatalytic activities of the as-synthesized photocatalysts were evaluated by degradation of a representative organic dye, methylene blue, under sunlight irradiation. A 250 mL Pyrex beaker was used for the photodegradation experiments. 50 mg of catalyst was added to 100 mL of an aqueous solution of 10 ppm MB dye and the mixture was initially stirred in the dark for 1 h to achieve adsorption–desorption equilibrium of the MB dye with the photocatalysts. After 1 h, the mixture was exposed to sunlight for photodegradation. At regulated intervals of time, 5 mL of reaction mixture was collected, centrifuged to remove the catalyst and then analysed using a UV-visible spectrometer. The progress of the reaction was monitored by observing changes in the intensity of the maximum absorption of the MB dye at 662 nm.
3 Results and discussion
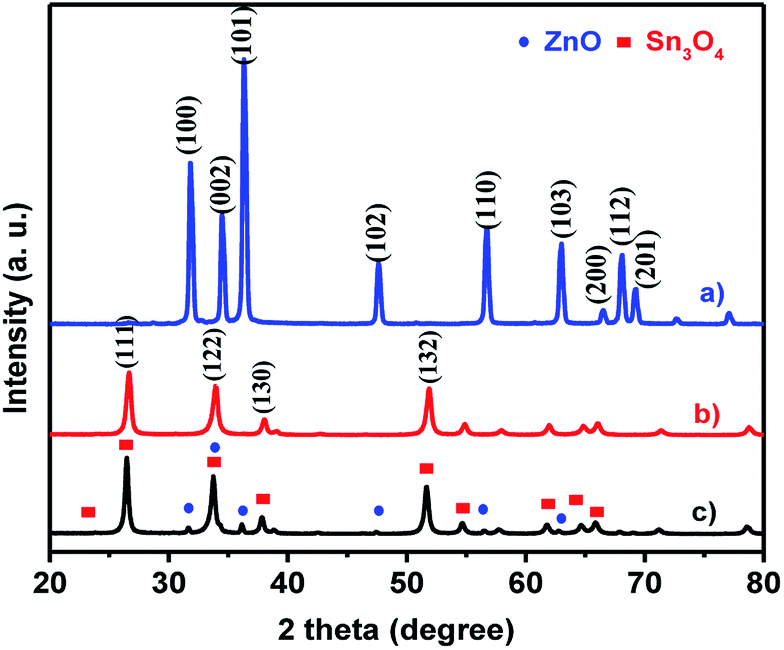
The crystal structures and phase purity of as-synthesized ZnO, Sn3O4 and ZnO@Sn3O4 products were examined by powder XRD, as shown in Fig. 1. Curve (a), the X-ray diffractogram of pristine ZnO, shows diffraction peaks at 31.2°, 34.4°, 36.6°, 47.2°, 56.4°, 62.7°, 66.3°, 67.5° and 69.2° corresponding to the (100), (002), (101), (102), (110), (103), (200), (112) and (201) planes of the hexagonal wurtzite phase of ZnO (JCPDS card no. 89-0510).32 Curve (b), the X-ray diffractogram of as-synthesized Sn3O4, corresponds to the (101), (111), (122), (130), and (132) reflection planes, attributed to the triclinic phase (JCPDS card no. 16-0737).23 The XRD pattern of the as-synthesized ZnO@Sn3O4 nano-heterostructure is depicted in Fig. 1(c). It can be seen that the XRD pattern exhibits diffraction peaks corresponding to both ZnO and Sn3O4, indicating the formation of a ZnO phase along with Sn3O4. No other impurity peaks were detected.
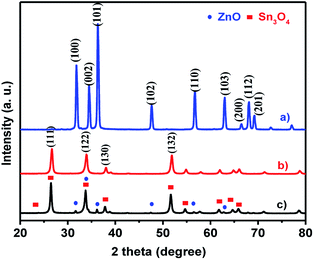 |
| | Fig. 1 X-ray diffraction patterns (XRD) of (a) ZnO, (b) Sn3O4 and (c) ZnO@Sn3O4. | |
The XRD data were further utilized for determining the crystallite size using the Debye–Scherrer formula. The estimated crystallite sizes were found to be 48.6, 36.2 and 34.4 for pristine ZnO, Sn3O4 and ZnO@Sn3O4 nanostructures, respectively.
