DOI:
10.1039/C9RA00965E
(Paper)
RSC Adv., 2019,
9, 7594-7600
Chemical and biological study of aplysiatoxin derivatives showing inhibition of potassium channel Kv1.5†
Received
5th February 2019
, Accepted 25th February 2019
First published on 6th March 2019
Abstract
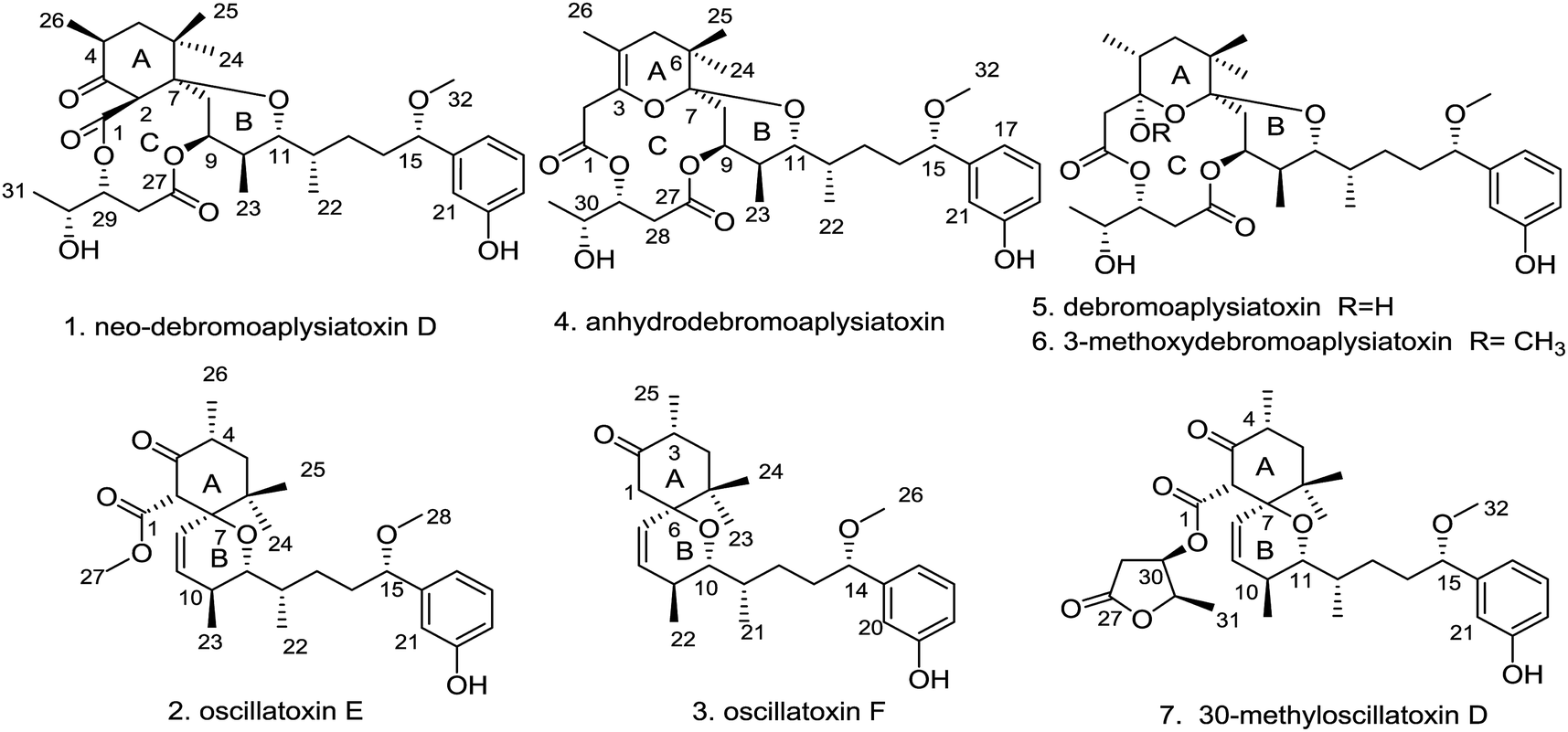
Three new aplysiatoxins, neo-debromoaplysiatoxin D (1), oscillatoxin E (2) and oscillatoxin F (3), accompanied by four known analogues (4–7), were identified from the marine cyanobacterium Lyngbya sp. Structural frames differ amongst these metabolites, and therefore we classified compounds 1 and 4–6 as aplysiatoxins as they possess 6/12/6 and 6/10/6 tricyclic ring systems featuring a macrolactone ring, and compounds 2, 3 and 7 as oscillatoxins that feature a hexane-tetrahydropyran in a spirobicyclic system. Bioactivity experiments showed that compounds 1 and 4–6 presented significant expression of phosphor-PKCδ whereas compounds 2, 5 and 7 showed the most potent blocking activity against potassium channel Kv1.5 with IC50 values of 0.79 ± 0.032 μM, 1.28 ± 0.080 μM and 1.47 ± 0.138 μM, respectively. Molecular docking analysis supplementing the binding interaction of oscillatoxin E (2) and oscillatoxin F (3) with Kv1.5 showed oscillatoxin E (2) with a strong binding affinity of −37.645 kcal mol−1 and oscillatoxin F (3) with a weaker affinity of −32.217 kcal mol−1, further supporting the experimental data.
Introduction
Cyanotoxins, otherwise known as poisonous metabolites yielded by cyanobacteria, can be classified into neurotoxins (anatoxin-a, β-methylamino alanine), hepatotoxins (microcystins, nodularins), cytotoxins (cylindrospermopsin) and dermatotoxins (lyngbyatoxin-a, aplysiatoxins) based on their mode of action.1 The aplysiatoxins (ATXs) are a class of biologically active dermatotoxins with anti-proliferative activity, tumour-promoting properties, proinflammatory actions and antiviral activity.2–6 Initially, aplysiatoxin and debromoaplysiatoxin were obtained from the sea hare Stylocheilus longicauda, whilst further research indicated that these compounds are metabolized by cyanobacteria.5,7 27 ATXs have been isolated from marine cyanobacteria so far.5–14 According to their structural characteristics, the early isolated ATXs were classified into three categories: aplysiatoxins possessing 6/12/6 tricyclic ring systems featuring a macrolactone ring (ABC ring) (e.g. aplysiatoxin, debromoaplysiatoxin, manauealides A–C and oscillatoxin A); oscillatoxins featuring a hexane-tetrahydropyran of a spirobicyclic system (AB ring) (oscillatoxin D and 30-methyloscillatoxin D); and nhatrangins featuring an opening chain (nhatrangins A and B). The main structural skeleton of ATXs (tricyclic ring systems) vary greatly, whilst their side chains, that contain aromatic rings, often remain unchanged. Our research group recently isolated two new ATXs that display rare carbon skeletons; neo-debromoaplysiatoxin A showing a 6/10/6 fused-ring system which we grouped as an aplysiatoxin and neo-debromoaplysiatoxin B with a 6/6/6 fused ring system as an oscillatoxin. In addition to this structural novelty, these compounds also exhibit excellent bioactivity with potent blocking action against potassium channel Kv1.5.14 Kv1.5 has been presumed to be a pivotal target for new treatment of atrial tachyarrhythmias with minimal side effects.15,16 In pursuit of additional novel Kv1.5 inhibitors, our group isolated three new aplysiatoxins, neo-debromoaplysiatoxin D (1) and oscillatoxin E and F (2 and 3), and four known aplysiatoxins (4–7) from the cyanobacterium Lyngbya sp. extracted from the South China Sea (Fig. 1). Due to their structural characteristics, compounds 1 and 4–6 can be classified as aplysiatoxins, with their corresponding ABC ring, and 2, 3 and 7 as oscillatoxins. This report details the separation, structural elucidation and biological activity of these metabolites. Herein, we demonstrate that compounds 1 and 4–6 expressed phosphor-PKCδ dramatically, but our oscillatoxins, 2, 3 and 7, lacked any activity. Importantly, selective blocking of Kv1.5 was seen to be significant for 2, 5 and 7 with IC50 values of 0.79 ± 0.032 μM, 1.28 ± 0.080 μM and 1.47 ± 0.138 μM, respectively.
