
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComparative study of hydroxyapatite, fluor-hydroxyapatite and Si-substituted hydroxyapatite nanoparticles on osteogenic, osteoclastic and antibacterial ability
Jing Suna,
Tao Wua,
Qihang Fana,
Qing Hua and
Bin Shi *ab
*ab
aThe State Key Laboratory Breeding Base of Basic Science of Stomatology (Hubei-MOST), Key Laboratory of Oral Biomedicine Ministry of Education, School & Hospital of Stomatology, Wuhan University, Wuhan, 430079, CN, China. E-mail: shibin_dentist@whu.edu.cn
bDepartment of Dental Implantology, School and Hospital of Stomatology, Wuhan University, China
First published on 22nd May 2019
Abstract
This study compared the effects of hydroxyapatite (HA), fluor-hydroxyapatite (FHA) and Si-substituted hydroxyapatite (SiHA) on osteogenic differentiation, osteoclastic activity and antibacterial properties. HA, FHA and SiHA were prepared via a sol–gel reaction and characterized by scanning electron microscopic analysis (SEM), transmission electron microscopic analysis (TEM), and X-ray photoelectron spectrometry. Cell proliferation was evaluated using an MTT assay and cytoskeletal morphology was observed by fluorescence microscopy. Osteogenic differentiation was evaluated using alkaline phosphatase activity and Alizarin red staining. Quantitative real-time PCR was used to evaluate the mRNA expression of runt-related transcription factor 2 (Runx2) and osteopontin (OPN). New bone formation was tested using μCT, haematoxylin and eosin staining and TRAP staining. The antibacterial actions against Porphyromonas gingivalis (P. g) were evaluated through plate counting and live-dead bacterial staining. The results demonstrated that HA, FHA and SiHA can promote proliferation of bone mesenchymal stem cells (BMSCs). ALP activity in FHA extract with a concentration of 625 μg mL−1 was the highest after 14 days osteogenic induction; similar results were observed for Runx2 and OPN mRNA expression. HA, FHA and SiHA decreased trabecular space in bone defects, but FHA reduced osteoclastic activity and inhibited P. g growth. In conclusion, FHA can promote osteogenic activity, reduce osteoclastic activity and enhance antibacterial effects.
Introduction
There are nearly 2.2 million patients worldwide who are going to receive bone grafting to repair bone defects resulting from trauma, infection, tumour resection or osteoradionecrosis before undergoing oral implant surgery.1 Bone augmentation is a surgical procedure that involves the repair of defective bone with a biological or synthetic bone graft. However, the source of the autologous bone graft is limited and necessitates secondary surgical incision. The development of synthetic bone substitutes is considered as being important in overcoming the intrinsic limitations of biological bone grafts.Hydroxyapatite [HA, Ca10(PO4)6(OH)2] is one of the most important inorganic minerals in biological hard tissue and nearly 60–70% of bone tissue is composed of HA.2,3 HA is also considered to be a suitable synthetic bone substitute or implant coating material owing to its biocompatibility and osteoconductivity.4–6 However, there remain several disadvantages of HA as a bone substitute. The mismatch of HA degradation rate and the new bone formation rate imposes restrictions on its' use.7 Meanwhile, as an implant coating material, the relatively high solubility results in the detachment of the coating layer, leading to implantation failure.8–11 However, HA in natural bone can be combined with other ions such as Mg2+, Zn2+, Sr2+, Ag+, Si4+ and F−12,13 to promote bone remodelling and tissue regeneration, impart magnetic properties, add antibacterial activity, enhance bio-integration, etc.
Fluoridated hydroxyapatite [FHA, Ca10(PO4)6(OH)2−xFx], is an inorganic substance in which a fluorine ion (F−) replaces the hydroxyl group (OH)in HA. Fluorine (F) is an important constituent of bone and teeth. An appropriate concentration of F (a fluoridation degree of 0.8–1.1 (the x value in Ca10(PO4)6(OH)2−xFx)) can promote the proliferation of the osteoblasts14–17 and accelerate new bone formation.7,18 Fluoride can promote osteoblast activity, which may be related to the Ras-Raf-MAPK mitogenic signal transduction pathway19,20 and the Wnt/β-catenin signalling pathway.21 F can also inhibit bacterial growth and protect teeth from caries.22,23 In addition, FHA possesses better chemistry and heat stability, better mechanical strength, better biocompatibility and a longer biodegradation period23–26 compared to HA.
Silicon-substituted hydroxyapatite [SiHA, Ca10(PO4)6−x(SiO4)x(OH)2] is an inorganic material with silicate (SiO4) substituting for phosphate (PO4) in HA. Silicon (Si) is another essential trace element involved in bone formation and calcification.27–29 Si4+ can promote calcification along with Ca2+ and enforce the attachment, proliferation and osteogenic differentiation of bone mesenchymal stem cells (BMSCs).27,30,31 Moreover, SiHA shows better osteoconductivity and osteoblast proliferation abilities than HA.32,33 It has been reported that osteoblasts on 0.8 wt% SiHA have the best bioactivity.34,35 With its structure a faster biodegradation rate, which matches the natural bone formation rate better than HA.34
Even though many studies have reported the physicochemical properties and biological characteristics of HA, FHA and SiHA, no studies to date have compared the effects of the three materials on bone formation in vitro and in vivo with reference to their use as a bone substitute. Therefore, the aim of this study was to systematically investigate the effects of HA, FHA and SiHA nanoparticles on cell proliferation, bone formation or resorption and antibacterial abilities in vitro and in vivo.
Material and methods
HA preparation
HA nanoparticles were prepared via sol–gel method by dropping an (NH4)2HPO4 aqueous solution into a stirred Ca(NO3)2 aqueous solution at 75 °C for 2 h and pH value was maintained at 11–12 by adding ammonium hydroxide solution. The completely mixed reactants were suspended for 24 h at room temperature (RT) and then was washed, filtered and dried overnight at 60 °C. The prepared HA nanoparticles were sintered at 1100 °C in order to improve their crystallinity.36FHA preparation
FHA nanoparticles were prepared through sol–gel method in a solution of Ca(NO3)2·4H2O and (NH4)2HPO4 and NH4F at 75 °C for 2 h and pH value was maintained at 10–11. The definitive reactants were suspended for 72 h at RT, then washed, filtered and repeated the above steps twice and finally dried overnight at 60 °C. The as-prepared FHA nanoparticles were heated at 1100 °C to improve their crystallinity.37–39SiHA preparation
SiHA nanoparticles were prepared via sol–gel method in a solution of Ca(NO3)2·4H2O, (NH4)2HPO4 and tetraethyl orthosilicate (TEOS) at 75 °C. The pH was also maintained at 10–11 as above. The processes of suspension, lavation and drying were the same with that of HA except that the temperature for sintering SiHA nanoparticles were set at 1200 °C for 2 h.35Characterization and mechanical test
In vitro cell culture work
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rcf at 4 °C for 10 min. Afterwards, the ALP activity and total amount of protein were determined by p-nitrophenyl phosphate method.41
000 rcf at 4 °C for 10 min. Afterwards, the ALP activity and total amount of protein were determined by p-nitrophenyl phosphate method.41| Primer | Sequence 5′–3′ |
|---|---|
| OPN | Forward: ACGAATCTCACCATTCCGAT |
| Reverse: AGGTCCTCATCTGTGGCATC | |
| Runx2 | Forward: ATCCAGCCACCTTCACTTACACC |
| Reverse: GGGACCATTGGGAACTGATAGG | |
| GAPDH | Forward: AGAAGGTGGTGAAGCAGGCGG |
| Reverse: ATCCTTGCTGGGCTGGGTGG |
In vivo study
000 IU mL−1) as an antibiotic prophylaxis for three days. Animals were sacrificed randomly at 4 and 8 weeks after material insertion operation, respectively.
 | ||
| Fig. 1 A 2.5 mm-diameter latero-lateral biocortical channel beneath the growth palate (black arrow) and perpendicular to the shaft axis on rat (a–c). | ||
Result
Characterization of HA, FHA and SiHA
SEM and TEM results illustrated that three kinds of nanoparticles were rod shaped but with different size. HA nanoparticles were 200 nm in length (Fig. 2a and d), while FHA nanoparticles were 700–800 nm in length (Fig. 2b and e) and SiHA nanoparticles were 400–500 nm in length (Fig. 2c and f). | ||
| Fig. 2 SEM images of HA (a), FHA (b) and SiHA (c); TEM images of HA (d), FHA (e) and SiHA (f). | ||
XRD analysis showed typical apatite peaks in all patterns (Fig. 3a–c). The peak intensity of FHA was a little greater than that of HA, and the peak width was a little narrower, indicating better crystallization and higher purity; the peak intensity of SiHA was a little less than that of HA, illustrating weaker crystallization. The results suggested that the adding of F ions in HA would form a more ordered structure with enhanced chemical and thermal stability and adding Si ions in HA nanoparticles was in opposite term.