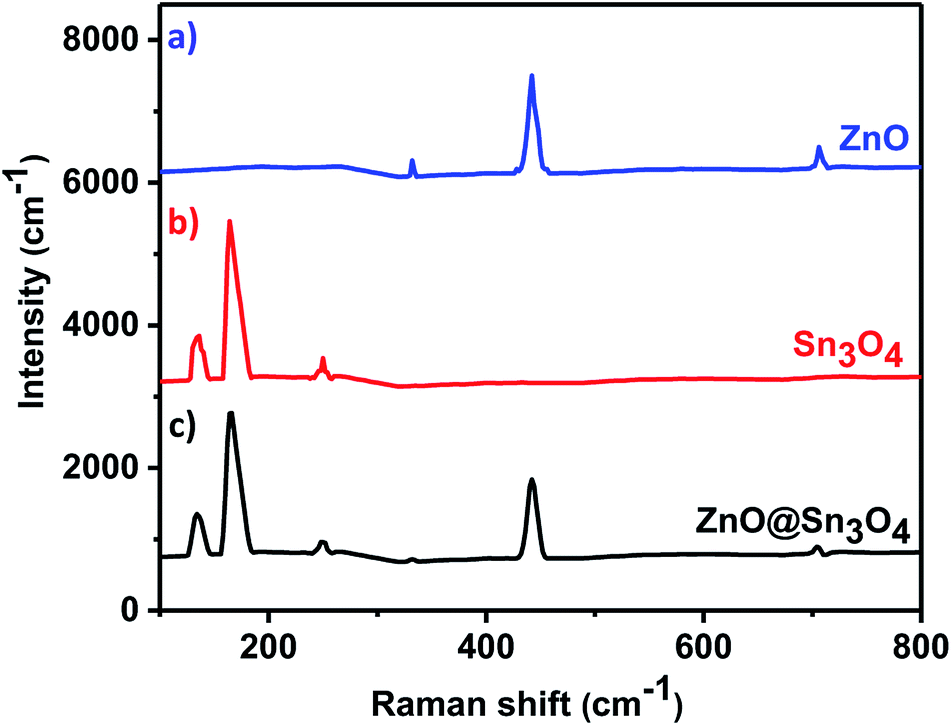
The as-synthesized ZnO, Sn3O4 and ZnO@Sn3O4 samples were further analyzed by Raman spectroscopy and the results are shown in Fig. 2. Curve (a) represents the Raman spectrum of pristine ZnO. From the spectrum, the vibrational peaks appearing at 332, 438 and 698 cm−1 for ZnO can be clearly seen. The most intense peak located at 438 cm−1 is characteristic of the E2 mode of the hexagonal wurtzite structure.33
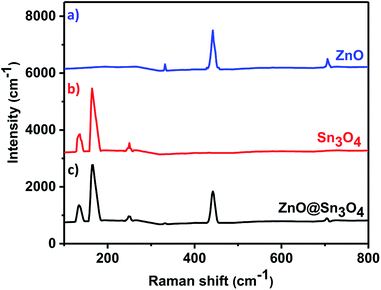 |
| | Fig. 2 Room temperature Raman spectra of (a) ZnO, (b) Sn3O4 and (c) ZnO@Sn3O4. | |
The other modes with frequencies of 332 and 698 cm−1 are due to multiphonon scattering processes and cannot be explained within the framework of bulk single phonon modes.33 For pristine Sn3O4 (curve (b)), the two sharp peaks at 135 and 168 cm−1 are characteristic peaks of triclinic Sn3O4. However, the weak peak at 243 cm−1 may be due to the oxidation of the sample.30 Curve (c) shows the Raman spectrum of ZnO@Sn3O4, which exhibits mixed peaks for Sn3O4 and ZnO. This confirms that all of the samples exhibit crystalline peaks and that these results are in good accordance with the XRD results.
Based on the above characterization features, a mechanism of the formation of ZnO@Sn3O4 has been proposed. Sn3O4 was well dispersed homogeneously in a 1:1 ethanol:water mixture. Zn2+ and OH− ions were provided by zinc acetate and sodium hydroxide, respectively, which reacted to form a stable Zn(OH)4 complex. Under sonochemical treatment, the sonolysis of water generates various radicals, such as ˙H, ˙OH, ˙OH2 and ˙O2−. The ˙O2− plays an important role in the formation of ZnO nanostructures. The formation reactions of ZnO are represented by eqn (1)–(3):34
| | |
Zn2+ + 4OH− → Zn(OH)42−
| (1) |
| | |
Zn(OH)42− → ZnO + H2O + OH−
| (2) |
| | |
Zn2+ + 2˙O2− → ZnO + 3/2O2
| (3) |
The morphological analysis of ZnO, Sn3O4 and ZnO@Sn3O4 nano-heterostructures was further analyzed by FESEM and the results are depicted in Fig. 3. From Fig. 3(a) and (b), it can be seen that pristine ZnO exhibits a hexagonal rod-like morphology with a width ranging from 50–180 nm to 200–400 nm in length. The FESEM analysis of pristine Sn3O4, as depicted in Fig. 3(c) and (d), reveals that it consists of irregularly-shaped sheets with a thickness of about 20 nm, with some flat nanocrystals deposited on them. Fig. 3(e) and (f) show that in the FESEM analysis of as-synthesized ZnO@Sn3O4 nano-heterostructures, the morphology was retained. However, sheet-like nanostructures with deposited ZnO nanocrystals of reduced size were observed for these composites. The reduced size of the nanosheets and deposited nanocrystals might be attributed to ultrasound irradiation during the sonochemical synthesis.
 |
| | Fig. 3 FESEM images of (a and b) ZnO, (c and d) Sn3O4, (e and f) ZnO@Sn3O4, respectively. | |
The crystallinity and heterojunction of the as-synthesized samples were confirmed using field emission transmission electron microscopy (FETEM). Fig. 4 shows the FETEM images of ZnO@Sn3O4. Fig. 4(a) and (b) reveal a sheet-like morphology with the thickness of the nanosheet less than 30 nm and some nanocrystals of ZnO present on the Sn3O4 nanosheets. Fig. 4(c) represents the SAED pattern of the sample, which clearly shows the polycrystalline nature of ZnO@Sn3O4. Further, from the HRTEM image of the ZnO@Sn3O4 (Fig. 4(d)), the close connection/binding of ZnO and Sn3O4 can be clearly seen. The lattice fringes obtained from different regions with lattice spacings of 0.33 and 0.25 nm can be assigned to the (111) plane of triclinic Sn3O4 and the (002) plane of wurtzite ZnO, respectively.