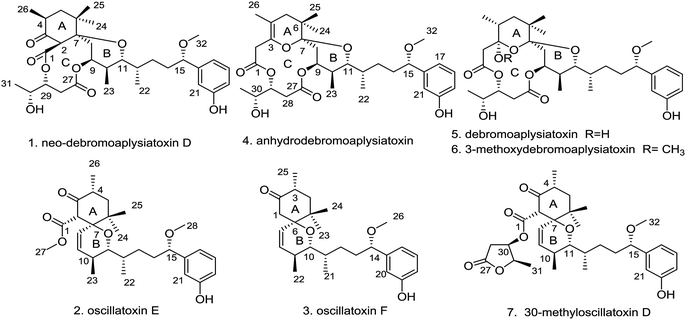 |
| | Fig. 1 Chemical structures of compounds 1–7. | |
Results and discussion
The cyanobacterium Lyngbya sp. was collected in the South China Sea and extracted with MeOH and CH2Cl2 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v). The extract was suspended in aqueous MeOH and extracted with petroleum ether, dichloromethane and ethyl acetate. To identify the main constituents of the extracts preliminary, a 1H-NMR method was utilized to the extracts and revealed the enrichment of aplysiatoxins in the dichloromethane extract. Sequential chromatography of dichloromethane extract on vacuum liquid chromatography (VLC) and octadecyl silica gel (ODS), followed by C18 reverse-phase HPLC, led to the isolation of seven aplysiatoxin derivatives including three new compounds, neo-debromoaplysiatoxin D (1), oscillatoxin E (2) and oscillatoxin F (3), and four known compounds (4–7). These four known structures were identified as anhydrodebromoaplysiatoxin (4), debromoaplysiatoxin (5), 3-methoxydebromoaplysiatoxin (6) and 30-methyloscillatoxin D (7) by comparing their spectroscopic data with the literature data.6,10,11
:1, v/v). The extract was suspended in aqueous MeOH and extracted with petroleum ether, dichloromethane and ethyl acetate. To identify the main constituents of the extracts preliminary, a 1H-NMR method was utilized to the extracts and revealed the enrichment of aplysiatoxins in the dichloromethane extract. Sequential chromatography of dichloromethane extract on vacuum liquid chromatography (VLC) and octadecyl silica gel (ODS), followed by C18 reverse-phase HPLC, led to the isolation of seven aplysiatoxin derivatives including three new compounds, neo-debromoaplysiatoxin D (1), oscillatoxin E (2) and oscillatoxin F (3), and four known compounds (4–7). These four known structures were identified as anhydrodebromoaplysiatoxin (4), debromoaplysiatoxin (5), 3-methoxydebromoaplysiatoxin (6) and 30-methyloscillatoxin D (7) by comparing their spectroscopic data with the literature data.6,10,11
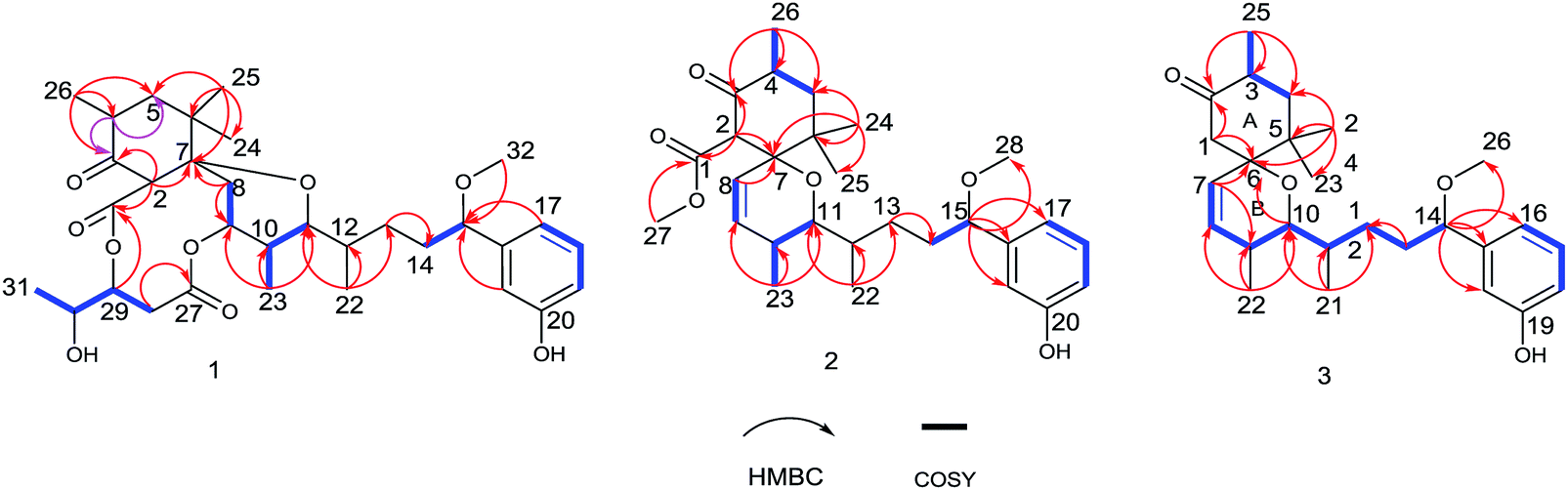
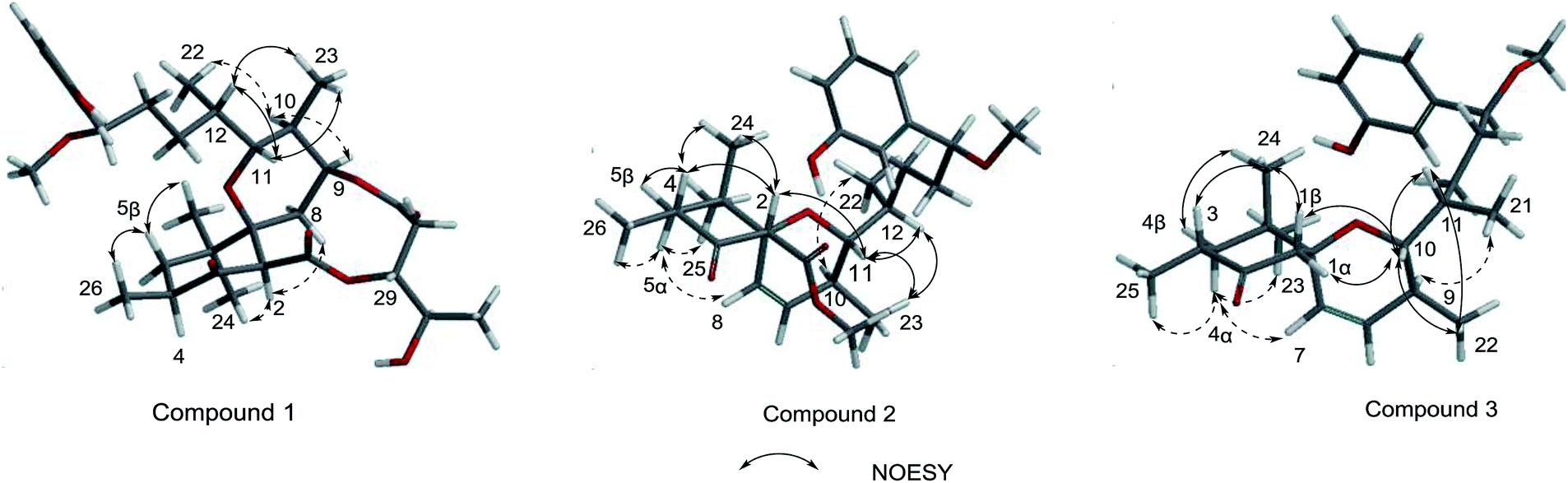
Neo-debromoaplysiatoxin D (1) was isolated as a colourless solid, with a molecular formula C32H46O9 indicating ten degrees of unsaturation was established by HRESIMS. The 1H NMR spectrum uncovered the appearance of a 1,3-disubstituted aromatic ring at δH 7.20 (t, J = 7.8 Hz), δH 6.99 (m), δH 6.88 (d, J = 7.6 Hz) and δH 6.75 (dd, J = 8.1, 2.5 Hz), four methyl doublets at δH 1.25, 1.10, 0.89 and 0.75 and three methyl singlets at δH 3.27, 1.02 and 0.79. The 13C and DEPT spectrum of 1 displayed seven quaternary carbons including one keto carbon (δC 