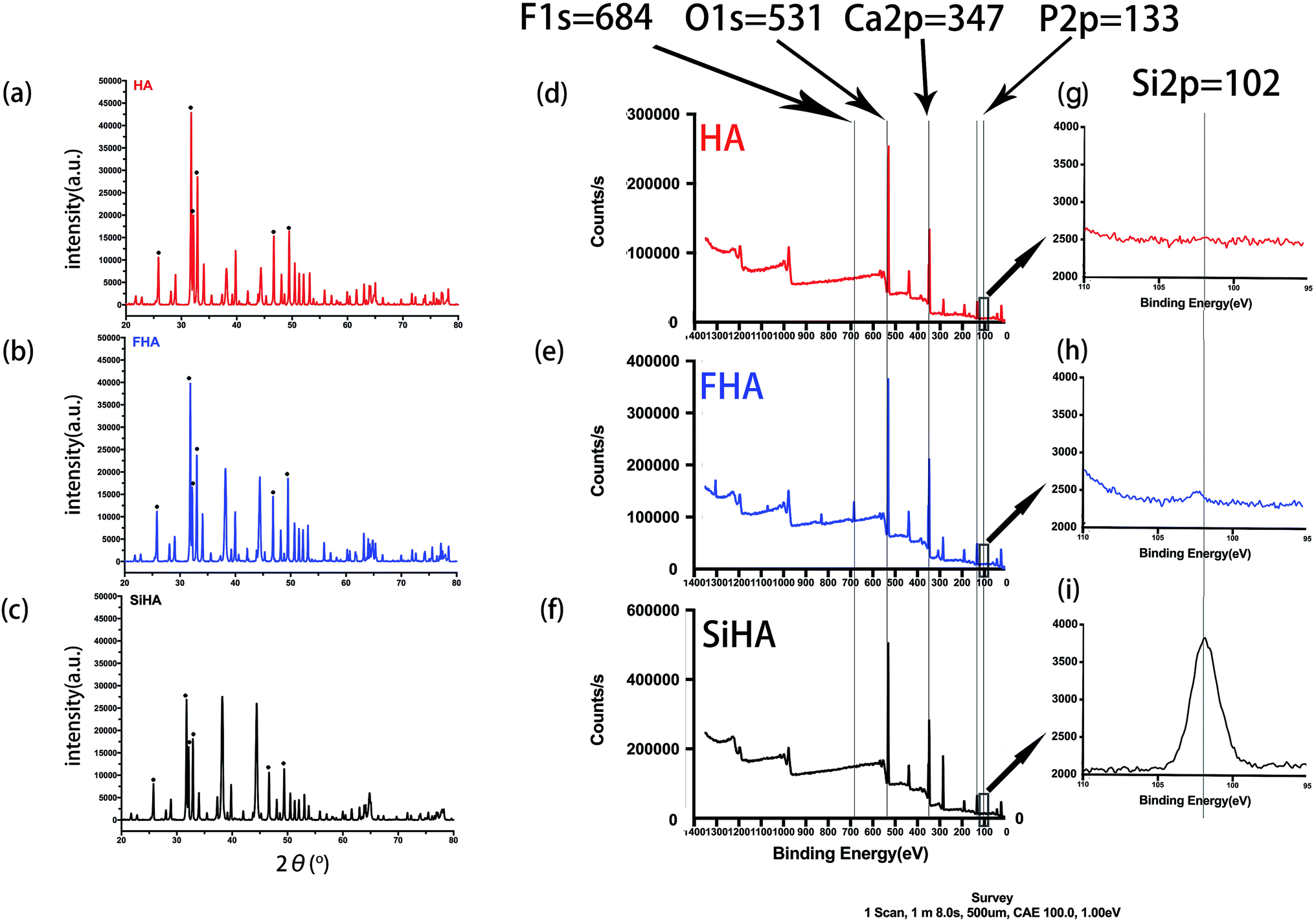 | ||
| Fig. 3 XRD patterns of sintered HA (a), FHA (b) and SiHA (c) nanoparticles; XPS spectra of HA (d), FHA (e) and SiHA (f); the curve which referred to Si 2p between 105 eV to 110 eV of HA (g), FHA (h) and SiHA (i). | ||
Fig. 3d–f shows the XPS spectra of HA, FHA and SiHA. For FHA, an F 1s peak was obvious in the wide scan at ∼684 eV, which is the fingerprint for fluorine in the FHA structure.44 An Si 2p peak was evident in the narrow scan at ∼102 eV, which is the fingerprint for silicon in the SiHA structure. Moreover, the contents of Ca, F, Si and P in the materials were determined by the ratio of the area under the respective elemental peak in the XPS narrow scan spectrum; the results are presented in Table 2. As demonstrated in Table 2, the measured Ca/P ratios of HA, FHA and SiHA were 1.66, 1.67 and 1.66, respectively; close to the stoichiometric value of 1.67. This indicates that HA with high purity and FHA with a fluoridation degree of 1.1 were formed. The wt% of Si in SiHA was 0.77 wt%, which is close to the theoretical weight of SiHA with 0.8wt% of Si.45
| Sample | Content atomic% | Atomic ration Ca/(P + Si) | Formula | |||
|---|---|---|---|---|---|---|
| Ca | P | F | Si | |||
| HA | 22.18 | 13.46 | — | — | 1.66 | Ca10(PO4)6(OH)2 |
| FHA | 21.44 | 12.84 | 5.28 | — | 1.67 | Ca10(PO4)6(OH)0.9F1.1 |
| SiHA | 21.12 | 12.10 | — | 0.58 | 1.66 | Ca10(PO4)5.724(SiO4)0.276(OH)2 |
The concentration of F ions and Si ions in extracts used in our experiment was illustrated in Table 3. The concentration of F ions in extracts with 625 μg mL−1 FHA was 0.3897 mg L−1. And the concentration of Si ions in extract with 625 μg mL−1 SiHA was 0.8235 mg L−1.