 |
| | Fig. 4 TEM (a and b), SAED pattern (c), HRTEM (d) images of ZnO@Sn3O4, respectively. | |
Fig. 5 shows the elemental mapping of the ZnO@Sn3O4 nanostructures. From the figure, the signals pertaining to Zn, Sn and O have a uniform spatial distribution. For further confirmation of the uniform distribution of Zn, Sn and O in ZnO@Sn3O4 nanosheets, spot EDS analysis was carried out and the results are shown in Fig. 5(b)–(f). In the figures, no peaks corresponding to any impurities can be observed and the stoichiometric ratios used for Zn and Sn were found to be in good agreement with those used in the synthetic architecture during the synthesis.
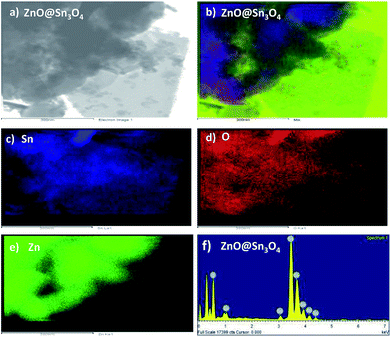 |
| | Fig. 5 (a) TEM image of ZnO@Sn3O4 for EDS; (b–f) EDS element mapping data of Zn, Sn and O elements for ZnO@Sn3O4, respectively. | |
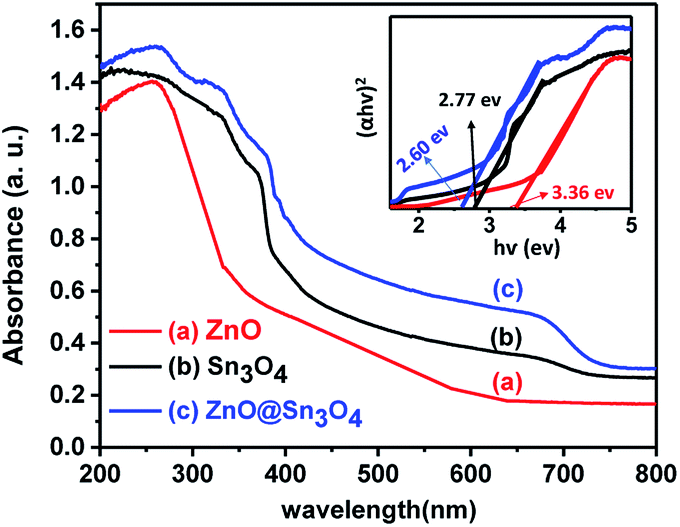
From Fig. 6, it can be seen that pristine ZnO (curve (a)) and Sn3O4 (curve (b)) exhibit absorption onsets at 391 nm and 445 nm, respectively. Whereas ZnO@Sn3O4 (curve (c)) exhibits absorption onsets at 392 nm and 460 nm. These can be attributed to the presence of both Sn3O4 and ZnO in the nanostructures.35 This data was further utilized for the determination of band gaps of the as-synthesized materials. The corresponding Tauc plot is shown in the inset of Fig. 6. From the figure the estimated band gap energies of ZnO, Sn3O4 and ZnO@Sn3O4 were found to be 3.36 eV, 2.77 eV and 2.60 eV, respectively. These results further demonstrated that the band gap of pristine Sn3O4 can be fine-tuned simply by coupling with ZnO.
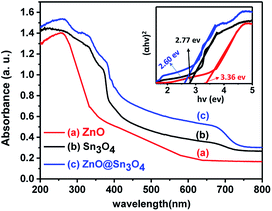 |
| | Fig. 6 UV-vis absorption spectra of (a) ZnO, (b) Sn3O4 and (c) ZnO@Sn3O4. The inset shows (αhν)2 vs. photon energy (hν) spectra for the calculation of optical band gap by extrapolation on the hν axis at α = 0. | |
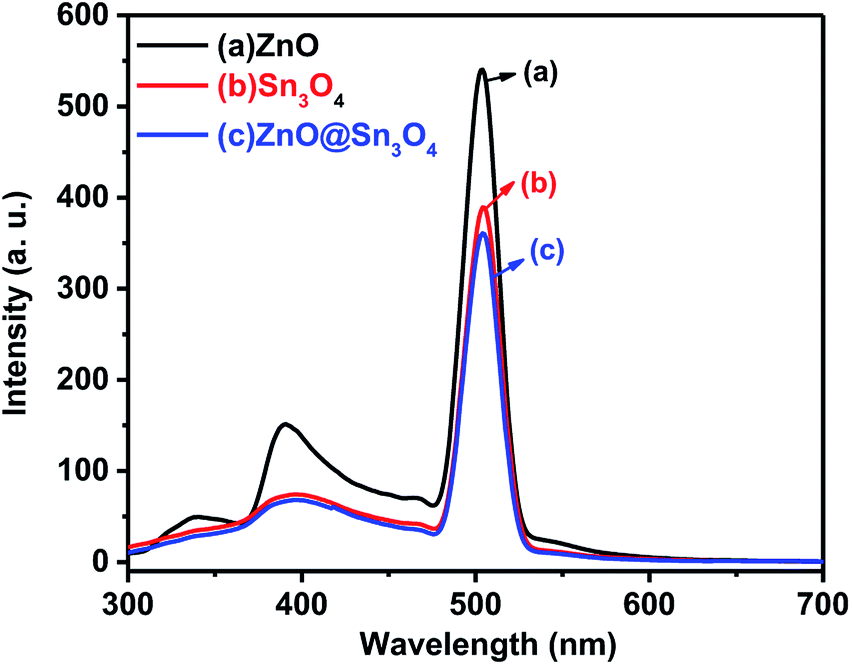