211.6), two carbonyl carbons (δC 170.9 and 169.8) and two aromatic carbons (δC 156.6 and 144.9), thirteen methine carbons including four aromatic methine carbons (δC 129.6, 116.9, 114.5 and 114.3), five methylene carbons and seven methyl carbons (Table 1). This NMR spectroscopic data indicated that 1 contained a tricyclic core and its' planar structure closely resembled neo-debromoaplysiatoxin A.14 Careful analysis of these two compounds' 1D and 2D NMR spectroscopic data uncovered that an oxygenated quaternary carbon resonance of δC 75.7 at C-4 in neo-debromoaplysiatoxin A was replaced by the methine resonances of δC 40.4/δH 3.29 in 1. The HMBC correlations from H-4 to C-3 and C-5 in 1 strongly support this assignment (Fig. 2). The NOESY experiments and vicinal coupling constants were utilized to establish its relative stereochemistry. The large coupling constants of H-5β (J = 13.4, 10.6 Hz) and the NOESY cross-peak of H-2/H3-24 discovered that H-2, 4, 5β and 24 were axially oriented.10,11 The NOESY correlations from H-5β to H3-25 and H3-26 suggested these protons were β-orientated, whereas the α-orientation was indicated at H-5α, H-2, H-4 and H3-24, supported by the NOESY correlations of H-2/H3-24 and the large coupling constants of H-5β (J = 13.4, 10.6 Hz).10,11 The coupling constants of H-11 (J = 10.8, 1.6 Hz) and the NOESY correlation of H-12/H-11/H3-23 indicated that H-10 and H-11 were anti orientated (JH-10, H-11 = 10.8 Hz and JH-11,H-12 = 1.6 Hz). The NOESY cross-peaks of H-11/H3-23 indicated these protons were co-facial, while the NOESY correlations of H-9/H-10 uncovered these hydrogens were the same side of the cyclohexane, the small coupling between H-12 and H-11 (JH-11,H-12 = 1.6 Hz) and the NOESY correlations of H-11/H-12/H3-23 and H3-22/H-10 established the stereochemistry of C-12 (Fig. 3). The NOESY correlations of H2-8/H-2 indicated that the ether oxygen at C-7 was attached axially to the ring A.11 Furthermore, taking note of the structural similarities of compounds 1–7, it is likely that these seven compounds have a common biosynthetic origin.13 The relative configuration of C-29 and C-30 in the partial structure of 3,4-dihydroxyvaleric acid were consistent with that of the known compounds 4–6 owing to their similar coupling constants between H-29 and H-30 (JH-29,H-30 = 4.1 Hz) and biosynthetic pathway.6,10,11 Interestingly, 1 was considered as a precursor to neo-debromoaplysiatoxin A in the plausible biosynthetic pathway of neo-debromoaplysiatoxin A (Scheme S1†),14 therefore, we proposed that 1 and neo-debromoaplysiatoxin A may have the same absolute configuration and tentatively assigned as 2R, 4S, 7S, 9S, 10S, 11R, 12S, 15S, 29R, 30R.