| Sample | Ion concentration (mg L−1) | |
|---|---|---|
| F | Si | |
| HA | — | — |
| FHA | 0.3897 | — |
| SiHA | — | 0.8235 |
Cell proliferation
A significant increase in the growth of BMSCs was observed in HA, FHA and SiHA material extracts. All samples showed similar proliferation patterns within 10 days of culture, except for the blank group which had limited proliferation from day 5 (p < 0.05) (Fig. 4a and b). Cells in the SiHA group exhibited relatively lower viability than the HA and FHA groups. Interestingly, a similar growth trend was observed in HA, FHA and SiHA group from day 0 to 7. After 7 days, the curve in the FHA group climbed constantly, the proliferation trend in the HA group ceased and even slightly declined (p > 0.05).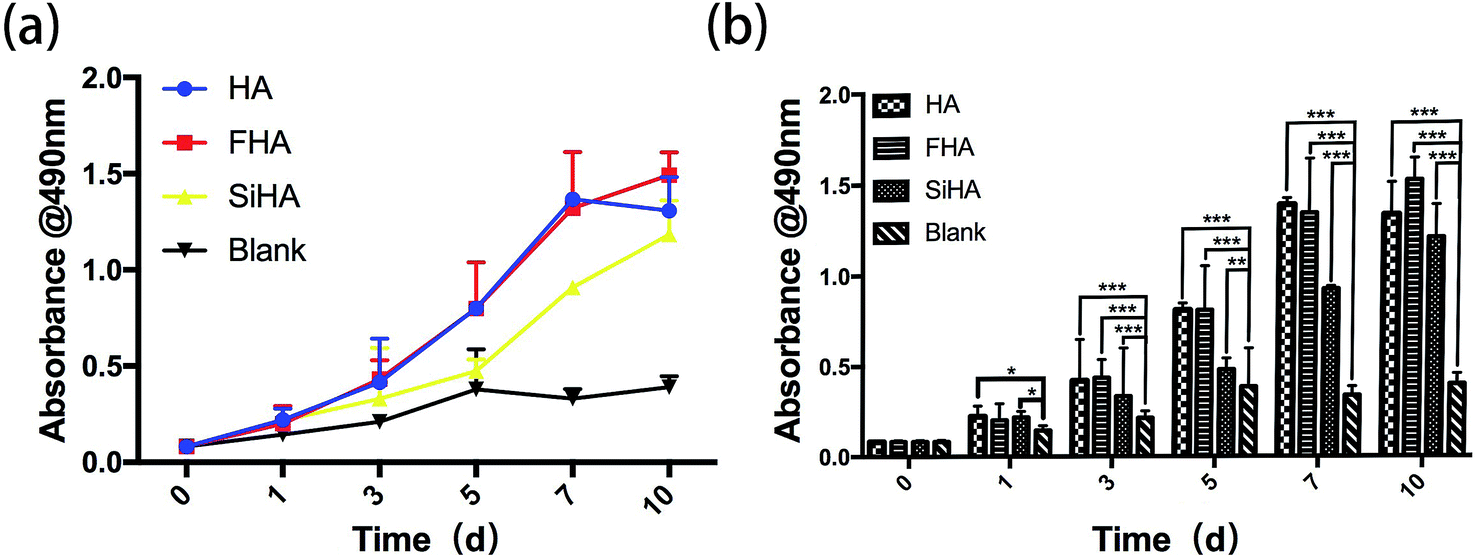 | ||
| Fig. 4 MTT assay: the cell proliferation image of BMSCs cultured in extracts of HA, FHA, SiHA and α-MEM culture medium from day 1 to day 9 (a and b) (n = 4 in each group; *p < 0.05). | ||
Cell morphology
Significant differences in the fluorescent microscopy photos were observed among the four groups (Fig. 5). On the one hand, the number of BMSCs in all groups increased over time. On the other hand, the morphology of the BMSCs changed differently during cell spreading and nuclear flattening. On day 1, there were no significant differences in the morphology and number of cells among the groups. On day 4, the number of cells was increased in all groups and the cell density in the SiHA group was higher than the other three groups. With respect to morphology, there was no obvious changes in the BMSCs in the blank group; BMSCs in this group had spread the most and tended to be spindle shaped and cells in the other three groups presented a clear outline and stereoscopic nucleolus. At day 7, there was a continuous increase in the cell density of all groups; cell density in the SiHA group was still the highest. Cell morphology in the SiHA and HA groups tended to be more elongated, while cells in the FHA group were smaller in size with an irregular shape; these cells appeared to have begun to shrink.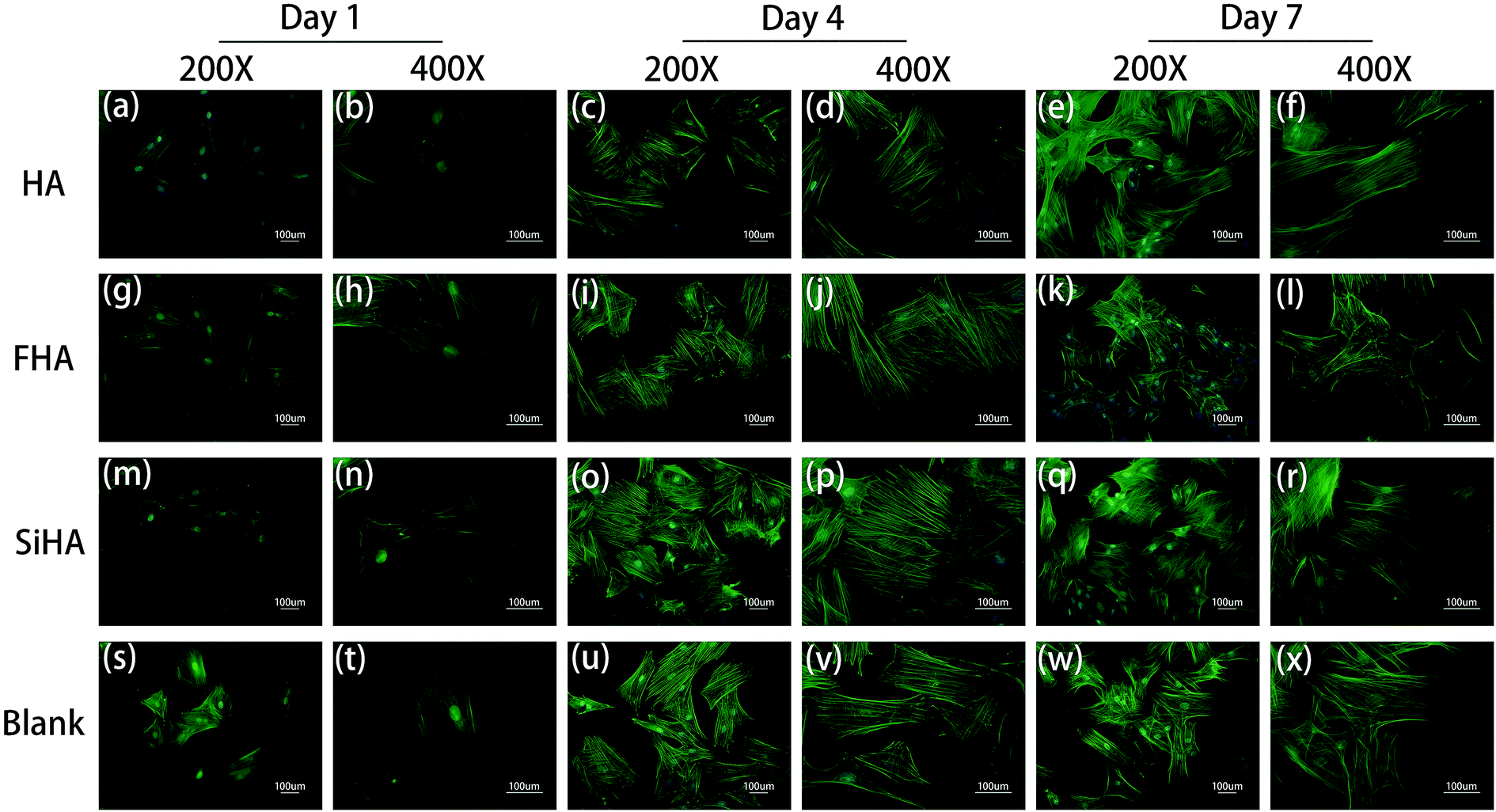 | ||
| Fig. 5 Cytoskeletal morphology of BMSCs cultured in HA extracts for 1 day (a and b), 4 days (c and d) and 7 days (e and f); cytoskeletal morphology of BMSCs cultured in FHA extracts for 1 day (g and h), 4 days (i and j) and 7 days (k and l); cytoskeletal morphology of BMSCs cultured in SiHA extracts for 1 day (m and n), 4 days (o and p) and 7 days (q and r); cytoskeletal morphology of BMSCs cultured in α-MEM culture medium for 1 day (s and t), 4 days (u and v) and 7 days (w and x). | ||
Osteogenic differentiation
Osteogenic differentiation ability was assessed by ALP activity assay and Alizarin red staining. In the ALP activity assay, there were no significant differences in BMSCs on day 7 among all four groups (p > 0.05). There were significantly higher levels of ALP activity in the HA, FHA and SiHA groups compared to the blank group (p < 0.05) on day 14; there were no significant differences among the HA, FHA and SiHA groups (p > 0.05). To further analyse cell mineralization, Alizarin red staining was performed after 7 day and 14 day osteogenic induction (Fig. 6a–h). A few mineralized nodules were formed in the HA, FHA and SiHA groups; no obvious mineralized nodules were noticed in the blank group at day 7. At day 14, obvious mineralized nodules were observed in all groups under a microscope, but the area of mineralization nodules in the FHA group was larger than that of the HA and SiHA groups; the blank group ranked last at the macro level (Fig. 6e–h). The results of Alizarin red staining were in accordance with those of the ALP activity assay.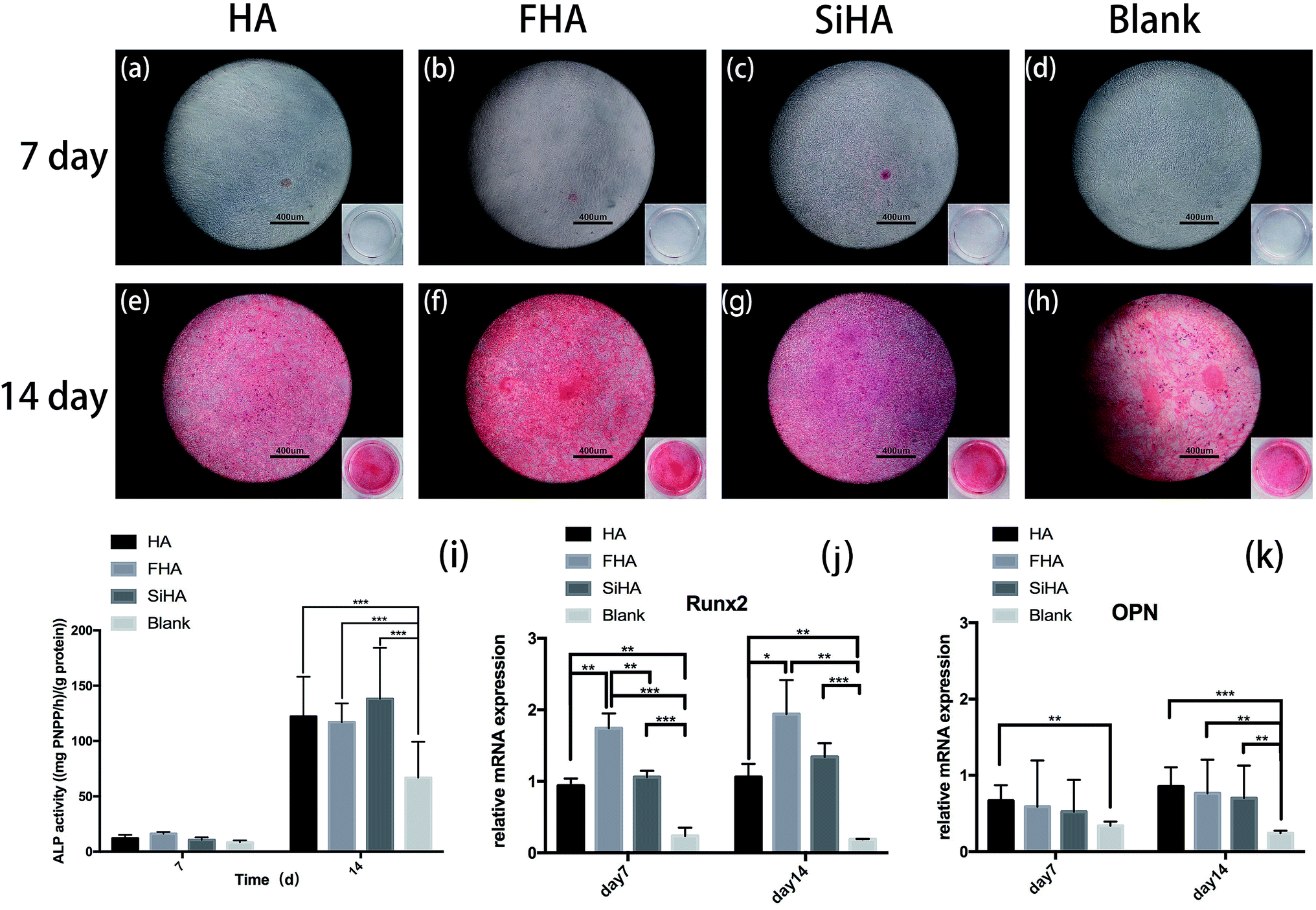 | ||