The PL spectra of the as-synthesized ZnO, Sn3O4 and ZnO@Sn3O4 samples were performed to understand the rate of photogenerated electrons and holes (Fig. 7). In general, a higher PL emission peak intensity arises due to higher recombination of photogenerated electron–hole pairs, whereas weak emission intensities indicate lower recombination rates and consequently the extended participation of photoexcited charge carriers in the photoreaction.36 In the PL spectrum of pristine ZnO, the weak emission peak at 398 nm is due to UV and the sharp emission peak at 498 nm corresponds to green emission.37 Pristine Sn3O4 exhibits a sharp emission peak at 510 nm, which can be attributed to the intrinsic luminescence of Sn3O4.30 The PL spectrum of ZnO@Sn3O4 also exhibits an emission peak at 510 nm. However, the PL peak intensity is considerably lowered. The significant decrease in the PL peak intensities of ZnO@Sn3O4 nanostructures clearly indicate the efficient inhibition of photoexcited electron–hole recombination.38
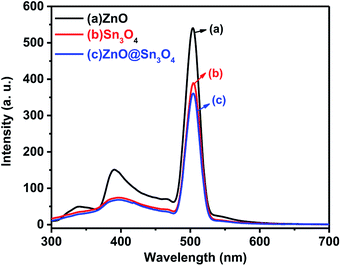 |
| | Fig. 7 PL spectra of (a) ZnO, (b) Sn3O4 and (c) ZnO@Sn3O4. | |
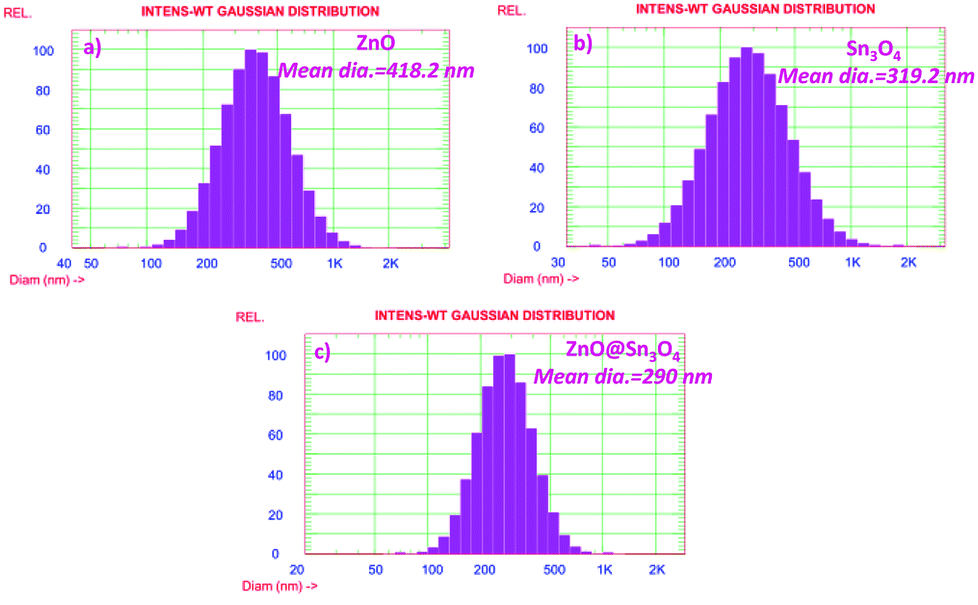
The particle size distribution results of as-synthesized ZnO, Sn3O4 and ZnO@Sn3O4 are shown in Fig. 8. From the figure, it can be seen that ZnO@Sn3O4 exhibits a lesser particle size compared to the pristine ZnO and Sn3O4 samples. The calculated mean Gaussian diameters observed for the ZnO, Sn3O4 and ZnO@Sn3O4 samples were 418.2, 319.2 and 290 nm, respectively. These results are in good agreement with the obtained FESEM results.
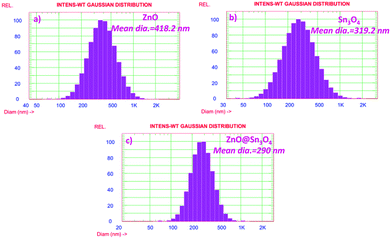 |
| | Fig. 8 Particle size distribution histogram of (a) ZnO, (b) Sn3O4 and (c) ZnO@Sn3O4, respectively. | |
Photocatalytic activity
Here in the present study, the photocatalytic activities of as-synthesized ZnO, Sn3O4 and ZnO@Sn3O4 nanostructures were evaluated by the photodegradation of methylene blue as a representative organic dye. Additionally, a direct comparison of the as-prepared samples with Degussa-P25 was also carried out. MB (C16H18ClN3S) is a well-known organic dyestuff and it is often used to a large extent for various purposes.