Table 1 1H (600 MHz) and 13C NMR (150 MHz) data for 1–3 in CDCl3 (δ in ppm, J in Hz)
| No. |
1 |
2 |
3 |
| δH (J in Hz) |
δC, type |
δH (J in Hz) |
δC, type |
δH (J in Hz) |
δC, type |
| 1α |
|
170.9, qC |
|
169.6, qC |
2.50, d (13.5) |
47.8, CH2 |
| 1β |
|
|
|
|
2.38, d (13.5) |
|
| 2 |
3.24, s |
62.8, CH |
3.84, s |
64.4, CH |
|
212.4, qC |
| 3 |
|
211.6, qC |
|
205.9, qC |
2.56, m |
41.4, CH |
| 4β |
3.29, m |
40.4, CH |
2.60, m |
41.2, CH |
1.71, dd (14.0, 6.8) |
44.3, CH2 |
| 4α |
|
|
|
|
1.31, dd (14.2, 11.5) |
|
| 5β |
2.33, dd (13.4, 10.6) |
43.8, CH2 |
1.65, dd (14.0, 6.8) |
43.7, CH2 |
|
38.6, qC |
| 5α |
1.13, m |
|
1.35, dd (14.0, 13.4) |
|
|
|
| 6 |
|
40.8, qC |
|
40.6, qC |
|
79.7, qC |
| 7 |
|
78.4, qC |
|
81.5, qC |
5.39, dd (10.3, 2.8) |
128.1, CH |
| 8 |
2.03, d (3.2) |
33.3, CH2 |
5.45, dd (10.4, 2.9) |
125.4, CH |
5.58, dd (10.3, 1.7) |
133.6, CH |
| 9 |
5.01, m |
73.0, CH |
5.74, dd (10.4, 1.7) |
134.2, CH |
2.12, m |
30.3, CH |
| 10 |
1.63, m |
33.1, CH |
2.09, m |
30.1, CH |
3.05, dd (9.5, 1.8) |
75.5, CH |
| 11 |
4.00, dd (10.8, 1.6) |
71.7, CH |
2.96, dd (9.5, 1.8) |
77.9, CH |
1.61, overlap |
33.7, CH |
| 12 |
1.48, m |
34.4, CH |
1.55, overlap |
34.1, CH |
1.33, m 1.27, m |
30.2, CH2 |
| 13α |
1.57, overlap |
31.6, CH2 |
1.43, m |
30.7, CH2 |
1.78, m |
36.1, CH2 |
| 13β |
|
|
1.23, m |
|
1.61, m |
|
| 14α |
1.85, m |
37.3, CH2 |
1.78, m |
35.9, CH2 |
3.99, t (6.6) |
84.4, CH |
| 14β |
1.57, overlap |
|
1.53, overlap |
|
|
|
| 15 |
4.08, m |
85.4, CH |
4.0, t (6.6) |
84.8, CH |
|
144.3, qC |
| 16 |
|
144.9, qC |
|
144.3, qC |
6.79, overlap |
119.5, CH |
| 17 |
6.88, d (7.6) |
116.9, CH |
6.84, d (7.6, 1.2) |
119.3, CH |
7.17, t (8.0) |
129.5, CH |
| 18 |
7.20, t (7.8) |
129.6, CH |
7.20, t (7.8) |
129.6, CH |
6.74,dd (8.1, 2.5) |
114.9, CH |
| 19 |
6.75, dd (8.1, 2.5) |
114.5, CH |
6.76, dd (8.0, 2.6) |
114.7, CH |
|
156.4, qC |
| 20 |
|
156.6, qC |
|
156.1, qC |
6.79, overlap |
113.5, CH |
| 21 |
6.99, m |
114.3, CH |
6.81, brs |
113.8, CH |
0.84, d (6.8) |
13.4, CH3 |
| 22 |
0.89, d |
13.5, CH3 |
0.85, d |
13.0, CH3 |
0.80, d (7.2) |
16.7, CH3 |
| 23 |
0.75, d |
13.9, CH3 |
0.82, d |
17.0, CH3 |
0.87, s |
24.9, CH3 |
| 24 |
0.79, s |
25.4, CH3 |
1.21, s |
22.5, CH3 |
1.07, s |
22.6, CH3 |
| 25 |
1.02, s |
25.1, CH3 |
0.88, s |
24.9, CH3 |
1.03, d (6.6) |
14.9, CH3 |
| 26 |
1.10, d |
15.3, CH3 |
1.04, d |
14.4, CH3 |
3.20, s |
56.7, CH3 |
| 27 |
|
169.8, qC |
3.56, s |
51.8, CH3 |
|
|
| 28β |
2.80, dd (15.6, 5.9) |
37.3, CH2 |
3, 21, s |
56.8, CH3 |
|
|
| 28α |
2.64, dd (15.6, 10.3) |
|
|
|
|
|
| 29 |
5.14, ddd (10.3, 5.8, 4.1) |
76.8, CH |
|
|
|
|
| 30 |
3.92, m |
69.8, CH |
|
|
|
|
| 31 |
1.25, d |
18.3, CH3 |
|
|
|
|
| 32 |
3.27, s |
57.4, CH3 |
|
|
|
|
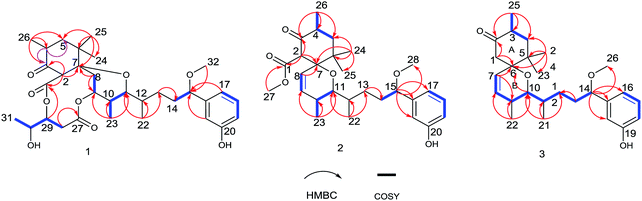 |
| | Fig. 2 1H– 1H key COSY correlations and HMBCs of 1, 2 and 3. | |
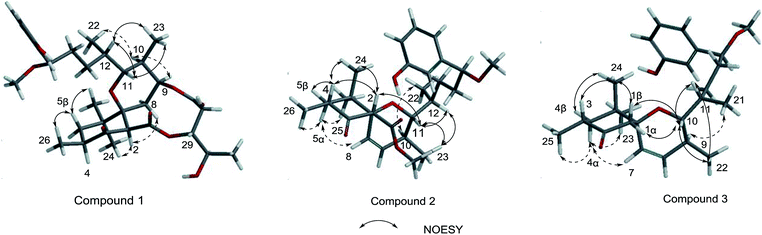 |
| | Fig. 3 Key NOESY correlations of 1, 2 and 3. | |
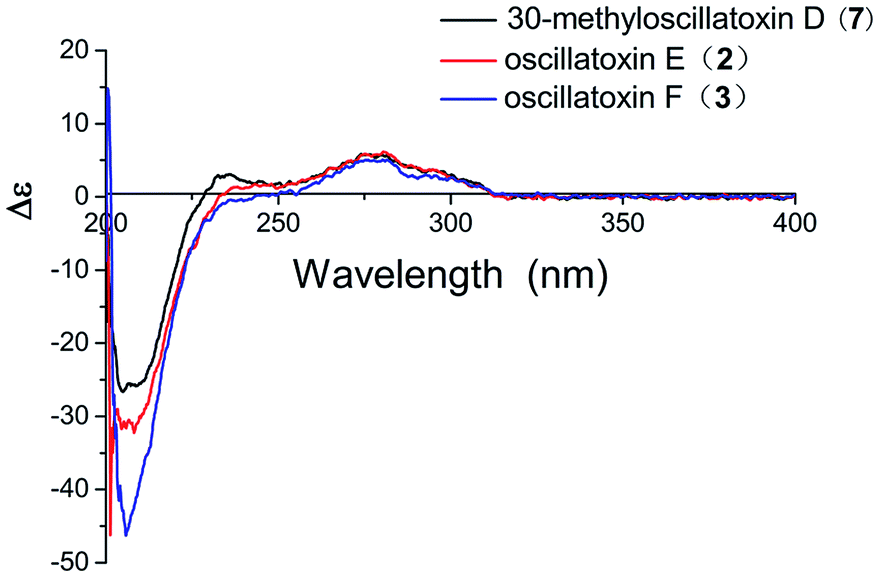
Oscillatoxin E (2) was obtained as a white solid, HRESIMS data (m/z 495.2732 [M + Na]+) assign its molecular formula as C28H40O6 with nine degrees of unsaturation. 28 carbon resonances can be observed in 13C and DEPT spectra and six quaternary carbons, twelve methines, three methylenes and seven methyls account for the 28 resonances. After counting one ketone group, one carbonyl carbon, one aromatic ring and two double bond carbons (Table 1), there are possible two additional rings left to finish nine degrees of unsaturation. The five partial structures, C26–C4–C5, C8–C9, C23–C10–C11, C14–C15 and C17–C18–C19, were established by the COSY correlations of H3-26/H-4/H2-5, H-8/H-9, H3-23/H-10/H-11, H2-14/H-15 and H-17/H-18/H-19 (Fig. 2). The ring A was closed by the HMBC correlations from H-2 to C-3 and C-7, from H3-26 to C-3, C-4 and C-5 and from H3-24 to C-5, C-6, C-7 and C-25. Moreover, the HMBC correlations from H3-27 and H-2 to C-1 positioned the function of –COOCH3 at C-2. The side chain at C-11 (C12–C21) was confirmed by the HMBC correlations from H3-22 to C-11, C-12 and C-13, H2-13 to C-14 and H-15 to C-16, C-17, C-21 and C-28. The ring B was established by the HMBC correlations of H-8/C7, H3-23/C-9, C-10 and C-11 and the implications of unsaturation degrees of this molecule completing the planar structure of compound 2 (Fig. 2). Interestingly, the planar structure of compound 2 was closely resembles a synthetic intermediate (22a) of the methyl ethers of 30-methyloscillatoxin D in Yoshihiko Nokura's total synthesis work.17 The relative configuration of 2 was identical with that of 22a by the NOESY correlations of H-5β/H-4/H-2/H3-24, H-5α/H-8, H3-25 and H3-26, H-10/H3-22, H-11/H-12, H3-23 and H-2 and H-12/H3-23 (Fig. 3). In addition, the cotton effects at 208 nm and 276 nm observed in ECD spectrum of compound 2 were consistent with those of 30-methyloscillatoxin D (7) (Fig. 4). Owing to the common biosynthetic origin of compounds 2 and 7, as well as the comparative optical rotation values of compound 2 and 22a (Fig. S37†), the absolute configuration of 2 was tentatively assigned as 2S, 4R, 7R, 10S, 11R, 12S and 15S.17,18