| Fig. 6 Alizarin red staining of BMSCs cultured in HA, FHA, SiHA extracts and α-MEM culture medium under osteogenic induction for 7 days (a–d) and 14 days (e–h). Quantitative ALP activity of BMSCs cultured in HA, FHA, SiHA and α-MEM culture medium with mineralized solution for 7 days and 14 days (i) (n = 4 in each group; *p < 0.05). Real-time PCR analysis of mRNA Runx2 and OPN in BMSCs cultured in HA, FHA, SiHA extracts and α-MEM culture medium under osteogenic induction for 7 days (j) and 14 days (k) (n = 4 per group; *p < 0.05). | ||
Runx2 is a key transcription factor that regulates bone development and maintenance of the extracellular matrix,46 and osteopontin (OPN) is an important marker in the later osteogenic differentiation period. mRNA expression of Runx2 and OPN is shown in Fig. 6j and k, as analysed by quantitative real-time PCR (RT-PCR). Runx2 mRNA expression was highest in the FHA group; there were significant differences between all groups (p < 0.01) except for the HA and SiHA groups at day 7 (Fig. 6j). Similar trends in Runx2 mRNA expression were observed at day 14; again, the FHA group exhibited the highest expression. However, mRNA expression of Runx2 increased faster in the SiHA group, resulting in no significant difference between the SiHA and FHA group (p > 0.05). OPN mRNA expression in the HA group was significantly higher than the blank group (p < 0.01) at day 7. At day 14, the blank group showed the lowest OPN mRNA expression; there were no statistically significant differences among the other three groups. In conclusion, the osteogenic differentiation results above indicate that the ability to promote osteogenesis can be ranked as follows: FHA > SiHA and HA > blank group.
In vivo study
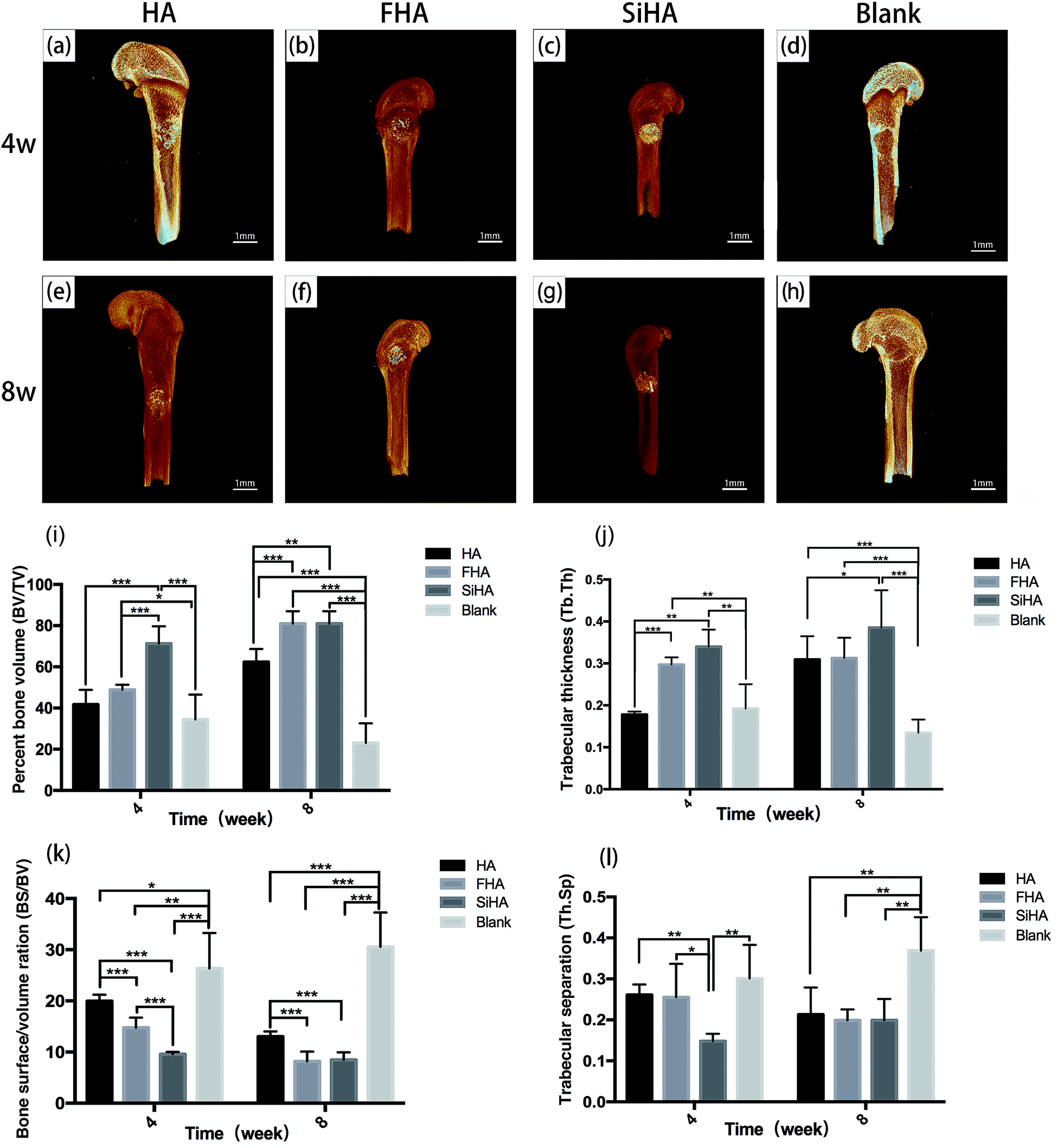 | ||
| Fig. 7 3D reconstruction μCT images of the femurs defects filled with HA (a and e), FHA (b and f), SiHA (c and g) and blank (d and h) 4 weeks and 8 weeks after surgery. Percent bone volume analysis (BV/TV) (i), trabecular thickness (Tb. Th) (j), bone surface/volume ration (BS/TV) (k) and trabecular separation (Tb. Ts) (l) of new bone formation in the defect areas filled with HA, FHA, SiHA for 4 weeks and 8 weeks (n = 4 per group; *p < 0.05). | ||
At week 4, the SiHA group exhibited the highest BV/TV and Tb. Th (p < 0.05), followed by the FHA and HA groups. There was no statistically significant difference in the BV/TV value between the FHA and HA groups, but the FHA group exhibited a better Tb. Th value than the HA group. The BS/BV value was ranked as follows: SiHA < FHA < HA < blank group (p < 0.05). The SiHA group exhibited the lowest Th. Sp value, but there were no statistically significant differences between the other three groups. At week 8, the BV/TV and Tb. Th values of the SiHA group were still the highest. BV/TV in the FHA group increased obviously and was significantly higher than the HA group (p < 0.05). The BS/BV value exhibited the opposite trend to the BV/TV value. For the Th. Sp value, all three bone substitute groups occupied a smaller space compared to the blank group (p < 0.05).