During the photodegradation experiments, 50 mg of photocatalyst was dispersed in 100 mL of 10 ppm MB aqueous solution and stirred in the dark to achieve adsorption–desorption equilibrium. After 1 h the MB dye solution containing the catalyst was exposed to sunlight. The progress of the photodegradation process was monitored using the change in intensities of the characteristic absorption of MB dye at 662 nm.
The experimental results pertaining to MB dye adsorbed and degraded at different time intervals under different experimental conditions are depicted in Fig. 9(a). For comparison, a blank experiment without any catalyst was also performed. In the absence of catalyst, MB dye was hardly degraded, but degradation of MB dye was achieved in the presence of a catalyst under sunlight irradiation.
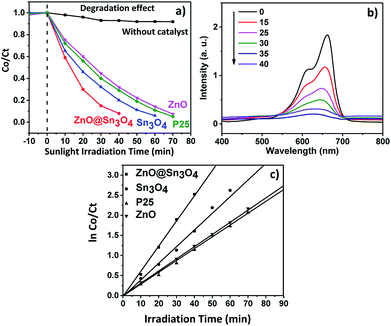 |
| | Fig. 9 (a) Plot of change in absorbance vs. irradiation time in the presence of ZnO, Sn3O4, P25 and ZnO@Sn3O4; (b) spectral changes of degradation of MB dye in the presence of ZnO@Sn3O4; (c) pseudo-first-order kinetic plots of ln(C0/Ct) vs. irradiation time for the degradation of methylene blue (MB) using ZnO, Sn3O4, P25 and ZnO@Sn3O4. | |
From Fig. 9(a) it can be seen that approximately 90% degradation of MB dye can be achieved in the presence of Sn3O4 and ZnO within 60 and 70 minutes, respectively, whereas P25 was able to degrade the same amount of MB dye within 65 min. However, the photodegradation efficiency is significantly enhanced in the presence of ZnO@Sn3O4 nanostructures. The spectral changes during the photodegradation of MB dye in the presence of ZnO@Sn3O4 under sunlight illumination are shown in Fig. 9(b). It can be clearly seen from the figure that more than 50% of the MB dye was degraded within the initial 20 min and complete degradation was achieved within a mere 40 min. This enhanced photoactivity of ZnO@Sn3O4 nano-heterostructures can be attributed to the narrow band gap and decreased e− and h+ recombination achieved due to the coupling of ZnO and Sn3O4.
Further, the experimental data was utilized for studying the photodegradation kinetics. The corresponding plots of lnC0/Ct vs. irradiation time are depicted in Fig. 9(c). The rate constants for the ZnO, P25, Sn3O4 and ZnO@Sn3O4 samples were found to be 0.0128, 0.0131, 0.0149 and 0.0178 min−1, respectively. Thus, the maximum photodegradation rates (i.e. 0.0178 min−1 with excellent linear correlation (Table 1)) were demonstrated by the ZnO@Sn3O4 photocatalyst, suggesting pseudo first-order kinetics.
Table 1 Summary of the pseudo-first-order kinetics for the photocatalytic degradation of methylene blue (MB) using ZnO, Sn3O4, P25 and ZnO@Sn3O4
| Catalyst |
R2 |
k (min−1) |
| ZnO |
0.973 |
0.0128 |
| P25 (TiO2) |
0.974 |
0.0131 |
| Sn3O4 |
0.986 |
0.0149 |
| ZnO@Sn3O4 |
0.998 |
0.0178 |
Considering the narrow band gap of as-synthesized ZnO@Sn3O4, these samples were also studied for photocatalytic hydrogen production via H2O splitting. Fig. 10 depicts a plot of hydrogen production from water with respect to time using the ZnO, Sn3O4 and ZnO@Sn3O4 nano-heterostructures both in the dark and under solar light. The maximum hydrogen evolution (98.2 μmol h−1/0.1 g) was obtained using the ZnO@Sn3O4 nanoparticles, and it was considerably higher than in previous reports.28,38 The results demonstrate that the ZnO@Sn3O4 nano-heterostructures could split water under sunlight conditions. These excellent characteristics of ZnO@Sn3O4 can be ascribed to the strong absorption of solar light due to the narrow band gap, which facilitates electron–hole pair formation and their transportation to the surface of the catalyst where adsorbed H2O can be split into H2 and O2.