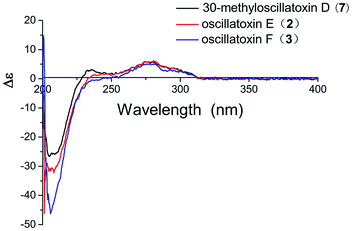 |
| | Fig. 4 Experimental ECD spectra of compounds 2, 3 and 7. | |
Oscillatoxin F (3) was obtained as white solid. The HRESIMS data assigned its molecular formula as C26H38O4 showing 58 mass fewer than compound 2. Comparison of 1D and 2D NMR spectroscope of compounds 3 and 2 discovered that the additional feature of –COOCH3 at C-2 in 2 was replaced by H-1α in 3, supported by the HMBC correlations from H-1α to C-2 and C-7 (Fig. 2 and Table 1). The relative stereochemistry of 3 was deduced from the NOESY spectrum and vicinal coupling constants. The large coupling constants of H-4α (J = 14.2, 11.5 Hz) and the NOESY correlation of H-4α/H-7 discovered that H-3, H-4α and the double bond at C-7 were axially orientated. The NOESY correlations of H-4α/H3-25, H3-23 and H-7 and H3-24/H-1β, H-3 and H-4β indicated that H-4α, H3-25, H3-23 and H-7 were positioned at the opposite side to H3-24, H-1β, H-3 and H-4β. In the ring B system, the coupling constants of H-10 (J = 9.5, 1.8 Hz) and the presence of the NOESY correlation of H-11/H-10/H3-22 indicated the H-10 and H-9 were anti relationship and H-10 and H-11 were gauche relationship (JH-10,H-9 = 9.5 Hz, JH-10,H-11 = 1.8 Hz). The NOESY correlations of H3-22/H-10/H2-1 showed that H-10, H2-1 and H3-22 were on the same side of the ring B, while the small coupling constant of H-10/H-11 (JH-10,H-11 = 1.8 Hz) and the NOESY correlations of H-10/H-11/H3-22 and H3-21/H-9 established the stereochemistry of C-11 (Fig. 3). Moreover, the stereochemistry at C-14 was referred to be S which was same as that of compounds 2 and 7 owing to their positive cotton effects at 275 nm and common biosynthetic origin.13,14 These spectroscopic data established a configuration of (3R*, 6S*, 9S*, 10R*, 11S*, 14S). The comparison of ECD spectra between 2, 3 and 7 suggested they share similar absolute configurations and the absolute configuration of 3 was tentatively assigned as 3R, 6S, 9S, 10R, 11S, 14S (Fig. 4).
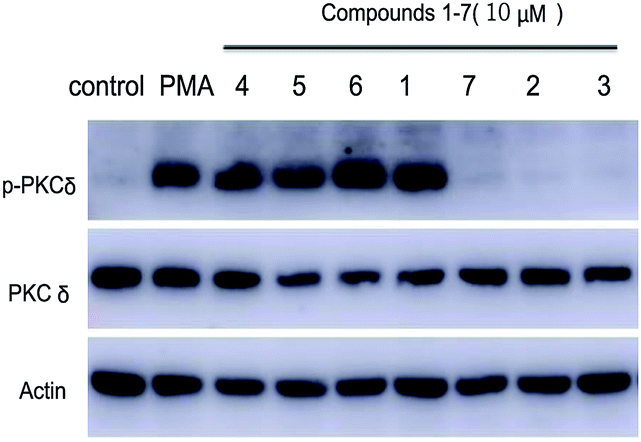
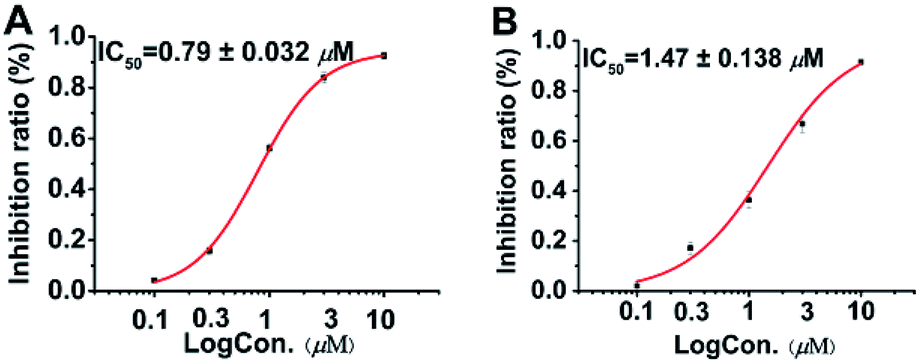
Following the protocol of previous research,14 it was found that all seven compounds did not show significant cytotoxicity at 10 μM. It has been well studied that aplysiatoxin and its derivatives are activators of protein kinase C (PKC).19,20 Therefore, we subsequently assessed compounds 1–7 on the expression of phosphor-PKCδ in HepG2 cells at 10 μM.21 Compounds 1 and 4–6 strongly up-regulated the expression of phosphor-PKCδ, while the compounds 2, 3 and 7 had no effect (Fig. 5). This difference is possibly due to compounds 2, 3 and 7 lacking a recognition domain which plays a vital role in intermolecular hydrogen bonding with the PKCδ C1B domain.19 Additionally, our previous research has highlighted the potential of aplysiatoxins as ion channel blockers, specifically the selective blocking of Kv1.5.14 Following this, we pre-screened our known aplysiatoxin compounds (5–7) for inhibitory activity on the shaker-related subfamily of voltage-gated channels (Kv1.1, Kv1.2, Kv1.3, Kv1.4 and Kv1.5). Our results showed that these three compounds all had significant inhibitory effect on Kv1.5 (Fig. S1.2.2.1†), therefore we expanded our experiment to test metabolites 1–3 and 5–7 for Kv1.5 inhibition, but we did not use compound 4 as the sample amount was unviable. Through this assessment, compounds 2, 5, 6 and 7 were seen to exhibit relatively strong inhibitory activity, whereas compounds 1 and 3 showed weak activities at 1 μM (Table S1.2.2†). Oscillatoxin E (2), debromoaplysiatoxin (5), 30-methyloscillatoxin D (7) were chosen to undertake a dose-response study to find their inhibitory value, the results showed that 2, 5 and 7 exhibited IC50 values of 0.79 ± 0.032 μM, 1.28 ± 0.080 μM and 1.47 ± 0.138 μM, compared to the control compound, acacetin, 5.96 ± 0.564 μM (Fig. 6, S1.2.2.2–S1.2.2.5†).