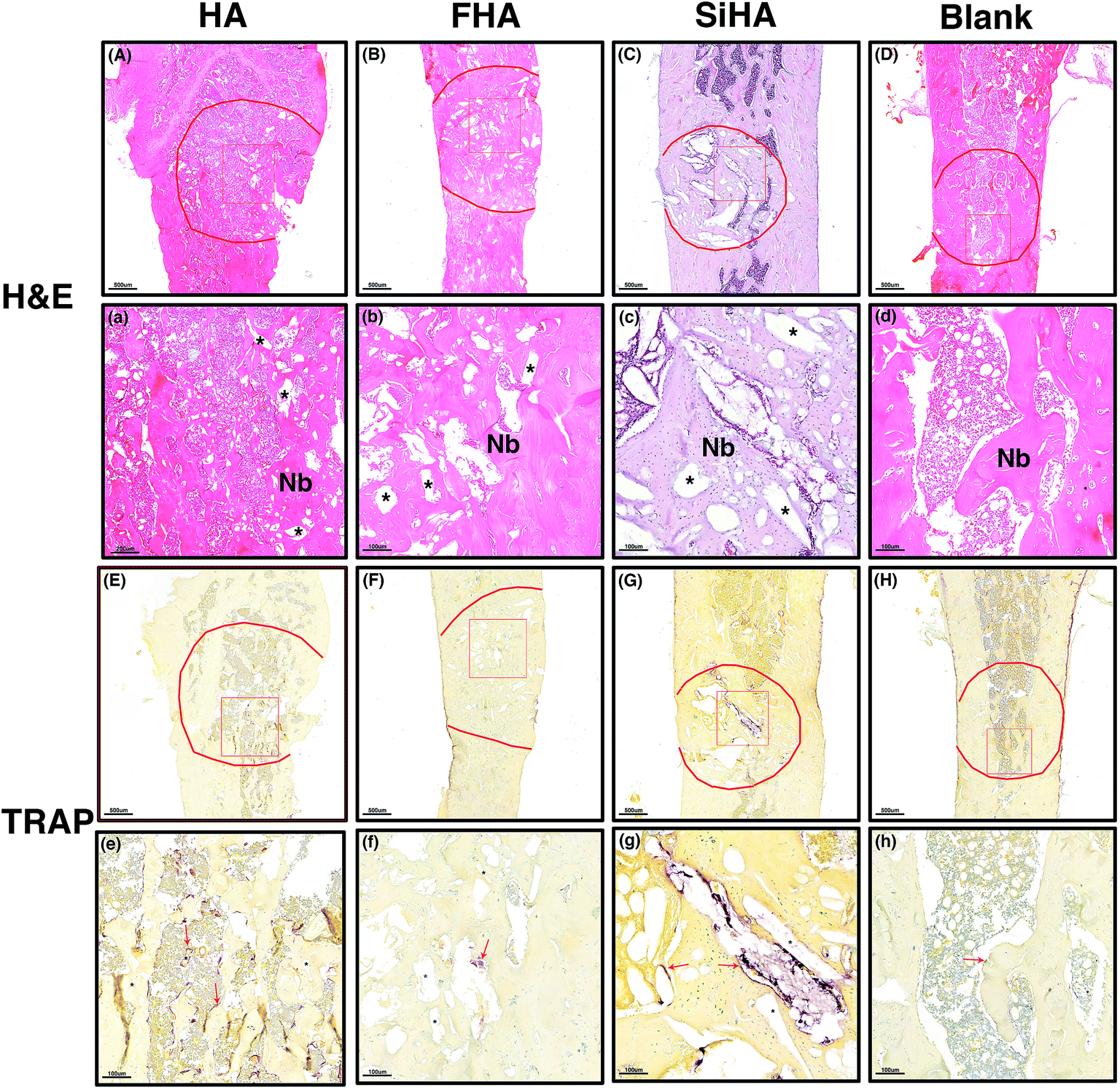 | ||
| Fig. 8 Hematoxylin- and eosin-stained histological morphology of the bone defects 8 weeks after filled with, HA (A and a), FHA (B and, b), SiHA (C and c) and blank (D and d). New bone (Nb) formation observed in all groups and the pore space (*) around Nb was no degraded material of HA, FHA, and SiHA. Trap staining of osteoclasts (red arrow) was observed around HA (E and e), FHA (F and f), SiHA (G and g) and blank (H and h), scale bar is 500 μm (A–H) and 100 μm (a–h). | ||
TRAP staining was used to study osteoclastic activity with osteoclasts in brownish-red staining. Most of the stained cells were distributed around the pore space and close to the material remnants; these had not degraded at that time point. The greatest number of stained osteoclasts were observed in the HA group (Fig. 8e) followed by the SiHA group (Fig. 8g). Less osteoclasts were observed in the other two groups (Fig. 8f and h).
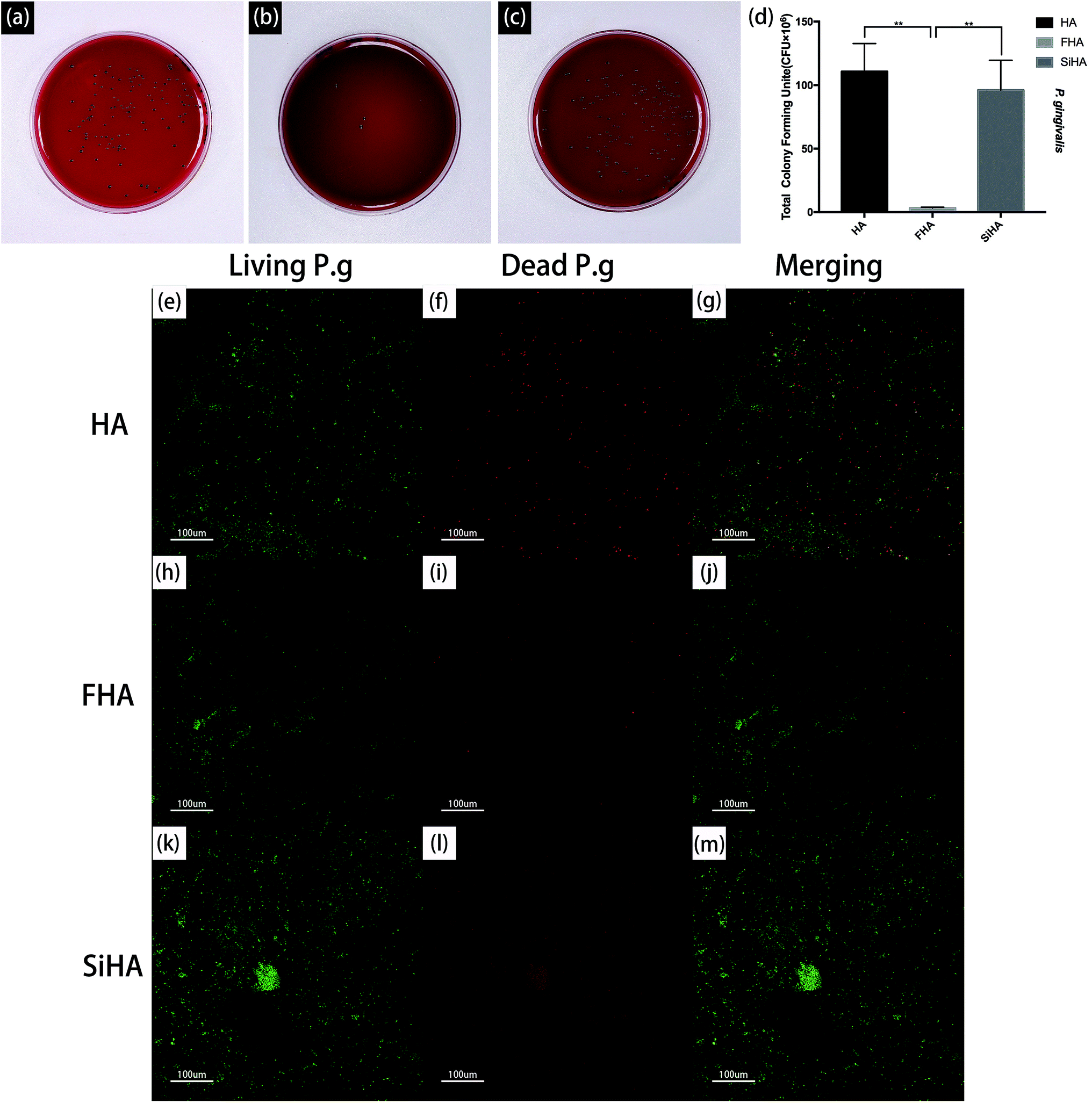 | ||
| Fig. 9 Photographs of Columbia blood agar media with colonies of P. g strains after incubation with HA (a), FHA (b) and SiHA (c) respectively and statistical results corresponding to the survivability of bacteria on three types of sample surfaces (d) n = 3 per group; *p < 0.05). Fluorescence microscopy images showing viability of the adherent P. gingivalis on the plates of three specimens respectively, as displayed by SYTO® 9 and PI dyes. HA (e, f and g), FHA (h, i and j) and SiHA plates (k, l and m). | ||
The viability of P. g on the three plates was also determined by fluorescence staining, as shown in Fig. 9e–m. After bacterial invasion for 48 hours, there were large amounts of viable bacteria (green fluorescence) on the HA plates and SiHA plates, and only small amounts of dead bacteria (red fluorescence) could be observed on the SiHA plates. In comparison, the amounts of living and dead bacteria on the FHA plates were both smaller than the other two kinds of nanoparticles.