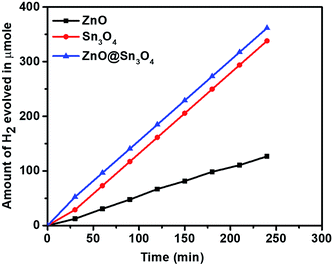 |
| | Fig. 10 Hydrogen production as a function of irradiation time in the presence of ZnO, Sn3O4 and ZnO@Sn3O4. | |
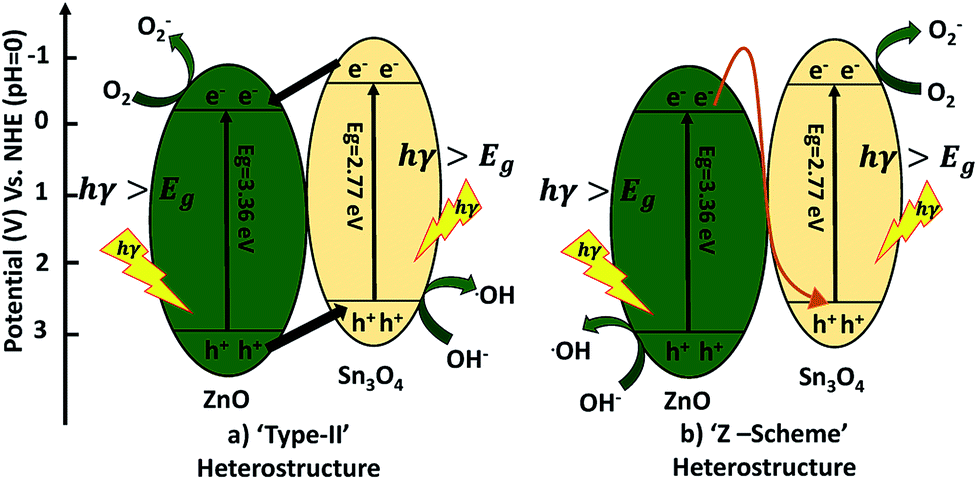
A possible mechanism for the degradation of methylene blue and hydrogen production by ZnO decorated Sn3O4 nano-heterostructures can be elucidated with the help of two probable schemes, as illustrated in Fig. 11. The band alignment in ZnO and Sn3O4 is suitable for a ‘type II’ heterostructure system.31,39,43 In the first probable mechanism, as shown in Fig. 11(a), in step (i), upon sunlight irradiation, photoexcitation of ZnO and Sn3O4 can take place, forming e−/h+ pairs. The electrons in the VB of both ZnO and Sn3O4 migrate to their respective CBs. In step (ii), as the CB of Sn3O4 is situated at a more negative energy potential (−0.58 eV) than the CB of ZnO (−0.39 eV), the photoinduced electrons migrate from the CB of Sn3O4 to the CB of ZnO. Whereas the VB of ZnO is situated at a more positive energy potential (+2.97 eV) than the VB of Sn3O4 (+2.74 eV), so the holes migrate from the VB of ZnO to the VB of Sn3O4. As a result, the photoinduced electrons accumulating in the CB of ZnO and the holes in the VB of Sn3O4 are well separated from each other. The potential energy value for the oxygen reduction reaction is −0.17 eV.
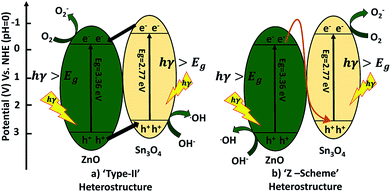 |
| | Fig. 11 Schematic diagram of electron transfer in the ZnO@Sn3O4. | |
In the second probable mechanism, the Z-scheme heterostructure40,41 can give a good interpretation of all the experimental results (Fig. 11(b)). For ZnO, the photoinduced e−/h+ in ZnO tend to recombine and few of them take part in the photocatalytic reaction, resulting in a relatively low activity. For the ZnO decorated Sn3O4 nano-heterostructures Sn3O4 can significantly prolong the light utilization from UV to visible light. Under sunlight irradiation, the photoinduced h+ are retained in the VB of ZnO, but the e− migrate from the CB of ZnO to the VB of Sn3O4. This mechanism can maintain at least a half of the photoinduced e−/h+ separated on opposite sides of the nano-heterostructure. Then, the e− originating in the CB of Sn3O4 (−0.58 eV) can easily reduce O2 to O2− since their potential is more negative than that of ZnO. Meanwhile, the h+ in the VB of ZnO (+2.97 eV) have a higher ability to oxidize OH− into ˙OH radicals. These photogenerated radicals can then bring about the degradation of MB dye molecules or H2O splitting. The overall mechanism is represented by following reactions.42,43
| | |
Sn3O4/ZnO (photocatalyst) → (h+)VB + (e−)CB
| (4) |
| | |
2H2O + (4h+)VB → 4H+ + O2
| (5) |
| | |
H2O + (h+)VB → OH˙ + H+
| (7) |
| | |
OH˙ + MB dye → degraded products
| (10) |
| | |
O2˙− + MB dye → degraded products
| (11) |
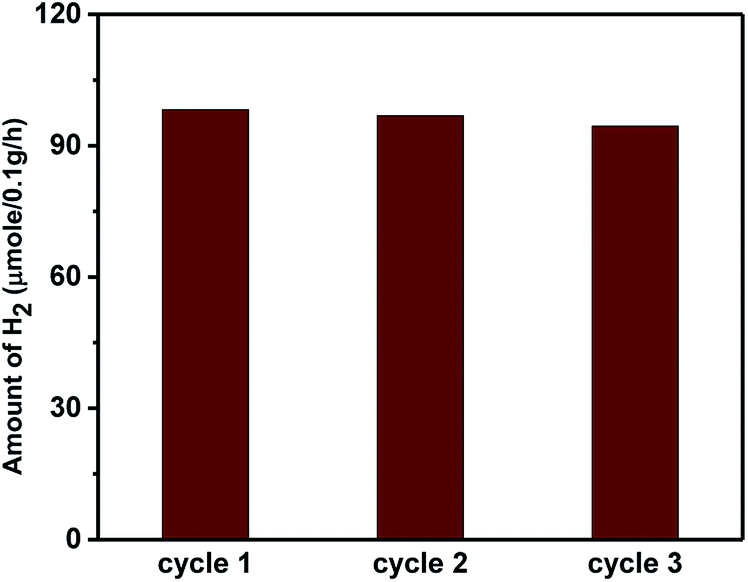
The higher activity of the nano-heterostructures is also evidenced by the decline in the PL peak intensity. This shows the inhibition of electron–hole recombination, which was responsible for enhanced photocatalytic activity. The synthesized ZnO, Sn3O4 and ZnO@Sn3O4 could not split water under dark conditions, either. However, under sunlight light irradiation a significant amount of H2 was produced from water splitting by the synthesized Sn3O4 and ZnO@Sn3O4. Further, the stability of ZnO@Sn3O4 was tested by repeating the experiments using the same ZnO@Sn3O4 photocatalyst after recycling (Fig. 12). In a nutshell, the ZnO@Sn3O4 nano-heterostructures were observed to be good stable photocatalysts under natural sunlight.