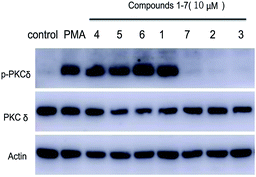 |
| | Fig. 5 Effect of compounds 1–7 on phosphor-PKCδ expression in HepG2 cells. | |
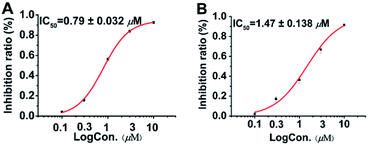 |
| | Fig. 6 Dose-response study of 2 and 7 with Kv1.5 expression in CHO cells at HP of −80 mV. Data points represent mean ± SEM of 3 to 5 measurements. Solid curve fits to the Hill equation. (A) Inhibitory effect of 2 showed IC50 value of 0.79 ± 0.032 μM. (B) Inhibitory effect of 7 showed IC50 value of 1.47 ± 0.138 μM. Acacetin with IC50 value of 5.96 ± 0.564 μM as positive control. | |
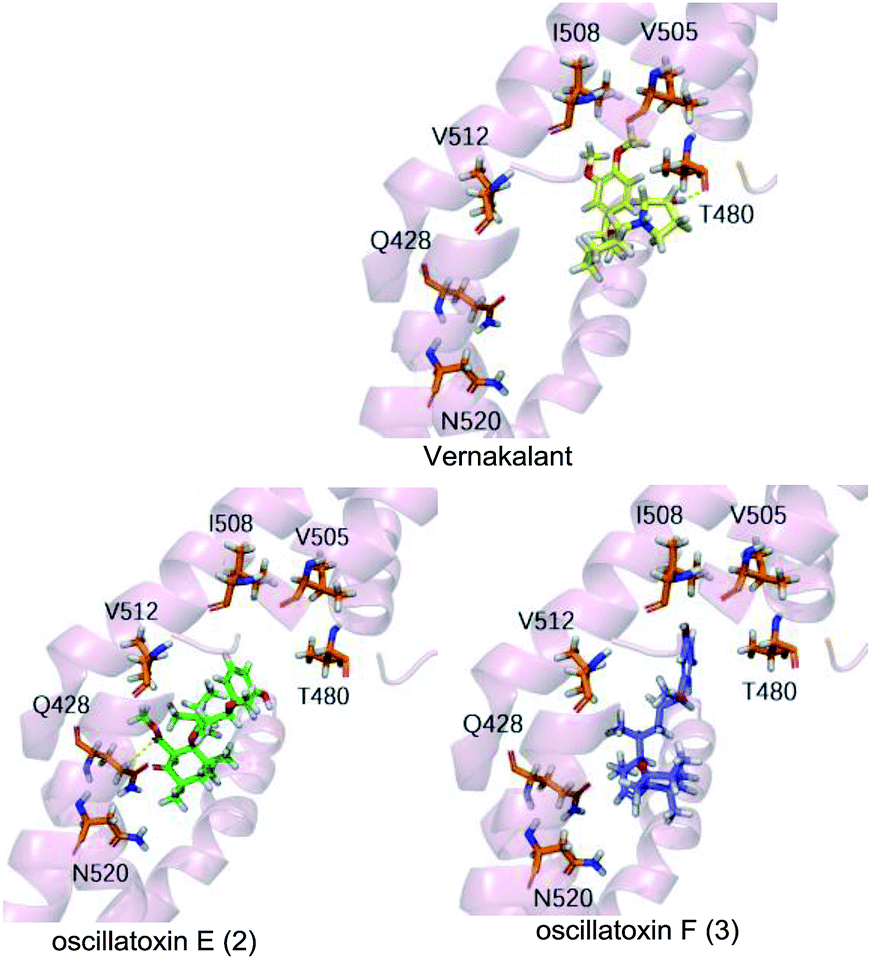
To supplement our knowledge of the interaction between our compounds and the Kv1.5 channel, we performed molecular docking computational analyses. Firstly, we generated a 3D homology model of Kv1.5 using the sequence of Kv1.2 which shares 90% similarity with our target, especially in the S6 helical domain. We selected resides 480–512 as the binding pocket as it matched the binding site for the known Kv1.5 channel blocker, vernakalant, which was our positive control.22 Vernakalant shows strong blocking activity against Kv1.5 and is approved in Europe and Canada as an antiarrhythmic agent for the rapid conversion of atrial fibrillation to sinus rhythm.23 The docking results showed that vernakalant and oscillatoxin E (2) had strong binding affinities of −37.374 kcal mol−1, −37.645 kcal mol−1 but oscillatoxin F (3) had relatively weaker binding at −32.217 kcal mol−1 (Table S1.1.1.1†). The difference between the compounds was most likely due to the fact vernakalant and oscillatoxin E (2) had key hydrogen binding interactions (vernakalant:T480, 2:Q428) however oscillatoxin F (3) did not (Fig. 7). This difference positively reflects our experimental results.