Discussion
Bone defects remain a challenge due to limitations associated with the sources of autogenous bone grafts. A good bone substitute must possess several properties such as biocompatibility, bioactivity, osteoconduction, osteoinduction, non-toxicity, etc.47 To better understand the application of HA, FHA and SiHA nanoparticles, we compared the osteoclastic activity, antibacterial abilities and the in vitro and in vivo osteogenesis of the three materials. We fabricated HA, FHA and SiHA using the sol–gel method,48 and analysed by SEM, TEM, XRD and XPS assay. The SEM results showed that all three nanoparticles had rod-like particles with approximate lengths of 200 nm, 700 nm and 450 nm for HA, FHA and SiHA, respectively. According to previous studies, we fabricated HA and FHA nanoparticles with molar Ca/P ratios approaching a value of 1.67 and SiHA nanoparticles with a molar Ca/(P + Si) ratio approaching 1.67. Further, FHA with a fluoridation degree of 1.1 was formed. The wt% of Si in SiHA was 0.77 wt%, which is close to the theoretical weight of SiHA with 0.8 wt%.34,45 And the concentration of F ions in extract with FHA was 0.3897 mg L−1, which is accordance to the proper level (ranging from 0.1 to 100 μM) and can promote proliferation and differentiation of BMSCs.14,21 And the concentration of Si ions in extract with SiHA was 0.8235 mg L−1, which is also at a proper level (under 5 mM) to promote BMSCs.49These characteristics indicate the successful fabrication of the three kinds of nanoparticles.
Many studies have reported that HA,4–6 FHA14–17 and SiHA32 possess the potential to promote cell proliferation and differentiation. In this study, cell proliferation potential was in accordance with former studies and the in vitro osteogenic enhancing capability of the three nanoparticles was ranked as follows: FHA > SiHA > HA; SiHA nanoparticles demonstrated the best osteogenic capability in vivo.
HA has received substantial research attention as a classic bone substitute that can accelerate bone formation via increasing cell proliferation and differentiation. The literature suggests that nanoparticles 100–250 nm in diameter have better repairing ability.50 The mechanism mainly can be summarized as follows. First, HA influences the metabolism of osteoblasts. When HA nanoparticles are absorbed in bone tissue, the released Ca and P ions promote ALP activity and stimulate bone formation.51 Second, HA takes parts in bone vascularization, which is key in the process of bone repair.52 HA can enhance cell responsiveness to vascular endothelial growth factor (VEGF) via NOS activity, causing microvascular endothelium to differentiate into capillary-like structures53 quickly.
With regard to FHA, F ion doping is an effective way to improve the biological properties of HA. Fluoride at a proper concentration, ranging from 0.1 to 100 μM, can significantly enhance both osteoblast proliferation and differentiation.14,21,54,55 The mechanism by which fluoride promotes cell proliferation and osteoblast activity is highly complicated and has not yet been clearly clarified. Studies suggest that fluoride promotes osteoblast proliferation by enhancing the effect of growth factor-mediated stimulation of the Ras-Raf-MAPK mitogenic signal transduction pathway.19,20 Another study suggested that fluoride may influence the Wnt/β-catenin signalling pathway in osteoblasts, which is known to play an important role in regulating cell proliferation, differentiation and morphogenesis.56,57 Fluoride was reported to induce the phosphorylation of Akt at serine 473 and subsequently activate the phosphorylation of glycogen synthase kinase-3β (GSK-3β) at serine 9, leading to inhibition of GSK-3β activity and activation of Wnt/β-catenin signalling.21 However, it has been shown that small doses of F ions can promote the proliferation and differentiation of osteoblasts7,58 while large doses of F ions can inhibit osteoblast proliferation and differentiation and induce cell apoptosis58 because of cell metabolism imbalance.17 In this study, although FHA was found to promote cell proliferation, the cell morphology revealed that cells in FHA group at day 7 showed shrinking morphology may be related to the accumulation of F ions (Fig. 5k and l).
Si ion doping is another important way to modify the biological properties of HA nanoparticles. Si ions can stimulate collagen type I synthesis and osteoblastic differentiation both in vivo and in vitro.59,60 Research has revealed that Si ions can activate the MAPK-ERK pathway in osteoblast-like cells61 and Si ions can enhance cellular differentiation and collagen production via the TGF-β pathway by the binding of TGF-β1 to its cognate receptors.62 Further, Si ions can significantly upregulate AXIN2, β-catenin, SHH and patched-1 (PTCH1) gene expression.49 AXIN2 and β-catenin are a critical members of the Wnt/β-catenin signalling pathway;63–65 SHH is one of the ligands of the Hedgehog-Gli signalling pathway and PTCH1 is a target gene of the SHH-Gli signalling pathway.66 All of these signalling pathways are involved in cell proliferation and differentiation.56,57,67–69 Beyond that, orthosilicic acid (SiO44−), which exists in SiHA, could regulate the expression of miR-146a to suppress the activity of NF-κB; this would influence both osteoblast differentiation and activity.70–73 It should be noted that when the concentration of Si ions is higher than 5 mM, which is much larger than that of FHA (0.1 to 100 μM), it may suppress cell proliferation. This could be due to the cytotoxicity of high concentrations of Si ions or the change in the pH and extracellular ionic environment.49
Bone formation and resorption appears to be a dynamic process. Aside from osteoblasts, osteoclasts are also involved in bone regeneration, especially during the degradation of materials.74–76 With regard to FHA, fluoride with concentration lower than 8 mg L−1 will decrease the activity of new formed osteoclasts because but will not affect osteoclasts formation, involving in inhibiting NFATc1 and downstream functional genes, such as DC-STAMP, c-Src, Atp6v0d2, MMP9, and CK; this leads to impaired capacity for osteoclast acidification and reduced secretion of proteolytic enzymes.77 When the concentration of F ions is between 0.5–1.0 mM, the number of resorption lacunae made by individual osteoclasts and the resorbed area per osteoclast is decreased.78 The pH value is also important for osteoclasts. Osteoclasts can be activated with pH values from 6.7 to 7.0,79 but the activity of osteoclasts is suppressed when pH > 7.0.80 The releasing F ion will increase the pH, resulting in less activity of osteoclasts. According to our results in Fig. 8E–H and e–h, the number of osteoclasts in the FHA group was significantly less than the other two groups. Several studies have reported that Si can also inhibit the activity of osteoclasts. Osteoprotegerin (OPG)/receptor activator for nuclear factor-κB ligand (RANKL) ratio plays a critical role in pre-osteoclast maturation and osteoclast activation. In normal physiological conditions, RANKL combines with receptor activator for nuclear factor-κB (RANK) which distributes on the surface of pre-osteoclast to promote the differentiation and activation of osteoclasts. Silica stimulates OPG transcription with unchanged RANKL;81,82 thus, OPG competes with RANKL to bind to (pre)osteoclastic receptor RANK, negatively affecting pre-osteoclast maturation and osteoclast activation.82,83 Accordingly, the SiHA group exhibited less osteoclasts in TRAP staining compared to the HA group, but more osteoclasts than the FHA group (Fig. 8).