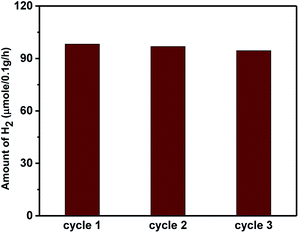 |
| | Fig. 12 Photocatalytic H2 evolution cycle of ZnO@Sn3O4 nanostructures under sunlight irradiation. | |
4 Conclusions
A novel, highly efficient ZnO@Sn3O4 nano-heterostructure photocatalyst obtained by a facile hydrothermal assisted sonochemical method has been demonstrated. Considering that the absorption edge of ZnO@Sn3O4 is well within the visible region, the photocatalytic degradation of methylene blue (MB) and hydrogen production from H2O under sunlight were performed. The prepared ZnO@Sn3O4 sample showed excellent photocatalytic activity for methylene blue degradation and for hydrogen production (98.2 μmole h−1/0.1 g) under natural sunlight. A suitable mechanism of the photocatalytic process was proposed. This study opens up a new possibility in the investigation of ZnO@Sn3O4 nanostructures and promotes their practical applications in environmental issues.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
SDB would like to thank Dr Abhay Pawar, Principal, D. Y. Patil college of Engineering, Pune, for encouragement and support. BBK is thankful to the Ministry of Electronics and Information Technology (MeitY), New Delhi & C-MET, Pune, for providing research facilities. PVA is grateful to ISRO, Bengaluru and VSSC, Thiruvananthapuram for financial support.
References
- H. Fang, K. V. S. Rao, S. Madapusi, R. Dharmarajan and N. Ravi, Appl. Catal., A, 2009, 359, 25–40 CrossRef.
- M. R. Hoffmann, S. T. Martin, W. Choi and D. W. Bahenemann, Chem. Rev., 1995, 95, 69 CrossRef CAS.
- S. Jian-Wen, C. Hao-Jie, Z. Xu, C. Shaohua, C. Jinsheng, X. Bin, Y. Weiya, L. Wang and F. Ming-Lai, Appl. Catal., A, 2012, 435, 86–92 Search PubMed.
- U. G. Akpan and B. H. Hameed, J. Hazard. Mater., 2009, 170, 520–529 CrossRef CAS PubMed.
- D. F. Ollis, Environ. Sci. Technol., 1985, 19, 480–484 CrossRef CAS PubMed.
- X. B. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891–2959 CrossRef CAS PubMed.
- J. Li, W. Ma, Y. Huang, X. Tao, J. Zhao and Y. Xu, Appl. Catal., B, 2004, 48, 17–24 CrossRef CAS.
- P. Xu, G. M. Zeng, D. L. Huang, C. L. Feng, S. Hu, M. H. Zhao, C. Lai, Z. Wei, C. Huang and G. X. Xie, Sci. Total Environ., 2012, 424, 1–10 CrossRef CAS PubMed.
- T. J. Liu, Q. Wang and P. Jiang, RSC Adv., 2013, 3, 12662–12670 RSC.
- M. Yasui, K. Katagiri, S. Yamanaka and K. Inumaru, RSC Adv., 2012, 2, 11132–11137 RSC.
- J. H. Li, W. Zhao, Y. Guo, Z. B. Wei, M. S. Han, H. He, S. G. Yang and C. Sun, Appl. Surf. Sci., 2015, 351, 270–279 CrossRef CAS.
- I. H. Chowdhury, S. Ghosh and M. K. Naskar, Ceram. Int., 2016, 42, 2488–2496 CrossRef CAS.
- D. Vione, C. Minero, V. Maurino, M. E. Carlotti, T. Picatonotto and E. Pelizzetti, Appl. Catal., B, 2005, 58, 79–88 CrossRef CAS.
- X. W. Zhang and L. C. Lei, Appl. Surf. Sci., 2008, 254, 2406–2412 CrossRef CAS.
- T. Soltani and M. H. Entezari, Ultrason. Sonochem., 2013, 20, 1245–1253 CrossRef CAS PubMed.
- S. Park, J. Park, R. Selvaraj and Y. Kim, J. Ind. Eng. Chem., 2015, 31, 269–275 CrossRef CAS.
- B. Qin, Y. B. Zhao, H. Li, L. Qiu and Z. Fan, Chin. J. Catal., 2015, 36, 1321–1325 CrossRef CAS.