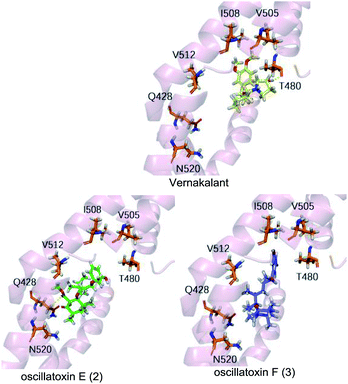 |
| | Fig. 7 Molecular docking analysis of between homology models Kv1.5 with Vernakalant, oscillatoxin E (2) and oscillatoxin F (3). Compounds shown as sticks, Homology Kv1.5 channel shown as cartoon, H-bonding interaction represented as yellow dashed line. | |
Conclusions
We successfully isolated several aplysiatoxin analogues which can be grouped into aplysiatoxins (1 and 4–6) and oscillatoxins (2, 3 and 7) based on their structural characteristics. The aplysiatoxins (1 and 4–6) showed strong up-expression of phosphor-PKCδ however the oscillatoxins did not, due to structural frames differences meaning the oscillatoxins lack the specific recognition domain seen in aplysiatoxins. All compounds showed selective blocking of potassium channel Kv1.5 through our preliminary screening assays, furthermore compounds 2 and 7 showed significant inhibitory effect on Kv1.5 with IC50 values of 0.79 ± 0.032 μM and 1.47 ± 0.138 μM. These results should provide helpful advice for researchers in search for new treatment of atrial tachyarrhythmias.
The differences in these compounds further suggest that the activity of voltage-gate potassium channels is modulated by two mechanisms: indirect ion channel modulation by protein phosphorylation and direct ion channel modulation by coupling the channels with intracellular signalling enzymes.15,16 Further experimental data is needed to compound this evidence and to discover the exact binding mechanism behind this inhibitors and Kv1.5.
Experimental section
General experimental procedures
An Autopol VI polarimeter manufactured by Rudolph Research Analytical, Hackettstown, NJ, USA and a Jasco J-810 spectropolarimeter were used for optical rotations and the ECD spectra, respectively. The NMR experiments were operated on a Bruker Avance 600 spectrometer. HRESIMS data were acquired on an ACQUITY™ UPLC & Q-TOF MS Premier spectrometer or a Waters Q-TOF micro YAO 19 mass spectrometer. HPLC purification was operated on a waters 1525 series instrument with waters xBridge Prep C-18 column (5 μm, 10 mm × 250 μm) and a 2998 photodiode array detector.
Material
The cyanobacterium Lyngbya sp. was collected from the South China Sea in June 2017 and identified by Prof. Bing-Nan Han (Zhejiang Sci-Tech University). A voucher specimen numbered as BNH-201706 has been well stored in Zhejiang Sci-Tech University.
Computational section
The detailed theoretical calculations of ECD and molecular docking were attached to the ESI.†24–26
Extraction and isolation
The cyanobacterium (70 g, dry weight) was soaked in MeOH and CH2Cl2 (1:1, v/v) accompanied by ultrasonic wave to obtained 25.9 g of extract, which was dissolved in 90% aqueous MeOH and extracted with petroleum ether repeatedly. Then the MeOH fraction was added distilled water to yield 60% aqueous MeOH which was partitioned against CH2Cl2 five times, the CH2Cl2-soluble fraction (3.4 g) was subjected to a VLC on silica gel with n-hexane–EtoAc (5:1, 2:1, 1:1, 1:2, 0:1, v/v) and yield nine fractions (VLC1-9). The fifth part (VLC5) was portioned into 21 subfractions on ODS (10–100%, MeCN/H2O, 180 min). The thirteenth and fourteenth fractions were further purified by RP-HPLC (YMC-Pack pro C18, 3 min L−1, UV detected at 195 nm). 4 (20.1 mg, tR = 26 min), 5 (50.0 mg, tR = 28 min), 6 (5.7 mg, tR = 29 min) and 1 (2.5 mg, tR = 25 min) were isolated from the thirteenth part (MeCN/H2O = 60:40) and 7 (3.4 mg, tR = 36 min), 2 (3.2 mg, tR = 40 min) and 3 (5.1 mg, tR = 43 min) were purified from the fourteenth fraction (MeCN/H2O = 70:30).
Neo-debromoaplysiatoxin D (1). Colorless solid; [α]25D +16.3 (c 0.1, MeOH); ECD (c 0.1, MeOH) λmax (Δε) 214 (−5.52), 275 (1.42) nm; 1H (600 MHz, CDCl3) and 13C NMR (150 MHz, CDCl3) data, Table 1; HRESIMS m/z 597.3040 [M + Na]+ (calcd for C32H46O9Na, 597.3040).
Oscillatoxin E (2). White solid; [α]25D −11.3 (c 0.1, MeOH); ECD (c 0.1, MeOH) λmax (Δε) 207 (−31.10), 248 (+1.74), 275 (5.86) nm; 1H (600 MHz, CDCl3) and 13C NMR (150 MHz, CDCl3) data, Table 1; HRESIMS m/z 495.2732 [M + Na]+ (calcd for C28H40O6Na, 495.2723).
Oscillatoxin F (3). White solid; [α]25D −6 (c 0.15, MeOH); ECD (c 0.1, MeOH) λmax (Δε) 206 (−46.27), 275 (5.03) nm; 1H (600 MHz, CDCl3) and 13C NMR (150 MHz, CDCl3) data, Table 1; HRESIMS m/z 437.2680 [M + Na]+ (calcd for C26H38O4Na, 437.2688).
Phosphor-PKCδ assay
The effects of compounds 1–7 on the phosphorylation of PKCδ were conducted in HepG2 cells referring to previous protocol.27–29 Sententiously, the cells were inoculated in 6-well plates. After 12 h, the cells were treated with compounds (10 μM) and PMA (1 μM) and incubated for 1 h. Then the cells were lysed with cell lysis buffer. Protein was extracted by BCA protein Assay Kit and determined using western blot.
Ion channel experiment
The Kv blocking activities of these compounds were determined in CHO cells (Sigma Chemical Co., St. Louis, MO, USA) referring to a reported method.30–32 Whole-cell patch-clamp technique was utilized to record the current, the intracellular fluid (KAspartate, 130 mM; MgCl2, 5 mM; EGTA, 5 mM; Hepes, 10 mM; Tris-ATP, 4 mM; pH, 7.2 (titrated by KOH)) was filled electrodes. The cells with density of 80% were digested by trypsin and translated into 35 mm Petri dish. The cells were cultured by DMEM/F 12 (10% FBS + P/S) culture medium in 37 °C incubator for 5% CO2. After 24 h, the cells were transferred to the perfusion tank and perfused with extracellular fluid (NaCl, 137 nM; KCl, 4 nM; CaCl2, 1.8 nM; MgCl2, 1 mM; HEPES, 10 mM; glucose 10 mM; pH, 7.4 (titrated by NaOH)). Compounds were dissolved in DMSO and then added into the extracellular. The holding voltage of cells was set at −80 mV and the depolarized to 0 mV to obtain currents. This procedure is repeated every 10 seconds. After stabilization, the cells were perfused with the extracellular fluid containing compounds at different concentration and the intensity of blocking was calculated. Data collection and analysis were conducted on pCLAMP 10 (Molecular Devices, Union City, CA).