It is important for bone substitutes to possess good osteogenic potential, but it is also important to prevent infection in the bacteria-filled microenvironment. Many studies have illustrated that fluoride can prevent dental caries. Further, fluoride can also inhibit pathogenesis in periodontal disease. P. g, a Gram-negative anaerobic bacterium implicated as a major pathogen associated with peri-implantitis and periodontitis,84 was used in this experiment to evaluate the antibacterial ability. The results demonstrated that P. g was obviously inhibited on FHA plates compared to the other two materials. The contact angle of FHA will increased along with the amount of F ions adding; this is associated with increased hydrophobicity, which reduces the initial adhesion of bacteria.85 Further, F ion released from FHA acts in several ways to affect bacteria metabolism. For instance, it can act as an inhibitor of the glycolytic enzyme, enolase.85 Further, fluoride complexes are responsible for fluoride-induced inhibition of proton translocating F-ATPases by mimicking phosphate to form complexes with ADP at the reaction centres of enzymes. ATPase plays an important role in the maintenance of intracellular pH by pumping out protons; inhibition of this enzyme disrupts bacterial metabolism and aciduric capability.86,87 Beyond that, fluoride ions inhibit acid-producing bacteria like Fusobacterium nucleatum (F. n) and Treponemas, which are periodontal pathogens; this helps to maintain a normal acid–base balance necessary for proper osteogenesis. Fluoride ions are released when bacterial metabolism begins and pH diminishes. The release of F ions can inhibit bacteria growth, stabilize the microenvironment and resistant the inflammatory process.88
Conclusion
HA, FHA and SiHA nanoparticles could promote osteogenesis and FHA and SiHA did better than HA. FHA and SiHA could both promote BMSCs proliferation, but FHA showed a better performance in osteogenesis in vitro, while SiHA did better in vivo. FHA suppressed osteoclasts activity and presented significant antibacterial properties. In conclusion, FHA and SiHA both can be a potential bone substitutes. However, FHA might be an ideal material in bone regeneration especially in infectious bone defects.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was supported by the Fundamental Research Funds for the Central University of China (No. 2042018kf0108).Notes and references
- J. Van der Stok, E. M. M. Van Lieshout, Y. El-Massoudi, G. H. Van Kralingen and P. Patka, Acta Biomater., 2011, 7, 739–750 CrossRef CAS PubMed.
- S. V. Dorozhkin and M. Epple, Angew. Chem., Int. Ed. Engl., 2002, 33, 3130–3146 CrossRef.
- S. F. Hulbert, F. A. Young, R. S. Mathews, J. J. Klawitter, C. D. Talbert and F. H. Stelling, J. Biomed. Mater. Res., 1970, 4, 433–456 CrossRef CAS.
- W. Suchanek and M. Yoshimura, J. Mater. Res., 1998, 13, 94–117 CrossRef CAS.
- D. W. Hutmacher, J. T. Schantz, C. X. F. Lam, K. C. Tan and T. C. Lim, J. Tissue Eng. Regener. Med., 2007, 1, 245–260 CrossRef CAS.
- A. S. Brydone, D. Meek and S. Maclaine, Proc. Inst. Mech. Eng., Part H, 2010, 224, 1329–1343 CrossRef CAS.
- Y. Wang, S. Zhang, X. Zeng, L. L. Ma, W. Weng, W. Yan and M. Qian, Acta Biomater., 2007, 3, 191–197 CrossRef CAS PubMed.
- T. S. B. Narasaraju and D. E. Phebe, J. Mater. Sci., 1996, 31, 1–21 CrossRef CAS.
- G. L. Darimont, B. Gilbert and R. Cloots, Mater. Lett., 2004, 58, 71–73 CrossRef CAS.
- S. Overgaard, M. Lind, K. Josephsen, A. B. Maunsbach, C. Bünger and K. Søballe, J. Biomed. Mater. Res., 1998, 39, 141 CrossRef CAS PubMed.
- I. Baltag, K. Watanabe, H. Kusakari, N. Taguchi, O. Miyakawa, M. Kobayashi and N. Ito, J. Biomed. Mater. Res., Part B, 2000, 53, 76 CrossRef CAS.
- M. Šupová, Ceram. Int., 2015, 41, 9203–9231 CrossRef.
- G. Graziani, M. Bianchi, E. Sassoni, A. Russo and M. Marcacci, Mater. Sci. Eng., C, 2016, 74, 219–229 CrossRef PubMed.
- J. R. Farley, J. E. Wergedal and D. J. Baylink, Science, 1983, 222, 330 CrossRef CAS.
- M. Kassem, L. Mosekilde and E. F. Eriksen, Eur. J. Endocrinol., 1994, 130, 381–386 CAS.
- B. Y. Reed, J. E. Zerwekh, P. P. Antich and C. Y. Pak, J. Bone Miner. Res., 1993, 8, 19 CrossRef CAS.
- D. Burgener, J. P. Bonjour and C. P. D. Joseph, J. Bone Miner. Res., 1995, 10, 164–171 CrossRef CAS PubMed.
- K. A. Bhadang and K. A. Gross, Biomaterials, 2004, 25, 4935–4945 CrossRef CAS PubMed.
- K.-H. W. Lau and D. J. Baylink, J. Bone Miner. Res., 1998, 13, 1660–1667 CrossRef CAS PubMed.
- K.-H. W. Lau and D. J. Baylink, J. Bone Miner. Res., 2003, 18, 1897–1900 CrossRef CAS.
- L. Pan, X. Shi, S. Liu, X. Guo, M. Zhao, R. Cai and G. Sun, Toxicol. Lett., 2014, 225, 34–42 CrossRef CAS.
- R. E. Marquis, Can. J. Microbiol., 1995, 41, 955–964 CrossRef CAS PubMed.
- H.-W. Kim, Y.-M. Kong, Y.-H. Koh, H.-E. Kim, H.-M. Kim and J. S. Ko, J. Am. Ceram. Soc., 2003, 86, 2019–2026 CrossRef CAS.
- M. Okazaki and R. Z. Legeros, Adv. Dent. Res., 1996, 10, 252 CrossRef CAS PubMed.
- H. W. Kim, H. E. Kim and J. C. Knowles, Biomaterials, 2004, 25, 3351 CrossRef CAS.
- H. W. Kim, Y. M. Kong, C. J. Bae, Y. J. Noh and H. E. Kim, Biomaterials, 2004, 25, 2919 CrossRef CAS PubMed.
- E. M. Carlisle, Science, 1970, 167, 279–280 CrossRef CAS.
- E. M. Carlisle, Science, 1972, 178, 619–621 CrossRef CAS.
- E. M. Carlisle, J. Nutr., 1980, 110, 1046–1056 CrossRef CAS PubMed.
- M. López-Álvarez, E. L. Solla, P. González, J. Serra, B. León, A. P. Marques and R. L. Reis, J. Mater. Sci.: Mater. Med., 2009, 20, 1131 CrossRef PubMed.
- S. R. K. Meka, S. Kumar Verma, V. Agarwal and K. Chatterjee, ChemistrySelect, 2018, 3, 3762–3773 CrossRef CAS.
- N. Patel, S. M. Best, W. Bonfield, I. R. Gibson, K. A. Hing, E. Damien and P. A. Revell, J. Mater. Sci.: Mater. Med., 2002, 13, 1199–1206 CrossRef CAS.
- S. R. K. Meka, V. Agarwal and K. Chatterjee, Mater. Sci. Eng., C, 2019, 94, 565–579 CrossRef CAS PubMed.
- K. A. Hing, P. A. Revell, N. Smith and T. Buckland, Biomaterials, 2006, 27, 5014–5026 CrossRef CAS PubMed.
- M. Honda, K. Kikushima, Y. Kawanobe, T. Konishi, M. Mizumoto and M. Aizawa, J. Mater. Sci.: Mater. Med., 2012, 23, 2923–2932 CrossRef CAS PubMed.
- L. Yubao, J. D. Wijn, C. P. A. T. Klein, S. V. D. Meer and K. D. Groot, J. Mater. Sci.: Mater. Med., 1994, 5, 252–255 CrossRef CAS.
- Y. Chen and X. Miao, Biomaterials, 2005, 26, 1205–1210 CrossRef CAS PubMed.
- G. Penel, G. Leroy, C. Rey, B. Sombret, J. P. Huvenne and E. Bres, J. Mater. Sci.: Mater. Med., 1997, 8, 271 CrossRef CAS.
- L. M. Rodríguez-Lorenzo, J. N. Hart and K. A. Gross, Biomaterials, 2003, 24, 3777–3785 CrossRef.