- T. K. Jana, A. Pal and K. Chatterjee, J. Alloys Compd., 2014, 583, 510–515 CrossRef CAS.
- J. Chen, W. Liao, Y. Jiang, D. Yu, M. Zou, H. Zhu, M. Zhang and M. Du, Nanomater. Nanotechnol., 2016, 6, 1–8 CrossRef CAS.
- A. Sadollahkhani, O. Nur, M. Willander, I. Kazeminezhad, V. Khranovskyy, M. O. Eriksson, R. Yakimova and P. O. Holtz, Ceram. Int., 2015, 41, 7174–7184 CrossRef CAS.
- K. Ravichandran, N. Chidhambaram and S. Gobalakrishnan, J. Phys. Chem. Solids, 2016, 93, 82–90 CrossRef CAS.
- S. Xu, L. Fu, T. S. H. Pham, A. Yu, F. Han and L. Chen, Ceram. Int., 2015, 41, 4007–4013 CrossRef CAS.
- M. T. Uddin, Y. Nicolas, C. Olivier, T. Toupance, L. Servant, M. M. Müller, H.-J. Kleebe, J. Ziegler and W. Jaegermann, Inorg. Chem., 2012, 51, 7764–7773 CrossRef CAS PubMed.
- J. Yang, X. Zhang, H. Liu, C. Wang and Y. Liu, Catal. Today, 2013, 201, 195–202 CrossRef CAS.
- M. Covei, P. Dana, C. Bogatu and A. Duta, Catal. Today, 2019, 321, 172–177 CrossRef.
- T. A. Gadhi, S. Hernández, M. Castellino, P. Jagdale, A. Hernández-Gordillo, T. Alberto and N. Russo, Catal. Today, 2019, 321, 135–145 CrossRef.
- M. Huang, J. Yu, B. Li, C. Deng, L. Wang, W. Wu, L. Dong, F. Zhang and M. Fan, J. Alloys Compd., 2015, 629, 55–61 CrossRef CAS.
- M. A. Behnajady and Y. Tohidi, Synthesis, Photochem. Photobiol., 2014, 90, 51–56 CrossRef CAS PubMed.
- M. Manikandan, T. Tanabe, P. Li, S. Ueda, G. V. Ramesh, R. Kodiyath, J. Wang, T. Hara, A. Dakshanamoorthy, S. Ishihara, K. Ariga, J. Ye, N. Umezawa and H. Abe, ACS Appl. Mater. Interfaces, 2014, 6, 3790–3793 CrossRef CAS PubMed.
- Y. He, D. Li, J. Chen, Y. Shao, J. Xian, X. Zheng and P. Wang, RSC Adv., 2014, 4, 1266 RSC.
- G. Chen, S. Ji, Y. Sang, S. Chang, Y. Wang, H. Pin, J. Claverie, H. Liu and G. Yu, Nanoscale, 2015, 7, 3117–3125 RSC.
- S. D. Balgude, Y. A. Sethi, B. B. Kale, N. R. Munirathnam, D. P. Amalnerkar and P. V. Adhyapak, RSC Adv., 2016, 6, 95663–95669 RSC.
- K. L. Foo, U. Hashim, K. Muhammad and C. H. Voon, Nanoscale Res. Lett., 2014, 9, 429 CrossRef PubMed.
- S. S. Warule, N. S. Chaudhari, B. B. Kale and M. A. More, CrystEngComm, 2009, 11, 2776–2783 RSC.
- P. V. Adhyapak, S. P. Meshram, D. P. Amalnerkar and I. S. Mulla, Ceram. Int., 2014, 40, 1951–1959 CrossRef CAS.
- P. Senthil Kumar, M. Selvakumar, S. Ganesh Babu, S. Induja and S. Karuthapandian, J. Alloys Compd., 2012, 701, 562–573 CrossRef.
- P. V. Adhyapak, S. P. Meshram, A. A. Pawar, D. P. Amalnerkar, U. P. Mulik and I. S. Mulla, Ceram. Int., 2014, 40, 12105–12115 CrossRef CAS.
- L. Xu, Y. Zhou, Z. Wu, G. Zheng, J. He and Y. Zhou, J. Phys. Chem. Solids, 2017, 106, 29–36 CrossRef CAS.
- X. Yu, L. Wang, J. Zhang, W. Guo, Z. Zhao, Y. Qin, X. Mou, A. Li and H. Liu, J. Mater. Chem. A, 2015, 3, 19129–19136 RSC.
- W. Yu, J. Chen, T. Shang, L. Chen, L. Gu and T. Peng, Appl. Catal., B, 2017, 15, 693–704 CrossRef.
- W. Yu, D. Xu and T. Peng, J. Mater. Chem. A, 2015, 3, 19936–19947 RSC.
- S. Balgude, Y. Sethi, B. Kale, D. Amalnerkar and P. Adhyapak, Mater. Chem. Phys., 2019, 221, 493–500 CrossRef CAS.
- M. A. Mahadadalkar, S. W. Gosavi and B. B. Kale, J. Mater. Chem. A, 2018, 6, 16064–16073 RSC.
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Dinesh P. Amalnerkarc and
Parag V. Adhyapak
a,
Dinesh P. Amalnerkarc and
Parag V. Adhyapak