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Special Fund for Agro-scientific Research in the Public Interest of Zhejiang province (LGN18C190011), the National Natural Science Foundation of China (Grants 81373321, U1605221), Science Foundation of Zhejiang Sci-Tech University (17042058-Y), Project for Jiaozhou Excellent Innovation Team (18-CX-1), and the National Key Research and Development Program of China (2016YFF0202300, 2018YFC0310900). In addition, we are very grateful to Lawrence Jordan Keen for English correction and revision.
Notes and references
- C. Moreira, V. Ramos, J. Azevedo and V. Vasconcelos, Appl. Microbiol. Biotechnol., 2014, 98, 8073–8082 CrossRef CAS PubMed.
- R. C. Yanagita, H. Kamachi, M. Kikumori, H. Tokuda, N. Suzuki, K. Suenaga, H. Nagai and K. Irie, Bioorg. Med. Chem. Lett., 2013, 23, 4319–4323 CrossRef CAS PubMed.
- M. Kikumori, R. C. Yanagita, H. Tokuda, N. Suzuki, H. Nagai, K. Suenaga and K. Irie, J. Med. Chem., 2012, 55, 5614–5626 CrossRef CAS PubMed.
- M. Suganuma, H. Fujiki, T. Tahira, C. Cheuk, R. E. Moore and T. Sugimura, Carcinogenesis, 1984, 5, 315–318 CrossRef CAS PubMed.
- J. S. Mynderse, R. E. Moore, M. Kashiwagi and T. R. Norton, Science, 1977, 196, 538–540 CrossRef CAS PubMed.
- D. K. Gupta, P. Kaur, S. T. Leong, L. T. Tan, M. R. Prinsep and J. J. Chu, Mar. Drugs, 2014, 12, 115–127 CrossRef PubMed.
- Y. Kato and P. J. Scheuer, J. Am. Chem. Soc., 1974, 5, 2245–2246 CrossRef.
- Y. Kato and P. J. Scheuer, Pure Appl. Chem., 1975, 41, 1–14 CAS.
- J. S. Mynderse and R. E. Moore, J. Org. Chem., 1978, 43, 2301–2303 CrossRef CAS.
- R. E. Moore, A. J. Blackman, C. E. Cheuk, J. S. Mynderse, G. K. Matsumoto, J. Clardy, R. W. Woodard and J. C. Craig, J. Org. Chem., 1984, 49, 2484–2489 CrossRef CAS.
- M. Entzeroth, A. J. Blackman, J. S. Mynderse and R. E. Moore, J. Org. Chem., 1985, 16, 1255–1259 CrossRef.
- H. Nagai, T. Yasumoto and Y. Hokama, J. Nat. Prod., 1997, 60, 925–928 CrossRef CAS PubMed.
- G. E. Chlipala, H. T. Pham, V. H. Nguyen, A. Krunic, S. H. Shim, D. D. Soejarto and J. Orjala, J. Nat. Prod., 2010, 73, 784–787 CrossRef CAS PubMed.
- B. N. Han, T. T. Liang, L. J. Keen, T. T. Fan, X. D. Zhang, L. Xu, Q. Zhao, S. P. Wang and H. W. Lin, Org. Lett., 2018, 20, 578–581 CrossRef CAS PubMed.
- J. Feng, B. Wible, G. R. Li, Z. Wang and S. Nattel, Circ. Res., 1997, 80, 572–579 CrossRef CAS PubMed.
- D. Fedida, J. Physiol., 1997, 499(Pt 3), 661–675 CrossRef CAS.
- Y. Nokura, Y. Araki, A. Nakazaki and T. Nishikawa, Org. Lett., 2017, 19, 5992–5995 CrossRef CAS PubMed.
- J. Wang, F.-R. Mu, W.-H. Jiao, J. Huang, L.-L. Hong, F. Yang, Y. Xu, S.-P. Wang, F. Sun and H.-W. Lin, J. Nat. Prod., 2017, 80, 2509–2514 CrossRef CAS PubMed.
- Y. Ashida, R. C. Yanagita, C. Takahashi, Y. Kawanami and K. Irie, Bioorg. Med. Chem., 2016, 24, 4218–4227 CrossRef CAS PubMed.
- F. H. Kong, Y. Kishi, D. Perez-Sala and R. R. Rando, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 1973–1976 CrossRef CAS.
- A. C. Newton, Chem. Rev., 2001, 101, 2353–2364 CrossRef CAS.
- J. Eldstrom, Z. Wang, H. Xu, M. Pourrier, A. Ezrin, K. Gibson and D. Fedida, Mol. Pharmacol., 2007, 72, 1522–1534 CrossRef CAS PubMed.
- D. Roy, B. H. Rowe, I. G. Stiell, B. Coutu, J. H. Ip, D. Phaneuf, J. Lee, H. Vidaillet, G. Dickinson, S. Grant, A. M. Ezrin and G. N. Beatch, J. Am. Coll. Cardiol., 2004, 44, 2355–2361 CrossRef CAS PubMed.
- H. Jiang, R. Deng, X. Yang, J. Shang, S. Lu, Y. Zhao, K. Song, X. Liu, Q. Zhang, Y. Chen, Y. E. Chinn, G. Wu, J. Li, G. Chen, J. Yu and J. Zhang, Nat. Chem. Biol., 2017, 13, 994–1001 CrossRef CAS PubMed.
- Q. Shen, F. Cheng, H. Song, W. Lu, J. Zhao, X. An, M. Liu, G. Chen, Z. Zhao and J. Zhang, Am. J. Hum. Genet., 2017, 100, 5–20 CrossRef CAS PubMed.
- J. Li, L. Sun, C. Xu, F. Yu, H. Zhou, Y. Zhao, J. Zhang, J. Cai, C. Mao, L. Tang, Y. Xu and J. He, Acta Biochim. Biophys. Sin., 2012, 44, 300–306 CrossRef CAS PubMed.
- H. Kim, R. Zamel, X.-H. Bai and M. Liu, PLoS One, 2013, 8, e64182 CrossRef CAS PubMed.
- H. Xiao, X. H. Bai, A. Kapus, W. Y. Lu, A. S. Mak and M. Liu, Mol. Cell. Biol., 2010, 30, 5545–5561 CrossRef CAS PubMed.
- B. Han, X. H. Bai, M. Lodyga, J. Xu, B. B. Yang, S. Keshavjee, M. Post and M. Liu, J. Biol. Chem., 2004, 279, 54793–54801 CrossRef CAS PubMed.
- J. Yu, M. H. Park and S. H. Jo, Eur. J. Pharmacol., 2015, 746, 158–166 CrossRef CAS PubMed.
- I. Jeong, S. H. Yoon and S. J. Hahn, Naunyn-Schmiedeberg's Arch. Pharmacol., 2012, 385, 707–716 CrossRef CAS PubMed.
- B. H. Choi, J.-S. Choi, S.-W. Jeong, S. J. Hahn, S. H. Yoon, Y.-H. Jo and M.-S. Kim, J. Pharmacol. Exp. Ther., 2000, 293, 634–640 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra00965e |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *d and
Bing-Nan Han
*d and
Bing-Nan Han