- N. O. Grad, P. Emoke and I. A. Mironiuc, Clujul Med., 2014, 86, 65–68 Search PubMed.
- C. Wu, R. Miron, A. Sculean, S. Kaskel, T. Doert, R. Schulze and Y. Zhang, Biomaterials, 2011, 32, 7068–7078 CrossRef CAS PubMed.
- H. Jing, Z. Yu and L. Li, J. Biomed. Mater. Res., Part A, 2008, 87, 33–37 CrossRef.
- W. K. Son, J. H. Youk, T. S. Lee and W. H. Park, Macromol. Rapid Commun., 2004, 25, 1632–1637 CrossRef CAS.
- M. A. Stranick and M. J. Root, Colloids Surf., 1991, 55, 137–147 CrossRef CAS.
- A. Aminian, M. Solati-Hashjin, A. Samadikuchaksaraei, F. Bakhshi, F. Gorjipour, A. Farzadi, F. Moztarzadeh and M. Schmücker, Ceram. Int., 2011, 37, 1219–1229 CrossRef CAS.
- J. H. Jonason, G. Xiao, M. Zhang, L. Xing and D. Chen, J. Dent. Res., 2009, 88, 693–703 CrossRef CAS.
- G. Aaron and P. P. Weitzel, Sports Med. Arthrosc. Rev., 2007, 15, 158 CrossRef PubMed.
- M. Esnaashary, M. Fathi and M. Ahmadian, Int. J. Appl. Ceram. Technol., 2014, 11, 47–56 CrossRef CAS.
- P. Han, C. Wu and X. Yin, Biomater. Sci., 2013, 1, 379–392 RSC.
- W. Zhu, J. Xiao, D. Wang, J. Liu, J. Xiong, L. Liu, X. Zhang and Y. Zeng, Int. Orthop., 2009, 33, 567–571 CrossRef.
- B. Chang-Jun, K. Hae-Won, K. Young-Hag and K. Hyoun-Ee, J. Mater. Sci.: Mater. Med., 2006, 17, 517–521 CrossRef PubMed.
- K. Woo Seob and K. H. Koo, J. Korean Med. Sci., 2005, 20, 479–482 CrossRef PubMed.
- S. Pezzatini, L. Morbidelli, R. Solito, E. Paccagnini, E. Boanini, A. Bigi and M. Ziche, Bone, 2007, 41, 523–534 CrossRef CAS.
- J. E. Wergedal, K. H. Lau and D. J. Baylink, Clin. Orthop. Relat. Res., 1988, 233, 274–282 CAS.
- W. J. Qu, D. B. Zhong, P. F. Wu, J. F. Wang and H. Bo, J. Bone Miner. Metab., 2008, 26, 328–334 CrossRef CAS PubMed.
- Y. Huang, X. Jin, X. Zhang, H. Sun, J. Tu and T. Tang, Biomaterials, 2009, 30, 5041–5048 CrossRef CAS PubMed.
- B. Zhang and J. Ma, Protein Cell, 2010, 1, 898–906 CrossRef CAS PubMed.
- C. Y. C. Pak, J. E. Zerwekh and P. Antich, Trends Endocrinol. Metab., 1995, 6, 229–234 CrossRef CAS.
- F. H. Nielsen and R. Poellot, J. Trace Elem. Exp. Med., 2004, 17, 137–149 CrossRef CAS.
- D. M. Reffitt, N. Ogston, R. Jugdaohsingh, H. F. Cheung, B. A. Evans, R. P. Thompson, J. J. Powell and G. N. Hampson, Int. Rev. Gesamten Hydrobiol. Hydrogr., 2003, 70, 27–46 Search PubMed.
- M. Y. Shie, S. J. Ding and H. C. Chang, Acta Biomater., 2011, 7, 2604–2614 CrossRef CAS PubMed.
- J. Li, L. Wei, J. Sun and G. Guan, J. Biomater. Appl., 2013, 27, 595–604 CrossRef CAS PubMed.
- Y. Hsiao-Man Ivy, J. Boris, S. Tzong-Jen, L. Bo, C. Frank, P. J Edward, B. Walter and H. Wei, Development, 2005, 132, 1995 CrossRef PubMed.
- E.-h. Jho, T. Zhang, C. Domon, C.-K. Joo, J.-N. Freund and F. Costantini, Mol. Cell. Biol., 2002, 22, 1172–1183 CrossRef CAS PubMed.
- N. Barker, Methods Mol. Biol., 2008, 468, 5 CrossRef CAS PubMed.
- M. Varjosalo and J. Taipale, Genes Dev., 2008, 22, 2454–2472 CrossRef CAS PubMed.
- P. N. De Aza, Z. B. Luklinska, A. Martinez, M. R. Anseau, F. Guitian and S. De Aza, J. Microsc., 2000, 197, 60–67 CrossRef CAS PubMed.
- T. Yusuke, I. Satoshi, A. V. Kaneshiro, E. Shigeyuki, J. E. Frencken and F. R. Tay, Dent. Mater., 2006, 22, 647–652 CrossRef PubMed.
- S. V. Prykhozhij, PLoS One, 2010, 5, e13549 CrossRef PubMed.
- J. Chang, Z. Wang, E. Tang, Z. Fan, L. Mccauley, R. Franceschi, K. Guan, P. H. Krebsbach and C. Y. Wang, Nat. Med., 2009, 15, 682–689 CrossRef CAS PubMed.
- Y. Masato, F. Hidefumi, S. Masashi, K. Takenobu, D. Takahiro, T. Tetsu and J. Eijiro, J. Biol. Chem., 2009, 284, 35987 CrossRef.
- L. Yan, L. Aimin, S. Karen, Z. Hongying, M. S. Nanes and W. M Neale, J. Bone Miner. Res., 2007, 22, 646–655 CrossRef PubMed.
- X. Zhou, F. M. Moussa, S. Mankoci, P. Ustriyana, N. Zhang, S. Abdelmagid, J. Molenda, W. L. Murphy, F. F. Safadi and N. Sahai, Acta Biomater., 2016, 39, 192–202 CrossRef CAS PubMed.
- K. Dennis, S. K. Wasim, H. Behrooz and M. David, Curr. Stem Cell Res. Ther., 2013, 8, 185–191 CrossRef.
- Z. Gu, W. Hao, L. Li, Q. Wang and X. Yu, Biomed. Mater., 2012, 7, 065007 CrossRef PubMed.
- S. Wenisch, J.-P. Stahl, U. Horas, C. Heiss, O. Kilian, K. Trinkaus, A. Hild and R. Schnettler, J. Biomed. Mater. Res., Part A, 2003, 67, 713–718 CrossRef CAS PubMed.
- J. Pei, B. Li, Y. Gao, Y. Wei, L. Zhou, H. Yao, J. Wang and D. Sun, Environ. Toxicol., 2014, 29, 588–595 CrossRef CAS.
- A. Okuda, J. Kanehisa and J. N. M. Heersche, J. Bone Miner. Res., 1990, 5, S115–S120 CrossRef CAS PubMed.
- I. R. Orriss and T. R. Arnett, Methods Mol. Biol., 2012, 816, 103–117 CrossRef CAS PubMed.
- T. R. Arnett and D. W. Dempster, Endocrinology, 1986, 119, 119–124 CrossRef CAS.
- F. Maehira, Y. Iinuma, Y. Eguchi, I. Miyagi and S. Teruya, J. Bone Miner. Metab., 2008, 26, 446 CrossRef CAS PubMed.
- M. Wiens, X. Wang, H. C. Schröder, U. Kolb, U. Schloßmacher, H. Ushijima and W. E. G. Müller, Biomaterials, 2010, 31, 7716–7725 CrossRef CAS.
- H. C. Schröder, X. H. Wang, M. Wiens, B. Diehl-Seifert, K. Kropf, U. Schloßmacher and W. E. Müller, J. Cell. Biochem., 2012, 113, 3197–3206 CrossRef.
- S. S. Socransky and A. D. Haffajee, J. Periodontol., 1992, 63, 322–331 CrossRef CAS PubMed.
- Nurhaerani, K. Arita, Y. Shinonaga and M. Nishino, Dent. Mater. J., 2006, 25, 684 CrossRef CAS.
- M. Yoshinari, Y. Oda, T. Kato and K. Okuda, Biomaterials, 2001, 22, 2043 CrossRef CAS.
- M. F. Hayacibara, O. P. Rosa, H. Koo, S. A. Torres, B. Costa and J. A. Cury, J. Dent. Res., 2003, 82, 267–271 CrossRef CAS.
- W. D. Nordquist, O. Hajima, K. Yutaka, K. Kazunari, O. Toshimitsu and D. J. Krutchkoff, J. Oral Implantol., 2011, 37, 35–42 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2019 |